

Abstract
A critical review of the state-of-the-art evidence in support of the mechanisms of glycosylation reactions is provided. Factors affecting the stability of putative oxocarbenium ions as intermediates at the SN1 end of the mechanistic continuum are first surveyed before the evidence, spectroscopic and indirect, for the existence of such species on the timescale of glycosylation reactions is presented. Current models for diastereoselectivity in nucleophilic attack on oxocarbenium ions are then described. Evidence in support of the intermediacy of activated covalent glycosyl donors is reviewed, before the influence of the structure of the nucleophile, of the solvent, of temperature, and of donor-acceptor hydrogen bonding on the mechanism of glycosylation reactions are surveyed. Studies on the kinetics of glycosylation reactions and the use of kinetic isotope effects for the determination of transition state structure are presented, before computational models are finally surveyed. The review concludes with a critical appraisal of the state of the art.
Graphical Abstract

1 Introduction
This review surveys the evidence in support of the mechanism(s) of “traditional” glycosylation reactions, namely those involving displacement of a leaving group from a glycosyl donor by a glycosyl acceptor usually with the assistance of a promoter (Scheme 1). It is therefore a review of the mechanism(s) of nucleophilic substitution at sp3 carbon with emphasis on the subset of electrophiles carrying an ether oxygen at the electrophilic site.
Scheme 1.

The Glycosylation Reaction
Nucleophilic substitution at sp3 carbon is usually considered in terms of two limiting mechanisms, the unimolecular, dissociative SN1 process involving a discrete carbenium ion intermediate, and the bimolecular SN2 process proceeding through an associative transition state in a single step. The gap between these two extremes is generally considered to be spanned by a continuum of more or less tightly associated ion pair mechanisms, and has been the focus of intense interest by physical organic chemists for more than half a century. Such studies typically involve the determination of reaction kinetics, and of stereoselectivity, the characterization of possible discrete reaction intermediates, and the investigation of substituent effects in both the electrophile and nucleophile, ideally in the form of linear free energy relationships. Much emphasis is placed on the identification and prediction of possible discontinuities in the continuum of ion pair mechanisms that might be regarded as representative of the switch between the two limiting mechanisms.1 Such information is obviously of practical importance as it enables the prediction of reaction conditions favoring a specific desired outcome.
Glycosylation is a microcosm of nucleophilic substitution at sp3 carbon in which one of the substituents at the carbon undergoing substitution is an ethereal oxygen atom, usually encapsulated within either a five (furanosyl) or six-membered (pyranosyl) ring that itself carries multiple C-O bonds.2 Glycosyl donors present themselves as one or the other of two possible diastereomers, known as anomers, or as a mixture of the two. The usual continuum mechanism for substitution consequently expands in two directions away from the oxygen-substituted carbenium ion, the oxocarbenium ion, that is the central intermediate in the limiting dissociative process, to include sets of diastereomeric ion pairs (Scheme 2). Such ion pair mechanisms for glycosylation were first described in the open literature by Rhind-Tutt and Vernon,3 and reiterated by various authors,4–6 including the seminal contribution of Lemieux and coworkers with its more graphical presentation.7
Scheme 2.

The Glycosylation Reaction with the Two Limiting Associative Mechanisms and the Central Dissociative Limiting Mechanism
As noted by Horenstein,8 while there has been much work on the mechanisms of glycosidic bond hydrolysis – chemical and enzymatic, physical organic studies on actual glycosidic bond forming reactions that go beyond the observance of frequently small changes in stereoselectivity as a function of substituents and conditions have been relatively limited until recently. This review, while certainly not neglecting the importance of stereoselectivity from both the mechanistic and practical standpoints, focuses on the detection and characterization of intermediates and on investigations into the kinetics of glycosylation. The reader is referred to several other books and reviews for complementary perspectives of the field.9–24 The concepts of stereodirecting neighboring group participation and anchimeric assistance in glycosylation are not covered in this review.
With the notable exception of the early workers, who provided clear kinetic evidence of associative mechanisms in several cases and of which more below,3,25–27 most practitioners of the art rationalize their stereochemical results in terms of dissociative mechanisms featuring oxocarbenium ion intermediates. Consistent with this mostly oxocarbenium ion centric universe, the review begins with a brief treatment of carbenium ions and the influence of substituents on them before moving on to the physical evidence in support of glycosyl oxocarbenium ions and other intermediates in glycosylation. It continues with an overview of models for the facial selectivity of nucleophilic attack on glycosyl oxocarbenium ions before going on to discuss the many studies that have identified and characterized activated covalent intermediates in glycosylation reactions. The effect of the acceptor, solvent, temperature, and hydrogen bonding are covered before consideration is given to the determination of reaction kinetics and of kinetic isotope effects. A final section reviews computational work in the field.
2. Carbenium Ions and Glycosyl Oxocarbenium Ions
Meerwein first reported the isolation of stable crystalline oxocarbenium ions formed by the reaction of triethyloxonium tetrafluoroborate with aldehydes and ketones in a series of papers beginning in 1937.28,29 Oxocarbenium ions, whose non-carbohydrate members have been reviewed,30 are resonance stabilized carbenium ions and, in many organic chemistry textbooks, serve as one of the prime examples of the concept of resonance stabilization (Figure 1). An alternative representation of oxocarbenium ions advocated by Deslongchamps uses Pauling’s concept of two bent tau bonds between the carbon and oxygen atoms rather than the standard model of a sigma and a pi bond (Figure 2).31 In this review we adopt the classical sigma and pi representation, referring only to the tau model in regard to the stereoselectivity of addition to oxocarbenium ions.
Figure 1.

Carbenium and Resonance Stabilized Oxocarbenium Ions
Figure 2.

The σ-π and τ Bond Models for Oxocarbenium Ions
The notion of the stability of oxocarbenium ions due to resonance delocalization is deeply ingrained in organic chemistry and nowhere more so than in carbohydrate chemistry where glycosyl oxocarbenium ions are widely drawn as reaction intermediates. Much less consideration, however, is given to the extent of this resonance stabilization and, most importantly in carbohydrate chemistry, the influence of β- rather than α-C-O bonds on carbenium ion stability. It is appropriate, therefore, to begin with a consideration of the magnitude of the influence of both α- and β-C-O bonds on carbenium ion stability and reactivity.
Relative carbenium ion stabilities can be estimated by comparison of their gas phase hydride ion affinities (HIAs). From the data of Keeffe and More O’Ferrall (Table 1),32 the methoxymethyl cation is approximately 2 kcal.mol−1 less stable than the tert-butyl cation, 3 kcal.mol−1 more stable than the benzyl cation, and 31 kcal.mol−1 more stable than the ethyl cation. Addition of one and then a second methyl substituent at the sp2 carbon increases the stability of the methoxymethyl cation in a comparable manner to the ethyl cation (Table 1). Thus, in the absence of electron-withdrawing effects from β- and more remote C-O bonds, simple oxocarbenium ions are clearly of comparable stability to similarly substituted benzylic and allylic carbenium ions. Indeed, this recognition recently prompted the Bennet and Withers laboratories to compare substituted cyclohexenyl mono- and dinitrophenyl ethers with the corresponding glycosides. It was found that the allylic ethers display kinetic parameters for their spontaneous hydrolysis that are closely analogous to those of the correspondingly substituted mono- and dinitrophenyl glycosides, and moreover that they are subject to hydrolysis by certain glycosidase enzymes for which they can also serve as covalent inhibitors.33,34
Table 1.
Computed Hydride Ion Affinities Relative to the tert-Butyl Cation (kcal.mol−1).32
| Carbocation | ΔHIA(g) |
|---|---|
| MeCH2+ | 33.8 |
| Me2CH+ | 14.7 |
| MeOCH2+ | 2.1 |
| MeOC(Me)H+ | −15.7 |
| MeOCMe2+ | −26.7 |
| PhCH2+ | 5.0 |
An alternative measure of the stability gained by delocalization of the positive charge in an oxocarbenium ion can be obtained from measurements of the barrier to rotation about the C-O bond. Such measurements were made by the Winstein group who determined a barrier of 18.4 kcal.mol−1 for the 7-methoxy-7-norbornyl cation by VT NMR spectroscopy in fluorosulfonic acid (Scheme 3).35 The barrier for syn-anti equilibration of the 2-methoxy-2-norbornyl cation was higher and estimated to be at least 19.5 kcal.mol−1.35 While there is considerable variation between the stabilization of ~30 kcal.mol−1 calculated from the hydride ion affinities and the barriers to rotation, on the whole they are in reasonable agreement with estimates of there being approximately 60% π-character in oxocarbenium ions derived by alkylation or protonation of aldehydes and ketones as judged by Olah and others on the basis of 17O, 1H, and 13C chemical shift measurements.36,37
Scheme 3.

Barrier to Rotation about the C-O π Bond in the 7-Methoxy-7-norbornyl Carbenium Ion Determined by VT NMR
When resonance stabilization is not possible, α-oxygen substitution of a carbenium ion is highly destabilizing. This is readily apparent from the relative rates of solvolysis of 1-adamantanyl tosylate and of its 2-oxa derivative, as determined by Meyer and Martin, which differ by a factor of more than one hundred (Figure 3).38 The stability of 2-oxa-3-cyano-1-adamantanyl triflate, a compound that is unchanged by filtration over alumina and which has a melting point of 94.5–97.5 °C,38 and of 7,7-di(trifloxy)norbornane, which can be purified by sublimation and has a melting point of 42 °C,39 underlines the electron-withdrawing nature of an α-oxygen substituent when resonance stabilization of the cation is prevented either by steric inhibition of resonance or removal of the electron density by a further electron-withdrawing substituent (Figure 4).
Figure 3.

Relative Rates of Solvolysis of 1-Adamantanyl Tosylate and 2-Oxa-1-adamantanyl Tosylate
Figure 4.

Stable α-Oxa Triflates
3 Influence of C-O Bonds and Other Substituents on the Formation of Oxocarbenium Ions
Carbohydrate-based oxocarbenium ions differ from simple aliphatic ones by the presence of multiple C-O bonds at the β- and more remote sites. As these C-O bonds are electron-withdrawing they exert considerable influence on the stability, lifetime, and reactions of oxocarbenium ions. A measure of the magnitude of these effects can be gained from the effect of β-C-O bonds on solvolysis reactions proceeding via simple carbenium ions; work by the Lambert and Kirmse laboratories is instructive. Thus, it was demonstrated that the acetolysis of 2-endo-norbornyl tosylate at 25 °C is retarded more than three thousand-fold in the presence of a synperiplanar acetoxy group in the 3-endo position.40 Similarly, the solvolysis of 2-endo-norbornyl brosylate at 25 °C was retarded some 105-fold by inclusion of a gauche oxygen in the norbornyl framework at the 6-position (Figure 5).41 Clearly, β-C-O bonds that are stereoelectronically not available for anchimeric assistance very strongly retard solvolysis reactions of simple alkyl sulfonates; it is not surprising that they retard glycosylation reactions similarly.
Figure 5.

Retardation of Relative Rates of Solvolysis by β-C-O Bonds
The influence of electron-withdrawing β-C-O bonds on the stability of glycosyl oxocarbenium ions is nicely illustrated by the chemistry of the phenylthiomethyl glycosides, which on activation with N-iodosuccinimide and triflic acid in dichloromethane at −30 °C in the presence of various alcohols afforded the corresponding alkoxymethyl glycosides in good yield (Scheme 4).42 Fragmentation of the anticipated intermediate trifloxymethyl glycoside to the corresponding glycosyl triflate and formaldehyde did not occur under these conditions as evidenced by the absence of formation of simple glycosides.
Scheme 4.

Substitution of a Phenylthiomethyl Glycoside without Fragmentation
Comparable results were reported for the acid catalyzed hydrolysis of isopropenyl α- and β-glucopyranosides, which occurs with exclusive cleavage of the vinyl-O bond as opposed to the glycosyl-O bond (Scheme 5).43,44 The use of protected isopropenyl glycosides as glycosyl donors is, however, more complex and affords either the acetal or the glycoside dependent on the promoter and solvent.45
Scheme 5.

Hydrolysis of Isopropenyl Glycosides Does not Involve Glycosyl Oxocarbenium Ions
Classically, studies on the acid-catalyzed hydrolysis of methyl glycosides provided useful information on the influence of C-O bonds (Table 2),46 but more extensive and informative data is provided by work from the Withers laboratory on the spontaneous hydrolysis of 2,4-dinitrophenyl glycosides (Table 3).47,48 It is noteworthy here that the 2-deoxy glucosyl system, lacking the β-C-O bond, could not be studied because of the instability of the dinitrophenyl glycoside. It is important to understand in passing that such studies of the hydrolysis of glycosides are typically carried out under pseudo-first order conditions in aqueous media that mask any concentration dependence of the nucleophile.
Table 2.
Selected Relative Rates of Acid-Catalyzed Hydrolysis of Methyl Glycosides in 2 N HCl at 58 °C.
| Glycoside | krel |
|---|---|

|
1 |

|
2090a |
| Glycoside | krel |
|---|---|

|
20 |

|
40 |
In 0.1 N HCl.
Table 3.
Selected Relative Rates of Spontaneous Hydrolysis of 2,4-Dinitrophenyl Glycosides at pH 6.5 in 25 mM Sodium Phosphate Buffer and 0.40 M KCl at 37 °C.
| Glycoside | krel |
|---|---|

|
1.0 |

|
4.0 |
| Glycoside | krel |
|---|---|

|
22.4 |

|
4.7 |
Similar studies have provided strong evidence that the imposition of the tg conformation49 on the carbohydrate side chain, either through use of a 4,6-O-acetal or in a designed bicyclic model system, maximizes the electron withdrawing effect of the C6–O6 bond (Table 4).50,51 Based on the retarded hydrolysis of a further trans-fused bicyclic model in which the electron-withdrawing O6 was replaced by a methylene group, it was suggested that this so-called benzylidene-effect is mainly due to torsional effects, as originally advanced by Fraser-Reid based on a comparative study of pentenyl glycoside activation and on computational work.52,53 This argument, however, does not take into account the extra torsional interactions introduced into the model by the C for O replacement.51
Table 4.
Relative Rates of Spontaneous Hydrolysis of Bicyclic Dinitrophenyl Glycosides at pH 6.5 at 37 °C.50,51
| Substrate (glu) | Conf | krel |
|---|---|---|

|
- | 11.1 |

|
gg | 3.6 |

|
gt | 2.1 |

|
tg | 1 |
| Substrate (gal) | Conf | krel |
|---|---|---|

|
- | 5.9 |

|
gg | 2.5 |

|
gt | 1.4 |

|
tg | 1 |
Extensive data from the Wong group on the relative reactivity values of a broad range of thioglycosides on activation under a standard set of conditions provide further information on the influence of C-O bonds on the reactivity of glycosyl donors, and extensive information on the modulation of the effects of C-O bonds by the appended protecting groups (Table 5).54,55 Similar work by the Ley group is also informative in this regard.56,57 Following earlier work by Oscarson and coworkers on the quantification of the relative reactivity of various alkyl and aryl thioglycosides,58 the Huang lab used a comparable method to assess the influence of substituents in the arylthioglycoside moiety on activation with NIS and TfOH (Table 6).59 It must be clear, however, that these extensive databases on the influence of C-O bonds and protecting groups do not distinguish between effects arising because of destabilization of the respective oxocarbenium ions and those due to reduction in the rates of reaction of the thioglycosides themselves with the promoter.
Table 5.
Selected Examples of Wong Relative Reactivity Values (RRVs) for the Activation of Thioglycosides by NIS-TfOH in Dichloromethane at Room Temperature.
| Substrate | RRV |
|---|---|

|
1 |

|
7.2 × 104 |

|
1.7 × 104 |

|
185.4 |

|
118.7 |

|
102.0 |

|
67.1 |

|
31.4 |
| Substrate | RRV |
|---|---|

|
28.9 |

|
24.1 |

|
17.6 |

|
13.1 |

|
5.7 |

|
2.7 |

|
1.7 |

|
1.3 |
Table 6.
Relative Reactivity Values (RRVs) of Substituted Aryl Thioglycosides Compared to Tolyl Per-O-acetyl-α-D-thiomannopyranoside under the Wong Conditions
| Substituents (R) | NO2 | Br | N3 | NHAc | OMe |
|---|---|---|---|---|---|

|
0.042 | 0.89 | 3.12 | 18.1 | 21.7 |

|
0.601 | 19.2 | 27.1 | 101 | 191 |

|
3.19 | 161 | 657 | 2482 | 3772 |
The effect of electron-withdrawing groups on the stability of oxocarbenium ions is most readily appreciated by consideration of the extensive efforts of several groups to characterize them by NMR spectroscopy. The NMR spectra of simple oxocarbenium ions, ie, ones lacking β-C-O bonds, have been available in superacidic media since the 1960’s and have been reviewed.17,30 More recently, Yoshida and coworkers were able to generate stable solutions of simple oxocarbenium ions in deuteriodichloromethane at low temperature in the presence of tetrabutylammonium tetrafluoroborate and record their NMR spectra.60 However, all attempts to extrapolate this method to the glycosyl oxocarbenium ions failed because of the inability to prevent reaction of the oxocarbenium ions with nucleophiles, including tetrafluoroborate and disulfides, in the reaction mixture.60–62 A report of the NMR spectroscopic detection of a glycosyl oxocarbenium ion generated by an electrochemical method, with a lifetime of 1 second at −78 °C in the presence of the non-nucleophilic tetrakis(pentafluorophenyl)borate anion and stabilized by co-ordination to a diaryl disulfide, is not supported by the necessary 13C chemical shift data.61
In posters presented at conferences in 2013 and 2014, Akien and Subramanian reported the NMR spectra of the per-O-methyl fructofuranosyl and per-O-methyl-2-deoxyglucopyranosyl oxocarbenium ions obtained by dissolving the corresponding methyl glycosides in superacidic media.63,64 Unfortunately, details of these experiments have yet to appear in the open literature. In an important advance, Blériot and coworkers reported in 2016 the successful recording of the 1H and 13C NMR spectra of per-O-acetyl 2-deoxy-glucopyranosyl and 2-bromo-2-deoxyglucopyranosyl oxocarbenium ions generated from the corresponding glycosyl acetates on dissolution in the superacidic HF/SbF5 medium.65,66 These oxocarbenium ions, which were stable to room temperature under the reaction conditions, were found to have all three acetoxy groups protonated and to exist predominantly in the 4E and 4H5 conformations, respectively (Scheme 6). Additionally, in the 2-bromo-2-deoxy case the oxocarbenium ion was stabilized by disymmetric bridging of the bromine atom. The 13C NMR spectra of the two oxocarbenium ions displayed anomeric chemical shifts of δ 229 and δ 198 in line with those of simple oxocarbenium ions in either superacids or dichloromethane. Attempts to generate the corresponding oxocarbenium ions from per-O-acetyl glucopyranose and per-O-acetyl-2-azido-2-deoxy glucopyranose failed due to the formation of the bridging dioxalenium ion in the former case, and because of lack of activation in the latter. Use of the corresponding per-O-acetyl 2-deoxy-2-fluoroglucopyranose precursor resulted in complex spectra.65
Scheme 6.

