

Abstract
Unlike normal B cells, and similar to fat cells, chronic lymphocytic leukemia (CLL) cells aberrantly express lipoprotein lipase (LPL), which contributes to free fatty acids (FFAs) metabolism. Here we show that, in CLL cells, the B cell receptor (BCR) inhibitor ibrutinib reduced LPL mRNA and protein levels and inhibited FFA metabolism in vitro. Likewise, in CLL cells from ibrutinib-treated patients, FFA metabolism was reduced and eventually stopped. Because ibrutinib disrupts CLL cells’ ability to use FFAs for energy production, and because various BCR-dependent cellular functions rely on a continuous supply of chemical energy, ibrutinib interrupts several pathways imperative for cellular function in CLL cells.
Keywords: CLL, Ibrutinib, metabolism, lipoprotein lipase
Introduction
Chronic lymphocytic leukemia (CLL) is characterized by the gradual accumulation of B lymphocytes that appear mature [1] and that aberrantly express cell surface proteins not present in normal B lymphocytes. One such protein, lipoprotein lipase (LPL), commonly detected in adipocytes and myocytes [2–4], is essential for CLL cells’ metabolism of free fatty acids (FFAs) [5,6] and provides CLL cells with energy and a survival advantage [5]. Because activation of the B cell receptor (BCR) signaling pathway increases LPL levels [7,8], and Ibrutinib, an inhibitor of the BCR signaling, is highly effective in patients with CLL, including those with high-risk or relapsed/refractory disease [9,10], we sought to determine whether Ibrutinib inhibits FFA metabolism in CLL cells.
Materials and Methods
Cell fractionation
After obtaining approval from The University of Texas MD Anderson Cancer Center’s Intitutional Review Board, we obtained peripheral blood (PB) cells from 18 patients with CLL treated at the Leukemia Clinic. These cells were fractionated using Ficoll-Hypaque 1077 (Sigma-Aldrich, St. Louis, MO). At first sampling, more than 90% of the peripheral blood fractionated lymphocytes obtained from these patients were CD19+/CD5+, as assessed by flow cytometry.
Measurement of cellular O2 consumption
Because fatty acid metabolism increases O2 consumption, we assessed the cells’ palmitic acid and oleic acid utilization by measuring dissolved O2 (dO2) levels as previously described [6]. This assay was performed only if at least 70% of the fractionated cells co-expressed CD5 and CD19. In each experiment, we used CLL cells at a concentration of 2 to 3 × 106 cells/mL. The cells were incubated at 37°C for 48 hours in minimum essential medium with Hank’s salts and L-glutamine (Life-Technologies, Carlsbad, CA). Cells were cultured in tightly sealed T25 tissue culture flasks (Corning, Tewksbury, MA) in the presence or absence of 80 mM palmitic acid, 2 mM oleic acid, or 80 mM palmitic acid and 80 mM oleic acid, with or without 0.5 or 1.0 µM of ibrutinib. The dO2 levels were measured in all flasks at 48 h of incubation. Measurements of dO2 were repeated at least 3 times for every data point using a SevenGo pro dissolved oxygen meter (Mettler Toledo, Columbus, OH). Results were analyzed by a one-way repeated-measure ANOVA using the GraphPad version 7 (San Diego, California) and SPSS (version 22, SPSS Inc., Chicago, IL).
Viability assay
To determine cell viability of CLL cells that were incubated for 48 h we used the trypan blue exclusion assay. Briefly, after cell density was assessed by using a hemocytometer, 0.1 ml of trypan blue (0.4% in 0.81% sodium chloride and 0.06% potassium phosphate dibasic solution; Gibco, Grand Island, NY) to 0.1 ml of cultured CLL cells. A total of 200 cells of each sample were counted and the percentage of non-viable (blue) and viable cells was calculated.
Western blot analys
Western blot analysis was performed as previously described [11]. Briefly, a lysates of CLL cell extract was mixed with 4x Laemmli sample buffer and denatured by boiling for 5 minutes. Forty micrograms of lysates were dissolved and separated using 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred to a nitrocellulose membrane. After blocking, the membrane was incubated with the following primary antibodies: monoclonal mouse anti-human STAT3 1 (BD Biosciences, San Jose, CA), monoclonal mouse anti-human LPL (Abcam, Cambridge, MA) and mouse anti-human β-actin (Sigma-Aldrich, St. Louis, MO).
