

ABSTRACT
Macroautophagy/autophagy is a highly regulated process involved in the turnover of cytosolic components, however its pivotal role in maintenance of bone homeostasis remains elusive. In the present study, we investigated the direct role of ATG7 (autophagy related 7) during developmental and remodeling stages in vivo using osteoblast-specific Atg7 conditional knockout (cKO) mice. Atg7 cKO mice exhibited a reduced bone mass at both developmental and adult age. The trabecular bone volume of Atg7 cKO mice was significantly lower than that of controls at 5 months of age. This phenotype was attributed to decreased osteoblast formation and matrix mineralization, accompanied with an increased osteoclast number and the extent of the bone surface covered by osteoclasts as well as an elevated secretion of TNFSF11/RANKL (tumor necrosis factor [ligand] superfamily, member 11), and a decrease in TNFRSF11B/OPG (tumor necrosis factor receptor superfamily, member 11b [osteoprotegerin]). Remarkably, Atg7 deficiency in osteoblasts triggered endoplasmic reticulum (ER) stress, whereas attenuation of ER stress by administration of phenylbutyric acid in vivo abrogated Atg7 ablation-mediated effects on osteoblast differentiation, mineralization capacity and bone formation. Consistently, Atg7 deficiency impeded osteoblast mineralization and promoted apoptosis partially in DDIT3/CHOP (DNA-damage-inducible transcript 3)- and MAPK8/JNK1 (mitogen-activated protein kinase 8)-SMAD1/5/8-dependent manner in vitro, while reconstitution of Atg7 could improve ER stress and restore skeletal balance. In conclusion, our findings provide direct evidences that autophagy plays crucial roles in regulation of bone homeostasis and suggest an innovative therapeutic strategy against skeletal diseases.
KEYWORDS: Autophagy, bone formation, bone mass, ER stress, osteoporosis
Introduction
Bone is a dynamic tissue that continuously undergoes modeling/remodeling process by balancing osteoblast-mediated bone formation and osteoclast-mediated bone resorption [1]. Imbalance between the activity of osteoblasts and osteoclasts during bone remodeling can lead to reduced bone mass, which underlies the pathogenesis of osteoporosis, the most common skeletal disease [2]. Regarding age-related bone loss, osteoblast number and activity decrease while osteoclast number and activity increase. Although extrinsic mechanisms such as reduced production of sex steroids or growth factors have been frequently reported to be the major causes of age-related osteoporosis, autophagy process associated with the survival of osteoblasts, osteoclasts, and chondrocytes within a hypoxic, and even hypertonic environment is now proposed to be a potential intrinsic mechanism for skeletal homeostasis [3]. However, physiological and pathological roles of autophagy in bone remain not well understood.
Autophagy is a highly regulated process involved in the turnover of cytosolic components, long-lived proteins, or damaged organelles. As a putative adaptive catabolic process, autophagy is activated during nutrient deprivation to promote cell survival and to increase amino acid retrieval [4]. Besides its physiological roles in the development and the maintenance of physiological function of normal tissues, dysregulated autophagy has been implicated in a number of aging and degenerative diseases such as Parkinson, and Alzheimer diseases [5]. Targeted disruption of autophagy genes in specific tissues induces β cell loss with resultant hyperglycemia [6], or neurodegeneration [5], which is probably due to accumulation of damaged molecules and organelles. Inhibition of autophagy would accelerate the process of aging, whereas stimulation of autophagy would have potent antiaging effects [7–9]. In fact, autophagy has been identified as a protective mechanism in normal cartilage against oxidative stress and other aging-related phenotypes and its aging-related loss was linked with cell death and osteoarthritis [10,11]. Our previous study also demonstrates that cartilage-specific autophagy deficiency contributes to growth retardation [12]. Moreover, in senile population, decreased activity of osteocyte autophagy with aging is proposed as an underlying cause of the bone loss [13]. While it is tempting to speculate that genetic suppression of autophagy in major bone cell types such as osteocytes, osteoblasts and osteoclasts mimics the effects of aging on bone mass, this relationship has not yet been fully established.
The significant relationship between autophagy pathway and osteoporosis has been highlighted in a genome-wide association study of wrist bone mineral density [14]. There is emerging evidence implying that autophagic mechanisms are active in osteocytes [13,15]. In preosteocyte-like murine cells, autophagy is upregulated following nutrient deprivation and hypoxic culture, stress conditions that osteocytes encounter in vivo [16]. Previous studies show that glucocorticoid excess decreases bone mass and stimulates osteocyte autophagy profoundly [17–19], whereas autophagy impairment aggravates the inhibitory effects of high glucose on osteoblast viability and function [20]. A recent study in mice, elegantly demonstrates that conditional deletion of and allele for Atg7 (autophagy related 7; an E1-like enzyme essential for autophagy) from osteocytes mimics skeletal aging with low bone mass, and decreased bone formation rate [15]. These findings clearly link the autophagy-related genes with skeletal homeostasis.
Indeed, the recent observations find that mice lacking Atg7 in osteoblast progenitors and their descendants have low bone mass and fractures associated with reduced numbers of osteoclasts and osteoblasts [21]. Of note, osteoblast-specific deletion of Rb1cc1/Fip200 (RB1-inducible coiled-coil 1) has no obvious developmental defects in major skeletal elements of newborn mice, but reveals an osteopenia phenotype at 1, 2, and 6 months after birth [22]. In addition to the activation of autophagy during osteoblast differentiation, autophagy-deficient osteoblasts exhibit reduced mineralization capacity and the elevation of TNFSF11/RANKL (tumor necrosis factor [ligand] superfamily, member 11) secretion [23], favoring generation of osteoclasts. However, the direct role of osteoblast-specific autophagy in skeletal maintenance in vivo remains unclear. In the present study, we investigated the pivotal role of autophagy on endogenous bone mass during developmental and remodeling stages using osteoblast-specific Atg7 conditional knockout (cKO) mice.
Results
Osteoblast-specific suppression of autophagy in atg7 cko mice
Both developmental and remodeling stages are crucial for understanding bone biology. Thus to directly address the role of autophagy in osteoblasts, we utilized 2 different stages, weanling (3 wk) and adult (22 wk), and generated mice conditionally lacking autophagy in osteoblasts through knocking out Atg7 using a tamoxifen ™-inducible Cre-loxP system under the control of the Col1a (collagen, type I, alpha 1) promoter. Both of mice positive and negative for TM-Cre (Atg7flox/flox and Cre+Atg7flox/flox) were administrated TM to control any effect of the TM treatment per se (Figure 1(a,b)). Analysis of genomic DNA revealed a particular gene excision in the calvaria from 22-wk Atg7 cKO (TM-treated Cre+Atg7flox/flox) mice but no changes in other tissues from the same mice or in calvaria from the controls (Figure 1(c)). As expected, direct analysis of Atg7 expression levels extracted from Atg7 cKO skulls by RT-qPCR revealed > 70% reduction both in the weanling and adult stages that did not occur in soft tissues (Figure 1(d,e)). Likewise, ATG7 expression was significantly lower in tibia of 3-wk Atg7 cKO mice when compared with that of controls by immunohistochemistry (Figure 1(f)). Furthermore, determination of MAP1LC3A/LC3A (microtubule-associated protein 1 light chain 3 alpha)-MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta) in Atg7 cKO skulls revealed decreased conversion from form I to form II both in the weanling and adult mice (Figure 1(g)). In contrast, accumulation of SQSTM1/p62 (sequestosome 1; which is degraded through the autophagy process) was significantly increased in Atg7 cKO bones (Figure 1(g)). Taken together, these results confirmed that autophagy was effectively inhibited in osteoblasts by ablation of Atg7 using the Col1a-Cre transgene.
Figure 1.
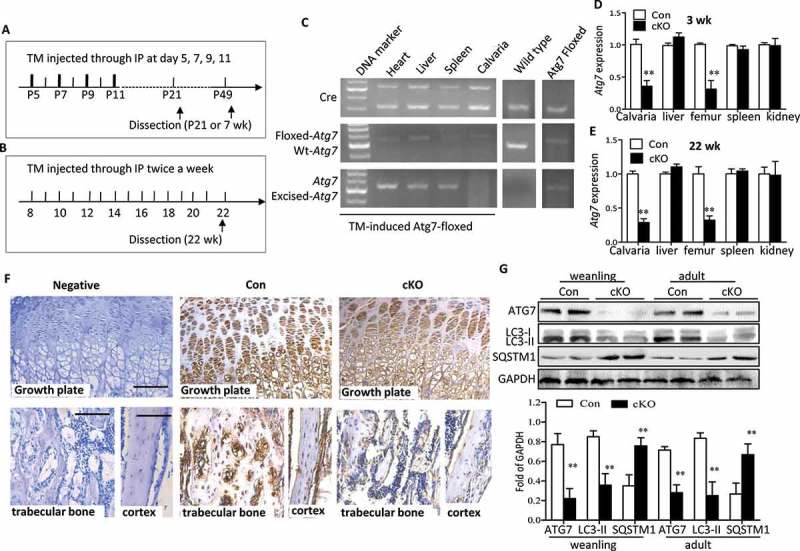
Ablation of Atg7 gene in TM-Atg7−/- (cKO) mice at P21- and 22-wks-old. (a and b) Scheme of the experimental design: In weanling mice, TM (0.2 mg/mouse) was injected intraperitoneally at day 5, 7, 9, 11 until dissection at P21. For adult stages, TM (50 mg/kg) was injected intraperitoneally twice a week from 8 wk until dissection at 22 wk. Both control mice (Cre−Atg7flox/flox) and cKO mice (Cre+Atg7flox/flox) received TM. (C) At 22 wk, PCR analyses of gDNA extracted from different tissues with primer sets for the Cre transgene, floxed-Atg7 allele, and sequences after gene excision (Atg7). (d and e) Quantitative RT-PCR shows reduced expression of Atg7 in femur and calvaria from P21 and 22-wk mice (n = 5 to 7/group) after TM administration. Results were presented as gene expression levels in all groups normalized to controls. (f) ATG7 protein in tibia from weanling mice assessed by immunohistochemistry. Scale bars: 100 μm. (g) Protein extracted from the calvaria of P21- and 22-wk-old mice was immunoblotted with antibodies directed to ATG7, LC3-I/-II and SQSTM1 (n = 3 or 4/group). Representative immunoblots and densitometric analysis are shown. *Statistically significant difference between Atg7 cKO and controls (mean ± SD, Student t test; **p < 0.01). Con: TM-Atg7flox/flox; cKO: TM-atg7−/-).