Generation of Glycosyl Oxocarbenium Ions in Superacidic Media
Exploiting the additional stabilization afforded by charge delocalization onto a second oxygen atom, Woerpel and coworkers generated simple monobenzyloxy tetrahydropyranosyl dioxacarbenium ions by alkylation of the corresponding lactones. NMR investigations in CD2Cl2, computational work, and single crystal X-ray diffraction studies converged on half chair-like ground state conformations in which the benzyloxy group adopts a pseudoaxial position (Figure 6).67 In contrast, the corresponding methyl substituted dioxacarbenium ions preferred the half chair conformation with the substituents pseudo-equatorial.67 Subsequent NMR spectroscopic studies revealed the dioxacarbenium ion derived by alkylation of 2-deoxy-3,4,6-tri-O-benzyl-D-glucuronolactone also adopts a half-chair conformation with all substituents pseudo-axial. Application of the lactone alkylation method to tetra-O-benzylmannonolactone, however, failed to provide a stable dioxacarbenium ion suggesting that even the presence of the additional second stabilizing C-O bond is insufficient to compensate for the destabilizing influence of a C-O bond at the 2-position.68 Overall, Woerpel’s findings are in agreement with extensive computational studies and suggest that pseudoaxial C-O bonds stabilize the oxocarbenium ions through space electrostatically,67,69–72 similar to the way in which axial C-O bonds stabilize piperidinium ions,73,74 of which more below. The contrast between the conformations of these systems and those reported by the Blériot team for the per-O-acetyl 2-deoxy and 2-bromo-2-deoxy glucopyranosyl oxocarbenium ions (Scheme 6), with pseudo-equatorial acetoxy groups, is best understood in terms of the repulsive electrostatic interactions between the protonated acetoxy groups and the oxocarbenium ion in the superacid media.65,66
Figure 6.

Structure and Preferred Conformations of 1-Alkoxyoxocarbenium Ions as a Function of the Substituent
It is widely appreciated that furanosides are less stable than pyranosides, and so that furanosyl donors are more reactive than pyranosyl donors, even if quantification is rare. Sinnott offers a benchmark and estimates that a ribofuranoside is hydrolyzed approximately an order of magnitude faster than the corresponding glucopyranoside of the same configuration.13 The clear implication is that a furanosyl oxocarbenium ion is more stable than the corresponding pyranosyl one, for which the underlying reasons are two-fold. First, a furanosyl oxocarbenium ion perforce contains one less electron-withdrawing C-O bond than the corresponding pyranosyl system. Second, the introduction of an sp2 center into a five-membered ring results in a significant loss of torsional strain whereas the opposite is true for six-membered rings.75 In spite of this, beyond the preliminary report of Akien and Subramanian, spectroscopic observation of furanosyl oxocarbenium ions has yet to be reported.63
Although oxocarbenium ions also are commonly written as intermediates in sialic acid chemistry, there have been no reports of the spectroscopic identification of such species in the literature. Indirect evidence for the existence of sialyl oxocarbenium ions in organic solution, however, was provided by De Meo and coworkers who studied the reactions of sialyl thioglycosides stereospecifically labelled with deuterium at the 3-position on activation with NIS and triflic acid.76 The results of these elimination reactions (Scheme 7) were interpreted as being consistent with the generation of oxocarbenium ions sufficiently long-lived for conformational equilibration and cleavage of the weaker C-H bonds from a pseudo-axial position (Scheme 7). Differences in the H/D elimination ratio between the acetamides and the corresponding trifluoroacetamides were taken as indicative of the influence of the electron-withdrawing ability of the amide group on the conformational equilibrium of the oxocarbenium ion. The preferential elimination of deuterium from the oxazolidinone was judged to be the result of the reduced conformational mobility of this bicyclic system (Scheme 8). The possibility of an alternative E2 mechanism for elimination from a set of rapidly equilibrating sialosyl triflates was considered unlikely by the authors but cannot be completely excluded.
Scheme 7.

Preferential Loss of Protium over Deuterium from Two Diastereomeric Sialyl Thioglycosides Suggestive of Equilibrating Oxocarbenium Ions
Scheme 8.

Loss of the Pseudoaxial Deuterium from a Conformationally Locked System
The inability to observe glycosyl oxocarbenium ions in all but superacidic media is indicative of oxocarbenium ion lifetimes that are short on the NMR timescale. Attempts to measure actual glycosyl oxocarbenium ion lifetimes date to the 1989 work of Amyes and Jencks using diffusion-controlled trapping by external azide in conjunction with the common ion effect as a clock reaction in the solvolysis of α-azido ethers. On this basis, the glucopyranosyl oxocarbenium ion was estimated to have “a short but significant lifetime in aqueous solution of 1 × 10−12 s” at 25 °C,77 revised by Bennet and coworkers in subsequent work to 2.5 × 10−12 s (Table 7),78 which was considered to be too short to allow complete diffusional equilibration with solute and even solvent molecules. It was further concluded that rapid internal return is likely to preclude the existence of contact ion pairs containing glycosyl oxocarbenium ions in water, and consequently that if such cations do exist they must be in the form of solvent-separated ion pairs. Extrapolating from this borderline existence in water, Sinnott boldly deduced that “if such intimate ion pairs of glycosyl cations and anions are too unstable to exist in water, a fortiori they have no real existence in organic solvents and mechanistic proposals, which invoke them are simply in error”.13
Table 7.
Oxocarbenium Ion Lifetimes in Water.



SSIP:M complex where M = 4-bromoisoquinoline.
Different lifetimes were reported for the two anomeric substrates leading to the range given
In subsequent work Bennet and coworkers, studying the hydrolyses of 2-deoxy glucopyranosyl pyridinium ions in the presence of added azide arrived at lifetimes of 1.4 × 10−11 s and 2.7 × 10−11 s for the ion pair encounter complexes originating from the β- and α-salts, respectively, and concluded that the 2-deoxyglucopyranosyl oxocarbenium ion is not fully solvent equilibrated in water (Table 7).79,80 Extrapolating the azide clock method to the solvolysis of CMP N-acetyl neuraminate, Horenstein and Brunner determined a lifetime of ≥3 × 10−11 s at pH 5 for the N-acetyl neuraminate oxocarbenium ion (Table 7), and concluded that the α-carboxylate stabilizes the oxocarbenium ion.81 This latter suggestion is in agreement with Horenstein’s early computational work in which it was found that the α-carboxylate stabilizes the oxocarbenium ion electrostatically by 110 kcal.mol−1 relative to the carboxylic acid in the gas phase, but only by 17 kcal.mol−1 in water.82
Crich and coworkers designed a series of cyclization reactions for use as unimolecular cation clock reactions in organic solution.83,84 Working in both the 4,6-O-benzylidene protected and the corresponding per-O-benzyl mannopyranosyl series, it was found that activation of either sulfoxide-based or trichloroacetimidate donors afforded mixtures of the cis- and trans-fused products from the cyclization reaction (Scheme 9). This result was interpreted in terms of formation of a covalent α-glycosyl triflate in equilibrium with a transient oxocarbenium ion triflate ion pair from which cyclization takes place. As the formation of the trans-fused product can only occur from the B2,5 conformation, the oxocarbenium ion must have sufficient lifetime to undergo conformational exchange in dichloromethane solution at the −72 °C of the reaction.
Scheme 9.

Equilibration and Cyclization of a Transient Mannopyranosyl Oxocarbenium Ion in CH2Cl2 at −20 °C
In the corresponding glucopyranose series, the use of sulfoxide-based donors was not successful owing to an intramolecular sulfenyl transfer to the allylsilane on activation,85 but the activation of the trichloroacetimidate afforded the cyclized product in the form of a single cis-fused isomer (Scheme 10).84 Activation of a cognate 2-O-(3-hydroxypropyl)glucopyranosyl sulfoxide at low temperature resulted in the formation of a bicyclic acetal demonstrated to have the β-configuration by NMR and crystallographic methods (Scheme 11).85 The reversal of stereoselectivity on switching from the allylsilane to the more nucleophilic alcohol is best rationalized in terms of a change in mechanism. With the alcohol, direct nucleophilic attack on the intermediate α-glucosyl triflate formed on activation of the sulfoxide rationalizes the observed product, whereas the intramolecular Sakurai product, arising from the weaker nucleophile, is the result of an SN1-type process on a transient oxocarbenium ion.84,85 The β-selectivity of the acetal-forming reaction, which contrasts with the α-selective intermolecular O-glycosylation reactions of benzylidene-protected glucopyranosyl triflates, is explained in terms of the high effective molarity of the nucleophile in the cyclization.85
Scheme 10.

Formation and Cyclization of a Transient Glucopyranosyl Oxocarbenium Ion in CH2Cl2 at −20 °C
Scheme 11.

trans-Selective Formation of a Bicyclic Acetal Indicative of Direct Displacement of an α-Triflate
In yet another series of cyclization reactions, Amarasekara and Crich observed differing diastereomeric ratios on activation and cyclization of two anomeric hydroxyl esters, albeit both favoring the formation of the β-(axial) C-O bond (Scheme 12).86 The implication is that mixed mechanisms are observed in which a pathway involving a common intermediate from both anomers, either a transient oxocarbenium ion or an α-glycosyl triflate, provides the background to competing SN2 displacement on the activated thioglycoside. The latter being accelerated when it leads directly to the formation of the axial C-O bond.
Scheme 12.

Intramolecular Sialidation Reactions
Overall, although lifetimes are too short for observation by conventional NMR methods, growing evidence supports the existence of glycosyl oxocarbenium ions in organic solution with lifetimes sufficient to permit conformational inversion and trapping by intramolecular nucleophiles.
4 Mass Spectrometric Methods for Probing the Influence of Substituents on Oxocarbenium Ion Generation
While most work aimed at characterizing glycosyl oxocarbenium ions and the influence of substituents and protecting groups on them has been conducted by NMR methods, mass spectrometry provides an alternative means of exploration. Building on a number of earlier studies, Denekamp and Sandlers used collision-induced fragmentation-ESI mass spectrometry to study the influence of anomeric stereochemistry on the fragmentation of the ammoniated peracetyl α- and β-D-glucopyranose ions. The observation that the β-isomer with the trans-disposition of the 1,2-acetoxy groups undergoes fragmentation to the oxocarbenium ion more readily was rationalized in terms of anchimeric assistance.87 It was also demonstrated that peracetyl galactopyranosides, with the axial acetoxy group at the 4-position undergo fragmentation more rapidly than their gluco-isomers.87 Subsequently, the same authors studied the influence of protecting groups at the 2- and 4-positions on the CID ESI mass spectra of a series of glycopyranosyl thioglycosides. On the basis of the ratios of the relative intensities of the ammoniated molecular ions to the oxocarbenium ions they concluded that the order of stabilization of the oxocarbenium ions in the gas phase by protecting groups at C2 and/or C4 are Bz > Ac > (CH3)3Si > alkyl,88 clearly showing the importance of neighboring group participation and even presaging the later studies described below on silyl protected superarmed donors.89,90
Crich and coworkers, adopting a poor man’s approach with a standard ESI mass spectrometer, determined the threshold cone voltage necessary for the anomeric fragmentation of a series of sialyl dibutyl phosphates. The data were correlated with the electron-withdrawing ability of the protecting group pattern at the 4- and 5-positions as suggested by the expected dipole moments (Scheme 13).91 In effect, the more electron withdrawing oxazolidinone and cyclic carbonates, with a single large dipole in the plane of the pyranose ring required the greatest energy for fragmentation. A caveat arises from the possible formation of the observed ion by McLafferty rearrangement rather than stepwise fragmentation and deprotonation. It was considered, however, that this would not negate the results as the fragmentation would nevertheless involve a substantial partial positive charge at the anomeric position. The use of thioglycosides as anomeric leaving groups in this study was thwarted by the preferential loss of an acetoxy group from the side chain.91 Indeed, in subsequent work on the CID-ESIMS of sialyl S- and O-glycosides, Chizhov and coworkers also found the loss of carboxylate groups to compete with that of the anomeric functionality.92,93
Scheme 13.

Threshold ESI Cone Voltages for the Fragmentation of Sialyl Phosphates as a Function of O4 and N5 Protecting Groups and Their Dipoles
5 Generation of Glycosyl Oxocarbenium Ions from Glycosylidene Carbenes
The chemistry of the glycosylidene carbenes studied by the Vasella laboratory presents an alternative approach to the generation of glycosyl oxocarbenium ions and provides unique insight into their reactivity and selectivity arising from their generation in the absence of a counter-ion other than the nucleophilic alkoxide.94,95 These ambiphilic to nucleophilic alkoxy carbenes are best generated by thermolysis or photolysis of glycosylidene diazirines. In the presence of alcohols they are protonated to generate oxocarbenium ion – alkoxide ion pairs (Scheme 14).96
Scheme 14.

Formation of an Oxocarbenium Ion by Protonation of a Glycosylidene Carbene
In the tetra-O-benzylglucopyranosyl series illustrated no facial selectivity is observed with weakly acidic alcohols in dichloromethane as solvent, whereas more acidic alcohols and phenols are somewhat β-selective;97 no selectivity is seen with a 2-deoxy system.98 It was argued that as protonation takes place in the σ-plane of the carbene and the subsequent nucleophilic attack occurs in the π* orbital, the alcohol that functions as proton donor is not the nucleophile. Rather, the initial alkoxide deprotonates a further molecule of acceptor that is positioned above or below the σ-plane so as to solvate the carbene. The lack of selectivity with typical alcohols suggests that collapse of the ion pair to afford the glycosides is essentially barrierless and so not subject to stereoelectronic control as subsequently argued by Woerpel for the reaction of oxocarbenium ions with nucleophilic alcohols.99 With phenols and more acidic alcohols, for which collapse of the ion pair is slower, other factors intervene. Thus, it was suggested that in such systems the oxocarbenium ion is stabilized by participation of the benzyloxy group at the 2-position resulting in preferential attack on the β-face. The absence of selectivity in the 2-deoxy series is then understood in terms of the absence of participation.98
6 Piperidinium Ions as Models for Substituent Effects in Glycosyl Oxocarbenium Ions
In an important series of papers the Bols laboratory has provided strong support for the hypothesis of through space electrostatic stabilization of oxocarbenium ions by pseudo-axial C-O bonds. The concept draws parallels between oxocarbenium ions and ammonium ions and employs data from pKa measurements on an extensive series of mono and bicyclic substituted piperidinium ions.73 A series of substituent constants were derived (Table 8) that clearly indicate axial substituents to be more stabilizing toward positive charge than the corresponding equatorial substituents, which was rationalized in terms of the greater spatial proximity of the axial electronegative atom to the center of positive charge resulting in greater electrostatic stabilization. Conversely, other than at the 2-position, equatorial substituents are more destabilizing. A strong correlation was found between the negative logarithm of the rate constant for spontaneous hydrolysis of a series of 2,4-dinitrophenyl glycosides and the summed stereoelectronic constants of the substituents for a given system (Figure 7) thereby validating the use of piperidinium ions as models for the influence of substituents on the developing positive charge at the anomeric center during hydrolysis.73
Table 8.
Stereoelectronic Substituent Effects on pKa of Piperidinium Ions in Water at 25 °Ca
| R group |

|

|

|

|
|---|---|---|---|---|
| OH | 1.3 | 0.5 | 0.6 | 0.2 |
| F | 2.3 | 1.5 | 1.0 | |
| COOMe | 1.2 | 0.2 | ||
| CONH2 | 1.5 | 1.3 | ||
| COO− | 0.5 | −0.2 | 0.2 | |
| CN | 2.8 | 3.0 | ||
| CH2OH | 0.4 | 0.5 |
σsvalues in pH units. The pKa of a piperidinium ion is decreased by the sum of the appropriate substituent values.
Figure 7.