After incubation with horseradish peroxidase–conjugated secondary antibodies (GE Healthcare, Buckinghamshire, UK) for 1 hour, blots were visualized with an enhanced chemiluminescence detection system (GE Healthcare).
Densitometry analysis was performed using an Epson Expression 1680 scanner (Epson America, Inc., Long Beach, CA). Densitometry values were normalized by dividing the numerical value of each sample signal by the density of the corresponding β actin protein, used as a loading control.
RNA purification and quantitative reverse transcription–polymerase chain reaction
RNA was isolated using an RNeasy purification procedure (QIAGEN Inc., Valencia, CA). RNA quality and concentration were analyzed with a NanoDrop spectrophotometer (NanoDrop Products, Wilmington, DE), and 500 ng of total RNA were used for one-step quantitative reverse transcription–polymerase chain reaction procedures that were performed according to the manufacturer’s instructions using TaqMan gene expression assays for BCL2, Cyclin D1, MCL1, c-Myc, p21, RELA, STAT3 and LPL (Applied Biosystems, Grand Island, NY).
Results
Ibrutinib disrupts FFA metabolism in vitro
To study the effect of ibrutinib on lipid metabolism, we obtained PB CLL cells from three ibrutinib-naïve CLL patients, cultured the cells in sealed flasks in the presence or absence of palmitic acid and measured the dO2 concentration after 48 hours. As previously reported, dO2 levels decreased when palmitic acid was added to cultured CLL cells [5], suggesting that the CLL cells consumed more oxygen because they metabolized FFA. However, when ibrutinib was added to the cell cultures, the delta dO2 values (the difference between dO2 with FFA and dO2 without FFA) were unchanged, suggesting that ibrutinib blocked FFA metabolism (Figure 1).
Fig 1.
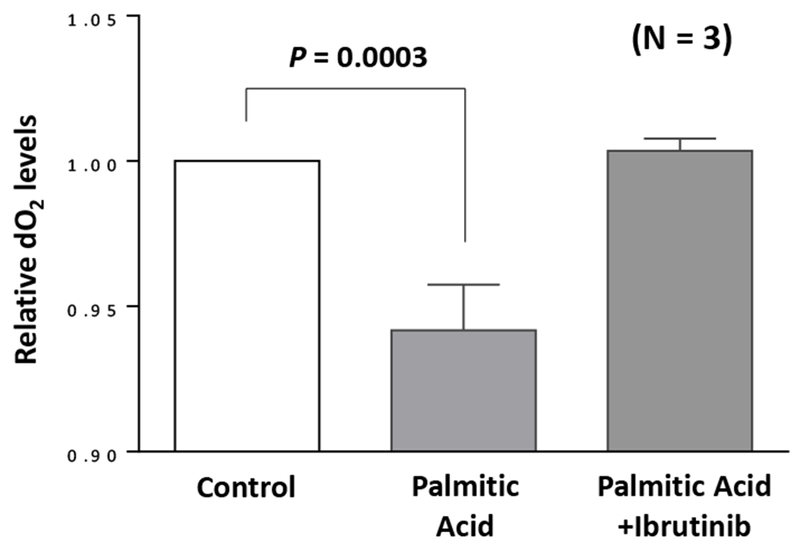
Ibrutinib inhibits palmitic acid-dependent metabolism of chronic lymphocytic leukemia (CLL) cells in vitro. CLL cells were incubated for 48 hours in sealed tissue culture flasks in the presence or absence of 80 mM palmitic acid with or without 1.0 µM ibrutinib. The dissolved oxygen concentration (dO2) was initially set at 1 (left bar), and the relative dO2 after 48 hours is depicted in the middle and right bars.
Ibrutinib disrupts FFA metabolism in vivo
To determine whether ibrutinib impairs CLL cells’ lipid metabolism, we obtained 2 to 5 (median: 4) samples of PB CLL cells from each of 14 CLL patients prior to and during ibrutinib treatment (median number of treatment days = 140; range, 9 – 165 days). Most patients (N=12, 80%) had received at least one previous treatment with ibrutinib (median, 1.5; range, 1 to 3). Patient and disease characteristics are shown in Table 1.
