

ABSTRACT
Viral infection causes many physiological alterations in the host cell, and many of these alterations can affect the host mitochondrial network, including mitophagy induction. A substantial amount of literature has been generated that advances our understanding of the relationship between mitophagy and several viruses. Some viruses trigger mitophagy directly, and indirectly and control the mitophagic process via different strategies. This enables viruses to promote persistent infection and attenuate the innate immune responses. In this review, we discuss the events of virus-regulated mitophagy and the functional relevance of mitophagy in the pathogenesis of viral infection and disease.
Abbreviation: ATG: autophagy related; BCL2L13: BCL2 like 13; BNIP3L/NIX: BCL2 interacting protein 3 like; CL: cardiolipin; CSFV: classical swine fever virus; CVB: coxsackievirus B; DENV: dengue virus; DNM1L: dynamin 1 like; FIS1: fission, mitochondrial 1; FUNDC1: FUN14 domain containing 1; HPIV3: human parainfluenza virus 3; HSV-1: herpes simplex virus type 1; IMM: inner mitochondrial membrane; IAV: influenza A virus; IFN: interferon; IKBKE/IKKϵ: inhibitor of nuclear factor kappa B kinase subunit epsilon; LUBAC: linear ubiquitin assembly complex; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MeV: measles virus; MAVS: mitochondrial antiviral signaling protein; MFF: mitochondria fission factor; NLRP3: NLR family pyrin domain containing 3; NDV: Newcastle disease virus; NR4A1: nuclear receptor subfamily 4 group A member 1; OMM: outer mitochondrial membrane; OPA1: OPA1, mitochondrial dynamin like GTPase; PRKN: parkin RBR E3 ubiquitin protein ligase; PINK1: PTEN induced putative kinase 1; PHB2: prohibitin 2; PRRSV: porcine reproductive and respiratory syndrome virus; PRRs: pattern-recognition receptors; RLRs: RIG-I-like receptors; ROS: reactive oxygen species; RIPK2: receptor interacting serine/threonine kinase 2; SESN2: sestrin 2; SNAP29: synaptosome associated protein 29; STX17: syntaxin 17; TGEV: transmissible gastroenteritis virus; TUFM: Tu translation elongation factor, mitochondrial; TRAF2: TNF receptor associated factor 2; TRIM6: tripartite motif containing 6; Ub: ubiquitin; ULK1: unc-51 like autophagy activating kinase 1; VZV: varicella-zoster virus
KEYWORDS: Disease, innate immunity, mitophagy, pathogenesis, virus
Introduction
As obligate intracellular parasites, viruses strategically regulate host cellular processes and subvert cellular antiviral defenses as part of their life cycle [1,2]. Mitochondria take part in a wide range of cellular processes, including the regulation of the innate immune system, and function as signaling platforms [3]. Consequently, dysfunctional mitochondrial activity leads to decreased ATP generation and increased production of ROS (reactive oxygen species) [4]. Therefore, some viruses can benefit from targeting mitochondria and disrupting the balance of the mitochondrial network to influence the host cells’ metabolism and physiology, which facilitates viral immune evasion [5]. In the past few years, the function of mitophagy during viral infections has been studied with several viruses. Viruses have developed different strategies to subvert and benefit from mitophagy. Some viruses induce intracellular events that trigger mitochondrial dysfunction and promote host PRKN (parkin RBR E3 ubiquitin protein ligase)-dependent or receptor-mediated mitophagy. Some viruses can directly trigger mitophagy via their own viral components. In some instances, viruses can affect every step of the mitophagy cycle to maximize persistent infection and facilitate pathogenesis.
Herein, we provide a brief overview of virus-induced mitophagy (Figure 1) and explore in depth how some viruses manipulate mitophagy to facilitate persistent viral infection and pathogenesis. Clearly, our knowledge of mitophagy during viral infection is in the initial stage; however, this review highlights details that should be clarified in future research.
Figure 1.
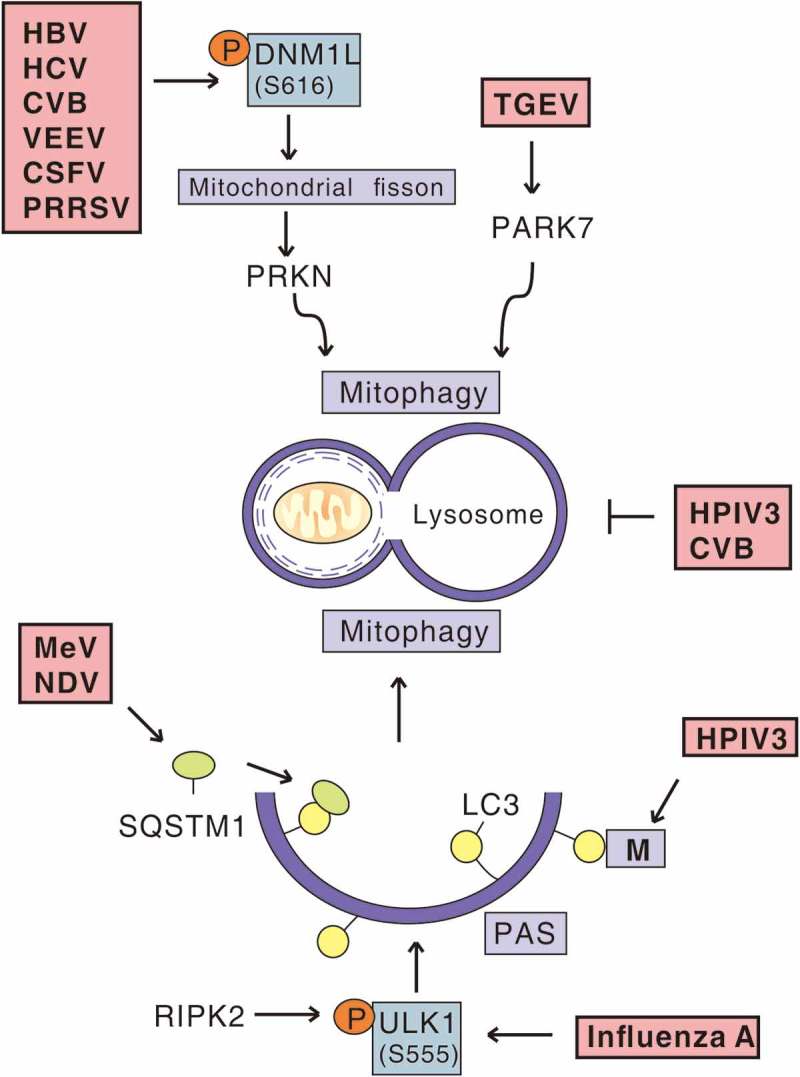
Viral infection and mitophagy. In the context of events of virus-induced mitophagy, HBV, HCV, CVB, VEEV, CSFV and PRRSV trigger DNM1L-mediated mitochondrial fission and subsequent PRKN-dependent mitophagy. TGEV promotes PARK7/DJ-1 upregulation and induces mitophagy. Furthermore, MeV and NDV trigger SQSTM1-mediated mitophagy. M of HPIV3 interacts with LC3 localized to phagophore assembly sites (PAS) to induce mitophagy. RIPK2 phosphorylates the inducer of mitophagy, ULK1, to mediate mitophagy during influenza A virus infection. HPIV3 and CVB induce occurrence of mitophagosomes, but block their fusion with lysosomes.