Plot of the Negative Logarithm of the Rate Constant for Spontaneous Hydrolysis of 2,4-Dinitrophenyl Glycosides versus the Sum of Stereoelectronic Constants for the Substrate
The general concept was subsequently extended to the study of the influence of protecting groups on the stabilization of positive charge at the anomeric center (Table 9).74 The results are consistent with the general trends evident in the Wong RRV series for the influence of protecting groups on glycosylation (Table 5),54,100 with the corresponding Ley series,56,57 and with the more limited earlier studies of the Fraser-Reid,101,102 Paulsen,103 and Glaudemans labs.104 Notably, plots of substituted piperidinium pKa values against the rate constants for the methanolysis of the corresponding glycosides on activation with NIS/TfOH and against the Wong RRV values were more or less linear indicating that the general trends apply to glycosylation as well as to glycoside hydrolysis.74 In yet a further study, Bols and coworkers investigated the influence of configuration on the basicity of a series of aminosugars and confirmed the enhanced ability of axial alcohols to stabilize positive charge as compared to their equatorial isomers.105 Finally, it was demonstrated that while the stabilizing ability of multiple axial hydroxyl groups for positive charge in piperidinium ions, and by extrapolation glycosyl oxocarbenium ions, is approximately additive, steric interactions between multiple axial groups eventually intervene to limit the effect.106
Table 9.
pKa Values of Piperidinium Ions as Models for the Influence of Protecting Groups on Oxocarbenium Ions
| Structure | pKa |
|---|---|

|
6.4 |

|
6.9 |

|
7.7 |

|
8.7 |

|
3.4 |

|
3.5 |






Measured in 65:35 THF:H2O.
Estimated from NMR spectroscopic titrations.
The stabilizing effect of axial and pseudoaxial C-O bonds on nascent positive charge at the anomeric center manifests itself in the so-called super-armed donors developed by Bols and coworkers90,107,108 in parallel with early demonstrations by the Yamada group.89,109 In these systems repulsive gauche interactions between vicinal diequatorial siloxy groups,110 perhaps assisted by attractive van der Waals interactions between 1,3-diaxial siloxy groups,110,111 cause per-silylated glycosyl donors to preferentially adopt inverted chair and twist boat conformations rich in axial and pseudo-axial C-O bonds,112,113 resulting in correspondingly high reactivity as glycosyl donors (Figure 8).
Figure 8.
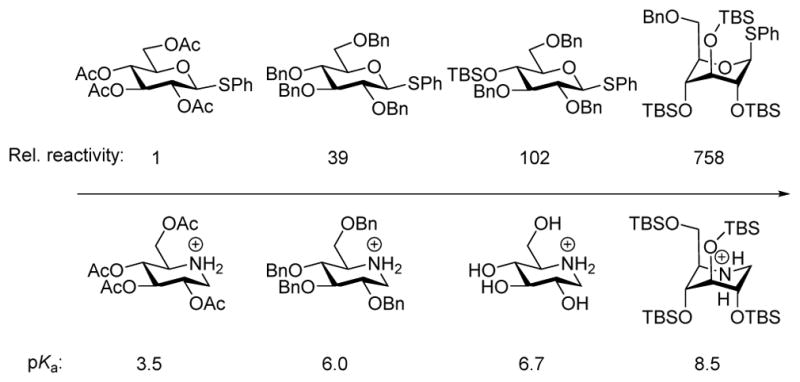
Comparison of the Relative Reactivity of Glycosyl Donors with the pKa of the Corresponding Piperidinium Ions
7 Facial Selectivity of Nucleophilic Attack on Putative Glycosyl Oxocarbenium Ions
The facial selectivity of nucleophilic attack on putative glycosyl oxocarbenium ions is governed by a number of factors, including their conformational equilibria, stereoelectronic factors, and steric effects. As discussed above most current thinking favors oxocarbenium ion conformations in which peripheral C-heteroatom bonds at all but the 2-position occupy pseudoaxial positions so as to provide electrostatic stabilization through space to the locus of positive charge. In the case of six-membered cyclic oxocarbenium ions taking up a half-chair conformation, and in the absence of pervading steric interactions, nucleophilic attack along the Burgi-Dunitz angle is then considered to take place preferentially in such a way as to provide the product directly in a chair-conformation with the newly introduced substituent in an axial position. Attack on the opposite face of the same half-chair conformer affords the product in an initial twist boat conformation and is, accordingly, considered less favorable.24,72,114 Indeed, the stereochemical outcomes of the reaction of the relatively weakly nucleophilic115 allylsilanes with partially substituted pyranosyl oxocarbenium ions conform to this model (Scheme 15).24,72,114
Scheme 15.

Facial Selectivity in the Reaction of Allylsilane with Per-O-benzyl Lyxopyranosyl Acetate
However, with more highly substituted systems such as the tetra-O-benzyl gluco- and mannopyranosyl ones steric effects can come to dominate. For example, nucleophilic attack on either face of the 3H4 conformer of the glucopyranosyl oxocarbenium ion is subject to steric impediment and reaction therefore takes place via the inverted 4H3 conformer with which it is in dynamic equilibrium (Scheme 16). Many substitution patterns have been studied by the Woerpel laboratory, and both Woerpel and van der Marel, Codée, and their coworkers have published extensive reviews to which the reader is referred for broader coverage of this important topic.24,68,72
Scheme 16.

Operation of Curtin Hammett-Type Kinetics in Nucleophilic Attack on the Gluco- and Mannopyranosyl Oxocarbenium Ions
Support for pseudoaxial attack on the α-face of the 4H3 conformer of the 2-deoxyglucopyranosyl oxocarbenium ion by weak nucleophiles is provided by the work of Blériot and coworkers. Thus, it was demonstrated that hydride transfer from perdeuteriocyclohexane to the experimentally determined proximal 4E conformation of the fully protonated tri-O-acetyl-2-deoxyglucopyranosyl oxocarbenium ion (Scheme 6) takes place with 98:2 selectivity on the α-face.65 It must be noted, however, that the type of Curtin Hammett kinetic scenario invoked in Scheme 16 becomes increasing unlikely as the energetic differences between the interconverting conformers increases, and also as the lifetimes of the intermediate oxocarbenium ions decrease with increasing numbers of electron-withdrawing C-O bonds.
When the nucleophile is a heteroatomic one the direct conversion of the oxocarbenium ion into a chair conformation with an axial C-X bond further benefits from the gain in stabilization due to the anomeric effect; a phenomenon that has come to be known as the kinetic anomeric effect.116,117 This latter effect has been challenged by Sinnott,118 whose views were endorsed by Cumpstey,119 on the grounds that it contravenes the Hammond postulate. Thus, nucleophilic attack on an oxocarbenium ion is considered to be highly exothermic and should have a correspondingly early transition state that is little influenced by the existence or not of anomeric effects in the product. This argument can be rebutted by consideration of the fact that the oxocarbenium ion is necessarily stabilized by the presence of a counterion and that product formation is accompanied by deprotonation, or related transformation, of the nucleophile, both of which reduce the exothermicity of the step and provide for a more advanced transition state.
The influence of the pyranosyl side chain conformation on the reactivity and selectivity of nucleophilic attack on glycosyl oxocarbenium ions has also been considered by Woerpel, using 1-alkoxyoxocarbenium ions as models. NMR spectra, backed up by computational modelling point to a 3H4 rich conformation for both the 2,3-dideoxy-4,6-di-O-benzyl-1-ethoxy and 2-deoxy-3,4,6-tri-O-benzyl-1-ethoxy glucopyranosyl oxocarbenium ions (Figure 9). In the less substituted system, the side chain adopts a conformation in which the C6–O6 bond projects over the face of the oxocarbenium ion and in doing so provides additional electrostatic stabilization to it. With the additional C-O- bond on the β-face at the 3-position steric effects cause the C5–C6 bond to adopt what is assigned as effectively a tg conformation.68,120 In a simpler system lacking C-O bonds on the pyranose ring the oxocarbenium ion adopts a half-chair conformation in which the side chain takes up a pseudoequatorial orientation and the gg-conformation about the C5–C6 bond (Figure 9).
Figure 9.

Dominant Solution Conformations of the 2,3-Dideoxy-4,6-di-O-benzyl-1-ethoxy, 2-Deoxy-3,4,6-tri-O-benzyl-1-ethoxy glucopyranosyl, and 1-Ethoxy-5-Benzyloxymethyltetrahydropyranosyl Oxocarbenium Ions with Emphasis on the Side Chain Conformation
In subsequent work, Kancharla and Crich found substantial differences in reactivity and selectivity in the glycosylation reactions of two sialyl donors differing simply in configuration at the 7-position. These differences were rationalized in terms of the differing and in this case competing steric and stereoelectronic influence of the side chain imposed by the change in configuration (Figure 10).121
Figure 10.

Influence of C7 Configuration on Reactivity and Selectivity in the Sialic Acids
The question of the influence of the side chain conformation was further probed by Dharuman and Crich who prepared a series of bicyclic mannopyranosyl donors with the C6–O bond locked in either the gg, gt, or tg conformations.122 The system with the enforced gg conformation was found to be the least β-selective (Figure 11) consistent with greater stabilization of incipient positive charge at the anomeric center by the spatially proximal axial C-O bond and with the earlier observation of the Bols and Crich groups on the relative rates of glycosidic bond hydrolysis in comparable systems (Table 4).50,51 In a study of the organocatalyzed addition of alcohols to glycals conformationally locked with the aid of a 3,4-di-O-tetraisopropyl disiloxane group Galan and coworkers found a 6-O-triisopropylsilyl protected system to be more α-selective than the corresponding 6-deoxy system. This observation was rationalized, with the help of a computational study, by the adoption of the gg-conformation in the siloxy system.123
Figure 11.

Enforced gg Conformation Reduces Equatorial Selectivity in Mannopyranosylation
Woerpel has studied the selectivity of nucleophilic addition to five-membered cyclic oxocarbenium ions, and has proposed a model whose central feature is attack on the inside face of an envelope conformation so as to minimize torsional strain in the transition state.124 When conflated with the propensity of a C-O bond at the 3-position to take up a pseudo-axial orientation so as to afford maximum electrostatic stabilization to the charge at the anomeric center, this results in high 1,3-cis-selectivity in the 2-deoxy series (Scheme 17).125 In the arabino-series, the combination of the syn-pentane interaction between the substituents at the 2- and 4-positions and the clash between the incoming nucleophile and the pseudo-axial 3-substituent are such that inside attack on the alternate envelope conformation is competitive and selectivity is lost (Scheme 17). When a cyclic protecting group spans the 3- and 5-positions selectivity is a function of the size of the appended ring and its nature.126,127
Scheme 17.

Woerpel’s Inside Attack Model for the Reactions of Furanosyl Oxocarbenium Ions
Considerable support for the Woerpel inside attack model was provided by Lowary and coworkers,128 and more recently by Filippov, Codée and coworkers who conducted a thorough experimental and computational study for the reaction of deuteriotriethylsilane with the conformationally unrestricted arabino-, lyxo-, ribo- and xylo-configurations of the oxocarbenium ion.129 In each case 1,2-cis selectivity was observed and rationalized on the basis of a full conformational analysis of the oxocarbenium ions, which were mostly found to react preferentially on the inside face of an envelope conformation as illustrated for the lyxose isomer (Scheme 18).
Scheme 18.

1,2-cis-Selective Attack on the Lyxofuranosyl Oxocarbenium Iona
aExperimental work employed benzyl ethers while computations were conducted with methyl ethers
A comparable inside attack model also has been used to rationalize the facial selectivity of simple nucleophiles for seven-membered cyclic oxocarbenium ions of possible relevance to the septanosides.130
8 tau Bond Model Interpretation of Stereoselective Addition to Oxocarbenium Ions
In the bent bond (τ) model advanced by Parent and Deslongchamps (Figure 2) oxocarbenium ions are formulated as two bent bonds above and below a nodal plane between the constituent C and O atoms (Figure 2). The equivalence of the two bonds in the parent system is perturbed by interaction with substituents at adjacent stereogenic centers such that the energy of a τ bond is reduced by the presence of an antiperiplanar electron-withdrawing C-X bond. Nucleophilic attack then is considered to take place antiperiplanar to the lowest energy τ-bond, and consequently syn to the electron-withdrawing C-X bond, and in such a manner as to generate the product in a staggered conformation with a lone pair at oxygen antiperiplanar to the newly formed C-nucleophile bond.31 Following studies with a number of simple bicyclic compounds, this model was used to explain the known stereoselectivity of O- and C-glycosylation of 4,6-O-benzylidene-protected gluco- and mannopyranosides. For the gluco-configuration, it is suggested that an intermediate oxocarbenium ion adopts the 4H3 conformation, which places the electron-withdrawing C2-O bond antiperiplanar to the τ-bond on the β-face thereby lowering its energy and directing attack to the α-face resulting overall in the axial product in a chair conformation (Scheme 19). Conversely, adopting current thinking, the corresponding mannosyl oxocarbenium ion is assigned to a B2,5-conformation with its C2-O bond antiperiplanar to the τ-bond on the α-face. The requirement for antiperiplanar attack of the nucleophile on the lowest lying bond therefore results in preferential reaction of the β-face and formation of the product in an initial 1S5 conformation (Scheme 19).31
Scheme 19.

tau-Bond Model for Nucleophilic Attack on the 4,6-O-Benzylidene-Protected Gluco and Mannopyranosyl Oxocarbenium Ions with Preferential Attack Antiperiplanar to the Lower Energy tau Bond (in Red)
9 Generation and Characterization of Covalently-Bound Activated Glycosyl Donors
In the simplest of all glycosylation reactions the initial glycosyl donor is displaced directly by the alcohol to give the glycosidic bond, however, such reactions are relatively rare. More commonly, the donor requires activation by a promoter into an activated form that then undergoes the glycosylation reaction. Two broad cases of this latter scenario can be envisaged. In the first and simplest case, the promoter simply increases the nucleofugacity of the initial anomeric leaving group. In the second, the initial leaving group is displaced by a new entity, derived either from the promoter itself or from an additive to the reaction mixture, affording a new and more reactive glycosyl donor in situ. In principle, activated donors of either kind can be detected spectroscopically when generated prior to the addition of the acceptor.
Studies of this type were initiated by the 1970 work of Igarashi and coworkers on the formation and isolation of glycosyl perchlorates on activation of glycosyl halides with silver perchlorate. Thus, it was demonstrated that an isolable perchlorate was obtained on reaction of either α- or β-per-O-acetyl glucopyranosyl chloride with silver perchlorate in either sulfur dioxide, diethyl ether, or toluene with the reaction rate correlating with solvent polarity (Scheme 20).131 Similarly, setting the precedent for subsequent observations,60,132,133 Igarashi and coworkers reported that activation of glycosyl chlorides by silver tetrafluoroborate resulted in the formation and isolation of glycosyl fluorides.132 Subsequent work by Schuerch and coworkers on the metathesis of glycosyl halides with silver sulfonates led to the formation and identification by NMR spectroscopy of a range of glycosyl sulfonates (Scheme 21),4,5,134–136 including the first glycosyl triflate although this latter was not sufficiently stable to permit spectroscopic characterization.137
Scheme 20.

Formation of 2-Chloro-2-deoxy-gluco- and Mannopyranosyl Perchlorates from the Corresponding Chlorides
Scheme 21.

Formation of a Glycosyl Toluenesulfonate by Metathesis with the Corresponding Bromide
However, it was not until the 1997 demonstration by Crich and Sun of the formation of glycosyl triflates from glycosyl sulfoxides on activation with triflic anhydride using low temperature NMR spectroscopy (Scheme 22)138 that such covalent intermediates came to be widely studied and accepted. The chemistry of the glycosyl sulfonates has been reviewed several times subsequently,139–146 and a comprehensive review of the detection of glycosylation intermediates by NMR methods was published in 2015147 to which the reader is referred for full coverage. Subsequent to the 2015 review the NMR spectroscopic characterization of a variety of activated glycosyl donors continues to be reported in the literature. Beyond numerous further glycosyl triflates,148–152 mesylates and other sulfonates (Scheme 23),153–155 these reports include the observation of a trans-fused bicyclic glycosyl thienium ion formed on activation of a 2-O-(2-thienylmethyl) trichloroacetimidate,156 other cis and trans-fused bicyclic glycosyl sulfonium ions,157 glycosyl oxyphosphonium salts (Scheme 24),158 and glycosyl oxysulfonium ions.148,150 In yet further studies NMR spectroscopy has identified the formation and subsequent displacement of glycosyl phosphates from glycosyl trichloroacetimidates in the presence of phosphoric acids,159 and of glycosyl fluorides from glycosyl trichloroacetimidates with retention of configuration on activation by BF3-etherate.133 The characterization and chemistry of the bicyclic glycosyl sulfonium ions and related species have been reviewed recently.160
Scheme 22.

Identification and Characterization of Glycosyl Triflates in the Mannopyranosyl Series
Scheme 23.

Formation and Characterization of Glycosyl Mesitylenesulfonates from Thioglycosides with Mesitylenesulfonyl Hydroxylamine
Scheme 24.