Table 1.
Patient and Disease Characteristics at Time of Diagnosis
| Pt. # | Age (years) | SEX | RAI staging | B2M | IgHv mutational status | FISH | ALC | % of leukemia cells |
|---|---|---|---|---|---|---|---|---|
| 1 | 60 | Mal | 4 | 9.6 | UM | Negative | 15.13 | 73 |
| 2 | 64 | F | 2 | 4.4 | M | Negative | 19.6 | 70 |
| 3 | 47 | F | 2 | 4.5 | UM | 17p | 182 | 93 |
| 5 | 77 | M | 2 | 2.7 | UM | Negative | 14.43 | 82 |
| 6 | 60 | M | 4 | 5.3 | UM | 11q,13q | 26.28 | 93 |
| 7 | 43 | M | 3 | 2.2 | UM | Negative | 38.5 | 89 |
| 8 | 60 | M | 3 | 3.8 | ND | 11q,13q | 79 | 95 |
| 9 | 49 | M | 3 | 2.6 | UM | 17p,11q,13q | 83.8 | 94 |
| 10 | 68 | M | 4 | 6.4 | M | 11q,13q | 53 | 91 |
| 12 | 44 | F | 3 | 4 | UM | Negative | 124 | 95 |
| 13 | 62 | M | 1 | 1.6 | M | 13q | 8.81 | 52 |
| 14 | 65 | M | 0 | 2.3 | M | del13q | 6.34 | 42 |
| 15 | 64 | M | 0 | 1.4 | M | T12 | 17.07 | 85 |
| 16 | 51 | M | 2 | 3.3 | UM | 11q,13q | 24 | 80 |
| 17 | 66 | M | 2 | 4.1 | UM | 11q | 158.3 | 93 |
| 18 | 54 | M | 2 | 2.2 | UM | 13q | 124 | 92 |
| 19 | 60 | M | 4 | 9.6 | UM | Negative | 15.13 | 72 |
Pt, patient; B2M, beta 2 microglobulin; IgHv, immunoglobulin heavy chain; FISH, Florescence in-situ hybridization; ALC absoloute lymphocyte counts; UM, unmutated; ND, not done; M, mutated;
As was the case with CLL cells from ibrutinib-naïve patients, adding FFA to the CLL cells from these patients prior to ibrutinib treatment induced a reduction (12%) in delta dO2, suggesting that the cells metabolized FFA. After a median of 4 days (range: 2 to 7 days) of ibrutinib treatment, the delta dO2 value was only 6%, and after a median of 147 days (range: 85 to 165) of ibrutinib treatment, there was no longer a change in the dO2 value (Figure 2, A to C). Remarkably, the number of viable cells at time of first sampling and after 48 hours of incubation was similar across all culture conditions, with and without FFA (Figure 2D). The diminished capacity of CLL cells from ibrutinib-treated patients to metabolize FFA was not associated with patient age, sex, white blood cell count, IgHv mutation status, cytogenetic abnormalities or clinical characteristics.
Fig 2.