Overview of macroautophagy and mitophagy
Macroautophagy/autophagy is a highly conserved homeostatic and multi-step process in which cellular components are captured by phagophores, precursors to double-membrane autophagosomes, and shuttled to lysosomes for degradation [6,7]. During the process, autophagosome formation is driven by 2 protein-protein and protein-lipid conjugation systems. The first conjugation system contributes to the coupling of ATG12 (autophagy related 12) to ATG5 (autophagy related 5) to form a covalently linked heterodimer, which then recruits ATG16L1 (autophagy related 16 like 1) [8]. The second conjugation system couples an Atg8-family protein, such as MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) or GABARAP (GABA type A receptor-associated protein), to the phospholipid phosphatidylethanolamine [9]. These two conjugation systems are involved in the formation of the phagophore and the maturation of the autophagosome. Finally, the autophagosome fuses with a lysosome to form an autolysosome for degradation [6]. Many proteins that are required for autophagosome-lysosome fusion, lysosomal acidification and lysosomal digestion coordinately contribute to degradation processes [10]. More details about the mechanism of autophagy can be seen from the review written by Parzych and Klionsky [11].
Autophagy was previously described as a non-selective process induced in starvation conditions, but now it is clear that autophagy can selectively eliminate damaged or dysfunctional organelles, aggregated proteins, or intracelluar pathogens [12]. Organelle-specific autophagy can clear mitochondria, peroxisomes, ribosomes, endoplasmic reticulum, and the nucleus. All these types of organelle-specific autophagy are initiated via a signal that induces downstream events triggering degradation cues for a specific target, and then the target is tagged by molecules as cargo that is sequestered and eliminated by autophagy-related components [12,13].
The selective degradation of mitochondria by autophagy is called mitophagy. Damaged or dysfunctional mitochondria are sequestered within autophagic membranes to form mitophagosomes and shuttled to lysosomes for degradation, which maintains mitochondrial dynamics and cellular homeostasis [14,15]. Mitophagy can be triggered by various stimuli, such as hypoxia, mitochondrial depolarization, and viral infection [2]. The process of mitophagy is initiated by effectors that bring together molecular signals on mitochondria and LC3 on phagophores, thereby allowing the autophagic machinery to come into proximity with the mitochondria [16]. The mechanisms of mitophagy in mammalian cells can be classified into two groups according to their dependence on the E3 ligase PRKN: PRKN-dependent and PRKN-independent mitophagy [14].
PRKN-dependent mitophagy is generally related to the alteration of mitochondrial transmembrane potential, which results in depolarized mitochondria and involves 2 key gene products: PINK1 (PTEN induced putative kinase 1) and PRKN [17] (Figure 2(a)). PRKN mutations have been linked with neurodegenerative diseases, including Parkinson disease, the second-most common neurodegenerative disorder worldwide [18]. In contrast, PRKN-independent mitophagy is generally required for processes that involve mitophagy receptors [14]. Several mitophagy receptors have been identified in mammalian cells, including BCL2L13 (BCL2 like 13) [19], BNIP3L/NIX (BCL2 interacting protein 3 like) [20–22], BNIP3 (BCL2 interacting protein 3) [23], and FUNDC1 (FUN14 domain containing 1) [24]. All these mitophagy receptors anchor to outer mitochondrial membranes and interact with LC3 via the LC3-interacting region motif, which promotes the engulfment of mitochondria by the autophagic system instead of relying on the PRKN-dependent system [25] (Figure 2(b)). Nevertheless, some inner mitochondrial components such as PHB2 (prohibitin 2) and cardiolipin (CL) may also be involved in the process under different conditions [26,27]. Interestingly, mounting evidence indicates crosstalk between PRKN-dependent and PRKN-independent mitophagy effectors. For example, BNIP3L/NIX promotes carbonyl cyanide m-chlorophenyl hydrazine (CCCP)-induced mitochondrial depolarization and contributes to mitochondrial priming by controlling the mitochondrial translocation of PRKN [28].
Figure 2.