Formation and Characterization of Glycosyloxyphosphonium Salts
Returning to the detection of glycosyl oxocarbenium ions in more typical glycosylation solvents, it is important to note that the anomeric 13C chemical shift of a glycopyranosyl triflate138 (or of related covalent species)147 is around 100–105 ppm in the region of typical acetal carbons, and much removed from those of typical oxocarbenium ions (200–250 ppm). Any dynamic equilibrium therefore very strongly favors the covalent form over the ion pair and argues against the possibility of detecting the oxocarbenium counterion pair below the coalescence temperature.
As also described in full in a 2015 review,147 the glycosyl sulfonium,161 pyridinium, and even phosphonium ions162 have been characterized as possible intermediates in glycosylation reactions. Most recent work has involved intramolecular participation by tethered sulfides,163 selenides,164 and pyridine groups165 and so is not formally part of this review. Work by the Turnbull group is noteworthy in so far as X-ray crystal structures of two bicyclic glycosyl sulfonium ions were obtained (Figure 12), in spite of which the glycosylation reactions were considered more likely to take place via the SN1 manifold on the basis of a study of substituent effects and DFT calculations.23,166 Notably this conclusion is at odds with the subsequent kinetics-based one of Boons and coworkers (vide infra),163 whereby related bicyclic sulfonium ions were found to react via SN2-like mechanisms with inversion. The importance of kinetic studies in distinguishing associative and dissociative glycosylation mechanisms, to which we return below, is thereby underlined. In addition to characterization by low temperature NMR spectroscopy, simple monocyclic glycosyl sulfonium ions have also been characterized by cold-spray mass spectrometry.167
Figure 12.

Crystallographically Established Structures of Bicyclic Glycosyl Sulfonium Ions
In yet a further study on the stereodirecting effects of bicyclic sulfonium ions, Boltje and coworkers identified a cis-fused bridged bicyclic sulfonium ion by low temperature NMR spectroscopy in the triflic anhydride-mediated α-selective mannosylation of a carboxybenzyl-type donor (Scheme 25).168 A related trans-fused cyclic sulfonium ion was observed as the predominant species at −20 °C on activation of a mannurono-3,6-lactone-based donor indicative of stereodirecting participation by this engineered group (Scheme 26). However, the cognate 2-O-benzyl ether was also highly β-selective, and further studies, including NMR work at lower temperatures, identified participation by the 4-O-benzyl ether and/or a glycosyl triflate as possible underlying causes of the selectivity (Scheme 27).168
Scheme 25.

NMR Characterization of a cis-Fused Bicyclic Sulfonium Ion
Scheme 26.

Observation of a trans-Fused Bicyclic Sulfonium Ion as a Possible but not Necessary Intermediate in β-Selective Mannosylation with a 3,6-Mannuronolactone-Based Donor
Scheme 27.

Alternative Intermediates Tentatively Identified by NMR Spectroscopy in β-Selective Mannosylation with a 3,6-Mannuronolactone-Based Donor
The impact of additives beyond counterions obligatorily generated in the activation process on the outcome of glycosylation reactions has long been known, and many covalent intermediates generated in this manner have been characterized. Thus tetramethylurea, introduced by Hanessian and Banoub as a mild base to scavenge triflic acid,169 is known to form glycosyl uronium salts, which themselves are subsequently displaced by acceptor alcohols (Scheme 28).170 N,N-Dialkyl acetamides and formamides behave similarly and numerous O-glycosyl imidates have been characterized by NMR spectroscopy.171–175 Glycosyloxy sulfonium salts were characterized by Gin and coworkers following activation of hemiacetals with combinations of a sulfoxide reagent and triflic anhydride.176 Glycosyloxy sulfonium salts are also formed by reaction of glycosyl triflates with sulfoxide reagents,176 and on activation of thioglycosides with diarylsulfoxides and triflic anhydride.177,178 The NMR detection and characterization of intermediates in glycosylation reactions was reviewed in 2015,147 and the use of additives in general in 2014.179
Scheme 28.

Formation, Characterization and Displacement of a Glycosyl Isouronium Salt
The additive tetraethylammonium bromide was employed by Lemieux and coworkers in their halide-ion catalyzed approach to axial glycosides from glycosyl bromides. In this seminal work, in what amounts to a demonstration of the Curtin-Hammett principle, the added bromide was considered to displace bromide from the initial axial donor to populate the less stable but more reactive equatorial bromide, which itself was subsequently displaced by the alcohol (Scheme 29).7,180 Guindon and coworkers extended the Lemieux in situ anomerization concept to the synthesis of nucleosides. On activation of furanosyl hemiacetals with dimethylboron bromide they observed the formation of equilibrating mixtures of anomeric bromides by NMR spectroscopy that, on subsequent addition of a silylated base, underwent SN2-like displacement to give the corresponding nucleosides (Scheme 30).181
Scheme 29.

The Lemieux Bromide Ion Catalysis Concept for the Synthesis of α-Glycosides
Scheme 30.

Equilibrating Furanosyl Bromides Identified by NMR Spectroscopy in the Synthesis of Nucleosides
The question of in situ anomerization of donors is one that is not limited to the anomeric halides and one that is frequently invoked in discussions of mechanism.180,182 Actual studies of the rate of anomerization as compared to the overall rate of glycosylation are however sparse. Recently, D’Angelo and Taylor provided a solution to this problem in the form of NMR exchange spectroscopy (EXSY).155 In this method the rate of anomerization is obtained by irradiation of the anomeric signal of one of the anomers, while observing the integral for the second anomer for a range of mixing times. Conducting the experiment with a pair of 2,3,4,6-tetra-O-benzyl glucopyranosyl mesylates in CDCl3 necessarily containing one equivalent of an ammonium mesylate at 25 °C, the authors obtained rate constants of 1.02 × 10−2 and 9.3 × 10−4 s−1 for the β→α and α→β conversions, respectively, consistent with the NMR-determined equilibrium ratio of α:β = 10:1 (Scheme 31). Working with added tetrabutylammonium mesylate, a linear relationship between mesylate concentration and rate of anomerization was determined, thereby establishing the SN2-like nature of the inversion process.
Scheme 31.

Rates of In Situ Anomerization of Glycosyl Mesylates Determined by EXSY
In a study of the triphenylbismuth ditriflate promoted reactions of ethyl 2,3,4,6-tetra-O-benzyl-β-D-thiogalactopyranoside Pohl and coworkers found that epimerization to the α-anomer preceded the actual glycosylation reaction. Moreover, albeit the actual mechanism of activation is unclear in view of the lengthy induction periods observed, it was found that the α-anomer was the more reactive of the two under the reaction conditions described,182 thereby drawing attention to the fact that equatorial glycosides are not necessarily more reactive than their axial counterparts in all instances. Indeed, a subsequent study by Zhu and coworkers comparing the relative reactivities of the two anomers of a set of glucopyranosyl thioglycosides under NIS/TMSOTf activation revealed an interesting albeit unexplained dependence on the protecting group pattern and configuration (Figure 13).183 The role of anchimeric assistance by esters at the 2-position in the activation of the 1,2-trans-thioglycosides is clear from this reaction order, as noted earlier by Crich and Li184 with subsequent commentary from Bols and coworkers.185 It is equally clear that for the 2-O-benzyl series, the relative reactivity of the two anomers is a function of the remaining protecting groups and, contrasting with the work of Pohl and coworkers, either the configuration at the 4-position or the promoter or both. The observation of Yoshida and co-workers on the greater reactivity of an authentic NMR-characterized α-glucosyl sulfonium ion as contrasted to the β-anomer, rationalized simply in terms of the higher ground state energy of the axial sulfonium ion, may be relevant (Figure 14).167 Thus, in so far as the activation of thioglycosides generally can be considered to take place via the formation of glycosyl sulfonium-like ions, more tightly-associated and consequently more sulfonium-like thioglycoside promoter pairs would be expected to display greater reactivity in the sterically disfavored axial anomer.
Figure 13.

Reactivity Sequence of a Series of Thioglycosides Toward Activation by N-Iodosuccinimide and Trimethylsilyl Triflate
Figure 14.

Relative Reactivity Order of Two NMR Characterized Glycosyl Sulfonium Ions
Perhaps the most widely used additive, frequently employed in the form of solvent, is acetonitrile, which promotes the formation of equatorial glycosides in the absence of neighboring group participation through the in situ formation of axial glycosyl nitrilium ions. Intermolecular trapping studies with benzoic acids by the Sinaÿ and Fraser-Reid labs, and related observations by Briner and Vasella, demonstrated the validity of this concept (Scheme 32).97,186,187 Subsequent work by Crich and Patel, in which the nitrilium ions were captured by the acceptor alcohol to afford imidates, established that such glycosyl nitrilium ions are also formed in the manno and rhamnopyranosyl series,188 even though the usual effect favoring equatorial glycoside synthesis is not seen in such configurations.189,190 Intramolecular trapping experiments 191–197 further support the formation of glycosyl nitrilium ions as does the isolation and characterization of a cyclic imidate formed on activation of a donor carrying a 2-O-(2-cyanobenzyl) ether followed by addition of chlorobenzoic acid (Scheme 33).198 Initial spectroscopic evidence for glycosyl nitrilium ions was provided by Sinaÿ and coworkers working in deuterioacetonitrile at −30 °C. On the basis of 15N chemical shifts and long range proton couplings associated with molecular mechanics calculations, it was determined that the tetra-O-benzyl-α-D-glucopyranosyl nitrilium ion adopted a conformation close to the °S2 twist boat (Figure 15).199 Turnbull and coworkers subsequently characterized an α-D-xylofuranosyl nitrilium ion by NMR spectroscopy in deuterioacetonitrile solution at room temperature and noted that it was stable for several days under those conditions (Figure 15).200
Scheme 32.

Intermolecular Trapping of a Glycosyl Nitrilium Ion
Scheme 33.

Intramolecular Trapping of a Glycosyl Nitrilium Ion
Figure 15.

Glycosyl Nitrilium Ions Characterized by NMR Spectroscopy
In the sialic acid series, acetonitrile or mixtures of acetonitrile with dichloromethane are the solvents of choice for equatorial glycoside synthesis and the axial glycosyl nitrilium ions are assumed as intermediates,189,201–203 albeit no direct evidence of either spectroscopic or chemical nature was available for many years. Recently, however, working with an intramolecular alcohol as nucleophile Amarasekara and Crich observed an apparent cyclic imidate by mass spectrometry on activation of a thiosialoside in the presence of acetonitrile. On work up hydrolysis took place to afford the corresponding N-sialyl acetamide (Scheme 34).86
Scheme 34.

Isolation of an N-Acetyl Sialyl N-glycoside Indicative of Intermediate Nitrilium Ion Formation
In contrast to the usual assumption of the greater stability of the axial nitrilium ion, using PM3-level semi-empirical calculations, Martichonok and Whitesides estimated the equatorial nitrilium ion in the per-O-acetyl-N-acetylneuraminic acid methyl ester series to be 1.46 kcal.mol−1 more stable than its axial isomer.204 However, as such low level semi-empirical calculations are unlikely to treat the anomeric effect effectively205 and, presumably, neither solvent nor counterion were included, this result should be treated with considerable caution.
While the generation of highly reactive glycosyl donors on activation of an initial donor in the presence of an additive and absence of the acceptor is well-established, the participation of such intermediates in glycosylation reactions when the activation is conducted in the presence of the acceptor continues to be controversial for all but the glycosyl nitrilium ions. At the heart of the problem is the ability of the additive to compete with the acceptor alcohol for the capture of a first formed transient oxocarbenium ion or related species, as suggested in the original mechanistic proposal of Crich and Sun for the 4,6-O-benzylidene-protected β-mannosylation reaction (Scheme 35). In that study it was found that working with a 2-O-tert-butyldimethylsilyl mannosyl sulfoxide in ether rich mixtures of ether and benzene at −78 °C preactivation with triflic anhydride prior to addition of primary acceptors gave β-selective reactions, whereas activation in the presence of the acceptor afforded mainly the α-mannoside.206 This pattern of reactivity was framed in terms of a hypothesis whereby the preactivation protocol leads to the generation of the α-mannosyl triflate, that is primed for SN2-like reaction with the alcohol when it is subsequently added resulting in the formation of the β-mannoside, whereas activation in the presence of the alcohol simply results in α-selective trapping of the oxocarbenium ion by the alcohol (Scheme 35).207,208 It was soon recognized that dichloromethane gave better selectivities than ether in the preactivation protocol and allowed the use of secondary alcohols as acceptors.15,207,208 Consequently preactivation in dichloromethane became the conditions of choice enabling the successful synthesis of numerous β-mannosides in the Crich laboratories and elsewhere.145,209,210 However, reports soon emerged from other laboratories on the observation of modest to good β-selectivity on activation of various benzylidene-protected mannosyl donors in dichloromethane in the presence of the acceptor alcohol, indicating, reasonably, that triflate is not the only viable leaving group and that the mechanism of Scheme 35, derived from reactions conducted in ether, might be an over-generalization.211–219 Pertinently, Fukase and coworkers reported the β-selective couplings of either anomer of a N-phenyl trifluoroacetimidate on activation with trimethylsilyl tetrakis(pentafluorophenyl)borate, thus in the complete absence of triflate (Scheme 36),220 while Bols and coworkers, reported β-selective mannosylation reactions on activation of 4,6-O-silylene and benzylidene-protected mannosyl thioglycosides in the presence of only catalytic triflic acid or on activation with silver perchlorate in the absence of triflate (Scheme 37).218,219
Scheme 35.

Hypothesis for the Selectivity of 4,6-O-Benzylidene-Directed Mannosylation Based on the Order of Mixing
Scheme 36.

β-Mannoside Formation in the Absence of Triflate with Trimethylsilyl Tetrakis(pentafluorophenyl)borate as Promotor
Scheme 37.

β-Mannoside Formation in the Presence of Only Catalytic Triflate or in the Presence of Perchlorate
As a result of their findings with a trichloroacetimidate-based donor activated with trimethylsilyl triflate Weingart and Schmidt earlier argued that the benzylidene-directed mannosylation is best explained by a β-selective attack on a twist boat conformation of an oxocarbenium ion imposed by the presence of the benzylidene acetal.212 Fukase and coworkers suggested a 4E conformation for the putative benzylidene-protected mannosyl oxocarbenium ion for their system (Scheme 36) on the basis of B3LYP/6–31** calculations,220 while Bols and coworkers on the other hand suggested their results to be best explained by the β-selective attack on an intermediate of a 4,6-O-benzylidene or silylene protected oxocarbenium ion in the B2,5-conformation (Scheme 37), akin to the one deduced by Crich and coworkers (Scheme 9) on the basis of their intramolecular Sakurai reaction.83,84,221 Thus, over the course of approximately fifteen years three different laboratories, two with the aid of computational modelling,218–220 suggested three different conformations for the benzylidene-protected and related mannopyranosyl oxocarbenium ion and the selective formation of β-mannosides. Such oxocarbenium ion centric proposals are, however, not consistent with the kinetics of the benzylidene-directed mannosylation as determined either by KIE measurements or cation clock methods (vide infra).83,84,222 Thus, the anomer-independent selectivity observed by Fukase (Scheme 36) can be rationalized by in situ anomerization of the imidates with direct displacement of the protonated α-imidate by the acceptor alcohol. In this respect the close analogy between the N-phenyltrifluoroacetamide byproduct (or its silylated derivative) of the imidate coupling and the 3,3-difluorooxoindole nucleophilic catalyst discovered later by the Demchenko group223 (vide infra) is pertinent. An alternative explanation for the Bols observation (Scheme 37), and indeed those of other authors working in dichloromethane in the presence of sub-stoichiometric triflate is that triflate is simply a better nucleophile than the acceptor alcohol resulting in the formation of the α-mannosyl triflate as key intermediate in the usual way. Reactions conducted in the presence of perchlorate in place of triflate can be adequately explained by the formation of an α-mannosyl perchlorate that is subsequently displaced by the acceptor. Indeed, there is considerable literature evidence that both triflate and perchlorate are powerful nucleophiles toward cation-like electrophiles.224,225 It was further noted by Bols and coworkers that activation of a benzylidene-protected α-mannosyl trichloroacetimidate with BF3.OEt2 and the acceptor alcohol gave none of the expected glycoside owing to competing fluoride abstraction by the activated glycosylating agent. Inclusion of lithium triflate in the reaction mixture, however, restored the formation of the β-mannosides, clearly indicating triflate to be a better nucleophile than the fluoride ion donor and prompting the authors to comment on the beneficial catalytic effect of the triflate anion (Scheme 38).219
Scheme 38.

Role of Lithium Triflate in Overcoming Fluoride Abstraction on Activation of a Mannosyl Trichloroacetimidate in the Presence of the Acceptor by Boron Trifluoride Etherate
The hypothesis of triflate as a catalytic nucleophile is supported by the work of Zhu and Yu who noted that activation of a β-alkynyl benzoate with triphenylphosphinogold (I) triflate in the presence of acceptor alcohols afforded the β-mannosides with excellent selectivity, whereas the use of gold complexes with non-co-ordinating anions (triflimide, hexafluoroantimonate, and tetrakis(pentafluorophenyl)borate) resulted in the formation of the α-mannoside by direct SN2-like displacement of the glycosyl isochromenylium complex (Scheme 39).226 When generated in the absence of triflate or acceptor the β-chromenylium complex underwent isomerization to its more stable α-anomer, which was both spectroscopically observable and afforded the β-mannoside on addition of the alcohol.
Scheme 39.

Counterion Sensitive Outcome of a Gold-Catalyzed β-Mannosylation Reaction
In a similar vein Kowalska and Pedersen reported the formation of α-mannosides on activation of a β-mannosyl trichloroacetimidate with TMS triflimide, and contrasted this with the β-selective mannosylation of a comparable β-mannosyl trichloroacetimidate on activation with TMSOTf in the presence of the acceptor alcohol reported by Schmidt (Scheme 40).227 Clearly, the counterion-dependent selectivity is best explained by displacement of the activated trichloroacetimidate by a double inversion process involving an intermediate α-mannosyl triflate when the activating agent is TMSOTf.
Scheme 40.