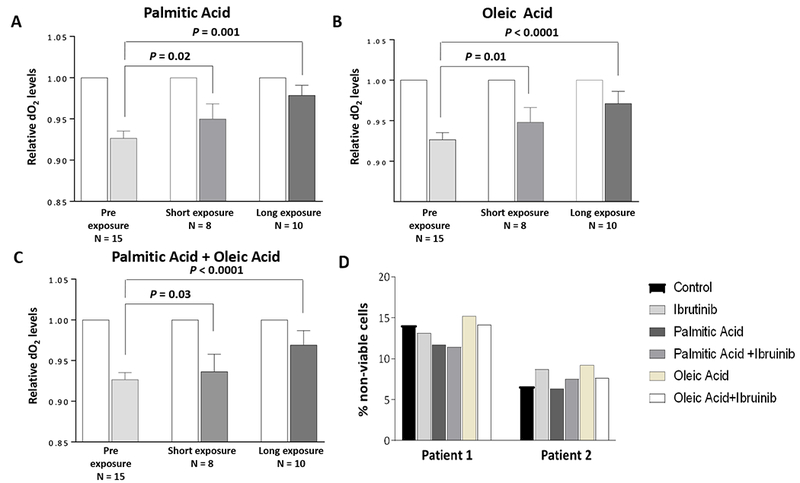
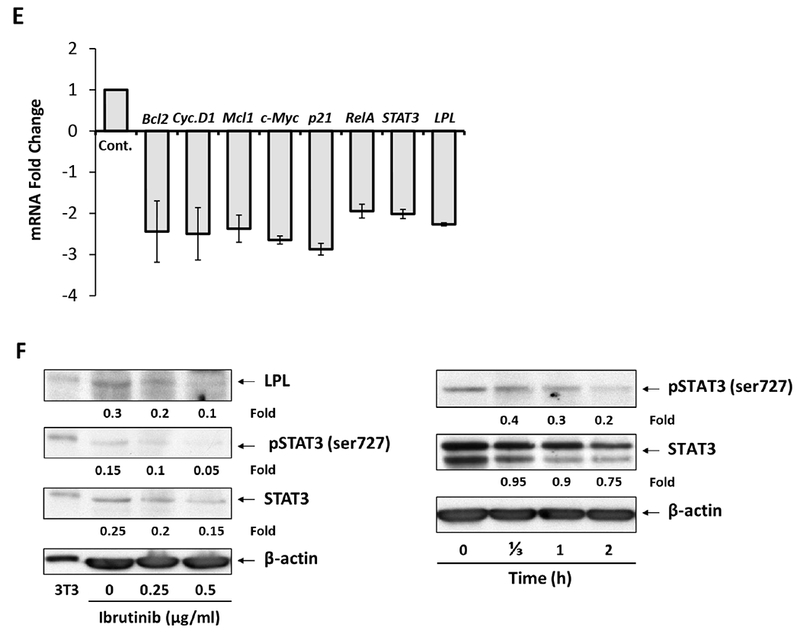
Ibrutinib inhibits free fatty acid metabolism of CLL cells in vivo. After a median of 4 days (short exposure range: 2 to 7) and 147 days (long exposure range: 85 to 165) of treatment with ibrutinib, peripheral blood (PB) CLL cells were obtained from CLL patients prior to treatment and cultured in the presence or absence of 80 mM (A) palmitic acid, (B) oleic acid or (C) palmitic acid plus oleic acid. The dO2 was initially set at 1 (white bars), and the relative concentration of dO2 after 48 hours is depicted after short (middle bars) and long (right bars) durations of ibrutinib treatment. (D) Viability of CLL cells incubated with palmitic or oleic acid with or without ibrutinib. (E) Ibrutinib downregulates LPL’s mRNA levels. CLL cells were incubated with 0.5 μM ibrutinib for 48 hours. Cellular RNA was extracted and subjected to quantitative reverse transcription–polymerase chain reaction testing using primers directed to detect the STAT3-regulated genes Bcl2, Cyclin D1, MCL1, c-Myc, p21, RelA, STAT3 and LPL. The experiment was repeated using PB CLL cells from 3 different patients. The means and standard errors of the means are depicted as fold change (fold reduction in the genes’ mRNA levels). As shown, ibrutinib reduced mRNA levels of STAT3-target genes, including LPL. (F) Ibrutinib downregulates STAT3, serine pSTAT3 and LPL levels in a time- and dose-dependent manner. Left panel: CLL cells were incubated without or with 0.25 μM or 0.5 μM ibrutinib for 24 hours. Western immunoblotting shows a dose-dependent decrease in levels of serine pSTAT3, STAT3 and LPL. For positive controls, we used 3T3 cells. Right panel: CLL cells were incubated with 0.5 μM ibrutinib and harvested at 4 time points (0 - 2h). Western immunoblotting shows a time-dependent decrease in serine pSTAT3 and STAT3 protein levels.