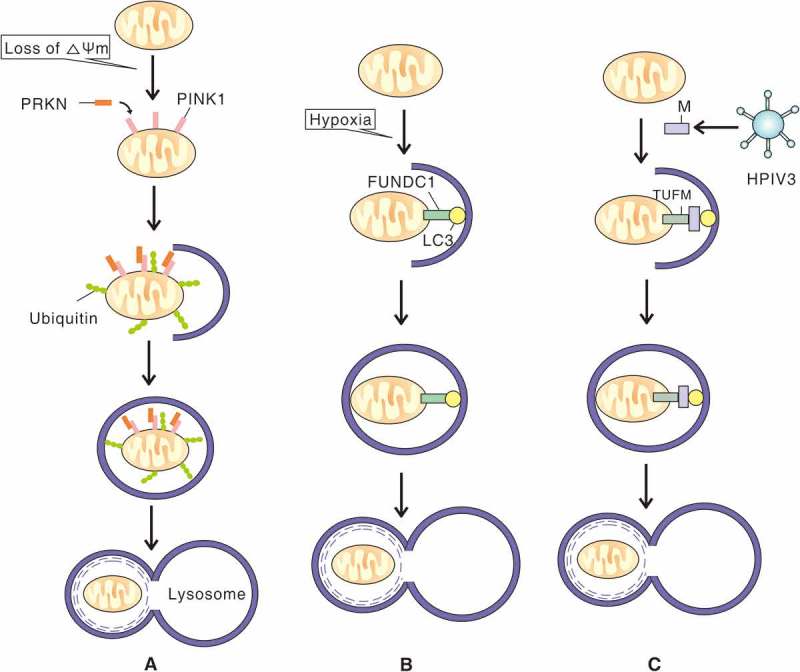
The mechanisms of PRKN-PINK1-, FUNDC1- and M of HPIV3-induced mitophagy. (A) When mitochondria lose membrane potential, PINK1 accumulates on the outer mitochondrial membrane and recruits PRKN to the damaged mitochondria. PRKN conjugates more ubiquitin chains to the mitochondria which promotes engulfment by a phagophore. The subsequently formed autophagosome then fuses with a lysososome. (B) In response to hypoxia, outer mitochondrial membrane protein FUNDC1 binds to LC3 through an LC3 interacting region motif and mediates sequestration of mitochondria into a phagophore. (C) Upon HPIV3 infection, M anchors to mitochondria via TUFM interaction and binds to LC3, which promotes sequestration of mitochondria into a phagophore and subsequent fusion of an autophagosome with a lysosome.
Events of virus-triggered mitophagy
Mitochondrial fission as a precursor for virus-induced mitophagy
Mitochondrial dynamics, involving mitochondrial fission and fusion, sustain mitochondrial homeostasis and are tightly regulated in response to alterations in cellular physiology [29]. Mitochondrial fission can be induced via a collapse of mitochondrial transmembrane potential (∆Ψm) and is modulated by outer mitochondrial membrane proteins, including FIS1 (fission, mitochondrial 1) and MFF (mitochondria fission factor). These proteins coordinate to recruit DNM1L/DRP1 (dynamin 1 like) to mitochondria [30,31]. Mitochondrial fusion involves the MFN1 (mitofusin 1) and MFN2 (mitofusin 2) proteins and the inner mitochondrial membrane protein OPA1 (OPA1, mitochondrial dynamin like GTPase) [32]. The mitochondrial dynamic process can be altered by viral infections, which disrupt cellular homeostasis. In dengue virus (DENV) infection, DENV NS4B (nonstructural protein 4B) can induce the elongation of mitochondria by inactivating DNM1L, which alters mitochondrial morphology to promote infection [33]. Moreover, pseudorabies virus infection disrupts mitochondrial dynamics for viral growth and spread [34].
In the context of virus-induced mitophagy, during the early process of viral infection, some viruses induce the collapse of mitochondrial transmembrane potential, which results in depolarized mitochondria by promoting mitochondrial fission/fragmentation. These viruses may modulate virus-induced mitochondrial fission as a precursor to the host mitophagic process, which can be triggered by PRKN or PRKN-independent mediators for persistent infection. Perhaps the earliest described example of this concept was seen with hepatitis viruses. Hepatitis B virus (HBV) and hepatitis C virus (HCV) stimulate the gene expression of DNM1L and MFF and promote DNM1L recruitment to mitochondria by stimulating the phosphorylation of DNM1L (Ser616), leading to mitochondrial fission. Virus-induced DNM1L-mediated mitochondrial fission is then followed by PRKN-dependent mitophagy [35–37]. These findings implicate mitophagy as a potential therapeutic target against HBV- and HCV-associated liver diseases.
Likewise, in Venezuelan equine encephalitis virus (VEEV)-infected cells, DNM1L, PRKN, and PINK1 are enriched in mitochondrial fractions as compared with uninfected cells, and mitophagy ensues in infected cells [38]. Similar findings were reported for coxsackievirus B (CVB), classical swine fever virus (CSFV), and porcine reproductive and respiratory syndrome virus (PRRSV) [39–41]. One obvious interpretation of these findings is that during viral infection, mitochondrial fission is an early event in the PRKN-dependent or PRKN-independent mitophagic process. The virus-induced mitochondrial fission and subsequent host mitophagy reveal a unique view of coordination between mitochondrial dynamics and mitophagy in the pathogenesis of viral infection, and provide new avenues for the development of antiviral strategies.
Viral protein-mediated mitophagy
Given that some viral proteins can translocate to the mitochondria (eg severe acute respiratory syndrome [SARS]-coronavirus [CoV] open reading frame [ORF]-9b) and interact with LC3 (eg influenza A virus [IAV] M2 protein) during viral infection [42,43], it is reasonable to speculate that apart from utilizing host mitophagic machinery to trigger PRKN-dependent or receptor-mediated mitophagy, some viruses can directly induce mitophagy via their own viral factors. Of note, a recent study revealed that M (matrix protein) of human parainfluenza virus 3 (HPIV3) is sufficient to induce mitophagy by interacting with TUFM (Tu translation elongation factor, mitochondrial) to translocate to the mitochondria and interacting with LC3 to mediate mitophagosome formation [44] (Figure 2(c)). This finding does not exclude the possibility that other mitochondrial proteins may also facilitate the recruitment of M to mitochondria, and unknown cellular proteins may bridge the interaction of M with LC3, thereby recruiting mitochondria to phagophores [44]. These observations provide the first demonstration that a viral protein can act as a mitophagy receptor by bridging phagophores and mitochondria. The identification of other specific viral pathogenic factors that act as mitophagy receptors will be important, and these viral components may represent a novel target for antiviral therapeutics.