Counterion Sensitive Outcome of a β-Mannosylation Reaction Employing β-Mannosyl Trichloroacetimidates as Donors
In a related work a combination of 1H and 19F diffusion ordered NMR spectroscopy (DOSY) was used to probe the activation of a glucopyranosyl trichloroacetimidate by TMSOTf, TMSNTf2, and BF3.OEt2.151 With TMSOTf in CD2Cl2 at −55 °C the imidate was rapidly converted to the α-glycosyl triflate, which was identified from its typical NMR signature and its DOSY characteristics, especially the incorporation of the glucosyl and triflate but not the trimethylsilyl moieties into a single molecular entity as judged by their diffusion coefficients. With TMSNTf2 on the other hand, under the same conditions the TMS and glucosyl residues, but not the NTf2 moiety, exhibited the same diffusion coefficients resulting in a species formulated as the N-silylated trichloroacetimidate in loose association with the triflimide counterion (Scheme 41). In the absence of acceptor alcohol the initially formed α-anomer was found to equilibrate to the β-anomer, which eventually predominated in the mixture (Scheme 41).151 When BF3.OEt2 was employed to activate the trichloroacetimidate in the absence of acceptor, the anomeric glycosyl fluorides were the only sugar-based substances identified.
Scheme 41.

Identification of Counter-ion Dependent Divergent Mechanistic Pathways by Diffusion Ordered NMR Spectroscopy
Sasaki and coworkers described the configuration dependent preparation of β-glycosides of mannurono-2,6-lactones,228 according to which the α-donors gave significantly greater β-selectivity than the corresponding β-anomers (Scheme 42). This observation held with trichloroacetimidate donors activated with a gold(III) chloride-thiourea combination or with iodide donors activated with triphenylphosphine oxide and Hunig’s base, and clearly suggests a high degree of association in the reactions of the α-donors as was confirmed by the concentration dependence of the selectivity.
Scheme 42.

Donor Configuration Dependent Selectivity in Glycosylation Reactions of 2,6-Mannuronolactone Based Donors
Further support for the action of perchlorate and triflate as nucleophiles in glycosylation reactions is provided by the work of Demchenko in which it was demonstrated that a somewhat disarmed 3,4,6-tri-O-benzoyl-2-O-benzyl-β-D-glucopyranosyl thioimidate and a comparable thioglucoside gave significantly greater α-selectivity when activated with silver perchlorate and silver triflate-based reagents than silver tetrafluoroborate or hexafluorophosphate ones (Scheme 43).229 The implication here is that reaction occurs through the higher energy β-glucosyl triflate and/or perchlorate in a Lemieux-type kinetic scheme. The reduced influence of the counterion with the analogous tetra-O-benzyl-protected donors simply arises from the more armed nature of this system and the consequent shift toward the more dissociative end of the mechanistic spectrum. Yet further examples of the influence of nucleophilic counterions were described by the Toshima and Poletti laboratories, this time for glycosylations in ionic liquids. Thus, it was demonstrated that the use of glycosyl fluorides and trichloroacetimidates was equatorially selective in imidazolium triflate, whatever the configuration of the initial donor, but depended heavily on the donor configuration in imidazolium hexafluorophosphates, tetrafluoroborates, and triflimides.230–232 Jensen and coworkers found enhanced β-selectivity in the glycosylation of simple alcohols by 4,6-O-benzylidene protected mannosyl 3,5-dinitrosalicylates under microwave heating in N-methylpyrrolidinone in the presence of added lithium triflate, suggestive of the possible intermediacy of a mannosyl triflate; no increase in selectivity was observed, however, with lithium perchlorate or with the corresponding trichloroacetimidate.233
Scheme 43.

Counterion Dependent Selectivity in Glucosylation Reactions
Demchenko and coworkers have employed 3,3-difluoro-2-oxindole (HOFox) as nucleophilic catalyst in BF3.etherate promoted reactions of glycosyl bromides. It was demonstrated that the rate of coupling of simple glycosyl bromides with acceptor alcohols was significantly enhanced in the presence of as little as 10 mol% of the catalyst, a feature that was attributed to the slow background reaction of the bromide with the alcohol as compared to its reaction with HOFox to give the more reactive OFox glycoside (Scheme 44).234
Scheme 44.
Dependence of Reaction Time on the Concentration of the Nucleophilic 3,3-Difluoro-2-oxindole Additive Suggestive of the Formation of a Covalent Intermediate
Overall, there is growing evidence for the participation of triflate, and other nucleophilic anions, through the formation of covalent glycosyl triflates and related species in glycosylation reactions even when they are present in only catalytic amounts. This is supported by the evidence of the nucleophilicity of triflate toward cation-like electrophiles in other branches of organic chemistry,225 and by a computational study pointing to the increased intrinsic reactivity of triflate as compared to methanol.235 Careful quantification of the nucleophilicity of the triflate and other anions and their inclusion in Mayr-type nucleophilicity scales115,236–238 would be an important contribution to the area. Also noteworthy in this respect is Mayr’s observation that DMSO is a significantly stronger nucleophile toward benzyhydryl cations than water or ordinary alcohols.1 This clearly points to the intervention of the reagent as nucleophile in systems activated by combinations of sulfoxides and triflic anhydride, and consequently of glycosyloxy sulfonium ions as intermediates even when the activation is conducted in the presence of the acceptor alcohol.177,239
A number of studies have focused on the use of chiral phosphoric acids, either alone or in combination with thioureas and other additives, as promoters of glycosylation reactions, and the field has been reviewed.240,241 Fairbanks and coworkers were the first to describe the use of binol-derived phosphoric acid catalysts for the activation of a per-O-benzyl α-D-galactopyranosyl trichloroacetimidate; significant changes in anomeric selectivity were observed as a function of the chirality of the catalyst pointing to the inclusion of the phosphate in the product determining step of the reaction (Scheme 45).242 In related work with a per-O-benzyl-α-D-glucopyranosyl trichloroacetimidate donor, Toshima and coworkers demonstrated that the anomeric selectivity of the reaction was a function of the combination of the absolute configuration of both the binol-derived phosphoric acid catalyst and the simple racemic alcohols employed as acceptors, the later enabling kinetic resolution of the acceptor. However the phenomenon did not extend directly to the use of the corresponding β-trichloroacetimidate, prompting the authors to suggest a three component cyclic transition state in which the phosphoric acid plays the dual role of Brønsted acid, activating the donor, and of Brønsted base, activating the acceptor (Figure 16), rather than the formation of covalent glycosyl phosphate intermediates or phosphate-based ion pairs with the oxocarbenium ion.243 Subsequent work by Bennett and coworkers with tri-O-benzyl 2-deoxyglucopyranosyl trichloroacetimidates further underlined the interdependence of the anomeric configuration of the donor with the absolute configuration of the binol-phosphate catalyst and thus the role of the chiral anion in the mechanism.244
Scheme 45.

Dependence of Selectivity on the Absolute Configuration of an Added Phosphoric Acid
Figure 16.

Three Component Transition State with Dual Function of the Phosphoric Acid
It was subsequently demonstrated by Nagorny and coworkers that the above kinetic resolutions of racemic alcohols by glycosyl trichloroacetimidates in the presence of chiral phosphoric acids could be extended to the regioselective glycosylation of macrolide-based polyols and to the desymmetrization of meso-diols, and in particular a protected version of 2-deoxystreptamine.245,246 A computational study of the mechanism of the polyol desymmetrization indicated a double inversion sequence to be lower in energy than one involving oxocarbenium ion phosphate ion pairs. Subsequent NMR experiments identified the formation of glycosyl phosphates from the trichloroacetimidate donors, and demonstrated that these phosphates themselves functioned as donors, with inversion of configuration, on addition of model diols (Scheme 46).245
Scheme 46.

Regioselective Polyol Glycosylation with Formation of an Intermediate Glycosyl Phosphate245
Nagorny and coworkers also conducted a detailed study of the mechanism of the spirocyclization of 1-(n-hydroxyalkyl)glycals under catalysis by chiral phosphoric acids. A Hammett study using a benzo-fused glycal demonstrated the build-up of considerable positive charge at the anomeric center at the transition state, while use of a 2-deuterio substrate revealed the syn-selective nature of the addition and ruled out the possible intermediacy of a long-lived fully-fledged oxocarbenium ion as intermediate. Computational studies then suggested an asynchronous concerted mechanism in which the phosphoric acid serves as a proton shuttle between the nucleophilic alcohol and the 2-position of the glycal (Scheme 47).159
Scheme 47.

Concerted Cyclization of a Tethered Alcohol onto a Glycal in the Presence of a Chiral Phosphoric Acid
10 Dual Acid-Base Catalysis in Glycosylation
In addition to the above described studies on the influence of chiral phosphoric acids on glycosylation, the concept of dual acid-base catalysis of glycosylation reactions is experiencing increased popularity. This approach, patterned on the eight-membered cyclic transition state invoked for the invertive substitution of glycosyl trichloroacetimidates by phosphoric acids247 and some carboxylic acids,248 has been explored by Schmidt,249 and others.241 Considerable work has been done with thioureas as catalysts both for the addition of alcohols to glycals resulting in the formation of 2-deoxyglycosides and in the activation of glycosyl donors, and the field has been reviewed recently.241,249 Multicomponent transition states have been drawn by several authors to account for the synergy exhibited by combinations of a thiourea and a second catalyst, sometimes a Brønsted acid, and sometimes a transition metal, but these are rarely supported by physical evidence and so are only briefly commented here.241,249 For example, Schmidt and coworkers proposed a four-component transition state to account for the co-operativity observed in the activation of trichloroacetimidates between a thiourea and a diaryl phosphoric acid (Figure 17).250 Surprisingly in view of the generally greater nucleophilicity of thioureas compared to ureas, and of the demonstrated participation by tetraalkyl ureas,170 glycosyl isothiouronium ions have yet to be advanced as potential intermediates in thiourea-catalyzed glycosylations.
Figure 17.

Hypothetical Four-component Transition State Accounting for the Roles of a Thiourea and a Phosphoric Acid in Organocatalyzed Glycosylation with a Trichloroacetiomidate Donor
One theme that does consistently emerge is the use of 2-deuterioglycals to probe the stereoselectivity of addition of alcohols as reported, inter alia, by Galan and coworkers, and again rationalized on the basis of a multicomponent transition state (Scheme 48).251
Scheme 48.

Use of a 2-Deuteriogalactal to Probe the Selectivity of Addition and a Hypothetical Four-Component Transition State
The most compelling work in this area, however, was described by the Jacobsen group and involves the use of a chiral macrocyclic bisthiourea to promote SN2-like glycosylations of armed axial glycosyl chlorides. Computational studies and kinetic isotope effect experiments were advanced in support of a mechanism in which one of the two thioureas serves to co-ordinate and activate the departing chloride, while the second functions as a hydrogen bond acceptor in such a way as to position and activate the acceptor alcohol (Figure 18).252,253
Figure 18.

Computationally Derived Transition State for Macrocyclic Bisthiourea Promoted SN2-Like Galactosylation252,253
Catalysis of various glycosylation reactions by gold(I) and (III) complexes has been described by several authors and reviewed recently,241 but mechanistic studies are sparse. Nevertheless, Peng and Schmidt have demonstrated by low temperature NMR spectroscopy that tetra-O-benzyl-α-D-glucopyranosyl trichloroacetimidate and gold(III) chloride form only a very loose interaction in CDCl3 at −55 °C, whereas greater changes were observed in the NMR spectrum of isopropanol on addition of AuCl3. This led to the suggestion of dual acid base catalysis of the glycosylation reaction with the metal salt activating both the donor and the acceptor and resulting in an SN2-like displacement with a seven-membered cyclic transition state. Gold(I) chloride catalysis of the same reaction was stated to follow a similar pattern.254
11 Influence of the Acceptor
The influence of the acceptor on the outcome of glycosylation reactions, evident to every practitioner of the art, and the source of Paulsen’s oft-quoted comment on every glycosylation reaction being different,255 is illustrated many times over in Barresi and Hindsgaul’s catalog of all glycosylation reactions published in the year 1994.256 Systematic studies, however, are more limited and begin with the 1991 publication of Spijker and van Boeckel comparing the silver triflate-promoted reaction of a pair of enantiomeric fucosyl bromides with a single enantiomerically pure acceptor (Scheme 49),257 for which the reversal in selectivity was interpreted in terms of Horeau’s concept of double diastereodifferentiation and matched and mismatched pairs.258,259 Further examples of the same phenomenon were reviewed through 2009 by Bohé and Crich,260 and the effect continues to be invoked even if not always rigorously established.261–265 It follows260 from the concept of double diastereodifferentiation that different glycosyl donors will exhibit differing regioselectivity in their coupling reactions with di- and polyols and, self-evidently, with different acceptor alcohols as commented in the reciprocal donor acceptor selectivity principle of Fraser-Reid and coworkers. 266,267
Scheme 49.

Double Diastereodifferentiation in Glycosylation
Beaver and Woerpel correlated diastereoselectivity with acceptor reactivity in the coupling reactions of a series of primary alcohols of varying nucleophilicity with 2-deoxy glucopyranosyl thioglycosides on activation with N-iodosuccinimide in acetonitrile (Table 10). The inverse correlation of selectivity with nucleophilicity was interpreted in terms of diffusion-controlled unselective trapping of intermediate oxocarbenium ions by the highly reactive ethanol gradually being replaced by stereoelectronic control with decreasing nucleophilicity of the acceptor.268 Similarly, trimethylsilyl cyanide reacts in a less selective diffusion-controlled manner with tetrahydropyranosyl oxocarbenium ions than the less nucleophilic allyltrimethylsilane.269
Table 10.
Influence of Acceptor Nucleophilicity as Judged by the Field Inductive Effect Parameter F on Glycosylation Stereoselectivity

| ||
|---|---|---|
| RCH2OH | F | α:β |
| CF3CH2OH | 0.38 | 83:17 |
| F2CHCH2OH | 0.29 | 67:33 |
| FCH2CH2OH | 0.15 | 56:44 |
| BrCH2CH2OH | 0.14 | 55:45 |
| ClCH2CH2OH | 0.13 | 56:44 |
| CH3CH2OH | 0.00 | 51:49 |
Codée and coworkers extended Woerpel’s concept to a broader range of glycosyl donors, examining the selectivity of benzylidene protected gluco and mannopyranosyl donors as well as mannuronic acid donors with alcohols of varying nucleophilicity.270 For each of the three systems the more reactive alcohols were found to afford greater β-selectivity than their less reactive counterparts (Table 11). These results were interpreted in terms of the more reactive alcohols being capable of SN2-like displacement of covalent glycosyl triflates in contrast to their less nucleophilic counterparts, which necessarily follow a more SN1-like mechanism. For each of the three donors, the shift from SN2-like to SN1-like mechanisms and selectivity occurred at a different point on the scale of alcohol nucleophilicity, consistent with the general pattern of stability of the glycosyl triflates. The same pattern of nucleophile dependent selectivity was subsequently observed with a series of 2-deoxy-2-azidofucopyranosyl donors,148 with a set of 4,6-O-benzylidene and 4,6-O-silylene-protected 2-deoxy-2-azido glucopyranosyl donors,150 and other systems.149 The latter two studies also included an investigation of the influence of the donor protecting groups on the selectivity, whose results were consistent with the overall picture. Thus, the more electron-withdrawing the donor protecting groups, the more stable the covalent glycosyl triflate or comparable species, and the greater the spectrum of alcohol nucleophilicity compatible with the associative reaction mechanism (Table 12).
Table 11.
Influence of Acceptor Nucleophilicity on Stereoselectivity with Different Donors
|
α:β, % yield |
 α:β, % yield |
 α:β, % yield |
|
|---|---|---|---|
| EtOH 7.4a, 0.01b |
1:5, 70 | 1:10, 68 | 1:8, 95 |
| FC2H4OH -a, 0.15b |
1:5, 86 | 1:3, 70 | 1:6, 70 |
| F2C2H3OH -a, 0.29b |
1:5, 90 | 5:1, 70 | 1:5, 87 |
| F3C2H2OH 1.11a, 0.38b |
1:4, 78 | >20: 1, 64 | 1:2.5, 85 |
| AllylTMS 1.68a, -b |
<1:20, 44 | >20:1, 42 | >20:1, 40 |
Mayr’s nucleophilicity parameter.
Field inductive effect parameter
Table 12.
Influence of Acceptor Nucleophilicity on Stereoselectivity as a Function of Donor Protecting Groups
 α:β, % yield |
 α:β, % yield |
 α:β, % yield |
|
|---|---|---|---|
| EtOH 7.4a, 0.01b |
1:10, 68 | <1:20, 65 | <1:20, 86 |
| FC2H4OH -a, 0.15b |
1:2.8, 70 | 1:5, 79 | 1:6.5, 83 |
| F2C2H3OH -a, 0.29b |
5:1, 70 | 2.7:1, 76 | 2.7:1, 84 |
| F3C2H2OH 1.11a, 0.38b | >20:1, 64 | >20:1, 82 | >20:1, 86 |
Mayr’s nucleophilicity parameter.
Field inductive effect parameter
The differences between the observations of the Woerpel (Table 10) and Codée (Tables 11 and 12) laboratories can be reconciled on the basis of the vastly different reactivities of the donors studied, the different solvents employed, and the associated shifts in the key equilibria between the covalently bound intermediates and the various ion pairs. Thus, the Woerpel system lacking the strongly electron-withdrawing C-O bond at the 2-position of the donor, and employing the more polar acetonitrile as solvent, is expected to be much closer to the SN1 end of the mechanistic spectrum.
While the tendency is to interpret the above correlations of selectivity with alcohol nucleophilicity in terms of a shift away from SN2 toward SN1 mechanisms with less nucleophilic alcohols, other explanations are possible. Thus, on the basis of the evidence provided it cannot be ruled that the more reactive alcohols are sufficiently nucleophilic to take part in SN2 reactions with the more stable and highly populated of the two equilibrating activated covalent anomeric donors, while the less reactive alcohols react through an SN2-like process with the more reactive, but less populated anomer of the activated donor. The shift of the selectivity inflection point from one donor to another (Tables 11 and 12) in such a Curtin-Hammett scenario is then related to the change in anomeric effect from one donor to another, which effectively changes the energy difference between the two anomers of the activated donor. For example, it is well known that the anomeric effect in mannopyranose is greater than in glucopyranose such that reaction through the higher energy anomer can be expected to be less important for a given mannose donor than its glucose counterpart. Equally, it has been demonstrated in several systems that for a given configuration of donor the protecting group system affects the magnitude of the anomeric effect.271,272 Distinctions between the two hypotheses can only be made on the basis of kinetic analyses or through the investigation of transition states by the measurement of kinetic isotope effects as discussed further below.
Pedersen, Bols, and coworkers prepared an extensive series of aminodeoxy sugars and determined the pKa value of the conjugate ammonium salts, some of which are presented in Table 13.105 It was found that acidity correlates with the number of vicinal C-O bonds, such that the most basic amines have fewer vicinal C-O bonds. For an equal number of vicinal C-O bonds the greatest reduction in amine basicity was found for an antiperiplanar relationship with the C-N bond. Taking amine basicity as a surrogate for alcohol nucleophilicity, and ignoring steric effects, this study provides a measure of understanding of the relative reactivity of acceptor alcohols that agrees with oft–quoted trends.255
Table 12.
pKa Values of Conjugate Acids of Aminosugars as Models for the Nucleophilicity of Hydroxy Groups on Sugar Rings
| Structure | pKa |
|---|---|