Atg7 cko mice showed a reduced bone mass phenotype
To determine the relevance of autophagy in osteoblasts within the skeletal system, we first examined the femur and tibia of 3-wk and 22-wk-old Atg7 cKO mice by H&E staining. As shown in Figures S1A and 2A, less trabecular bone was present in femurs and tibias of 3-wk and 22-wk-old Atg7 cKO mice, compared to controls, respectively. Morphologically, Atg7 cKO mice had no overt development defects in terms of body weight or body length both at 3 wk and 22 wk compared with controls in either sex (Figures S2A to C). At 22 wk, Atg7 cKO mice of both sexes displayed the hallmarks of osteopenia, manifested by a 16.5% and 11.1% reduction in bone mineral density (BMD) at femur and spine, compared to the controls, respectively (Figure 2(b)). There was no significant difference between females and males, so the data for the sexes at approximately equal distribution in each group are combined. Further bone structure and architecture analysis by microcomputed tomography (μCT) imaging revealed that the distal femurs and the L4 vertebra of 22-wk-old Atg7 cKO mice displayed significantly underdeveloped trabecular regions compared to those of controls, which is shown by the 3D reconstruction (Figure 2(c,d)). In distal femora trabecular bone of 22-wk-old Atg7 cKO mice, in addition to a significant increase in trabecular spacing (Tb.Sp), bone volume per tissue volume (BV/TV), trabecular number (Tb.N) and trabecular thickness (Tb.Th) decreased about 40.3%, 23.9% and 22%, respectively, suggesting a reduction in trabecular bone volume (Figure 2(e-h)). Consistent with these changes in femur, similar results were observed in vertebra of 22-wk-old Atg7 cKO mice (Figure 2(e-h)). As seen in older mice, BMD measurement in 3-wk-old mice revealed a lesser decrease in tibia and spine in male conditional knockout mice (Figure S1B). However, no change in BMD at any site was found in female mice at this age (data not shown). Likewise, bone mass in tibia of 3-wk-old Atg7 cKO mice was decreased in a smaller amount with respect to BV/TV (decreased), Tb.Sp (increased), Tb.Th (decreased), and Tb.N (decreased) than that of the controls as noted in Figures S1C to F. Similarly, in vertebra bones of 3-wk-Atg7 cKO mice, these differences are also apparent except for Tb.Th (Figures S1C to F). Moreover, no statistical difference was observed in femoral or tibial length of Atg7 cKO mice when compared with controls at any age (Figures S2D and E). Consequently, overall data suggest that autophagy deficiency in osteoblasts reduced bone mass during both the early modelling and later remodeling stages of bone formation.
Figure 2.
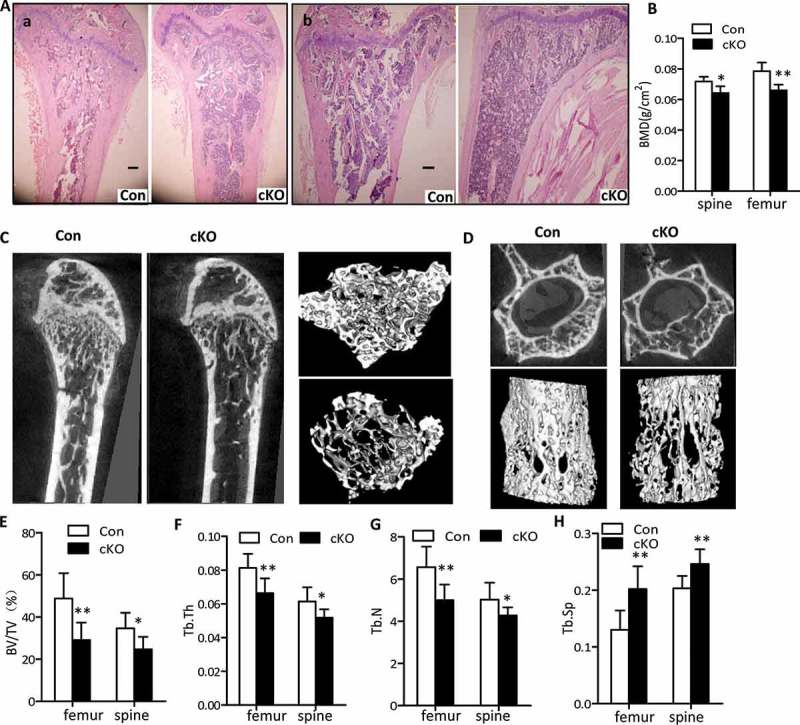
Lower bone density in osteoblast-specific Atg7 knockout mice at 22 wk. (a) H&E staining of femur (a) and tibia (b). Bars: 200 µm. (b) BMD was determined by μCT using lumbar (L4) and femur (n = 5 to 7/group). (c and d) Reconstructions of bone structure from μCT. Trabecular bone in femur (c) and lumbar vertebrae (L4) (d) are shown. Quantification of trabecular bone volume and architecture (n = 5 to 7/group): (e) BV/TV; (f) Tb.Th, trabecular thickness; (g) Tb.N, trabecular number; (h) Tb.Sp, trabecular spacing. *Statistically significant difference between control and cKO (mean ± SD, Student t test; *P < 0.05, **P < 0.01).
Effects of conditional deletion of atg7 on bone formation and resorption
To investigate the cellular basis underlying the bone loss in Atg7 cKO mice, we performed static and dynamic bone histomorphometric analysis in sections of undecalcified distal femurs at 3 and 22 wk, respectively. Consistent with a low bone mass phenotype by BMD and μCT analysis, Von Kossa staining in distal femur confirmed a reduction in bone volume in 3-wk-old mutants as well ((Figure 3(a)). Calcein and xylenol orange double labeling, which allows an assessment of dynamic bone formation parameters, confirmed a remarkably decreased bone formation rate (BFR) associated with Atg7 deficiency in 22-wk-old animals (Figure 3(b)). Mineral apposition rate (MAR, an index of individual osteoblast activity), wall thickness, and bone formation rate/bone surface (BFR/BS is determined from the number and the function of osteoblasts) were remarkably decreased in Atg7 cKO mice compared with controls, respectively (Figure. 3(c-e)). Histomorphometric analysis of the percentage of bone surface occupied by osteoblasts (Ob.S/BS) and the osteoblast number per bone perimeter showed an obvious reduction by 34% and 31%, respectively, in 22-wk-old Atg7 cKO mice compared with controls (Figure 3(f,g)). All these results suggest that the decreased bone density in Atg7 cKO mice can be possibly attributed to a decrease in the bone-forming activity of osteoblasts. Since osteoblasts regulate osteoclastogenesis by producing TNFSF11, an osteoclast differentiation factor, and TNFRSF11B/OPG (tumor necrosis factor receptor superfamily, member 11b [oteoprotegerin]; a decoy receptor for TNFSF11), we next tested whether reduced osteoblast activity would result in changes in osteoclast function. Histomorphometric analysis revealed that bone resorption parameters, the extent of bone surface occupied by osteoclasts (Oc.S/BS) and osteoclast number per bone perimeter were significantly increased in femora of 22-wk-old Atg7 cKO mice (Figure 3(i,j)). A further extensive histochemical analysis of osteoclasts revealed a marked elevation in the number of ACP5/TRAP (acid phosphatase 5, tartrate resistant)-positive multinucleated osteoclasts, a readout for osteoclast activity, in femoral sections of 22-wk-old Atg7 cKO mice (Figure 3(h)). Changes in bone always correlated with alterations in serum biomarkers. In this regard, further evidence for bone formation and resorption includes a reduction in amino-terminal propeptide of type I collagen (PINP), a circulating marker of bone formation, and an increase in bone resorption marker C-terminal telopeptide of type I collagen (CTX) in serum from 22-wk-old Atg7 cKO mice (Figure 3(k,l)). Similar results were seen in 3-wk-old Atg7 cKO mice (Figure S3).
Figure 3.
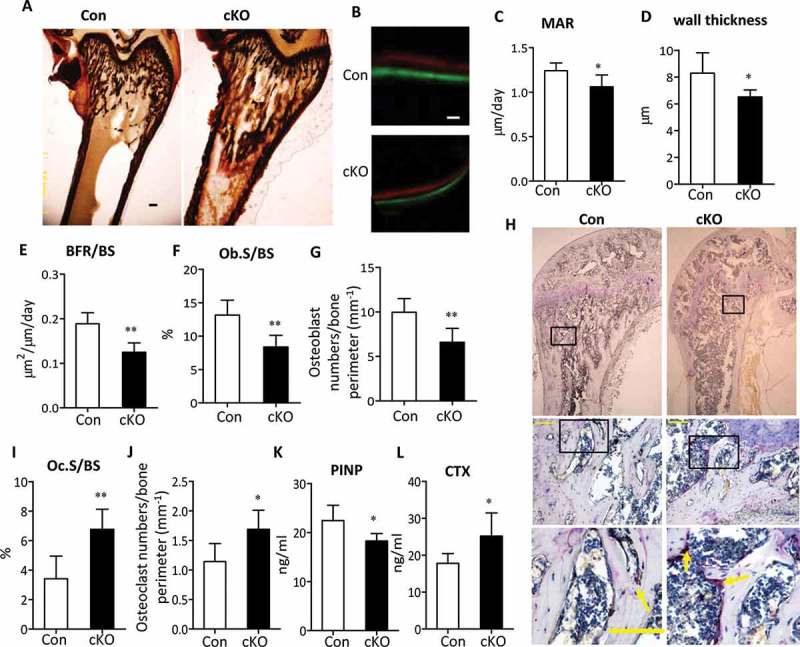
Decreased bone formation and increased bone resorption in Atg7 mutant mice. (a) Von Kossa staining of the distal femur sections of 3-wk Atg7 mutant (right) mice. Bars: 100 μm. (b) Representative Calcein and xylenol orange double-label staining images in femur sections (10 days apart between 2 labelings) of 22-wk-old male mice. Bars: 10 µm. Analysis of mineral apposition rate (MAR) (c), wall thickness (d), BFR/BS (e), Ob.S/BS (f) and Osteoblast numbers/bone perimeter (g) in 22-wk-old male animals. (h) Representative histological femur sections of 22-wk-old male mice stained for ACP5. Scale bars: 100 µm. Analysis of Oc.S/BS (i) and Osteoclast numbers/bone perimeter (j), and serum PINP and CTX levels (k and l) in 22-wk-old male animals. Data are presented as mean ± SD (n = 5 to 7/group, Student t test; *P < 0.05, **P < 0.01).