Viral infection-induced incomplete mitophagy
The last step of complete mitophagy is the fusion of mitophagosomes with lysosomes to form degradative mitolysosomes [14]. In the absence of this step, mitophagosomes accumulate in the cell, which is termed “incomplete mitophagy” [45]. Mitophagosomes are cytosolic double-membraned vesicles, and some RNA viruses replicate their genomes in association with cytosolic membranes [46,47]. Therefore, these viruses may use mitophagosomes for their own replication. However, if mitophagosomes fuse with lysosomes, the viral contents are degraded. Consequently, some viruses have developed strategies to block the fusion of mitophagosomes and lysosomes and induce incomplete mitophagy. For example, during infection, CVB localizes to mitochondria, induces mitophagy, and subsequently disperses from the cell in a mitophagosome-virus complex. However, the exact mechanism by which CVB-induced mitophagosomes are released from the cell rather than being degraded by lysosomes remains to be delineated [39]. A similar anti-degradative phenomenon has been also observed in HPIV3 infection, and the viral phosphoprotein (P) is necessary and sufficient to inhibit mitophagosome degradation. P binds to SNAP29 (synaptosome associated protein 29) and compromises its interaction with STX17 (syntaxin 17), thereby preventing these 2 host SNAP receptor proteins from mediating mitophagosome-lysosome fusion. The HPIV3-induced incomplete mitophagy increases extracellular viral production but does not affect viral protein synthesis [44,48].
The virus-induced incomplete mitophagy indicates a complicated relationship between viruses and host cell degradation vesicles. Viruses manipulate every step of the mitophagic process for their own use. To date, there is no other published evidence indicating the induction of incomplete mitophagy during viral infection, and further work to learn if other viruses manipulate incomplete mitophagy for replication or to uncover other novel roles of virus-induced mitophagosomes will help our understanding of mitophagy regulation in general.
Virus-induced mitophagy and pathogenesis
Virus-triggered mitophagy inhibits apoptosis for viral replication
Apoptosis is a cell death pathway that is distinct from cell necrosis and can be triggered by a wide range of stimuli [49]. Mitochondrial damage leads to ROS accumulation, the release of CYCS (cytochrome c, somatic), and the collapse of mitochondrial transmembrane potential, all of which are pro-apoptotic stimuli [50]. Apoptosis can confer advantages to virally infected hosts by preventing cell-to-cell spread, which would otherwise result in the death of infected host cells and allow viral infection to continue. Therefore, the suppression of host cell apoptosis may be beneficial to the virus [51]. For example, varicella-zoster virus (VZV) ORF 12 protein inhibits apoptosis and promotes viral spread from cell to cell, thereby favoring the virus instead of the host [52]. Similarly, herpes simplex virus type 1 (HSV-1) blocks apoptosis by precluding mitochondrial CYCS release in a caspase-independent manner, which increases the likelihood of diseases in the host cell [53].
The functional relationship between apoptosis and autophagy is complex. Generally, autophagy can block the induction of apoptosis, and apoptosis-associated caspase activation inhibits the autophagic process [54,55]. In the context of viral infection, several studies have suggested that viruses trigger mitophagy to prevent apoptosis, thus facilitating the propagation and pathogenesis of viral infection and disease (Figure 3). This mechanism of preventing apoptosis may be the most widely studied functional aspect of mitophagy during viral infection. This concept was most notably described in measles virus (MeV). During measles virus vaccine strain Edmonston B (MV-Edm) infection, MV-Edm induces mitophagy, leading to decreased CYCS release, which blocks the pro-apoptotic cascade in non-small cell lung cancer cells. The inhibition of apoptosis by mitophagy favors viral replication and promotes the pathogenesis of viral infection [56]. Similar findings have been reported with HBV, HCV, VEEV, CSFV, PRRSV, Newcastle disease virus (NDV), and transmissible gastroenteritis virus (TGEV) [35,37,38,40,41,57,58]. These examples share 2 concurrent phenomena: (1) upon infection, viruses induce mitophagy via different mechanisms, and (2) subsequent mitophagy mitigates apoptosis to promote viral infection.
Figure 3.
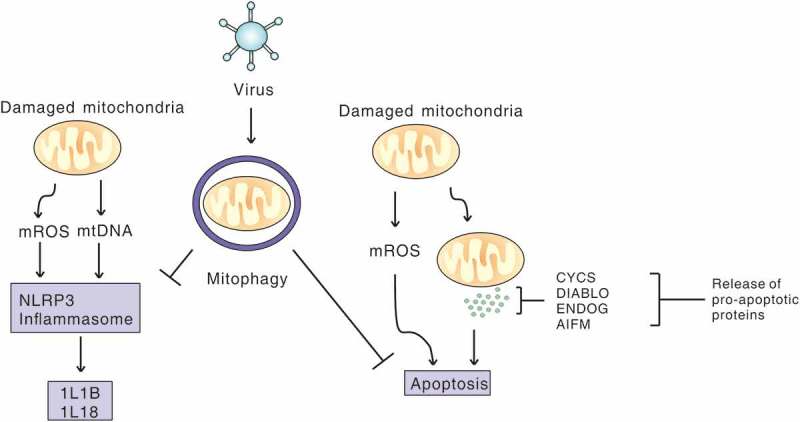
Viral infection induces mitophagy to inhibit apoptosis and NLRP3 inflammation. Virus-induced mitophagy limits NLRP3 inflammation and apoptosis by removing damaged mitochondria that release mitochondrial ROS (mROS) and mitochondrial DNA (mtDNA), and pro-apoptosis proteins, to promote viral propagation.
Additional research is needed to determine whether apoptosis can counteract mitophagy during viral infection and to precisely characterize the interplay among mitophagy, apoptosis, and viral infection.
Viruses induce mitophagy to mitigate type i interferon responses
Following infection, invading microorganisms are sensed by pattern-recognition receptors (PRRs) of the innate immune system in the host cells [59]. PRRs that bind conserved pathogen-associated molecular patterns (PAMPs) trigger immune signaling pathways. PRRs can be categorized into 4 groups: Toll-like receptors (TLRs), nucleotide oligomerization domain-like receptors (NLRs), C-type lectin receptors (CLRs), and RIG-I-like receptors (RLRs) [5].