|
7.5 |

|
7.8 |

|
6.8 |

|
8.9 |
| Structure | pKa |
|---|---|

|
7.9 |

|
8.0 |

|
7.3 |

|
8.9 |
| Structure | pKa |
|---|---|

|
7.2 |

|
8.1 |

|
7.2 |

|
9.0 |
The regioselective glycosylation of polyols with the aid of catalysts designed to enhance the nucleophilicity of one among two or more hydroxyl groups is a field of considerable current interest with the most elegant advances made by the Taylor and Toshima laboratories. Most importantly, Taylor and coworkers provided kinetic evidence for the involvement of borinate complexes of diols in the rate determining step of two distinct glycosylation reactions, thus pointing to SN2-like mechanisms, one heterogeneous and one homogeneous (Schemes 50 and 51).155,273 The latter case employing the glycosyl mesylates is particularly revealing. Thus, in the absence of the catalyst the reaction was first order in acceptor and selective for formation of the α-anomer of the product indicating an SN2-like glycosylation in which the relatively weak nucleophile displaces the leaving group from the more reactive of the two equilibrating glycsoyl mesylates. On addition of the catalyst the reaction was found to be zero order in alcohol but first order in catalyst and to be β-selective suggesting that the more nucleophilic catalyst-acceptor complex preferentially rapidly displaces the leaving group from the more concentrated of the two equilibrating mesylates (Scheme 51).
Scheme 50.

Complete Kinetic Analysis of a Borinate-Catalyzed Heterogeneous Glycosylation Indicating an Associative Mechanism
Scheme 51.

Complete Kinetic Analysis of a Borinate-Catalyzed Homogeneous Glycosylation Indicating an Associative Mechanism (PMP = pentamethylpiperidine)
Work from the Toshima laboratory employed arylboronic or diarylborinic acids to promote the regio and stereoselective coupling of diols with 1,2-anhydrosugars. Boronate or borinate esters serve as Lewis acids to activate the epoxide and in doing so also both activate and position the diol for coupling to the transient oxocarbenium ion. The general concept is illustrated for the formation of a β-mannoside (Scheme 52).274–276 It is noteworthy that, in contrast to the enormous majority of glycosylation reactions, the success of this method depends on the inclusion of several equivalents of water to hydrolyze the bridging boronate ester that is the immediate product of the reaction and so facilitate turnover. In the complete absence of water this bridging boronate itself catalyzes off-cycle glycosylations resulting in complex reaction mixtures.
Scheme 52.

Boronic Acid Catalyzed β-Mannosylation of with a 1,2-Anhydromannopyranose Derivative
The extensive work of the Vasella laboratory on the determination of intramolecular hydrogen bonding networks in carbohydrate polyols and the relative acidities of the various individual hydroxyl groups should prove of considerable use as the field of selective polyol glycosylation advances in the coming years.94,277–279 Also of interest in this context is an extensive study by Schmidt and coworkers on the reasons underlying the regioselective monobenzoylation of polyols by benzoyl cyanide,280 which can be summarized as selective activation of the most acidic hydroxyl group by the weakly basic cyanide ion.
12 Influence of Solvent
As with any reaction, solvents exert a significant influence on the outcome of glycosylation reactions.281,282 With regard to the general glycosylation reaction this influence can be readily understood in terms of the differing ability of solvents to stabilize positive and negative charge. Unfortunately, with the exception of nitrile-based solvents discussed above, few studies extend beyond the observation of solvent-based changes in glycosyl stereoselectivity. Participation by ethereal solvents in the form of O-glycosyl oxonium ions is frequently invoked in explanation of anomeric stereoselectivity phenomena, but actual physical organic evidence for the existence of such species has yet to be provided.190
In general, the greater the ability of the solvent to support charge separation and/or to stabilize oxocarbenium ions and their associated anions through solvation, the closer the mechanism to the SN1 end of the spectrum. Taken to the extreme, superacidic solvents enable the direct NMR spectroscopic observation of at least a limited range of glycosyl oxocarbenium ions as discussed above.65 Conversely, lower polarity, less solvating systems should promote more SN2-like glycosylations. Such was demonstrated by Crich and Dudkin who observed improved β-mannosylation of a tetrabutylammonium dialkyl phosphate in toluene at −78 °C compared dichloromethane (Scheme 53).283 The use of such non-polar solvents to promote SN2-like reactions, however, is limited by their ability to dissolve most common glycosyl donors, acceptors, and promoters in sufficiently high concentrations. A more extensive study of solvent effects on C- and O-glycosylation selectivity later was conducted by Woerpel and coworkers who concluded that trichloroethene is the optimum solvent for intended SN2-like glycosylations at low temperature in view of its low polarity, low freezing point, and ability to dissolve typical substrates.284
Scheme 53.

Influence of Solvent in the Formation of a Mannosyl Phosphate
A recent study disclosed the use of supercritical CO2 as solvent for glycosylation of alcohols by glycosyl halides. The influence of pressure, temperature, protecting and leaving groups on these reactions, which were conducted in the absence of external promotor, were noted but a satisfactory explanation awaits further work.285
The conventional wisdom on concentration and solvent effects in glycosylation reactions has been challenged by Kononov and coworkers who advanced the concept of supramolecular aggregates, or supramers, in support of their observations.286–289 The supramer hypothesis adopts the notion of concentration-dependent mesoscale structuring of solvent and solute according to which mesoscale inhomogeneities and microemulsions influence reaction outcome. The concept has found broadest application in sialidation reactions where intermolecular hydrogen bonding networks between amide residues afford a mechanism for mesoscale organization. As presented in a recent review, the reactivity and selectivity of a given donor or acceptor vary according to the extent to which it is involved in a hydrogen bonded network. Reasonably, the degree of association is considered to vary with solvent and concentration, and can be disrupted by the inclusion of tertiary imides, such as N-methyl acetimide, capable of accepting but not donating hydrogen bonds, with supporting evidence provided by IR measurements. The observation of non-linear effects and discontinuities in correlations of concentration with physical phenomena of the bulk solution (IR absorptions and specific rotations) and with reaction outcomes (yield and selectivity) resulted in the proposal of mixed supramers. While the concept originated, and continues,286,287,290 with hydrogen-bonding as the organizing motif for supramer formation, it has recently been extended to other systems. Thus, the reactivity and selectivity of an arabinofuranosylation sequence were demonstrated to vary in a non-linear fashion, with the optimal concentration correlating loosely with a discontinuity in the concentration dependence of specific rotation of a solution of donor (Figure 19).291
Figure 19.

Correlation of Anomeric Selectivity and Specific Rotation of the Solution in an Arabinofuranosylation Reaction
13 Influence of Temperature
Consideration of the general glycosylation mechanism (Scheme 2) suggests that associative SN2-like glycosylation mechanisms, with their greater entropic penalty, will be favored over dissociative SN1-like mechanisms at lower temperatures. It will also be noted that the various equilibria of Scheme 2 are shifted toward the covalent donors and away from the ion pairs at lower temperatures because of the reduced entropy, again favoring the associative SN2-like mechanisms. Application of these fundamental principles simply suggests that selectivity for the SN2-like mechanisms can be optimized by lowering the reaction temperature. Unfortunately, while there are many reports on the influence of reaction temperature on the outcomes of glycosylation reactions, few are systematic. A good example of this effect, however, is given by the influence of temperature on the selectivity of sialidation reactions conducted with several 5-azido-5-desacetamido-sialyl donors. Thus it was reported by several groups over a number of years that such donors only gave good yields of the desired equatorial glycosides in couplings to primary alcohols.292–296 This led to the general impression that azido protected sialyl donors were poorly selective with secondary alcohols and their consequent abandonment in most synthetic schemes.297 It was subsequently found, however, that if a suitably reactive donor is used and activated at −78 °C, as opposed to the earlier temperatures of ≥-40 °C excellent equatorial selectivities are obtained in the 5-azido-5-desacetamido sialoside series.298
Notwithstanding the above example, it must be understood that the use of lower reaction temperatures necessitates the use of longer reaction times such that a compromise must frequently be found. The complexity of the problem is nicely illustrated by a recent study from the Kononov lab, which revealed a discontinuity in the Arrhenius-type plot of selectivity against reciprocal temperature in the above mentioned arabinofuranosylation (Figure 19).291 Such break points are indicative of changes in the predominant reaction mechanism with temperature, and are most readily understood in terms of the temperature dependence of the SN1-SN2 threshold. Kononov has pointed out, however, that alternative explanations exist, most notably the influence of temperature on the mesoscale structuring of the solution.291
Variable temperature NMR spectroscopy can be used to determine the influence of temperature on the activation of glycosyl donors and on the stability of activated intermediates. Thus, Lowary and coworkers used VT NMR spectroscopy to investigate the activation of isomeric 2,3-anhydrofuranosyl sulfoxides by triflic anhydride, resulting in the identification of −40 °C as the optimal reaction temperature below which formation of the critical glycosyl triflate was in efficient (Scheme 54).299
Scheme 54.

Use of VT NMR Spectroscopy to Identify the Minimum Temperature for Clean Activation
VT-NMR has been widely applied to determine decomposition temperatures of numerous glycosyl triflates.138,147,148,150,300 Such studies, carried out under standard conditions of solvent and concentration, provide valuable information on the role of protecting groups on the stability of activated glycosyl donors. They should not, however, be interpreted as absolute measures of the stability of a given system as it follows from the general mechanism and the common ion effect that operating in the presence of added triflate anion will increase the decomposition temperature of any given glycosyl triflate (and retard substitution at any given temperature).
14 Hydrogen Bonding and H-Atom Transfer
The notion that acceptor-donor hydrogen bond formation plays a critical role in many glycosylation reactions is attributed to Whitfield and co-workers who observed such interactions in their computational studies of alcohols with oxocarbenium ions,301 leading them to suggest that such interactions can be important factors in the stereoselectivity of glycosylation reactions. For example, it was suggested that β-mannosylation is directed by a hydrogen bond from the acceptor to O-3 of the oxocarbenium ion in a 1S5 twist boat conformation, whereas α-glucosylation is facilitated by hydrogen bonding to O-2 of the oxocarbenium ion (Figure 20).301 Comparable hydrogen bonds were subsequently found by Crich, Pratt and coworkers in their computational study of SN2-like transition states for the 4,6-O-benzylidene-directed β-mannosylation and α-glucosylation reactions.222
Figure 20.

Computed Methanol Adducts with the 4,6-O-Benzylidene Protected Manno- and Glucopyranosyl Oxocarbenium Ions Including Donor-Acceptor Hydrogen Bonds
The importance of the computationally revealed hydrogen bonds in the 4,6-O-benzylidene-directed β-mannosylation and α-glucosylation was questioned early by Crich and Sharma who pointed out that C-mannosylation and C-glucosylation of the same donors with typical carbon-based nucleophiles exhibits the same sense of facial selectivity as O-glycosylation albeit without the possibility of hydrogen bonding.302 However, subsequent kinetic evidence revealing the mechanistic divergences of the C- and O-mannosylations and glucosylations in questions enables reconciliation of the two opposing viewpoints.84 Thus, it is possible that the SN2-like β-O-mannosylation is facilitated by acceptor-O3 hydrogen bonding, whereas the SN1-like β-C-mannosylation on the B2,5-conformation of the oxocarbenium ion has no such requirement.
An important conclusion drawn from the computational studies of Whitfield on acceptor-donor hydrogen bonding concerns the timing of proton transfer in glycosylation. The authors found that early proton transfer is associated with bimolecular SN2-like reactions, whereas late proton transfer is a feature of monomolecular SN1 reactions.303–306
The role of acceptor-donor hydrogen bonding has gained broader acceptance since Demchenko’s discovery of the stereodirecting effects of remote hydrogen bond-accepting picolyl and picolinyl groups in the donor as illustrated, for example, by the β-mannosylation presented in Scheme 55.307–310 It must be noted that, while the effect is most readily understood in terms of positioning of the nucleophilic alcohol for attack on one face of an oxocarbenium ion (Scheme 55), it is equally consistent with proximity-induced acceleration of an SN2-like glycosylation. In other words acceptor-donor hydrogen bonding serves to provide a high effective molarity of the acceptor on one face of the system and so accelerates associative displacement from the appropriately configured covalent donor.
Scheme 55.

Stereodirecting Donor-Acceptor Hydrogen Bonding Aided by the Presence of a Picolyl Group in the Donor
Subsequent work in the Demchenko and other groups307–312 has extended the central idea to a number of other donor systems, and it is clear that stereodirecting donor-acceptor hydrogen bonding is a concept that is ripe for further exploitation, both in terms of the development of new systems, and of simplifying explanations to previous experimental observations. In this latter vein, Boons and coworkers computationally located a hydrogen bond from the incoming acceptor to the carbonyl group of an equatorial ester at O3 in their α-glucosylation system (Figure 21) thereby providing a possible explanation for the greater selectivity observed with the ester protected systems,163 and, incidentally, the high levels of α-selectivity previously observed by numerous investigators with donors carrying esters at the 3-position.313–316 As physical organic studies beyond the computational work have yet to be conducted and as the area has been recently reviewed306 it is not covered further here.
Figure 21.

Computationally-Located Transition State for the SN2-Like Opening of a Bicyclic Sulfonium Ion Reveals a Key Hydrogen Bond to the O3 Carboxylate
15 Kinetic Measurements
Despite the many studies catalogued above on the detection and characterization of glycosylation intermediates, a true distinction between associative and dissociative glycosylation mechanisms can only be made with the help of kinetic measurements.
Rhind-Tutt and Vernon carried out kinetic studies of the reactions of 2,3,4,6-tetra-O-methyl-α-D-glucopyranosyl and α-D-mannopyranosyl chlorides with two acceptors of different nucleophilic strength (methanol and thiophenoxide).3 The methanolyses of both donors were found to follow first order kinetics indicative of SN1-like reactions. However, whereas an almost completely inverted product was obtained from the glucosyl donor, the mannosyl isomer led to a ~1:1 mixture of anomeric glycosides. The almost complete inversion in the unimolecular glucosylation reaction was attributed to the shielding effect of the departing anion in an initial ion-pair (CIP). The addition of external chloride to the methanolysis of the glucosyl donor resulted in a sharp increase in the proportion of the α-glycoside, which was rationalized in terms of chloride competing with methanol for the initial ion pair, resulting in the formation of an intermediate β-glucosyl chloride with subsequent rapid displacement by methanol leading to the α-glucoside (Scheme 56). This important but often overlooked paper therefore contains the first published application of the Winstein ion pair theory to glycosylation and the elements of what would later become Lemieux’s halide-ion catalyzed approach (Scheme 29) to the synthesis of axial glycosides. The lack of selectivity observed with the mannosyl chloride was attributed to the involvement of a relatively free oxocarbenium ion.3
Scheme 56.