We next examined bone formation and resorption by measuring the expression levels of a panel of osteoblast lineage-, and osteoclast-related mRNAs extracted from ribs of weanling mice or calvaria of adult mice by RT-qPCR. In adult calvaria, in line with reduced bone formation, measurable reductions in the expression of formation markers Runx2 (runt related transcription factor 2), Bglap/Ocn(bone gamma carboxyglutamate protein), Sp7/osterix (Sp7 transcription factor 7) and Alpl (alkaline phosphatase, liver/bone/kidney) were observed in mutants, although no statistical changes were detected in Spp1/Opn (secreted phosphoprotein 1) expression (Figure 4(a)). Moreover, Atg7 cKO mice displayed remarkable impaired expression of Tnfrsf11b mRNAs in calvarial bone during adult stage but a significant enhanced expression of Tnfsf11, leading to an increase of the ratio of Tnfsf11 to Tnfrsf11b (140.9%) that favors the increased osteoclastogenesis (Figure 4(b)). Similarly, the same changes in serum levels of TNFSF11 and TNFRSF11B were observed (Figure 4(d,e)), although changes in the expression of Bmp4 (bone morphogenetic protein 4) and Tgfb1 (transforming growth factor, beta 1) were not significant (Figure 4(b)). Resorption markers Mmp9 (matrix metallopeptidase 9), Ctsk (cathepsin K), and Acp5, were markedly increased ~ 20% as well (Figure 4(c)), in line with the elevated osteoclast number stained by ACP5. Likewise, in weanling ribs where decreased Runx2 expression was observed (Figure 4(f,g)), Ctsk, Tnfsf11 and Spp1 mRNA expression were unchanged; however, Acp5 increased by 23% and Tnfrsf11b significantly reduced by 32% (Figure 4(f)). To confirm the effects of Atg7 deletion in osteoblasts on osteoclastogenesis through the TNFSF11-TNFRSF11B pathway in vivo, we next set up a tissue culture system using calvaria derived from uninduced newborn mice (Cre+Atg7flox/flox), which were bisected and then cultured with or without 4-OH TM (1 μM). The efficiency of Atg7 knockout using this method was confirmed by a notable reduction of Atg7 mRNA expression in Atg7 cKO (4-OH TM+) compared with controls (4-OH TM-) (Figure S4A). The cultured calvarial bones exhibited elevated expression levels of resorption markers Ctsk, Acp5, and Mmp9 in Atg7 cKO compared with controls (Figure S4B). Notably, in contrast with the increased Tnfsf11 expression, Tnfrsf11b expression was reduced (Figure S4C), accompanied by the corresponding changes in medium levels of TNFSF11 and TNFRSF11B (Figure S4D), thus resulting in a marked elevation of the ratio of TNFSF11 to TNFRSF11B in the treated group, which corroborates in vivo results.
Figure 4.
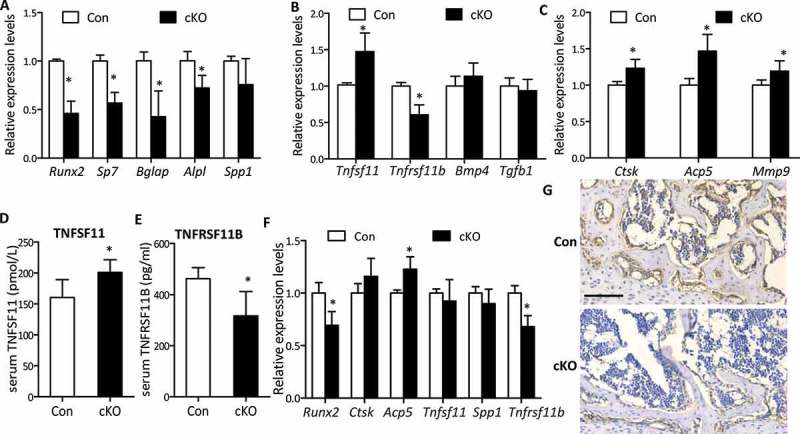
Expression levels of bone formation and resorption markers in osteoblast-specific Atg7 knockout mice during adult and weanling stages. RNA was extracted from ribs of P21 mice for weanling stages and calvaria of 22-wk for adult stage. (a) RT-qPCR for bone formation markers (Runx2, Spp1, Bglap, Alpl, and Sp7) in mice during adult stage. (b) mRNAs coding for Tnfrsf11b, Tnfsf11, Tgfb1, and Bmp4 expressed by osteoblasts during adult stage. (c) RT-qPCR for bone resorption markers expressed by osteoclasts (Mmp9, Ctsk, and Acp5) during adult stage. (d and e) Serum TNFSF11 and TNFRSF11B levels during adult stage. (f) RT-qPCR for Runx2, Spp1, Ctsk, Acp5, Tnfrsf11b and Tnfsf11 in P21 mice. (g) RUNX2 protein in tibia from weanling mice assessed by immunohistochemistry. Scale bars: 100 µm. *Statistically significant difference between control and cKO (n = 4 to 6/group), and results were presented as gene expression levels in all groups normalized to controls (mean ± SD, Student t test; *P < 0.05).
Autophagy deficiency impeded osteoblast mineralization and promoted apoptosis and ER stress in cultured primary osteoblasts
To determine the impact of conditional deletion of Atg7 on osteoblast function ex vivo, we examined osteoblastic differentiation and gene expression profiles in primary osteoblast cultures derived from control and Atg7 cKO calvarial bones. In vitro, primary Atg7−/- calvarial osteoblasts displayed impaired osteoblastic differentiation and maturation, as evidenced by lower ALPL activity compared with controls (Figure 5(a)). This inhibitory effect of defective autophagy was further observed in a mineralizing nodule assay (Figure 5(b,c)). Accordingly, expression of characteristic osteoblast markers including ALPL, bone sialoprotein (Ibsp), BGLAP and RUNX2 examined by western blot or RT-qPCR was diminished in Atg7−/- osteoblasts compared with controls (Figure 5(d,e)). Considering the regulatory role of autophagy on apoptosis, we next explored whether the compromised differentiation was due to apoptosis in Atg7 cKO osteoblasts by in situ cell death. We found that the number of TUNEL-positive cells was increased 163.6% in Atg7 cKO osteoblasts at the end of 7 days culture as compared to controls (Figure 5(f,g)), suggesting the enhanced apoptosis may be partly responsible for compromised mineralization.
Figure 5.
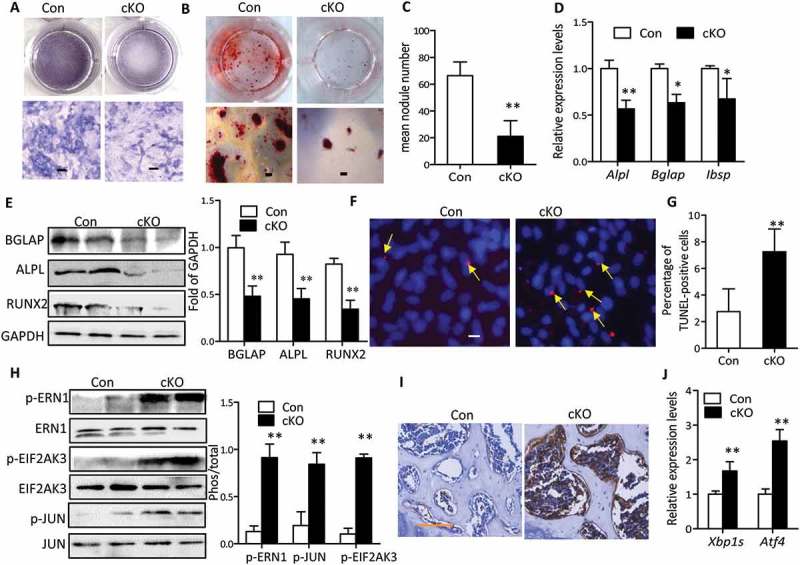
Autophagy deficiency inhibited osteoblasts mineralization and promoted apoptosis and ER stress. (a) Representative images for ALPL staining of primary osteoblasts derived from control and Atg7 cKO mice and cultured in differentiation medium for 7 days. Scale bars: 100 µm. (b) Representative images for Alizarin Red S staining of osteoblasts differentiated for 14 days. Scale bars: 200 µm. Statistical analysis of mineralization nodule numbers per well is shown in (c). (d) RT-qPCR for Alpl, Bglap, and Ibsp in primary osteoblasts differentiated for 14 days. (e) Representative western blot of BGLAP, RUNX2, and ALPL expression in primary osteoblasts induced for 14 days. Quantification is shown on the right. (f) Representative TUNEL labeling images of primary osteoblasts differentiated for 7 days. Bars: 10 µm. Statistical analysis of TUNEL-positive cell numbers is shown in (g). The data in (c to g) expressed as mean ± SD in each bar group represents the average of 3 independent experiments (Student t test; *P < 0.05, **P < 0.01). (h) Representative immunoblots of p-ERN1, p-JUN, p-EIF2AK3) in calvaria from 22-wk-old cKO mice (n = 3/group). Quantification is shown on the right (mean ± SD, Student t test; **P < 0.01). (i) Representative Immunohistochemical images of DDIT3 in tibia of 22-wk-old mice (n = 3/group). Scale bars: 100 µm. (j) Relative expression of Xbp1s and Atf4 mRNA in calvaria from 22-wk-old mice (n = 4 to 6/group) (mean ± SD, Student t test; **P < 0.01).
It has been shown that defective autophagy promotes endoplasmic reticulum (ER) stress and then renders cells vulnerable to apoptosis. These reports and our present results raise the possibility that retarded bone mineralization during autophagy deficiency might be mediated by ER stress. As expected, we observed an elevation of phosphorylated EIF2AK3/PERK (eukaryotic translation initiation factor 2 alpha kinase 3) and ERN1/IRE1α (endoplasmic reticulum [ER] to nucleus signaling 1) in calvaria of 22-wk-old Atg7 cKO compared with control littermates (Figure 5(h)). MAPK8/JNK1 (mitogen-activated protein kinase 8) activity, indicated by JUN (jun proto-oncogene) phosphorylation, was also dramatically elevated in the Atg7 cKO mice (Figure 5(h)). Immunohistochemistry analysis also revealed an increased expression of DDIT3/CHOP (DNA-damage inducible transcript 3) in tibia of 22-wk-old Atg7 cKO mice (Figure 5(i)). Moreover, RT-qPCR of the spliced or processed form of Xbp1 (X-box binding protein 1; Xbp1s) and Atf4 (activating transcription factor 4) further confirmed these changes in calvaria of 22-wk-old Atg7 cKO mice (Figure 5(j)). Similar results were seen in 3-wk-old Atg7 cKO mice (data not shown). Considering the in vitro observation that defective autophagy also triggered ER stress in primary osteoblasts derived from Atg7 cKO mice, we next asked whether restoration of autophagy activity by adenoviral overexpression of Atg7 could rescue ER stress and the defects in bone mineralization. After adenoviral delivery, we verified that restoration of ATG7 expression resulted in significant attenuation in autophagy deficiency-induced ER stress as detected by western blot (Figures S5A and B), as well as restoration of osteoblastic gene expression, ALPL activity and mineralization (Figures S5C and D).