Upon viral infection, many viral PAMPs are sensed by RLRs, which then activate MAVS (mitochondria antiviral signaling protein) located on the outer mitochondrial membrane and trigger IFN (interferon) responses. Mitophagy plays critical roles in regulating mitochondrial quality. Consequently, in the absence of mitophagy, mitochondria accumulate, and subsequent mitochondrial fusion within the cells can increase MAVS levels to amplify RLR signaling [60] (Figure 4). Accordingly, viruses have developed several strategies to disable MAVS signaling and interrupt RLR signaling. SARS-CoV ORF-9b localizes to mitochondria and targets the MAVS signalosome by usurping PCBP2 (poly[rC] binding protein 2) and ITCH/AIP4 (itchy E3 ubiquitin protein ligase) for degradation of MAVS [42]. In CVB3-infected cells, MAVS is cleaved by CVB3 2Apro independently of caspase or proteasome activities, and this cleavage inhibits RLR signaling [61].
Figure 4.
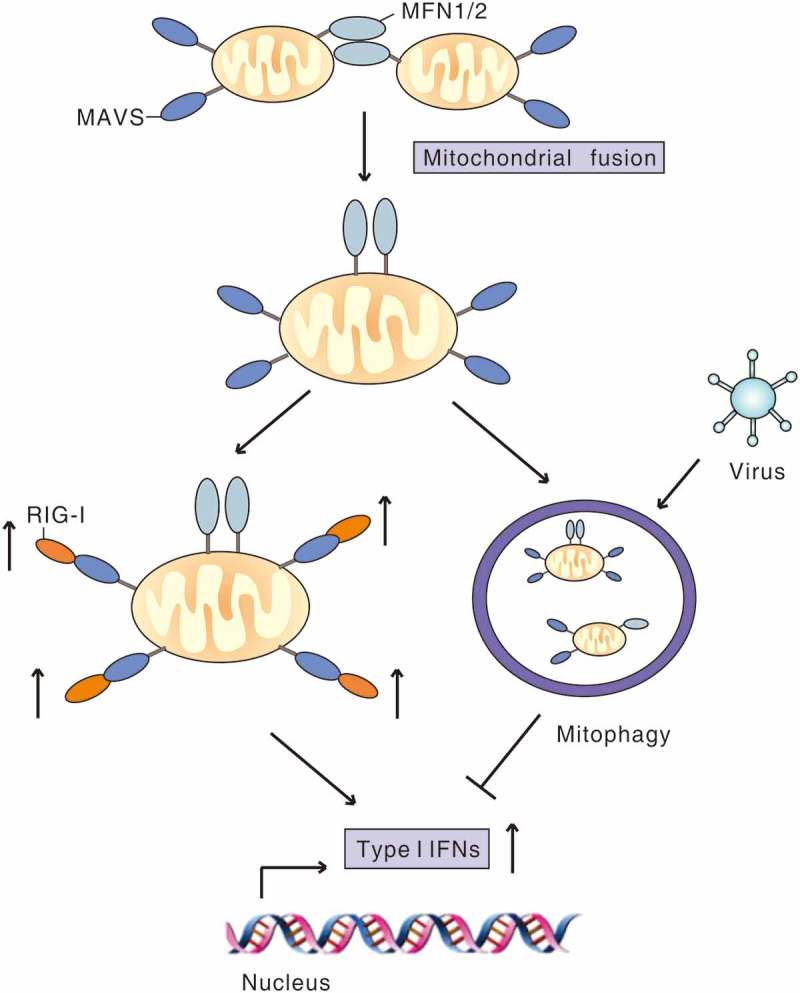
Virus-induced mitophagy attenuates RLR signaling. MFN1 and MFN2 promote fusion of mitochondria, which increases MAVS interaction with downstream signaling molecules to amplify RLR signaling and the type I IFN response. During infection, virus induces mitophagy to mitigate RLR signaling by degradation of MAVS or other strategies to favor viral replication.
Complete mitophagy can lead to the degradation of innate immune signaling proteins such as MAVS. Some viruses that induce mitophagy can benefit from decreased MAVS levels or disrupted MAVS signalosome that blocks its downstream signaling for inhibiting a type I IFN response. Evidence of mitophagy-induced degradation of innate immune signaling proteins stems largely from a study done with oncolytic MeV. Infection with MeV of the Edmonston strain causes mitochondrial dysfunction and induces SQSTM1 (sequestosome 1)-mediated mitophagy to degrade MAVS, thus attenuating RLR signaling to promote viral replication [62]. Silencing SQSTM1 expression inhibits mitophagy and restores MAVS expression [62]. This strategy is simple and effective, but the more detailed mechanism remains to be elucidated. Furthermore, HBV induces PRKN-dependent mitophagy, and the virus-induced PRKN can recruit the linear ubiquitin assembly complex (LUBAC) to mitochondria and disrupt the MAVS signalosome to abrogate IFNB1/IFN-β synthesis [63]. A similar phenomenon has also emerged with HPIV3 infection. The M protein of HPIV3 can mitigate RLR signaling, thereby inhibiting a mitophagy-dependent type I IFN response [44]. Moreover, instead of triggering mitophagy, the M protein of Nipah virus can target TRIM6 (tripartite motif containing 6) to inhibit the IKBKE/IKKϵ (inhibitor of nuclear factor kappa B kinase subunit epsilon)-mediated IFN response [64]. Apart from promoting viral assembly and budding during the viral life cycle, matrix proteins of some RNA viruses may negatively regulate antiviral immunity via different mechanisms, but additional evidence is needed to confirm this hypothesis. Future studies should focus on diverse viral proteins encoded by numerous viruses that can negatively regulate antiviral innate immunity, and explore additional viruses that can mitigate RLR signaling via mitophagy, as well as determine the precise mechanisms involved in this process.