Kinetics Derived Early Depiction of the Application of Winstein’s Ion Pair Theory to Glycosylation
The reaction of the glucosyl donor with thiophenoxide ion in propanol was found to follow second-order kinetics and to take place with clean inversion of configuration clearly supportive of an SN2 mechanism. However, with the mannosyl donor the predominant product was a mixture of propyl mannosides with no significant rate increase observed in the presence of thiophenoxide. The difference in behavior of the two donors was attributed to steric factors, according to which steric repulsion between the incoming acceptor and the axial 2-methoxy-group disfavored the formation of an associative transition state from the mannosyl donor.3
Overend and coworkers carried out kinetic studies of the displacement of halide from a series of acetyl glycosyl halides by lithium thiophenoxide.25 Three tetra-O-acetyl-hexopyranosyl bromides (α-D-gluco, α-D-galacto and α-D-manno), two chlorides (α-D-gluco, β-D-gluco), three tri-O-acetyl-6-deoxy homologues (α-D-gluco, α-L-manno and 6-iodo-α-D-gluco) and three per-O-acetylpentopyranosyl bromides (α-D-xylo, β-L-arabino and β-D-ribo) were reacted with lithium thiophenoxide in n-pentanol-toluene mixtures at 22°C. In all cases but one second order kinetics were observed indicative of a bimolecular mechanism. The exception to the general rule was provided by the reaction of thiophenoxide with tetra-O-acetyl-α-D-mannosyl bromide when the rate of consumption of the acceptor was slower than that of the donor, leading to the conclusion that, under the reactions conditions studied, bimolecular substitution of tetra-O-acetyl-α-D-mannosyl bromide was retarded by steric hindrance, recalling the conclusion of Rhind-Tutt and Vernon.3
Lemieux and Hayami carried on the halide-ion catalyzed anomerization of the tetra-O-acetyl-D-glucopyranosyl chlorides in acetonitrile. The employment of 36Cl-labelled chloride and the determination that the process was first order in chloride revealed these reactions to proceed by a bimolecular substitution mechanism with inversion of configuration (Scheme 57).26
Scheme 57.

Bimolecular Displacement of Chloride from Acetochloroglucose by Chloride as Revealed by Isotopic Labelling and Kinetics
In a similar vein Paulsen and coworkers studied the displacement of bromide from variously protected 2-azido-2-deoxy-α-D-pyranosyl bromides with tetraethylammonium chloride in acetonitrile.103 The inverted β-chlorides were shown to be the kinetic products, but were slowly transformed to their α-anomers with longer reaction times. In both the gluco- and the galacto configurations the rate of formation of the β-chloride was found to be a function of the concentration of both the substrate and the tetraethylammonium chloride leading the authors to propose an SN2-like mechanism.103 Moreover, in the gluco-series the rate of the inversion reaction was dependent on the nature of the protecting groups at the 3- and 4-positions, being most rapid for the dibenzyl ether and slowest for diesters indicating that the SN2-reaction at the anomeric center is retarded by electron-withdrawing groups (Table 14). However, no such correlation was found in the galacto-series.103
Table 14.
Second Order Rate Constants for Bromide Ion Displacement by Chloride
| Structure | ka |
|---|---|

|
9.3 |

|
4.6 |

|
4.0 |

|
2.7 |

|
1.7 |

|
1.55 |
| Structure | ka |
|---|---|

|
11.6 |

|
10.9 |

|
9.0 |

|
3.4 |

|
3.2 |
Second order rate constants (L.mol−1min−1) for reactions conducted in acetonitrile at room temperature
Johnson and coworkers studied the alcoholysis of tetra-O-acetyl-α-D-glucopyranosyl bromide with four primary (methanol, ethanol, propanol and butanol) and two secondary alcohols (isopropanol and cyclohexanol), and determined initial rate constants and activation energies for isopropanol and the four primary alcohols.27 The lower activation energy of the propan-2-olyses as compared with the primary alcoholyses (12.3 kcal mol−1 vs 19–21 kcal mol−1) and the large increase in rate observed on addition of bromide ion suggested an SN2 mechanism with inversion at the anomeric center for the formation of the secondary glycosides. A time-dependent increase in the fraction of the α-glucopyranoside was attributed to the anomerization of α-glucosyl bromide and subsequent alcoholysis of the β-D-bromide.
Eby and Schuerch studied the reactions of two 1-O-tosyl-D-glucopyranose derivatives (2,3,4,6-tetra-O-benzyl- and 2,3,4-tri-O-benzyl-6-O-(N-phenylcarbamoyl)-) with several alcohols at various concentrations in diethyl ether.4 At the lowest alcohol concentrations (donor:alcohol = 1:1), the rates of reaction were lower for the more hindered alcohols, but at higher alcohol concentrations (donor: alcohol ≤ 1:5) the rates were almost independent of the alcohol size, thus disfavoring a bimolecular substitution mechanism. The lack of clear dependence of the reaction rate on the structure of the alcohol and the absence of second-order kinetics led the authors to advance an SN1-like mechanism with the intermediacy of an oxocarbenium ion and a series of interchanging tight ion-pairs intermediates reminiscent of Rhind-Tutt and Vernon’s proposal (Scheme 56).3 To account for the anomeric ratios observed at high concentrations of alcohol it was suggested that reaction of the β-ion pair must be faster than that of the α-ion pair under the conditions employed. The enhanced fraction of α-glycosides observed at low alcohol concentrations was considered to be the result of rapid equilibration of the ion pairs coupled with rapid invertive substitution of the β-ion pair (Scheme 58).
Scheme 58.

Early Ion Pair Mechanism for the Reactions of Glycosyl Sulfonates
Wallace and Schroeder studied the mechanism of the mercury(II) cyanide-promoted reactions of 2,3,4,6-tetra-O-methyl-α-D-glucopyranosyl bromide with cyclohexanol in benzene-nitromethane mixtures (1 : 1 v/v) in the temperature range of 2 °C to 20 °C, with predominant formation of cyclohexyl 2,3,4,6-tetra-O-methyl-β-D-glucopyranoside.6 First-order kinetic dependences on the glucosyl bromide and mercury(II) cyanide concentrations were observed, but the rates were independent of cyclohexanol concentration. Nevertheless, the selectivity for formation of the β-glucoside increased with increased alcohol concentrations and with lower reaction temperatures. These facts suggested a mechanism involving the Hg(II)-assisted heterolysis of the C-Br bond to form an oxocarbenium ion intermediate in the rate-determining step. The influence of alcohol concentration on the subsequent product-determining step suggested that the initial α-contact ion pair suffered alcohol dependent substitution giving the β-product in competition with equilibration with a solvent separated ion pair that was the source of the minor α-isomer of the product.
Wulff and Röhle studied the mechanism of the Koenigs-Knorr reaction of 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide with alcohols in the presence of silver carbonate in diethyl ether and found the reaction rate to depend on the concentration of both donor and acceptor, leading them to propose an associative transition state for glycoside formation that occurs on the catalyst surface (Figure 22).317
Figure 22.

Kinetics-Derived Associative Transition State for the Reaction of Acetobromoglucose with Alcohols and an Insoluble Silver Salt.
Banait and Jencks investigated the reaction of α-D-glucopyranosyl fluoride with anionic nucleophiles in water at 30 °C, and found the first-order rate constants for the disappearance of the donor to depend linearly on the concentration of the nucleophile.318 Further, the products of the reactions with azide and acetate ions, β-D-glucopyranosyl azide and 1-O-acetyl-β-D-glucose, respectively, showed almost complete inversion of anomeric configuration, all of which supported concerted bimolecular SN2-type mechanisms. The alternative bimolecular reaction of an ion pair was ruled out as not significant in good ionizing solvents, such as water, where the rate of ion pair separation is expected to be more rapid than diffusion controlled attack by the dilute nucleophile.
Tan and coworkers studied the stereoinvertive methanol-catalyzed epoxide-opening and spirocyclization of substituted 1-aryl-1,2-anhydro sugars, which showed a second order dependence on added methanol concentration. Low-temperature NMR studies in combination with a Hammett analysis suggested an SN2 or SN2-like mechanism of spirocyclization in which the transition state is supported by two molecules of methanol, one serving to activate the epoxide oxygen and a second serving as an organizing bridge between the ring and nucleophilic oxygens (Scheme 59).319
Scheme 59.

Associative Mechanism for Cyclization of a 1-(2-Hydroxymethylphenyl)-1,2-Anhydro Sugar Requires Assistance from Two Molecules of Methanol as Revealed by Kinetic Analyses
Boons and coworkers studied the mechanism of glycosylation of trifluoroacetimidate donors carrying their 2-O-[(1S)-phenyl-2-(phenylsulfanyl)ethyl] group and leading selectively to 1,2-cis-α-glycosides via in situ generated bicyclic (trans-decalin like) sulfonium ions. A kinetic study showed that the initial rate of glycosylation increases with increased acceptor concentration and fit a second order rate equation, suggesting that nucleophilic attack of the acceptor alcohol at the bicyclic sulfonium ion follows a bimolecular SN2-like mechanism. A computational study gave more insight into the reaction path indicating the initial formation of a hydrogen-bonded complex between the bicyclic sulfonium ion and the acceptor, with both O2 and the 3-O-acetyl group serving as H-bond acceptors in a complex that ideally positioned the acceptor for an SN2-like attack leading to an α-glycoside (Figure 21).163
Taylor and coworkers developed the borinic acid-catalyzed regioselective monoglycosylation of vicinal and 1,3 diols and investigated the kinetics of these reactions.320 A first study273 of the Koenigs-Knorr reactions of glycosyl halide donors with polyhydroxylated glycosyl acceptors highlighted the efficiency of the borinate ester derived from diphenylborinic acid and ethanolamine as a catalyst, and revealed first order kinetics with respect to the glycosyl bromide, the acceptor, and the catalyst, indicative of an SN2-like mechanism (Scheme 51). A subsequent kinetic and computational study of the monotosylation of cyclohexane 1,2-diol in homogeneous media and using the same catalyst supported the role of cyclic borinate esters as key intermediates in the turnover limiting step of the catalytic cycle.321
More recently, in a study of the regio- and stereoselective couplings of diols and triols with glycosyl mesylates, D’Angelo and Taylor studied uncatalyzed and diaryl boronic acid-catalyzed reactions. Both the uncatalyzed and catalyzed glycosylations show kinetics consistent with associative SN2-like mechanisms leading to products generated by inversion of configuration of the mesylate (Scheme 51).155 It was further suggested that the modest preference for the α-configured glycoside product in the absence of catalyst appears to reflect the higher reactivity of the minor β-mesylate, consistent with the Curtin–Hammett principle; in the presence of the catalyst the displacement of the α-mesylate is accelerated, leading to the selective formation of the β-glycoside.
16 Kinetic Isotope Effect Measurements
An alternative to the measurement of actual kinetic data is the determination of kinetic isotope effects by NMR methods, ideally at natural abundance by the Singleton method.322 Crich and Chandrasekera, using a mannosyl sulfoxide partially enriched in deuterium at the anomeric position and in the benzylidene acetal, studied the formation of a β-mannosyl linkage to a relatively unreactive glucopyranosyl alcohol via the corresponding α-mannosyl triflate in deuteriodichloromethane. They determined a secondary α-deuterium KIE of 1.16–1.21 at −78 °C, corresponding to 1.10 at 25 °C.323 This value is in good agreement with a subsequent calculated secondary α-deuterium KIE of 1.12 for the coupling of the same α-mannosyl triflate to isopropanol and is consistent with a loosely associative mechanism for this displacement (Figure 23).222 Subsequently, working at natural abundance with an 800 MHz NMR spectrometer Crich and coworkers measured primary 13C KIEs for the reaction of 4,6-O-benzylidene protected α-manno and glucopyranosyl triflates with isopropanol in deuteriodichloromethane leading to the α- and β-glycosides (Figure 23).222 Three of the four cases gave results close to the range (1.03–1.08) expected for a bimolecular reaction324 and consistent with computed KIEs for associative mechanisms proceeding via exploded associative transition states. Thus, the formation of the β-manno- and glucosides are understood as direct displacements on the α-triflates, whereas the α-glucoside is viewed as formed by associative displacement of a minor but more reactive β-glucosyl triflate in dynamic equilibrium with its α-anomer. The α-mannoside, however, displayed a 13C primary KIE (1.005 ± 0.002) that is distinct from that calculated (1.023) for the bimolecular mechanism, but within the range (1.00–1.01) expected for a monomolecular SN1-type mechanism.324 The problem of spontaneous ion pair collapse, discussed further below, prevented the computation of an SN1-like transition state.
Figure 23.

Experimental and Computed 13C Primary Kinetic Isotope Effects for the 4,6-O-Benzylidene Directed Manno- and Glucopyranosylation and an Experimental and Computed Secondary Deuterium KIE for β-Mannoside Formation
Working with a donor partially deuteriated at the anomeric position and in the acetate group as internal standard, Gervay-Hague and coworkers measured secondary α-deuterium KIEs of 1.16 and 1.19 for the formation of the β- and α-glycosides in the reaction of oxetane with a 6-O-acetyl-2,3,4-tri-O-benzyl-α-mannosyl iodide in deuteriobenzene at room temperature (Scheme 60).325 These values were compared with computed transition states for associative displacements leading to the suggestion that both reactions proceeded via exploded SN2-like transition states, albeit the formation of the α-glycoside displayed a significantly longer partial C-I bond than the β-anomer. Clearly this result, with both anomers of the product formed via SN2-like transition states with inversion of configuration, requires the rapid in situ anomerization of the α-mannosyl iodide used as substrate. While no experimental values were measured, transition states with secondary α-deuterium KIEs of 1.01 and 1.31 were also computed for the reaction 4,6-O-methylidene protected α-and β-mannosyl iodides, respectively, and taken as suggestive of a looser process for the formation of the α-anomer.325 It is noteworthy that in an actual reaction 4,6-O-benzylidene-2,3-di-O-benzyl-α-D-mannopyranosyl iodide gave exclusively the β-mannoside on reaction with oxetane suggesting, by comparison with the results of Scheme 60, that the presence of the cyclic acetal suppresses in situ anomerization of the iodide.
Scheme 60.

Secondary Deuterium KIEs Reveal Associative Mechanisms for the Formation of α- and β-Mannosides from Mannosyl Iodides
Using a 19F NMR spectroscopic method,326 in conjunction with the more classical polarimetric quasi-racemate method327 and a series of isotopically enriched substrates, Bennet and coworkers measured a suite of primary and secondary KIE values for the invertive substitution of α-D-glucopyranosyl fluoride by azide in aqueous buffer at pH 6. The full suite of experimental KIEs were used to inform a computed transition state for the displacement, which is best considered as an exploded associative transition one (Table 15).328 Knowledge of this mechanism enabled Miller, Schepartz, and their coworkers to develop a method for the O-glycosylation of simple alcohols from unprotected glycosyl fluorides using calcium salts as promotors.329
Table 15.
Experimental and Computed KIEs for the Displacement of Fluoride from α-D-Glucopyranosyl Fluoride by Azide

| ||
|---|---|---|
| Isotope | KIE by 19F NMR | KIE (cald) TS |
| α-D | 1.192 ± 0.006 | 1.197 |
| β-D | 1.046 ± 0.007 | 1.041 |
| γ-D (C5) | 0.987 ± 0.004 | 0.986 |
| α-13C | 1.024 ± 0.006 | 1.034 |
| α-18O | 0.981 ± 0.003 | 1.011 |
Zimmerman, Nagorny and their coworkers investigated the mechanism of the stereoselective spiroketalizations of substituted cyclic enol ethers (including glucals) catalyzed by chiral non-racemic phosphoric acids. Using deuteriated substrates the reaction was found to be syn-selective (Scheme 47). A Hammett study with a substituted system indicated the development of positive charge in the transition state but less than expected for a SN1 mechanism involving an intermediate oxocarbenium ion. Finally, a secondary deuterium kinetic isotope effect of 0.85 was measured leading to the proposal of an asynchronous concerted mechanism with a relatively short-lived polar transition state and highlighted the dual function of the phosphoric acid, as a Brønsted acid catalyst and a Lewis base in deprotonation following ketalization (Scheme 47). Quantum mechanical calculations further supported this hypothesis.159
Using a modification of the Singleton method with use of a DEPT sequence to enhance sensitivity, Jacobsen and coworkers have examined the reaction of 2,3,4,6-tetra-O-methyl-α-D-galactopyranosyl chloride with benzyl alcohol with catalysis by a tailored chloride-complexing thiourea.253 Operating in toluene at room temperature in the presence of isobutylene oxide to trap the expelled chloride, the experimental 13C KIEs were consistent with loose SN2-like transition state with a high degree of oxocarbenium ion character (Table 16 and Figure 18).
Table 16.
Computed and Experimental KIE Values Supporting a Loose Associative Transition State for the Displacement of Chloride from Per-O-methyl-α-D-galactopyranosyl Chloride Promoted by a Macrocyclic Bisthiourea (Figure 18).
| method | C1 | C4 | C5 | C6 |
|---|---|---|---|---|
| HF/cc-pVQZ | 1.033 | 1.03 | 1.036 | 1.039 |
| MP2/cc-pVTZ | 1.015 | 1.016 | 1.036 | 1.019 |
| CCSD/cc-pVDZ | 1.024 | 1.022 | 1.019 | 1.029 |
| B3LYP/cc-pVQZ | 1.023 | 1.018 | 1.022 | 1.028 |
| experimental | 1.022 | 1.019 | 1.024 | 1.028 |
In the corresponding mannose series, starting from the axial chloride, secondary α-deuterium KIEs of 1.15 and 1.25 were measured for both the β- and α-anomers of the product using isotopically enriched samples. The results are consistent with a loose asynchronous associative transition state for the formation of the β-isomer and an SN1-like mechanism for the α-anomer (Scheme 61).252
Scheme 61.