Ibrutinib inhibits STAT3 transcription and reduces LPL protein levels in CLL cells
We have previously reported that STAT3 induces the transcription of LPL and inhibition of STAT3 significantly reduces LPL levels and decreases FFA metabolism in cells. Because FFA metabolism in CLL cells correlated with STAT3-induced aberrant expression of LPL and ibrutinib inhibits FFA metabolism [5] we asked whether ibrutinib inhibits the transcription of STAT3. We incubated CLL cells with 0.5 μM ibrutinib for 48 hours and found that ibrutinib downregulated mRNA levels in STAT3-regulated genes, including STAT3 and LPL (Figure 2E). Furthermore, by using Western immunoblotting, we found that ibrutinib downregulated STAT3 and LPL protein levels in a dose- and time-dependent manner (Figure 2F).
Discussion
Unlike normal B cells, CLL cells utilize FFA for their metabolic needs [5]. Here we show that ibrutinib inhibits this metabolic pathway both in vitro and in PB CLL cells from ibrutinib-treated patients. Lipid metabolism in CLL cells is maintained by the transcriptional activity of the nuclear receptor peroxisome proliferator-activated receptor-α [12] and the nuclear factor (NF)-κB/STAT3 pathways [6]. A previous report demonstrated that ibrutinib inhibits NF-κB by blocking the BCR signaling pathway [13], which depends on a continuous supply of chemical energy in the form of ATP. Here we show that in-vitro, that ibrutinib inhibited STAT3 and, as a result, the transcription of LPL. Whether ibrutinib reduced STAT3 mRNA levels in CLL cells of ibrutinib-treated patients was not tested. However given that more than 93% of the drug is bound to plasma protein [14] and 4 hours after administration ibrutinib is covalently bound to more than 95% of the BTK [15], it is reasonable to assume that ibrutinib reduces STAT3 mRNA levels in CLL cells of ibrutinib-treated patients.
It is likely that both effects result in the inhibition of FFA metabolism in CLL cells. Whether or how inhibition of LPL transcription contributes to ibrutinib-induced inhibition of lipid metabolism is currently unknown. LPL has catalytic and non-catalytic functions. It catalyzes the hydrolysis of triglycerides into FFA and, in addition, induces a non-catalytic effect [16]. A previous report suggested that LPL is catalytically inactive in CLL cells [17]. We found that the metabolic activity of CLL cells is LPL-dependent even in the presence of FFA, circumventing the need for triglyceride hydrolysis [5], suggesting that LPL affects the lipid metabolism CLL cells through its non-catalytic activity.
Consistent with our previous report [5] we found that lipid metabolism was active in CLL cells from all patients regardless of stage, IgHv mutation status or cytogenetic abnormalities, suggesting that the cells utilize this metabolic pathway from early stages of the disease. Similarly, in line with a recent study, LPL protein levels were found to be increased in all CLL patients regardless of the severity of their disease [18]. Yet, whether LPL-dependent metabolism contributes to CLL cells’ survival is still a matter of debate. We and others have shown that inhibition of LPL activity or LPL knock-down increased cellular apoptotic rate [5,19]. Conversely, another group reported that LPL-siRNA did not increase CLL cell apoptosis rates likely because low levels of transfection efficacy (a median knock-down efficiency of only 33%) [18].
Ibrutinib inhibits a diverse plethora of cellular functions such adhesion [20], migration [21] and proliferation [22]. By inhibiting the capacity of CLL cells to utilize FFA, likely through downregulation of LPL levels, ibrutinib disrupts a metabolic pathway that that is imperative for cellular function.
Acknowledgements:
We thank Laura Russel for editing our manuscript
Financial Support:
This study was supported by grants from the CLL Global Research Foundation and the Cancer Center Support Grant from the NIH/NCI, P30 CA016672 and by the generous philanthropic contributions to The University of Texas MD Anderson Chronic Lymphocytic Leukemia Moon Shots Program.
Footnotes
Disclosure statement
Alessandra Ferrajoli, Jan Burger, Phillip Thompson, Nitin Jain,William Wierda, Michael J. Keating and Zeev Estrov received honoraria from pharmacyclics.
Uri Rozovski, David M. Harris, Ping Li, Zhiming Liu, Preetesh Jain report no conflicts of interest.