Viruses subvert mitophagy to attenuate inflammation activation
Damaged mitochondria can release damage-associated molecular patterns that contribute to the activation of the NLRP3 (NLR family pyrin domain containing 3) inflammasome, which can trigger the secretion of inflammatory cytokines that are needed for innate immunity and the proper activation of the adaptive immune response [65,66]. The NLRP3 inflammasome consists of pro-CASP1 (caspase 1), the adaptor protein PYCARD/ASC (PYD and CARD domain containing), and the sensor protein NLRP3 [67].
The NLRP3 inflammasome can be activated by a number of dangerous stimuli, including damaged mitochondria releasing mitochondrial ROS, and damaged mitochondrial DNA [68–70]. Mitophagy can remove damaged mitochondria, so this process is important in regulating NLRP3 inflammasome activation [71]. A recent study revealed an anti-inflammatory mechanism of mitophagy in which celastrol promotes NR4A1 (nuclear receptor subfamily 4 group A member 1) translocation from the nucleus to mitochondria, where it is ubiquitinated by TRAF2 (TNF receptor associated factor 2). Ubiquitinated NR4A1 then interacts with SQSTM1, leading to mitophagy and the alleviation of inflammation [72]. Consistent with this finding, SESN2 (sestrin 2), a stress-inducible protein, suppresses prolonged NLRP3 inflammasome activation by clearing damaged mitochondria via the induction of mitophagy in macrophages [73]. These results indicate that mitophagy is one of the self-limiting systems that can inhibit inflammation (Figure 3).
Many reports have shown that viral infection can trigger an NLRP3 inflammasome-initiated inflammatory response [74]; conversely, some viral proteins can also inhibit NLRP3 inflammasome activation to help viruses escape host antiviral immune defenses. Typically, MeV infection in THP-1 cells triggers the release of IL1B (interleukin 1 beta) via the NLRP3 inflammasome, but the MeV non-structured V protein can reduce IL1B production. Furthermore, IL1B production in V protein-lacking mutant MeV-infected THP-1 cells is higher than that in wild-type MeV-infected cells [75].
Given that mitophagy acts as a brake on inflammasome signaling by removing dysfunctional mitochondria, and the attenuated inflammatory response may help viruses escape host antiviral immune defenses, it is reasonable to deduce that viruses may attenuate NLRP3 inflammasome activation via mitophagy, which parallels the notion that viruses mitigate RLR signaling via mitophagy. During IAV infection, IAV triggers RIPK2 (receptor interacting serine/threonine kinase 2)-mediated mitophagy in a kinase-dependent manner by phosphorylating the mitophagy inducer ULK1 (unc-51 like autophagy activating kinase 1). The virus-induced mitophagy can suppress NLRP3 inflammasome activation, consistent with a defect in mitochondrial quality control. ripk2−/− bone marrow-derived dendritic cells exhibit an accumulation of dysfunctional ROS-producing mitochondria [76]. The mechanism revealed in this study can likely be extrapolated to other viruses; however, to date, little other published evidence has linked virus-induced mitophagy and inflammasome signaling. To characterize the mechanism and function of virus-induced mitophagy and inflammasome signaling accurately, more in vitro and in vivo studies are required.
Conclusions and perspectives
Several recent studies have expanded the scope of the role of mitophagy in viral infection, from biogenesis to involvement in mitochondrial signaling. Virus-induced mitophagy disturbs mitochondrial dynamics, impedes apoptosis, and modulates innate immune signaling pathways by attenuating RLR signaling and inflammasome activation for persistent infection and disease pathogenesis. Despite many efforts in the past several years to understand the implication of viral infection in the regulation of mitophagy, many questions remain unanswered. Future research should reveal additional ways that viruses initiate and regulate mitophagy, especially the crosstalk between viral factors and host mitophagy-specific proteins. In addition, a recent study demonstrated that BNIP3- and BNIP3L-mediated mitophagy promotes the survival of virus-specific natural killer cells during the contraction phase to promote memory [77], so exploring virus-induced mitophagy at an adaptive immunity level at such an early stage in this research area could be exciting. Thus, in-depth characterizations of the relationship between mitophagy and viral infection and its relevance to pathogenesis may increase our understanding of pathogenic processes. In short, a better grasp of mitophagy regulation in viral infection and the intrinsic molecular mechanisms of this process will provide new avenues for the development of therapeutic strategies to combat viral infections and associated diseases.
Funding Statement
This work was supported by the National Natural Science Foundation of China (NSFC) [81471939]; The natural science foundation of hubei province innovation group [2017CFA022]; National Natural Science Foundation of China (NSFC) [31630086].