Amelioration of ER stress abrogated atg7 ablation-mediated effects on osteoblast differentiation and bone formation
Considering the negative role of ER stress in apoptosis and its involvement in osteoblast differentiation, we reasoned that ER stress triggered by Atg7 deletion might be responsible for the apoptosis and compromised osteoblast differentiation. To test this hypothesis, primary osteoblasts derived from Atg7 cKO mice were cultured in the presence or absence of 10 mM phenylbutyric acid (PBA), a chemical chaperone known to attenuate ER stress. Indeed, PBA reversed Atg7 ablation-induced expression of ER stress makers, as assessed by reduced expression of DDIT3 and phosphorylation of EIF2AK3, ERN1 and JUN (Figure 6(a,b)). Interestingly, PBA also restored the expression of Alpl, Runx2 and Bglap, thus partially neutralizing such effects on ALPL activity at day 7 and mineralization by Alizarin red staining at day 14 induced by autophagy deficiency (Figure 6(c,d)). Likewise, there was a significant decrease in apoptosis by TUNEL assay in Atg7 cKO osteoblasts exposed to PBA at day 7 (Figure 6(e,f)). To further confirm whether the inhibitory effects of Atg7 ablation on osteoblast function are mediated by ER stress, PBA (5 mg/kg) was daily intraperitoneal injected to 3-wk-old Atg7 cKO mice for 4 wks. By the end of the 4-wk treatment period, PBA reversed Atg7 ablation-induced expression of ER stress makers as expected (Figure 7(a,b)). Furthermore, PBA partially neutralized the inhibitory effects of Atg7 ablation on bone formation (markers of RUNX2, ALPL, Ibsp, BGLAP and Sp7 assessed by RT-qPCR or western blot and the bone formation rate determined by calcein and xylenol orange double labeling) (Figure 7(c-f)).
Figure 6.
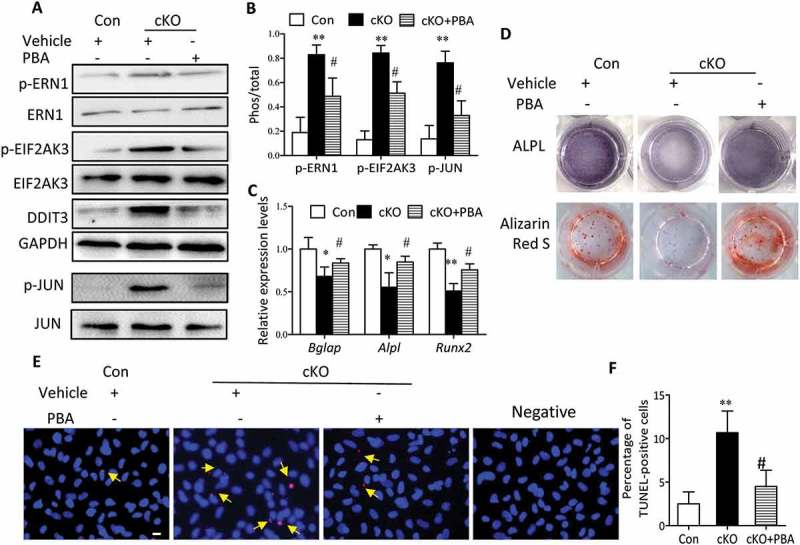
Amelioration of ER stress by PBA abrogates Atg7 ablation-mediated inhibitory effects on osteoblast mineralization in vitro. Primary osteoblasts derived from control and Atg7 cKO mice were cultured with or without 10 mM PBA. (a and b) The expression of ER stress markers were detected by western blot, and the relative phosphorylation of p-ERN1, p-JUN and p-EIF2AK3 was analyzed using ImageJ software. (c) Relative expression of Bglap, Alpl and Runx2 mRNA. (d) Representative images for ALPL and Alizarin red S staining. (e and f) Representative TUNEL labeling images of primary osteoblasts cultured with or without PBA; representative TUNEL-positive cells are indicated by the arrows (quantified in f), and bars: 10 μm. Data are mean ± SD (one-way ANOVA, *P < 0.05, **P < 0.01 cKO vs. Con, # P < 0.05 cKO + PBA vs. cKO).
Figure 7.
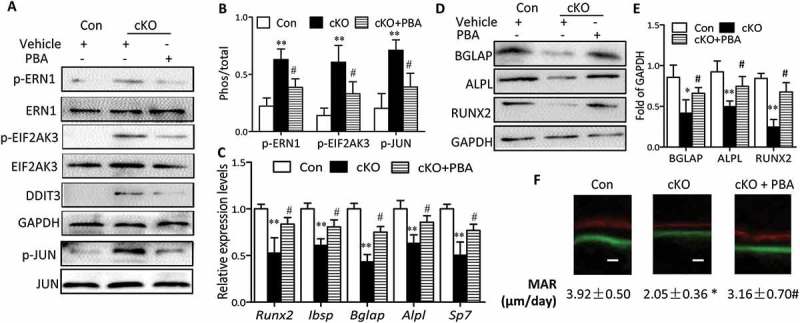
Administration of PBA partially reversed the suppression of bone formation by Atg7 deletion in vivo. PBA (5 mg/Kg) or vehicle was daily intraperitoneal injected to 3-wk-old cKO or control mice for 4 wks. By the end of 4-wk treatment period, total protein and RNA was extracted from calvaria (n = 3/group). (a and b) Expression of the ER stress markers (p-ERN1, p-JUN, DDIT3 and p-EIF2AK3). (c) Relative expression of Runx2, Ibsp, Bglap, Alpl, and Sp7 mRNA normalized to controls. (d and e) Expression of bone formation markers (BGLAP, ALPL and RUNX2). (f) Representative images of the double label staining in femur section with calcein and xylenol orange. Scale bars: 10 µm (mean ± SD, one-way ANOVA, *P < 0.05, **P < 0.01 cKO vs. Con, # P < 0.05 cKO + PBA vs. cKO).
Osteoblast-specific atg7 ablation impairs osteoblast function partially in a DDIT3- and MAPK8-SMAD1/5/8 dependent manner
EIF2AK3 activation results in phosphorylation of α subunit of eukaryotic initiation factor 2, leading to the induction of DDIT3, which promotes ER stress–induced apoptosis [24]. There is evidence that DDIT3 forms heterodimers with CEBPB (CCAAT/enhancer binding protein [C/EBP], beta) and inhibits the DNA-binding activity as well as RUNX2-binding activity of CEBPB, resulting in an impairment of osteoblast-specific gene transcription [25]. To explore whether DDIT3 plays regulatory roles in the inhibition of osteoblast differentiation in Atg7 cKO osteoblasts, we next examined the response of osteoblast derived from cKO mice to Ddit3 siRNA transfection and its efficiency was validated by western blot (data not shown). As expected, DDIT3 knockdown partially restored the Atg7 deletion-induced suppression of BGLAP, Sp7, Ibsp, or ALPL determined by RT-qPCR or western blot (Figure 8(a,b)). Of note, it has previously been reported that BGLAP is a direct target of RUNX2 [26], thus restoration of BGLAP by Ddit3 siRNA in Atg7 cKO osteoblasts might be induced by RUNX2. However, no statistical changes were observed in RUNX2 expression (Figure 8(a,b)). These results suggest that DDIT3 might modify the activity of RUNX2 through separation from CEBPB instead of directly regulating RUNX2 expression. Moreover, DDIT3-deficient Atg7 cKO osteoblasts revealed increased ALPL activity and the formation of mineralization nodules (Figure 8(c)). Furthermore, we also observed that the activity of MAPK8 was activated by ERN1, consistent with previous findings [27]. Because the crosstalk between MAPK8 and BMP2-SMAD1/5/8 signaling had functional contribution to osteoblast differentiation, we next examined the response of Atg7 cKO osteoblast to specific inhibitors of MAPK8 (SP600125). As shown in Figure 8(d,e), the expression of osteoblast markers was partially reversed by SP600125 as determined by western blot or RT-qPCR. These effects were further confirmed by ALPL staining (Figure 8(f)). Notably, we observed a significant reduction of SMAD1/5/8 phosphorylation, BMP2 expression, and nuclear translocation of SMAD4 in Atg7 cKO osteoblasts, whereas treatment of SP600125 partially abrogated such inhibitory effects (Figure 8(d,g)). Collectively, all these data further support the notion that ER stress contributed to autophagy deficiency-induced impaired osteoblast differentiation partially via DDIT3- and MAPK8-SMAD1/5/8 signaling.
Figure 8.
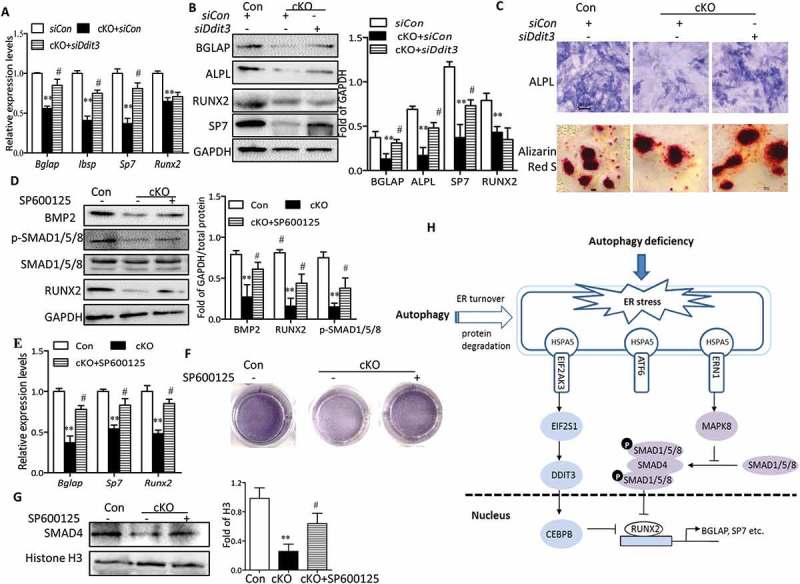
Autophagy deficiency impedes osteoblast differentiation partially via DDIT3- and MAPK8-SMAD1/5/8-dependent signaling. Relative expression of Bglap, Sp7, Ibsp and Runx2 mRNA (a), and BGLAP, ALPL, SP7 and RUNX2 proteins (b) were examined by RT-qPCR or western blot in primary osteoblasts transfected with or without siDdit3. Values expressed as mean ± SD of 3 independent experiments (one-way ANOVA, **P < 0.01 cKO + siCon vs. siCon, # P < 0.05 cKO + siDdit3 vs. cKO + siCon). (c) Representative images for ALPL and Alizarin red S staining in osteoblasts. Scale bars: 100 µm. Primary osteoblasts were pretreated with SP600125 (10 µM) for 2 h and then cultured in differentiation medium for 7 days. Representative western blot of BMP2 and RUNX2 and phosphorylation of SMAD1/5/8 (d), RT-qPCR for Bglap, Sp7 and Runx2 mRNA (e), representative images for ALPL staining (f), and representative western blot of nuclear SMAD4 expression (g) in osteoblasts, representative of 3 experiments (mean ± SD, one-way ANOVA, *P < 0.05 cKO vs. Con, # P < 0.05 cKO + SP600125 vs. cKO). (h) Model depicting the mechanism through which autophagy deficiency-mediated ER stress leads to inhibited osteoblast differentiation.