References
- 1.Chiorazzi N, Hatzi K, Albesiano E. B-cell chronic lymphocytic leukemia, a clonal disease of B lymphocytes with receptors that vary in specificity for (auto)antigens. Ann N Y Acad Sci 2005;1062:1–12. [DOI] [PubMed] [Google Scholar]
- 2.Heintel D, Kienle D, Shehata M, et al. . High expression of lipoprotein lipase in poor risk B-cell chronic lymphocytic leukemia. Leukemia 2005;19:1216–1223. [DOI] [PubMed] [Google Scholar]
- 3.Klein U, Tu Y, Stolovitzky GA, et al. . Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med 2001;194:1625–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenwald A, Alizadeh AA, Widhopf G, et al. . Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med 2001;194:1639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rozovski U, Grgurevic S, Bueso-Ramos C, et al. . Aberrant LPL Expression, Driven by STAT3, Mediates Free Fatty Acid Metabolism in CLL Cells. Mol Cancer Res 2015;13:944–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rozovski U, Hazan-Halevy I, Barzilai M, Keating MJ, Estrov Z. Metabolism pathways in chronic lymphocytic leukemia. Leuk Lymphoma 2016;57:758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreno P, Abreu C, Borge M, et al. . Lipoprotein lipase expression in unmutated CLL patients is the consequence of a demethylation process induced by the microenvironment. Leukemia 2013;27:721–725. [DOI] [PubMed] [Google Scholar]
- 8.Abreu C, Moreno P, Palacios F, et al. . Methylation status regulates lipoprotein lipase expression in chronic lymphocytic leukemia. Leuk Lymphoma 2013;54:1844–1848. [DOI] [PubMed] [Google Scholar]
- 9.Byrd JC, Furman RR, Coutre SE, et al. . Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burger JA, Tedeschi A, Barr PM, et al. . Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N Engl J Med 2015;373:2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrajoli A, Faderl S, Van Q, et al. . WP1066 disrupts Janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res 2007;67:11291–11299. [DOI] [PubMed] [Google Scholar]
- 12.Rakhshandehroo M, Knoch B, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res 2010;2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herman SE, Mustafa RZ, Gyamfi JA, et al. . Ibrutinib inhibits BCR and NF-kappaB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood 2014;123:3286–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bose P, Gandhi VV, Keating MJ. Pharmacokinetic and pharmacodynamic evaluation of ibrutinib for the treatment of chronic lymphocytic leukemia: rationale for lower doses. Expert Opin Drug Metab Toxicol 2016:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Advani RH, Buggy JJ, Sharman JP, et al. . Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013;31:88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mead JR, Irvine SA, Ramji DP. Lipoprotein lipase: structure, function, regulation, and role in disease. J Mol Med (Berl) 2002;80:753–769. [DOI] [PubMed] [Google Scholar]
- 17.Mansouri M, Sevov M, Fahlgren E, et al. . Lipoprotein lipase is differentially expressed in prognostic subsets of chronic lymphocytic leukemia but displays invariably low catalytical activity. Leuk Res 2010;34:301–306. [DOI] [PubMed] [Google Scholar]
- 18.Porpaczy E, Tauber S, Bilban M, et al. . Lipoprotein lipase in chronic lymphocytic leukaemia - strong biomarker with lack of functional significance. Leuk Res 2013;37:631–636. [DOI] [PubMed] [Google Scholar]
- 19.Pallasch CP, Schwamb J, Konigs S, et al. . Targeting lipid metabolism by the lipoprotein lipase inhibitor orlistat results in apoptosis of B-cell chronic lymphocytic leukemia cells. Leukemia 2008;22:585–592. [DOI] [PubMed] [Google Scholar]
- 20.de Rooij MF, Kuil A, Kater AP, Kersten MJ, Pals ST, Spaargaren M. Ibrutinib and idelalisib synergistically target BCR-controlled adhesion in MCL and CLL: a rationale for combination therapy. Blood 2015;125:2306–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaitseva L, Murray MY, Shafat MS, et al. . Ibrutinib inhibits SDF1/CXCR4 mediated migration in AML. Oncotarget 2014;5:9930–9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng S, Ma J, Guo A, et al. . BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia 2014;28:649–657. [DOI] [PubMed] [Google Scholar]