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (grant 31630086 and 81471939) and the Natural Science Foundation of Hubei Province Innovation Group (2017CFA022).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Deretic V, Levine B.. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009. June 18;5(6):527–549. PubMed PMID: 19527881; PubMed Central PMCID: PMC2720763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Khan M, Syed GH, Kim SJ, et al. Mitochondrial dynamics and viral infections: A close nexus. Biochim Biophys Acta. 2015. October;1853(10 Pt B):2822–2833. PubMed PMID: 25595529; PubMed Central PMCID: PMC4500740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].West AP, Shadel GS, Ghosh S.. Mitochondria in innate immune responses. Nat Rev Immunol. 2011. June;11(6):389–402. PubMed PMID: 21597473; PubMed Central PMCID: PMC4281487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nickel A, Kohlhaas M, Maack C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol. 2014;73:26–33. [DOI] [PubMed] [Google Scholar]
- [5].Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006. February 24;124(4):783–801. PubMed PMID: 16497588. [DOI] [PubMed] [Google Scholar]
- [6].Tanida Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal. 2011. June;14(11):2201–2214. [DOI] [PubMed] [Google Scholar]
- [7].Shibutani ST, Saitoh T, Nowag H, et al. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. 2015. October;16(10):1014–1024. PubMed PMID: 26382870. [DOI] [PubMed] [Google Scholar]
- [8].Hanada T, Noda NN, Satomi Y, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007. December 28;282(52):37298–37302. PubMed PMID: 17986448. [DOI] [PubMed] [Google Scholar]
- [9].Fujita N, Itoh T, Omori H, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008. May;19(5):2092–2100. PubMed PMID: 18321988; PubMed Central PMCID: PMC2366860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Reggiori F, Ungermann C. Autophagosome maturation and fusion. J Mol Biol. 2017. February 17;429(4):486–496. PubMed PMID: 28077293. [DOI] [PubMed] [Google Scholar]
- [11].Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014. January 20;20(3):460–473. PubMed PMID: 23725295; PubMed Central PMCID: PMC3894687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014. June;16(6):495–501. PubMed PMID: 24875736. [DOI] [PubMed] [Google Scholar]
- [13].Anding AL, Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell. 2017. April 10;41(1):10–22. PubMed PMID: 28399394; PubMed Central PMCID: PMC5395098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011. January;12(1):9–14. PubMed PMID: 21179058; PubMed Central PMCID: PMC4780047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007. June 15;462(2):245–253. PubMed PMID: 17475204; PubMed Central PMCID: PMC2756107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2014;7(3):279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kondapalli C, Kazlauskaite A, Zhang N, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012. May;2(5):120080 PubMed PMID: 22724072; PubMed Central PMCID: PMC3376738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lu H, Li G, Liu L, et al. Regulation and function of mitophagy in development and cancer. Autophagy. 2013. November 1;9(11):1720–1736. PubMed PMID: 24091872. [DOI] [PubMed] [Google Scholar]
- [19].Otsu K, Murakawa T, Yamaguchi O. BCL2L13 is a mammalian homolog of the yeast mitophagy receptor Atg32. Autophagy. 2015;11(10):1932–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008. July 10;454(7201):232–235. PubMed PMID: 18454133; PubMed Central PMCID: PMC2570948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007. December 4;104(49):19500–19505. PubMed PMID: 18048346; PubMed Central PMCID: PMC2148318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009. July;16(7):939–946. PubMed PMID: 19229244; PubMed Central PMCID: PMC2768230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hanna RA, Quinsay MN, Orogo AM, et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012. June 1;287(23):19094–19104. PubMed PMID: 22505714; PubMed Central PMCID: PMC3365942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012. January 22;14(2):177–185. PubMed PMID: 22267086. [DOI] [PubMed] [Google Scholar]
- [25].Birgisdottir AB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci. 2013. August 1;126(Pt 15):3237–3247. . PubMed PMID: 23908376. [DOI] [PubMed] [Google Scholar]
- [26].Wei Y, Chiang WC, Sumpter R Jr. et al. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017. January 12;168(1–2):224–238 e10. PubMed PMID: 28017329; PubMed Central PMCID: PMC5235968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013. October;15(10):1197–1205. PubMed PMID: 24036476; PubMed Central PMCID: PMC3806088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ding WX, Ni HM, Li M, et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-SQSTM1-mediated mitochondrial priming. J Biol Chem. 2010. September 3;285(36):27879–27890. PubMed PMID: 20573959; PubMed Central PMCID: PMC2934655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liesa M, Palacín M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009. July;89(3):799–845. [DOI] [PubMed] [Google Scholar]
- [30].Loson OC, Song Z, Chen H, et al. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013. March;24(5):659–667. PubMed PMID: 23283981; PubMed Central PMCID: PMC3583668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013. May;1833(5):1256–1268. . PubMed PMID: 23434681. [DOI] [PubMed] [Google Scholar]
- [32].Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012. August 31;337(6098):1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chatel-Chaix L, Cortese M, Romero-Brey I, et al. Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe. 2016. September 14;20(3):342–356. PubMed PMID: 27545046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kramer T, Enquist LW. Alphaherpesvirus infection disrupts mitochondrial transport in neurons. Cell Host Microbe. 2012. May 17;11(5):504–514. PubMed PMID: 22607803; PubMed Central PMCID: PMC3358700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kim SJ, Khan M, Quan J, et al. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013;9(12):e1003722 PubMed PMID: 24339771; PubMed Central PMCID: PMC3855539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kim SJ, Syed GH, Siddiqui A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013. March;9(3):e1003285 PubMed PMID: 23555273; PubMed Central PMCID: PMC3610669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kim SJ, Syed GH, Khan M, et al. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci U S A. 2014. April 29;111(17):6413–6418. PubMed PMID: 24733894; PubMed Central PMCID: PMC4035934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Keck F, Brooks-Faulconer T, Lark T, et al. Altered mitochondrial dynamics as a consequence of Venezuelan Equine encephalitis virus infection. Virulence. 2017. November 17;8(8):1849–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sin J, McIntyre L, Stotland A, et al. Coxsackievirus B escapes the infected cell in ejected mitophagosomes. J Virol. 2017. December 15;91(24). PubMed PMID: 28978702; PubMed Central PMCID: PMC5709598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gou H, Zhao M, Xu H, et al. CSFV induced mitochondrial fission and mitophagy to inhibit apoptosis. Oncotarget. 2017;8(24):39382–39400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li S, Wang J, Zhou A, et al. Porcine reproductive and respiratory syndrome virus triggers mitochondrial fission and mitophagy to attenuate apoptosis. Oncotarget. 2016;7(35):56002–56012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shi CS, Qi HY, Boularan C, et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunology. 2014. September 15;193(6):3080–3089. PubMed PMID: 25135833; PubMed Central PMCID: PMC4179872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Beale R, Wise H, Stuart A, et al. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe. 2014. February 12;15(2):239–247. PubMed PMID: 24528869; PubMed Central PMCID: PMC3991421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ding B, Zhang L, Li Z, et al. The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe. 2017. April 12;21(4):538–547 e4. PubMed PMID: 28407488. [DOI] [PubMed] [Google Scholar]