Discussion
In this study, we have shown that inducible osteoblast-specific autophagy ablation in vivo causes bone loss by induction of ER stress both during developmental and remodeling stages, and impairs osteoblast function in DDIT3- and MAPK8-SMAD1/5/8 dependent manner in vitro. At the skeletal early rapid growth stage, Atg7 cKO mice exhibited bone loss, lower bone formation rate and relative milder increase of bone resorption. While at the peak bone mass attainment stage, deficiency of autophagy causes lower bone formation rate concomitant with greater increased bone reabsorption, resulting in a more pronounced bone loss. Thus, the present study provides several lines of direct evidence that autophagy is required for osteoblast differentiation and bone homeostasis.
Although several studies have recently reported that autophagy regulates osteoblast and osteoclast function, and bone homeostasis [15,22], the impact of autophagy deficiency in bone remodeling has not been fully addressed and much remains to be elucidated. Our study indicated the pivotal regulation of autophagy in coupling bone formation and bone resorption and maintaining normal postnatal bone homeostasis. As osteoblasts are responsible for synthesis and secretion of matrix, and its subsequent mineralization, autophagy in osteoblasts would be expected to play critical role support protein synthesis and prevent secretion of misfolded matrix protein [28]. Indeed, in our study, targeted deletion of Atg7 in osteoblast caused low bone mass at both young and adult age in mice. This phenotype of Atg7 cKO attributed to decreased osteoblast formation, reduced matrix mineralization, accompanied with an increased osteoclast number and the extent of the bone surface covered by osteoclasts. In addition to our findings, one recent study shows that osteoblast-targeted deletion of Rb1cc1, part of the RB1CC1-ATG13-ULK1 complex essential for initiation of autophagosome formation, in a mouse model causes severe osteopenia due to reduced bone formation [22]. The other studies also suggest that suppression of autophagy by deletion of Atg7 in targeted osteocytes causes decreased bone mass and bone remodeling [15], and supports a direct role of autophagy in the osteoblast intracellular mineralization process, as well as an indirect effect on bone remodeling through the stimulation of osteoclastogenesis [23]. These studies indicate an involvement of the process of autophagy in bone formation, however the importance of autophagic flux for bone matrix secretion remains unknown.
Considering that autophagy is potentially essential for osteoblast differentiation which occurs during embryonic development and eventually affects bone formation and development, we choose to use TM-inducible Col1a-cre mice, which allow us to examine the function of autophagy on bone physiology and homeostasis after birth, especially on the specific stages of rapid bone mass increase and steady-state of bone remodeling (a pattern similar to clinical patients). Since TM is known as a selective estrogen receptor modulator, which may have similar effects to other selective estrogen receptor modulators reported to increase bone mass both in human and in mice [29,30], we optimized the dosage and timing of TM administration to exclude any potential ‘off-target’ effect of TM on skeletal tissue. We confirmed that in the current study conditions, TM has a minor effect on skeletal tissue while its effect in osteoblasts is sufficient to activate Col1a-CreERT2. Consistently, a previous study using the same TM inducible Col1a-creERT2 system reports effective target gene inactivation with no major side effect of TM global administration on skeletal tissue [31]. More recent literature further supports our strategy in that 10 mg/kg/day TM for 4 consecutive days does not significantly affect bone turnover or bone mass, but still activates Cre to a comparable level to the 100 mg/kg/day dose of TM [32]. It is noticeable that there is still a residual ATG7 present in the osteoblasts, as a trace protein expression of ATG7 was observed. This is not unexpected, as recombination mediated by an inducible promoter is generally mosaic and its efficiency is dependent on tissue types [33]. For example, it was reported that in a TM-inducible COL2/collagen type 2-CreER targeted chondrocyte system, 90.8% recombinase-positive cell were observed in chondrocytes, instead of the theoretical 100% as expected [34]. Similarly, it was also reported that frequency of the deletion of Sp7/Osterix exon 2 ranged from 60% – 90% of the osteoblasts in Sp7/Osterixflox/-;Col1a-CreERT2 by Cre recombinase after administration of TM [31,35], which again did not reach the theoretical 100% efficiency.
Bone remodeling is constantly renewed by the balanced action of osteoblastic bone formation and osteoclastic bone resorption, which are precisely coordinated to maintain the skeletal homeostasis. The ratio of TNFSF11 to TNFRSF11B, a TNFSF11 decoy receptor that inhibits osteoclast differentiation, is a major mechanism for controlling osteoclast differentiation from macrophage precursors in the hematopoietic lineage [36]. In our study, a striking phenotype in osteoblast-specific Atg7 cKO mice is that the trabecular bone volume is significantly lower than that of controls at 5 months of age, followed by the enhanced osteoclast bone resorption. Consistently, we found that deletion of Atg7 in osteoblasts altered the Tnfsf11:Tnfrsf11b ratio and the expression of Tnfrsf11b is greatly reduced, which eventually contributed to an elevated rate of osteoclast differentiation. These findings are similar to a previous report that autophagy-deficient osteoblasts induced by Atg5 ablation exhibit increased secretion of TNFSF11 [23]. Another independent study has failed to detect any changes in Tnfsf11 and Tnfrsf11b mRNA expression in autophagy-defective osteocytes induced by conditional deletion of Atg7 [15]. However, in this study, it is somewhat surprising that Tnfsf11 expression and secretion was increased in the context of ER stress triggered by Atg7 deletion, since protein translation would be suppressed to prevent the generation of more unfolded proteins under ER stress [37]. Our results indicated that the in vivo effects of Atg7 deletion on the TNFSF11-TNFRSF11B axis expression may be much more complicated than anticipated. Such result might be due to an unconventional secretion of proteins (such as IL1B [interleukin 1 beta] and interleukin 18), which is referred to as the autophagy-based exocytic process other than the classical ER/Golgi-dependent pathway [38]. In support of this idea, IL1B, a potential substrate for autophagy-based unconventional secretion, has been shown to be negatively regulated by autophagy [39–41]. Meanwhile, Marie et al. [23] also speculate that this abnormal autophagy-based unconventional secretion of proteins possibly contribute to the repressive osteoblast mineralization involving secretion of various vesicles, leading to the suppression of bone matrix formation. Therefore, loss of Atg7 led to impaired autophagy process in osteoblasts and further impeded osteoblast mineralization and bone formation.
As a putative adaptive catabolic process, autophagy, which can be induced under stressful conditions to remove damaged organelles and misfolded proteins for cell survival [28], has been shown to be linked with ER at many levels and is likely to be a critical component of normal ER function [42]. In our study, although defective autophagy induced by conditional Atg7 ablation appeared causal to remarkable bone loss, a definitive link could be established if the restoration of autophagy could rescue skeletal balance. In fact, we found that reconstitution of Atg7 can restore impaired functions and improve ER stress during bone abnormality, which may suggest a reciprocal functional interaction between autophagy and ER stress. Indeed, deletion of Atg7 in this study could induce ER stress and then potentially result in the accumulation of misfolded proteins in osteoblasts, whereas improvement of ER stress by PBA could alleviate ER dilation as well as synthesis and folding of ER [43], thus preventing such accumulation and restore normal ability of the osteoblasts to produce and deposit collagen matrix. Similarly, a recent study reports that mice deficient of Atg7 in chondrocytes exhibit ER cisternae dilation and accumulation of procollagen II, as well as defective formation of collagen II, implicating a crosstalk between induced ER stress and autophagy [44]. In contrast, other research demonstrates that although mutation-caused accumulation of misfolded of type I collagen induces pronounced ER dilation and malfunction of osteoblasts, no upregulation of binding immunoglobulin protein is expected in the unfolded protein response. Whereas enhancement of autophagy can prevent such accumulation, eliminate ER dilation, and restore normal ability of the cells to produce and deposit collagen matrix [45]. These observations indicate that the metabolic impact of autophagy disruption on ER stress may vary depending on the features of the stress, including its duration and severity.
The 3 major transducers of the unfolded protein response are EIF2AK3, ERN1 and ATF6, which sense the presence of the unfolded proteins in the ER lumen and transduce signals to the cytosol or nucleus. DDIT3 is a member of the C/EBP family and plays a role in osteoblastic and adipocytic cell differentiation [46–48]. Our study also revealed that suppression of DDIT3 in osteoblasts could partly restore Atg7 ablation-mediated attenuation of osteoblast differentiation and mineralization. We also observed that expression of BGLAP, Ibsp, and SP7 was partially restored by DDIT3 downregulation in Atg7-deficient cells. However, we did not detect significant changes in RUNX2 expression in Atg7 cKO cells treated with DDIT3 inhibitor. One possibility is that DDIT3 forms heterodimers with CEBPB and accordingly inhibits the RUNX2-binding activity to CEBPB as identified in previous studies [25,48]. In addition, we also found that suppression of MAPK8 activity partially abrogated the inhibitory effects of autophagy deficiency on BMP2 expression and nuclear translocation of SMAD4. We speculated that autophagy deficiency coupled with ER stress might inhibit BMP-SMAD1/5/8 pathway in a MAPK8-dependent manner and thereby suppress RUNX2 expression. In agreement with our findings, it has been reported that inhibition of MAPK8 expression and activity enhances BMP2-induced osteoblastic differentiation via RUNX2 phosphorylation [49]. Overall our results support the hypothesis that Atg7 ablation triggered ER stress in a DDIT3- and MAPK8-dependent manner that might impair the SMAD1/5/8-SMAD4 complex, thus repressing its nuclear translocation. As a result, these disturbances contribute to the diminished expression of RUNX2, BGLAP, ALPL and mineralization observed in Atg7 cKO mice (Figure 8(i)).
The causative mechanisms of autophagy defects implicated in skeletal abnormality could be diverse. Despite ER stress, several other possibilities might also be taken into consideration such as the intervention mode (inducible or not) and stage of autophagy deletion (prenatal or postnatal), the duration of ER stress and the chronic effects secondary to ER stress, and the possible Smad4 deregulation in postnatal bone homeostasis. Smad4 are primary cytoplasmic signal transducers both for TGFB/ACTIVIN and BMPs [50]. It has been shown that targeted ablation of Smad4 in differentiated osteoblasts results in growth retardation and BMD reduction, indicating an important role of SMAD4 in control of osteoblast function and regulation of bone mass [50]. Given that the phenotypes of the Smad4-deficient mice were more similar to the mice with reduced BMP signaling than those with diminished TGFB signal [50], it is plausible in our study that the differentiated osteoblasts at later stages might be less sensitive to TGFB due to the impaired Smad4 function in Atg7 cKO mice. Thus, further investigation should be warranted in the absence of other chronic changes to fully understand this adaptive response to autophagy regulation during bone remodeling.
In conclusion, our findings suggest the direct evidence that ATG7 mediated a pivotal role in maintaining bone homeostasis. Although further studies are warranted to address the adaptive role of autophagy in skeletal metabolism, our findings provide new insights into the functional basis of autophagy in metabolic homeostasis and stress response during bone modeling/remodeling, and suggest an innovative strategy to develop novel therapeutic agents against skeletal abnormality.
Materials and methods
Animal studies
B6. Cg-Tg (Col1a-cre/ERT2)1Crm/J mice were purchased from Jackson Laboratories (016241). Atg7flox/flox mice (strain name: B6; Cg-Atg7tmltchi) were kindly provided by the RIKEN Bio Resource Center [51]. The experimental animals used in most of the studies described here were obtained using a 2-step breeding strategy. Hemizygous Col1a-Cre/ERT2 transgenic mice were crossed with homozygous Atg7-flox mice to generate heterozygous Atg7-flox offspring with and without a Col1a-Cre allele. These offspring were then intercrossed to generate the following offspring: Col1a-CreERT2+Atg7flox/flox and Atg7flox/flox. The Cre transgene was detected by PCR with a thermal cycler (BIO-RAD, PTC-1148, Singapore) using the primers: Transgene Forward, 12,249, 5′-TCCAATTTACTGACCGTACACCAA-3′, Transgene Reverse, 12,250, 5′-CCTGATCCTGGCAATTTCGGCTA-3′, Internal Positive Control Forward, oIMR7338, 5′-CTAGGCCACAGAATTGAAAGATCT-3′, and Internal Positive Control Reverse, oIMR7339, 5′-GTAGGTGGAAATTCTAGCATCATCC-3′. The 500-base pair (bp) PCR product was detected as transgene, and the 324-bp PCR product as an internal positive control. The sequences of PCR primers for genotyping Atg7flox/flox mice are as follows: 2 forward primers, Hind-Fw, 5′-TGGCTGCTACTTCTGCAATGATGC-3′, Atg7-WT-forward, 5′-ATTGTGGCTCCTGCCCCAGT-3′, and a reverse primer, Pst-Rv, 5′-CAGGACAGAGACCATCAGCTCCAC-3′. In wild-type mice only the 460-bp PCR product can be detected, and in homozygous Atg7flox/flox mice only the 550-bp PCR product can be detected. Both 460-bp and 550-bp PCR products were detected in heterozygous mice (Atg7flox/WT). The resulting Col1a-CreERT2+Atg7flox/flox mice were viable and fertile in the absence of TM. The mice were maintained with good ventilation and a 12-h light/dark cycle, and were kept feeding and drinking ad libitum.
TM administration
TM (Sigma-Aldrich, T5648) was dissolved in a small volume of ethanol, diluted with corn oil (Sigma-Aldrich, C8267) at a concentration of 20 mg/ml, and stored at −20°C until use. In weanling mice, TM (0.2 mg/mouse) was injected intraperitoneally at day 5, 7, 9, 11 until dissection at P21. For adult stages, TM (50 mg/kg) was injected intraperitoneally twice a week from 8 wk until dissection at 22 wk. From now on, we will refer to Col1a-CreERT2+atg7−/-mice as Atg7 cKO mice and the Atg7flox/flox control littermates as controls.
Micro-computed tomography (μct)
For trabecular bone mass evaluation in mice, a desktop μCT system (eXplore Locus SP, GE Healthcare, USA) was employed as previously described [52]. At sacrifice, the right femora and tibia and the vertebrae (L4) were isolated and fixed in 4% paraformaldehyde for 24 h, and then switched to 70% ethanol. Briefly, the distal femoral and proximal tibia metaphysis were scanned at a resolution of 8 μm, a voltage of 80 kV, and a current of 80 μA. The region selected for analysis was defined from 0.1 mm to 2.6 mm away from the epiphysis. Trabecular region of interest from the L4 vertebrae is manually segregated from cortical bone by splining an areal region of interest at multiple levels and then interpolating among these to generate a volume of interest for the trabecular bone. Data were analyzed with the Micview V2.1.2 software. The interesting region segmented by a fixed threshold was reconstructed into 3-dimensional images. Quantification of trabecular bone was performed using parameters of BMD, BV/TV, Tb.Th, Tb.N, and Tb.Sp.
Bone histology and histomorphometry
Femurs and tibia from 3-wk and 22-wk-old mice were fixed in 4% paraformaldehyde (HEART Biological Technology, IS014), decalcified with 10% ethylenediaminetetraacetic acid (EDTA; HEART Biological Technology, BF041), and embedded in paraffin. Paraffin sections were cut in sagittal planes at 5 µm. For examination of osteoblast, and osteoclast and bone resorption, hematoxylin-eosin (H&E) staining and ACP5 (LEAGENE, DE0013) staining was respectively performed, as stated previously [53]. Quantification was performed using the ImageJ 1.47 software from at least 5 microscopy fields. Immunochemistry was performed using rabbit polyclonal antibodies against ATG7 (Beyotime, AA829-1), DDIT3 (Santa Cruz Biotechnology, sc-166,682), and RUNX2 (Santa Cruz Biotechnology, sc-101,145). Subsequently, an ABC kit (Santa Cruz Biotechnology, sc-2018; Zhongshan Biotechnologies, SP-9001) and 3, 3-diaminobenzidine tetrahydrochloride exposure were used for detection.
At 12 days and 2 days prior to sacrifice, mice received calcein (10 mg/kg body weight; Sigma-Aldrich, C0875) in 2% NaHCO3 intraperitoneally and xylenol orange (90 mg/kg; Sigma-Aldrich, 52,097) intraperitoneally respectively. At sacrifice, the left femora were isolated, fixed in 80% ethanol, and embedded in methyl methacrylate without decalcification. The specimens were sagittal sectioned into 30-μm sections using a hard tissue slicing machine (Leica, SP1600, Wetzlar, Germany) away from light. Double-labeled surfaces were evaluated by a fluorescence microscope (Nikon Eclipse Ti-S, Tokyo, Japan) and quantification was performed based on at least 5 images as previously described [52,53].
Serum measurement
Blood samples were collected from Atg7 cKO and control mice. Markers of bone resorption (RatLaps™ [CTX-I] EIA; Immunodiagnostic Systems, AC-06F1), bone formation (Rat and Mouse PINP EIA; Immunodiagnostic Systems, AC-33F1), TNFSF11 (Sangon Biotech, D720269) and TNFRSF11B (Sangon Biotech, D720234) were detected according to the instructions of manufacturer. TNFSF11 and TNFRSF11B were also detected for their concentrations in conditional media of calvaria cultures ex vivo.
RNA isolation and quantitative real-time PCR (rt-qpcr)
Total RNA was extracted from calvaria and ribs of weanling and adult mice (Invitrogen, 15,596–026) according to the manufacturer’s protocol. RT-qPCR (Bio-Rad, PTC-0148, Singapore) analysis was carried out as described previously [54]. Briefly, The recovered RNA was further processed using RevertAid First Strand cDNA Synthesis kit (Thermo Fisher Scientific, K1621) to produce cDNA for RT-qPCR by using SYBR Premix Ex Taq II (Takara Bio INC, RR820A). Taqman primers and probes used in this study were as follows: Atg7, 5′-ACTGTGCTGGTCTCCTTGCT-3′, 5′-CAGGGTGCTGGGTTAGGTTA-3′; Bglap, 5′-AGGAGGGCAATAAGGTAGTG-3′ and 5′-TGTAGGCGGTCTTCAAGC-3′; Alpl, 5′-GGTAGATTACGCTCACAACAAC-3′ and 5′-CAGGCACAGTGGTCAAGG-3′; Acp5, 5′-AGCAGCCAAGGAGGACTAC-3′ and 5′-CAGCACATAGCCCACACC-3′; Ctsk, 5′-GTGTTGGTGGTGGGCTATG-3′ and 5′-GCAGGCGTTGTTCTTATTCC-3′; Tnfrsf11b, 5′-GACGATGATGACGATGATGATG-3′ and 5′-CGACTGTAGGGACGATTGG-3′; Tnfsf11, 5′-GAAATGCCTCCGCTGTTATG-3′ and 5′-TTCTGTCTGTGCCTTCTTGG-3′; Runx2, 5′-TACTTCGTCAGCATCCTATCAG-3′ and 5′-CAGCGTCAACACCATCATTC-3′; Sp7, 5′-GGCAAGGCTTCGCATCTG-3′ and 5′-CTCAAGTGGTCGCTTCTGG-3′; Mmp9, 5′-GCCACCACAGCCAACTATg-3′ and 5′-TGCCCAGGAAGACGAAGG-3′; Tgfb1, 5′-ATTCCTGGCGTTACCTTGG-3′ and 5′-CCTGTATTCCGTCTCCTTGG-3′; Bmp4, 5′-GGAGGAGGAGGAGGAAGAG-3′ and 5′-GCTGCTGAGGTTGAAGAGG-3′; Spp1, 5′-GACGATGATGACGATGATGATG-3′ and 5′-CGACTGTAGGGACGATTGG-3′; Ibsp, 5′-ACAATCCGTGCCACTCACT-3′ and 5′-TTTCATCGAGAAAGCACAGG-3′; Gapdh (glyceraldehyde-3-phosphate dehydrogenase), 5′-TCAACGGCACAGTCAAGG-3′ and 5′-ACTCCACGACATACTCAGC-3′. The gene expression levels were normalized by the average levels of the mean-centered housekeeping gene Gapdh. All measurements were performed in triplicate and analyzed using the 2−△△Ct method.