- [45].Richards AL, Jackson WT. How positive-strand RNA viruses benefit from autophagosome maturation. J Virol. 2013. September;87(18):9966–9972. . PubMed PMID: 23760248; PubMed Central PMCID: PMC3754026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013. January;20(1):31–42. PubMed PMID: 22743996; PubMed Central PMCID: PMC3524633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mackenzie J. Wrapping things up about virus RNA replication. Traffic. 2005. November;6(11):967–977. PubMed PMID: 16190978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ding B, Zhang G, Yang X, et al. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe. 2014. May 14;15(5):564–577. PubMed PMID: 24832451. [DOI] [PubMed] [Google Scholar]
- [49].Marino G, Niso-Santano M, Baehrecke EH, et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014. February;15(2):81–94. PubMed PMID: 24401948; PubMed Central PMCID: PMC3970201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sinha K, Das J, Pal PB, et al. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013. July;87(7):1157–1180. [DOI] [PubMed] [Google Scholar]
- [51].Kennedy PG. Viruses, apoptosis, and neuroinflammation–a double-edged sword. J Neurovirol. 2015. February;21(1):1–7. PubMed PMID: 25604493. [DOI] [PubMed] [Google Scholar]
- [52].Liu X, Li Q, Dowdell K, et al. Varicella-Zoster virus ORF12 protein triggers phosphorylation of ERK1/2 and inhibits apoptosis. J Virol. 2012. March;86(6):3143–3151. PubMed PMID: 22238304; PubMed Central PMCID: PMC3302337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Aubert M, Pomeranz LE, Blaho JA. Herpes simplex virus blocks apoptosis by precluding mitochondrial cytochrome c release independent of caspase activation in infected human epithelial cells. Apoptosis. 2007. January;12(1):19–35. . PubMed PMID: 17080326; PubMed Central PMCID: PMC2799008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Neumann S, El Maadidi S, Faletti L, et al. How do viruses control mitochondria-mediated apoptosis? Virus Res. 2015. November 2;209:45–55. PubMed PMID: 25736565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Biol. 2007;8(9):741–752. [DOI] [PubMed] [Google Scholar]
- [56].Xia M, Meng G, Jiang A, et al. Mitophagy switches cell death from apoptosis to necrosis in NSCLC cells treated with oncolytic measles virus. Oncotarget. 2014;5(11):3907–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Meng G, Xia M, Wang D, et al. Mitophagy promotes replication of oncolytic Newcastle disease virus by blocking intrinsic apoptosis in lung cancer cells. Oncotarget. 2014;5(15):6365–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhu L, Mou C, Yang X, et al. Mitophagy in TGEV infection counteracts oxidative stress and apoptosis. Oncotarget. 2016;7(19):27122–27141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu Y, Olagnier D, Lin R. Host and viral modulation of RIG-I-Mediated antiviral immunity. Front Immunol. 2016;7:662 PubMed PMID: 28096803; PubMed Central PMCID: PMC5206486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tal MC, Sasai M, Lee HK, et al. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009. February 24;106(8):2770–2775. PubMed PMID: 19196953; PubMed Central PMCID: PMC2650341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Feng Q, Langereis MA, Lork M, et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J Virol. 2014. March;88(6):3369–3378. PubMed PMID: 24390337; PubMed Central PMCID: PMC3957915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Xia M, Gonzalez P, Li C, et al. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J Virol. 2014. May;88(9):5152–5164. PubMed PMID: 24574393; PubMed Central PMCID: PMC3993837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Khan M, Syed GH, Kim SJ, et al. Hepatitis B virus-induced parkin-dependent recruitment of Linear Ubiquitin Assembly Complex (LUBAC) to mitochondria and attenuation of innate immunity. PLoS Pathog. 2016. June;12(6):e1005693 PubMed PMID: 27348524; PubMed Central PMCID: PMC4922663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bharaj P, Wang YE, Dawes BE, et al. The matrix protein of nipah virus targets the E3-ubiquitin Ligase TRIM6 to inhibit the IKKepsilon kinase-mediated Type-I IFN antiviral response. PLoS Pathog. 2016. September;12(9):e1005880 PubMed PMID: 27622505; PubMed Central PMCID: PMC5021333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kuballa P, Nolte WM, Castoreno AB, et al. Autophagy and the immune system. Annu Rev Immunol. 2012;30:611–646. PubMed PMID: 22449030. [DOI] [PubMed] [Google Scholar]
- [66].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Reviews Immunol. 2013. October;13(10):722–737. PubMed PMID: 24064518; PubMed Central PMCID: PMC5340150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Schroder K, Tschopp J. The inflammasomes. Cell. 2010. March 19;140(6):821–832. PubMed PMID: 20303873. [DOI] [PubMed] [Google Scholar]
- [68].Yu JW, Lee MS. Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch Pharm Res. 2016. November;39(11):1503–1518. 10.1007/s12272-016-0827-4. PubMed PMID: 27600432. [DOI] [PubMed] [Google Scholar]
- [69].Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011. January 13;469(7329):221–225. PubMed PMID: 21124315. [DOI] [PubMed] [Google Scholar]
- [70].Gurung P, Lukens JR, Kanneganti TD. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. 2015. March;21(3):193–201. PubMed PMID: 25500014; PubMed Central PMCID: PMC4352396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kim M-J, Yoon J-H, Ryu J-H. Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Reports. 2016;49(10):529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Hu M, Luo Q, Alitongbieke G, et al. Celastrol-Induced Nur77 Interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol Cell. 2017. April 6;66(1):141–153 e6. PubMed PMID: 28388439; PubMed Central PMCID: PMC5761061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kim MJ, Bae SH, Ryu JC, et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy. 2016. August 2;12(8):1272–1291. PubMed PMID: 27337507; PubMed Central PMCID: PMC4968237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Chen IY, Ichinohe T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015. January;23(1):55–63. PubMed PMID: 25456015. [DOI] [PubMed] [Google Scholar]
- [75].Komune N, Ichinohe T, Ito M, et al. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1beta secretion. J Virol. 2011. December;85(24):13019–13026. PubMed PMID: 21994456; PubMed Central PMCID: PMC3233149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lupfer C, Thomas PG, Anand PK, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013. May;14(5):480–488. PubMed PMID: 23525089; PubMed Central PMCID: PMC3631456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].O’Sullivan TE, Johnson LR, Kang HH, et al. BNIP3- and BNIP3L-Mediated mitophagy promotes the generation of natural killer cell memory. Immunity. 2015. August 18;43(2):331–342. PubMed PMID: 26253785; PubMed Central PMCID: PMC5737626. [DOI] [PMC free article] [PubMed] [Google Scholar]