Ex vivo culture of calvaria bone and primary osteoblasts culture
Calvaria explants were established from 1- to 3-day-old untreated cKO mice (Col1a-CreERT2+Atg7flox/flox), then bisected at the sagittal suture and cultured in α-MEM (HyClone, SH30265.01) supplemented with ascorbic acid (50 μg/ml, Sigma, A4544), 10mM β-glycerophosphate (Sigma-Aldrich, G9422) and 10% fetal bovine serum (Gibco Invitrogen, 16,000,044) for the first 24 h in culture. One hemi-calvaria from each pair was treated with 4-OH TM (1 μM; Sigma-Aldrich, T176) for 5 days. Primary calvaria osteoblast isolation and culture was performed as described previously [22]. Briefly, Primary calvaria osteoblasts were isolated from 1- to 3-day-old transgenic neonates by 4 sequential collagenase A (Roche Diagnostics, 10,103,586,001)/dispase II (Roche Diagnostics, 04942078001) digestion. Parietal bones of calvaria were incubated in α-MEM containing 1 mg/ml collagenase A and 2 mg/ml dispase II at 37°C on a rocking platform. The first digestions were discarded, and subsequent 3 digestions were collected. Cells released from digestion were plated in α-MEM culture medium containing 10% fetal bovine serum and 1% penicillin/streptomycin (HyClone, SV30010) and differentiated with 50 µg/ml ascorbic acid and 10 mM β-glycerophosphate (Sigma, G5422). Osteoblasts were infected with adenoviral expression vectors for mouse Atg7 (Ad-Atg7) or an adenoviral control vectors (Ad-GFP) at a multiplicity of infection of 400. Adenoviral expression vector was designed, synthesized and constructed by Genechem Co. (Shanghai, China). Osteoblasts were transfected with pools of small interfering RNA targeted for Ddit3 (siDdit3; Invitrogen, 1,320,001) using Lipofectamine 2000 (Invitrogen, 11,668–019), according to the procedure recommended by the manufacturer. For Alizarin red S (LEAGENE, DS0072) staining, cells were fixed at day 14 of culture with 4% formaldehyde and stained for 10 min. For ALPL (Beyotime, C3206) assay, cells were fixed 4% formaldehyde and subjected to ALPL staining on day 7.
TUNEL assay
Apoptotic primary osteoblasts derived from Atg7 cKO and controls were identified by an In Situ Cell Death Detection Kit (Roche Diagnostics, 12,156,792,910), according to the instructions of manufacturer. Nuclei were counterstained with 4, 6-diamidino-2-phenylindole/DAPI. TUNEL-positive cells were stained by red fluorescence. The percentage of TUNEL-positive cells was calculated as the number of TUNEL-labeled cells per grid divided by the total number of cells per grid.
Western blot analysis
Protein extracts were prepared from calvaria, and primary calvaria osteoblasts isolated from neonatal mice. Cells and tissues were lysed using RIPA buffer (50 mM Tris-HCl, pH7.9, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40 [Fluka, 74,385], 0.1 mM PMSF [Beyotime, ST506]). Proteins were separated on 10% – 15% sodium dodecylsulfate polyacrylamide gels; separated proteins were then electronically blotted onto polyvinylidene fluoride membranes. The membranes were then blocked and subsequently incubated with designated primary antibodies overnight at 4°C. Antibodies to EIF2AK3 (sc-377,400), p-EIF2AK3 (sc-32,577), SMAD4 (sc-7966), RUNX2 (sc-101,145), p-JUN (sc-822), SMAD1/5/8 (sc-6031), H3 (sc-10,809), DDIT3 (sc-166,682), and GAPDH (sc-365,062) were purchased from Santa Cruz Biotechnology. Antibodies to LC3A/B (4108), ERN1 (3294), SQSTM1 (5114), and p-SMAD1/5/8 (9511P) were purchased from Cell Signaling Technology. Antibodies to JUN (AF1612), BMP2 (AF0075), and ALPL (AF1030) were purchased from Beyotime Biotechnology. Antibodies to ATG7 (ab133528), BGLAP (ab93876), and p-ERN1 (ab48187) were purchased from Abcam. On the following day, samples were washed 3 times with TBS-T (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% Tween 20 [T8220, Solarbio]) solution for 10 min each, and subsequently incubated with the secondary antibodies (Santa Cruz Biotechnology, sc-2004 and sc-2005) corresponding to the primary antibodies for 60 min. At last, the blots were visualized with an enhanced chemiluminescence (ECL, Millipore, WBKLS0100) detection system.
Statistics
Statistical analyses were performed using SPSS 17.0 software. Data are means ± S.D. Statistical analysis was performed using an unpaired, two-tailed Student t test between the 2 groups or one-way ANOVA followed by Tukey post hoc tests for more than 2 groups. Differences were considered significant when the P value was < 0.05.
Study approval
All procedures were performed in accordance with the recommendations in the NIH Guide for the Care and Use of Laboratory Animals. The Institutional Animal Ethics Committee of Xi’an Jiaotong University reviewed and approved the protocol.
Funding Statement
This work was supported by the National Excellent Young Scientist Program [81222026];National Natural Science Foundation of China (NSFC) [Nos. 81672221, 81472038, 81370899, 81670734, and 81600621];New Century Excellent Talents from the Ministry of Education of China [NCET-11-0437];National Institutes of Health Grant [P20GM104937];
Abbreviation
- ACP5/TRAP
acid phosphatase 5, tartrate resistant
- ALPL
alkaline phosphatase, liver/bone/kidney
- ATF4
activating transcription factor 4
- ATG
autophagy related
- BGLAP/OCN
bone gamma carboxyglutamate protein
- BMD
bone mineral density
- BMP4
bone morphogenetic protein 4
- BV/TV
bone volume per tissue volume
- BFR
bone formation rate
- BS
bone surface
- IBSP
integrin binding sialoprotein
- CEBPB
CCAAT/enhancer binding protein (C/EBP), beta
- cKO
conditional knockout
- Col1a
collagen, type I, alpha 1
- Ctsk
cathepsin K
- CTX
C-terminal telopeptide of type I collagen
- DDIT3/CHOP
DNA-damage inducible transcript 3
- EIF2AK3/PERK
eukaryotic translation initiation factor 2 alpha kinase 3
- ERN1/IRE1α
endoplasmic reticulum (ER) to nucleus signaling 1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IL1B
interleukin 1 beta; JUN, jun proto-oncogene
- MAR
mineral apposition rate
- MAP1LC3A/B (LC3A/B)
microtubule-associated protein 1 light chain 3 A/B
- MAPK8/JNK1
mitogen-activated protein kinase 8
- Mmp9
matrix metallopeptidase 9
- PBA
phenylbutyric acid
- PINP
amino-terminal propeptide of type I collagen
- RB1CC1/FIP200
RB1-inducible coiled-coil 1
- RUNX2
runt related transcription factor 2
- SP7/osterix
Sp7 transcription factor 7
- SPP1/OPN
secreted phosphoprotein 1
- SQSTM1/p62
sequestosome 1
- Tb.N
trabecular number
- Tb.Sp
trabecular spacing
- Tb.Th
trabecular thickness
- Tgfb1
transforming growth factor, beta 1
- TM
tamoxifen
- TNFRSF11B/OPG
tumor necrosis factor receptor superfamily, member 11b (osteoprotegerin)
- TNFSF11/RANKL
tumor necrosis factor (ligand) superfamily, member 11
- μCT
microcomputed tomography
- XBP1
X-box binding protein 1
Supplemental data
Supplemental data for this article can be accessed here.
Acknowledgments
We thank Professor Masaaki Komatsu, Tokyo Metropolitan Institute for Medical Science, RIKEN BioResource Center for providing the Atg7flox/flox mice. We appreciate advice of Prof. Ling Qin from the Chinese University of Hong Kong for improving the presentation of test. We appreciate the technical support of Professor Zhuojing Luo and Liu Yang from Xijing hospital, Fourth Military Medical University. This work was supported by the programs from the National Natural Science Foundation of China (Nos. 81672221, 81670734, 81600621, 81472038, and 81370899), National Excellent Young Scientist Program (No. 81222026), and the New Century Excellent Talents from the Ministry of Education of China (NCET-11-0437) and National Institutes of Health Grant P20GM104937.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Harada S, Rodan GA.. Control of osteoblast function and regulation of bone mass. Nature. 2003. May 15;423(6937):349–355. PubMed PMID: 12748654. [DOI] [PubMed] [Google Scholar]
- [2].Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007. July;13(7):791–801. PubMed PMID: WOS:000247902800025; English. [DOI] [PubMed] [Google Scholar]
- [3].Shapiro IM, Layfield R, Lotz M, et al. Boning up on autophagy: the role of autophagy in skeletal biology. Autophagy. 2014. January;10(1):7–19. PubMed PMID: 24225636; PubMed Central PMCID: PMC4028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011. May 04;13(5):495–504. PubMed PMID: 21531332; PubMed Central PMCID: PMC3099265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Beau I, Mehrpour M, Codogno P. Autophagosomes and human diseases. Int J Biochem Cell Biol. 2011. April;43(4):460–464. PubMed PMID: 21256243. [DOI] [PubMed] [Google Scholar]
- [6].Ebato C, Uchida T, Arakawa M, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008. October 8;(4):325–332. DOI: 10.1016/j.cmet.2008.08.009 PubMed PMID: WOS:000259897200009; English. [DOI] [PubMed] [Google Scholar]
- [7].Martinez-Lopez N, Athonvarangkul D, Singh R. Autophagy and Aging. Adv Exp Med Biol. 2015;847:73–87. PubMed PMID: WOS:000361797200004; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tan CC, Yu JT, Tan MS, et al. Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol Aging. 2014. May;35(5):941–957. PubMed PMID: WOS:000332308300001; English. [DOI] [PubMed] [Google Scholar]
- [9].Perluigi M, Di Domenico F, Butterfield DA. mTOR signaling in aging and neurodegeneration: at the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis. 2015. December;84:39–49. PubMed PMID: WOS:000367029300004; English. [DOI] [PubMed] [Google Scholar]
- [10].Sacitharan PK, Zarebska J, Nechyporuk-Zloy V, et al. Loss of sirt1 impairs autophagy in chondrocytes and is associated with accelerated cartilage aging and experimental osteoarthritis. Osteoarthr Cartilage. 2016. April;24:S145–S145. PubMed PMID: WOS:000373538800265; English. [Google Scholar]
- [11].Sasaki H, Takayama K, Matsushita T, et al. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis and Rheumatism. 2012. June;64(6):1920–1928. PubMed PMID: 22147463. [DOI] [PubMed] [Google Scholar]
- [12].Kang X, Yang W, Feng D, et al. Cartilage-specific autophagy deficiency promotes er stress and impairs chondrogenesis in perk-atf4-chop-dependent manner. Journal of Bone and Mineral Research: the Official Journal of the American Society for Bone and Mineral Research. 2017. March 17 DOI: 10.1002/jbmr.3134 PubMed PMID: 28304100. [DOI] [PubMed] [Google Scholar]
- [13].Chen K, Yang YH, Jiang SD, et al. Decreased activity of osteocyte autophagy with aging may contribute to the bone loss in senile population. Histochem Cell Biol. 2014. September;142(3):285–295. PubMed PMID: WOS:000340584000005; English. [DOI] [PubMed] [Google Scholar]
- [14].Zhang LS, Guo YF, Liu YZ, et al. Pathway-based genome-wide association analysis identified the importance of regulation-of-autophagy pathway for ultradistal radius BMD. J Bone Mineral Res. 2010. July;25(7):1572–1580. PubMed PMID: WOS:000280395900013; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Onal M, Piemontese M, Xiong JH, et al. Suppression of autophagy in osteocytes mimics skeletal aging. J Biol Chem. 2013. June 14;288(24):17432–17440. PubMed PMID: WOS:000320380600035; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zahm AM, Bohensky J, Adams CS, et al. Bone Cell Autophagy Is Regulated by Environmental Factors. Cells Tissues Organs. 2011;194(2–4):274–278. PubMed PMID: WOS:000293842500034; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Piemontese M, Xiong J, Fujiwara Y, et al. Cortical bone loss caused by glucocorticoid excess requires RANKL production by osteocytes and is associated with reduced OPG expression in mice. American Journal of Physiology. Endocrinology and Metabolism. 2016. September 1;311(3):E587–93. PubMed PMID: 27460899; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kenanidis E, Potoupnis ME, Kakoulidis P, et al. Management of glucocorticoid-induced osteoporosis: clinical data in relation to disease demographics, bone mineral density and fracture risk. Expert Opin Drug Saf. 2015. June;14(7):1035–1053. PubMed PMID: 25952267; English. [DOI] [PubMed] [Google Scholar]
- [19].Xia X, Kar R, Gluhak-Heinrich J, et al. Glucocorticoid-induced autophagy in osteocytes. J Bone Mineral Res. 2010. November;25(11):2479–2488. PubMed PMID: 20564240; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bartolome A, Lopez-Herradon A, Portal-Nunez S, et al. Autophagy impairment aggravates the inhibitory effects of high glucose on osteoblast viability and function. Biochem J. 2013. November 1;(455):329–337. . PubMed PMID: WOS:000326664100007; English. [DOI] [PubMed] [Google Scholar]
- [21].Piemontese M, Onal M, Xiong JH, et al. Low bone mass and changes in the osteocyte network in mice lacking autophagy in the osteoblast lineage. Sci Rep. 2016. April 11;6 DOI: 10.1038/Srep24262 PubMed PMID: WOS:000373765500001; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu F, Fang F, Yuan H, et al. Suppression of autophagy by FIP200 deletion leads to osteopenia in mice through the inhibition of osteoblast terminal differentiation. Journal of Bone and Mineral Research: the Official Journal of the American Society for Bone and Mineral Research. 2013. November;28(11):2414–2430. PubMed PMID: 23633228; PubMed Central PMCID: PMC3805719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nollet M, Santucci-Darmanin S, Breuil V, et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy. 2014;10(11):1965–1977. . PubMed PMID: 25484092; PubMed Central PMCID: PMC4502694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000. November;6(5):1099–1108. PubMed PMID: 11106749. [DOI] [PubMed] [Google Scholar]
- [25].Shirakawa K, Maeda S, Gotoh T, et al. CCAAT/enhancer-binding protein homologous protein (CHOP) regulates osteoblast differentiation. Mol Cell Biol. 2006. August;26(16):6105–6116. PubMed PMID: 16880521; PubMed Central PMCID: PMC1592788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ducy P, Zhang R, Geoffroy V, et al. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997. May 30;89(5):747–754. PubMed PMID: 9182762. [DOI] [PubMed] [Google Scholar]
- [27].Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002. November 21;420(6913):333–336. PubMed PMID: 12447443. [DOI] [PubMed] [Google Scholar]
- [28].Hocking LJ, Whitehouse C, Helfrich MH. Autophagy: A new player in skeletal maintenance? J Bone Mineral Res. 2012. July;27(7):1439–1447. PubMed PMID: WOS:000305297000001; English. [DOI] [PubMed] [Google Scholar]
- [29].Manolagas SC, O’Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol. 2013. December 9;12:699–712. PubMed PMID: WOS:000327364000005; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Starnes LM, Downey CM, Boyd SK, et al. Increased bone mass in male and female mice following tamoxifen administration. Genesis. 2007. April;45(4):229–235. PubMed PMID: WOS:000246019800009; English. [DOI] [PubMed] [Google Scholar]
- [31].Baek WY, De Crombrugghe B, Kim JE. Postnatally induced inactivation of Osterix in osteoblasts results in the reduction of bone formation and maintenance. Bone. 2010. April;46(4):920–928. PubMed PMID: WOS:000276009400006; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhong ZDA, Sun WH, Chen HY, et al. Optimizing tamoxifen-inducible Cre/Ioxp system to reduce tamoxifen effect on bone turnover in long bones of young mice. Bone. 2015. December;81:614–619. PubMed PMID: WOS:000365372800072; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Elefteriou F, Yang X. Genetic mouse models for bone studies–strengths and limitations. Bone. 2011. December;49(6):1242–1254. PubMed PMID: 21907838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nagao M, Cheong CW, Olsen BR. Col2-Cre and tamoxifen-inducible Col2-CreER target different cell populations in the knee joint. Osteoarthritis Cartilage. 2016. January;24(1):188–191. PubMed PMID: 26256767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kim JE, Nakashima K, De Crombrugghe B. Transgenic mice expressing a ligand-inducible cre recombinase in osteoblasts and odontoblasts: a new tool to examine physiology and disease of postnatal bone and tooth. Am J Pathol. 2004. December;165(6):1875–1882. PubMed PMID: 15579432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annu Rev Pathol. 2008;3:457–484. PubMed PMID: 18039135. [DOI] [PubMed] [Google Scholar]
- [37].Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002. November;110(10):1383–1388. PubMed PMID: 12438433; PubMed Central PMCID: PMC151821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Deretic V, Jiang S, Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012. August;22(8):397–406. PubMed PMID: 22677446; PubMed Central PMCID: PMC3408825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Harris J, Hartman M, Roche C, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011. March 18;286(11):9587–9597. PubMed PMID: 21228274; PubMed Central PMCID: PMC3058966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012. January 29;13(3):255–263. PubMed PMID: 22286270; PubMed Central PMCID: PMC4116819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008. November 13;456(7219):264–268. PubMed PMID: 18849965. [DOI] [PubMed] [Google Scholar]
- [42].Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010. March 19;140(6):900–917. PubMed PMID: 20303879; PubMed Central PMCID: PMC2887297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006. August 25;313(5790):1137–1140. PubMed PMID: 16931765; PubMed Central PMCID: PMC4741373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cinque L, Forrester A, Bartolomeo R, et al. FGF signalling regulates bone growth through autophagy. Nature. 2015. December 10;528(7581):272 PubMed PMID: WOS:000366053300043; English. [DOI] [PubMed] [Google Scholar]
- [45].Mirigian LS, Makareeva E, Mertz EL, et al. Osteoblast malfunction caused by cell stress response to procollagen misfolding in alpha 2(i)-g610c mouse model of osteogenesis imperfecta. J Bone Mineral Res. 2016. August;31(8):1608–1616. PubMed PMID: WOS:000383717200017; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Barone MV, Crozat A, Tabaee A, et al. CHOP (GADD153) and its oncogenic variant, TLS-CHOP, have opposing effects on the induction of G1/S arrest. Genes Dev. 1994. February 15;8(4):453–464. PubMed PMID: 8125258. [DOI] [PubMed] [Google Scholar]
- [47].Batchvarova N, Wang XZ, Ron D. Inhibition of adipogenesis by the stress-induced protein CHOP (Gadd153). The EMBO Journal. 1995. October 02;14(19):4654–4661. PubMed PMID: 7588595; PubMed Central PMCID: PMC394562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pereira RC, Delany AM, Canalis E. CCAAT/enhancer binding protein homologous protein (DDIT3) induces osteoblastic cell differentiation. Endocrinology. 2004. April;145(4):1952–1960. PubMed PMID: 14684614. [DOI] [PubMed] [Google Scholar]
- [49].Huang YF, Lin JJ, Lin CH, et al. c-Jun N-terminal kinase 1 negatively regulates osteoblastic differentiation induced by BMP2 via phosphorylation of Runx2 at Ser104. Journal of Bone and Mineral Research: the Official Journal of the American Society for Bone and Mineral Research. 2012. May;27(5):1093–1105. PubMed PMID: 22247071. [DOI] [PubMed] [Google Scholar]
- [50].Tan XH, Weng TJ, Zhang JH, et al. Smad4 is required for maintaining normal murine postnatal bone homeostasis. J Cell Sci. 2007. July 1;120(13):2162–2170. PubMed PMID: WOS:000247732100004; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005. May 9;169(3):425–434. PubMed PMID: WOS:000228995800009; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hu CH, Sui BD, Du FY, et al. miR-21 deficiency inhibits osteoclast function and prevents bone loss in mice. Sci Rep. 2017. February 27;(7):43191 . PubMed PMID: 28240263; PubMed Central PMCID: PMC5327426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dempster DW, Compston JE, Drezner MK, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. Journal of Bone and Mineral Research: the Official Journal of the American Society for Bone and Mineral Research. 2013. January;28(1):2–17. PubMed PMID: 23197339; PubMed Central PMCID: PMC3672237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Li H, Zhou B, Xu L, et al. Circulating PGRN is significantly associated with systemic insulin sensitivity and autophagic activity in metabolic syndrome. Endocrinology. 2014. September;155(9):3493–3507. PubMed PMID: 24971611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.