DKIE-Supported Transition State for the Formation of a β-Mannoside from the Corresponding α-Mannosyl Chloride on Promotion by a Macrocyclic Bisthiourea
Using the Singleton NMR method322 and a substrated partially enriched in deuterium Takahashi, Toshima and coworkers measured primary 13C and secondary α-deuterium KIEs of 0.9999 and 1.055, respectively, for the regio- and stereospecific boronic acid-catalyzed coupling of a 1,2-anhydroglucopyranose to polyols.276 Based on DFT calculations, and the fact that the couplings are run in the presence of several equivalents of water without competing hydrolysis, the authors favored a highly dissociated SNi mechanism (Scheme 62). Computational analysis of the evolution of the relevant bond orders over the course of the reaction revealed that cleavage of the C1-O2 bond very significantly preceeds formation of the glycosidic bond and cleavage of the nucleophilic B-O bond suggesting the mechanism to be close to the SN1 end of the mechanistic spectrum. Indeed, the experimental 13C primary KIEs of 0.9999 is closer to the 1.005 measured by Crich and coworkers for the SN1-like 4,6-O-benzylidene-directed α-mannosylation (Figure 23) than to the closest computed value of 1.009 for the SNi reaction. The absence of hydrolysis in the presence of several equivalents of water would then be explained simply by the high effective molarity of the intramolecular nucleophile. Computational analysis of the competing SNi transition state leading to the formation of the regioisomeric 1→6-linked product revealed it to be more sterically congested and some 3 kcal.mol−1 higher in energy. With the 1,2-anhydromannopyranose donor (Scheme 52) the opposite regioselectivity is observed in the glycosylation of 4,6-diol, which is again explained in terms of a less congested transition state. This switch in regioselectivity with configuration of the donor effectively constitutes another example of the match and mismatch phenomenon.260
Scheme 62.

Experimental and Computational Primary and Secondary KIEs Supporting an Exploded SNi Mechanism for the Boronic Acid-Catalyzed Glycosylation of a Polyol by a 1,2-Anhydroglucopyranose
Finally, using material partially enriched in deuterium at the anomeric position and in the remote acetate as internal standard, Lowary and coworkers determined the secondary α-deuterium KIE for the coupling of the α-2,3-anhydrolyxofuranosyl thioglycoside to benzyl alcohol (and to a primary carbohydrate-based alcohol) on activation with molecular sieves (or copper(II) triflate).128 The results were consistent with migration of the arylthio moiety to give furanosyl oxocarbenium ion, rather than an episulfonium ion, as rate determining step (Scheme 63). Computational work was in agreement with this conclusion and pointed to a preferred 3E conformation for the oxocarbenium with trapping from the inside face consistent with the Woerpel model124,330 and the subsequent studies of Filippov and Codée.129
Scheme 63.

Deuterium Kinetic Isotope Effects Support the Intermediacy of an Oxocarbenium Ion in Furanosylation with a 2,3-Anhydro Furanosyl Thioglycoside
17 Use of Cation Clocks to Determine Molecularity
Inspired by the Jencks azide clock77 and the use of radical cyclizations as clocks for intermolecular reactions,331 Crich and coworkers developed a cation clock method to determine the molecularity of glycosylation reactions. In this approach a unimolecular reaction (cyclization) is set up in competition with the glycosylation such that a plot of the glycosylation/cyclization product ratio against acceptor concentration reveals any dependence of the glycosylation product on concentration.83–86 A marked increase in the ratio of glycosylation to cyclization products with increasing acceptor concentration reveals an SN2-like associative mechanism. Alternatively, a glycosylation to cyclization ratio showing low acceptor concentration dependence is characteristic of an SN1-like dissociative mechanism. This method allows one to localize the mechanism of a given reaction in the SN1–SN2 continuum of mechanisms of glycosylation17,20 by a simple product study. The concept is illustrated (Scheme 64) for 4,6-O-benzylidene-directed β-mannosylation using the intramolecular Sakurai clock examined in more detail in Scheme 9 above.
Scheme 64.

The Cation Clock Concept
The method was employed to probe the molecularity of C- and O-glycosylations in the mannopyranosyl and the glucopyranosyl series using diversely protected trichloroacetimidate and sulfoxide donors.84 In the case of the 4,6-O-benzylidene protected mannosyl donors it was evident from the glycoside to cyclized product ratios (Figure 24) that β-O-mannosylation displayed a much stronger concentration dependence on the acceptor than α-O-mannosylation, whereas β-C-mannosylation was still less dependent. These results were interpreted as consistent with an SN2-like associative path for formation of the β-O-glycosides and with a much more dissociative SN1-like mechanism for that of the α-anomers. These results were in excellent agreement with the previously discussed kinetic isotope effects.222 The very low concentration dependence of the C-mannosylation reaction was interpreted as consistent with a highly dissociative mechanism proceeding via an oxocarbenium ion only loosely associated with the counterion. In the corresponding benzylidene-protected glucopyranose series formation of both the α- and β-O-glycosides shows significant dependence on the concentration of the acceptor, suggesting both to proceed via SN2-like mechanisms,84 again consistent with the earlier KIE studies.
Figure 24.

Plot of Mannoside to Cyclization Ratio vs Acceptor Concentration for Mannosylation Following Scheme 64.
While cation clock methods provide much valuable information on the molecularity of glycosylation reactions, an estimation of the influence of concentration on selectivity can also be obtained simply by studying selectivity as a function of concentration. In the course of their investigation into their supramer concept, numerous such studies were reported by Kononov and coworkers for various sialidation reactions.286–290 A curious inverse dependence on acceptor concentration in certain sialidation reactions was reported by Xing and coworkers and attributed to a change on supramer constitution.332 Yet further examples have been provided by Kononov who showed, inter alia, the influence of donor concentration on the stereochemical outcome of the reaction of an arabinofuranosyl bromide with dibutyl phosphate (Scheme 65).333
Scheme 65.

Dependence of Selectivity in an Arabinofuranosylation on Donor Concentration
18 Computational Studies
The considerable body of computational work on the structure of glycosyl oxocarbenium ions published prior to 2009 has been reviewed.205 The broad availability of DFT calculations has meant that computational work now complements many experimental studies, as presented in the relevant sections above, to the extent that no attempt has been made to cover computational work in a comprehensive manner in this review. Nevertheless, some conceptually important studies are discussed below.
The tendency for spontaneous collapse of oxocarbenium ion – anion pairs in quantum calculations results in the enormous majority of computational work either excluding the anion altogether or resorting to artifact to prevent collapse. Computations that exclude counterions focus only on intermediacy of oxocarbenium ions and consequently on selectivity in the SN1 wing of the general mechanism. While such studies provide much needed insight into the conformations and reactivity of free oxocarbenium ions they exclude from consideration any alternative associative mechanism and so necessarily fail to give a complete appraisal of the full range of possibilities.
In an early attempt to circumvent the problem and include the necessary counter ion Whitfield and coworkers incorporated a lithium ion at a fixed distance from the anomeric center so as to neutralize the charge on the departing triflate and prevent spontaneous collapse of any ion pairs in computations directed at the reaction of tetra-O-methyl α- and β-glucopyranosyl triflates with methanol.303 Later, in a QM/MD study of the reactions of tetra-O-methyl-D-glucopyranosyl triflate in various solvents (acetonitrile, ether, dioxane, toluene) and in the gas phase, Satoh, Hünenberger, and their co-workers employed distance constraints to prevent the collapse of ion pairs.334 Their MD simulations showed that in acetonitrile the oxocarbenium ion preferentially adopts a OS2 or, to a lesser extent, a 3H4 conformation, with the counter ion loosely associated with the anomeric carbon and distributed nearly equally between the α- and β-faces. In contrast, in lower polarity solvents, the oxocarbenium ion was found to adopt a 3H4 conformation with the counter ion tightly associated with the anomeric carbon and predominantly on the β-face (Figure 25). Overall, the results suggested that increased solvent polarity leads to weaker association of the triflate ion with the oxocarbenium ion and to a progressive shift of its preferential positioning from the β- to the α-face of the ring. Selected configurations of the oxocarbenium-counterion complex were extracted from the simulations in acetonitrile and dioxane and subjected to QM geometry optimization ultimately leading the authors to formulate their “conformer and counter ion distribution hypothesis”, according to which stereoselectivity is dictated by the conformational properties of the reactive oxocarbenium-counter ion complex as a whole.
Figure 25.

Schematic Representation of the Change in the Per-O-methylmannopyranosyl Oxocarbenium Ion Conformation and Counterion Location with Solvent Dielectric Constant When ion Pair Collapse is Prevented
SN2-like transition states for the displacement of triflate from covalent α- and β-gluco and mannopyranosyl triflates carrying 4,6-O-formylidene acetals and methyl ethers at the 2- and 3-positions, including α-deuterium kinetic isotope effects, were first computed by Li,335 and were in general agreement with the subsequent calculations of Pratt, Crich and coworkers on closely related systems (Figure 23).222 A common theme in transition state computations that include the leaving group is the finding of associative transition states, albeit with relatively long partial bonds to both the nucleophile and the leaving group reminiscent of the exploded transition states favored by the mechanistic enzymologists.
An important aspect of QM calculations of glycosylation reactions is the provision of theoretical kinetic isotope data to corroborate experimental measurements. Thus, the primary 13C-KIEs computed by Pratt, Crich and coworkers for the associative displacement of triflate from 4,6-O-benzylidene protected α-mannopyranosyl and α- and β-glucopyranosyl triflates were in good agreement with the experimental values and so provided strong support for SN2-like mechanisms for these reactions. The computed primary 13C KIE for the associative formation of the corresponding α-mannoside from the β-mannosyl triflate was, however, significantly different from the experimental value causing the authors to conclude that this latter reaction was more SN1-like in nature (Figure 23).222 The direct computation of KIEs for the dissociative transition was not possible because of the collapse of the necessary ions pairs in the calculation.
The problem of spontaneous ion pair collapse was eventually solved by Hosoya and coworkers by the solvation of the triflate anion with three explicit molecules of dichloromethane through C-H---O hydrogen bonding and a further molecule of solvent to stabilize the positive charge on the oxocarbenium ion. In this manner it proved possible to compute the relative energies of a series of permethylated gluco and mannopyranosyl triflates as well as the corresponding contact and solvent-separated ion pairs for the first time without recourse to artifact as illustrated for the gluco series in Figure 26.336,337 The method was subsequently extended to the 4,6-O-alkylidene protected manno- and glucopyranosyl triflates and the corresponding ion pairs leading to the conclusion that the cyclic acetal stabilizes a manno α- contact ion pair to a greater extent than in the gluco-configuration where a solvent separated ion pair is indicated.338 Importantly, while differing in method and detail, both the Hünenberger and Satoh,334 and Hosoya models338 underline the importance of the counter ion and its location on the relative energy and conformation of the associated oxocarbenium ion.
Figure 26.

Computed Per-O-Methyl Glucopyranosyl Oxocarbenium Ion Pairs with Explicit Dichloromethane-Solvation
In their experimental and theoretical study of the organocatalyzed displacement of chloride from various glycosyl donors, Jacobsen and coworkers measured a series of secondary deuterium KIE values, which were found to be small and normal for the formation of the β-glycoside from per-O-methyl-α-D-mannopyranosyl chloride, consistent with an associative mechanism (Figure 18). The formation of the minor α-anomer from the same reaction was found to be accompanied by a larger normal secondary deuterium KIE indicative of a dissociative mechanism.252 Subsequently, 1H to 13C polarization transfer was used to enhance signal to noise ratios and so reduce acquisition times in a Singleton-type322 natural abundance NMR determination of 13C primary and secondary KIEs for the thiourea-catalyzed displacement of per-O-methyl-α-D-galactopyranosyl chloride by benzyl alcohol.253 QM calculations were used in the normal manner to obtain the anticipated KIEs for the asynchronous SN2-like displacements, which showed reasonable agreement with the experimental values. In an interesting twist, however, and following the work of Berti on hydrolytic reactions,339 the authors argued that the equilibrium isotope effect is a satisfactory model for the KIE of the dissociative mechanism enabling them to calculate primary and secondary 13C KIEs for the competing SN1 reaction.253
In the furanoside series Filippov, Codée and coworkers129 carried out a computational and experimental study to gain insight into the stereoelectronic effects that govern the highly stereoselective formation of 1,2-cis glycosides regardless of the configuration of the furanosyl donor at C2 (Scheme 18). For each of the arabino, lyxo, ribo, and xylo configurations the relative energies of a total of two hundred and forty three ring and side chain conformations were computed for the per-O-methyl oxocarbenium ions, and correlated with the experimentally observed selectivities for reduction of the furanosyl acetates with deuteriotriethylsilane in the presence of trimethylsilyl triflate. Overall, the experimental stereoselectivities were adequately explained by inside attack of the acceptor on the lowest energy conformer of the corresponding oxocarbeniums consistent with the Woerpel model.72,124,125
DFT studies of the effect of the rotation of the C2–O2 bond on the conformation of ether-protected glycosyl oxocarbenium ions by Whitfield indicated a strong syn preference for the configuration of the C2–H2 and the O2–R bonds, where R is the O2 ether, as revealed by the H2-C2–O2–R torsion angles < 20°.205 This conformational preference was attributed to hyperconjugative interaction between a lone pair on O2 and the LUMO of the C1–O5 double bond.340 Realizing that covalent intermediates and contact ion pairs (CIPs) are not necessarily subject to the same conformational restrictions, Whitfield and Kumar hypothesized that the dependence of glycosylation selectivity on the configuration of a chiral O2 protecting group would be indicative of an oxacarbenium ion intermediate.341 In the event the change in selectivity observed between the R- and S-isomers on coupling of both armed and disarmed 2-O-[R,S-1-(cyclopropyl)ethyl]-protected D-glucopyranosyl donors with various alcohols was not sufficient to allow meaningful conclusions to be drawn.
With regard to the influence of substituents on the reactivity of glycosyl donors, computational work was performed in support of an experimental study showing that a 3-O-(α-mannopyranosyl) moiety on p-tolyl 2,4-O-benzyl-6-O-triisopropylsilyl-α-D-thiomannopyranoside is more disarming than an acetyl group at the same position. On the basis of the DFT calculations it was suggested that the presence of the bulky 3-O-glycosyl moiety increases the torsional strain engendered in the O2-C2-C3-O3 system as the system deforms from the 4C1 chair (60 °) of the donor to the 4H3 (45 °) of the putative oxocarbenium ion as compared to smaller groups at O3, resulting in a higher activation energy.342
19 Conclusions
While NMR spectroscopic evidence for glycosyl oxocarbenium ions has been provided in superacid media in recent years, it is still lacking in organic solvents owing either to decomposition or combination with the counterion to form covalent species. In this latter regard it is well to recall that the anomeric 13C chemical shift of a glycopyranosyl triflate is around 100–105 ppm in the region of typical acetal carbons and much removed from the chemical shifts measured for oxocarbenium ions (200–250 ppm). Any dynamic equilibrium therefore very strongly favors the covalent form, which argues against the possibility of detecting the oxocarbenium counterion pair below the coalescence temperature. Regardless, the isolation of trans-fused products in intramolecular Sakurai reactions in the benzylidene protected mannose series requires a sufficient lifetime to allow conformational inversion of the oxocarbenium ion. In contrast, the spectroscopic evidence for the formation of a wide variety of activated covalent glycosyl intermediates under preactivation conditions, typified but by no means limited to the glycosyl triflates, is extensive. Moreover, the evidence is mounting that nucleophilic counterions play an important role in glycosylation through the formation of covalent intermediates, even when used in substoichiometric quantities. Another important advance that bears on the glycosylation mechanism is the study of the nucleophilicity of the alcohol in relation to selectivity.
Ultimately, the question of whether a particular glycosylation reaction hews more to the SN1 than the SN2 end of the mechanistic continuum, or vice-versa, can only be answered by kinetic measurements. Early workers readily established that substitutions of several glycosyl halides by strong anionic nucleophiles proceeded by SN2-like mechanisms, but early work with model alcohols as nucleophiles was mostly conducted under pseudo-first order solvolytic conditions or in polar solvents resulting in an apparent preference for dissociative SN1-like mechanisms for glycosylation reactions. More recent work, with either indirect cation clock methods or direct kinetic methods, conducted in more typical solvents such as dichloromethane has revealed a strong concentration dependence of several O-glycosylation reactions on nucleophile concentration pointing to SN2-like processes. The use of kinetic isotope effects to probe transition state structure, an area where preparative carbohydrate chemistry lags far behind enzymatic glycosylation, has also provided strong evidence for associative mechanisms in some modern preparatively useful glycosylation reactions. Further kinetic and kinetic isotope effect studies are needed to inform the mechanisms of a broader spectrum of modern glycosylation reactions and so provide preparative chemists with the information needed to optimize the selectivity of current processes and rationally design improved ones.
Computational studies of glycosylation reactions, which for many years provided a very one sided oxocarbenium ion centric picture of events because of the unavoidable collapse of ion pairs in quantum calculations, are undergoing a transformation with new methods for the stabilization of ion pairs through the explicit solvation of the counterion. Such techniques, which will doubtless be subject to further improvement, promise to be a more reliable companion for the experimental chemist.
All told, studies on the mechanisms of glycosylation reactions have evolved considerably in the last twenty years but the range of reactions that have been subjected to careful scrutiny, especially from the critical kinetic view point, remains limited. Such studies are of great importance in the design and elaboration of new, improved and more robust glycosylations suitable for routine use by a broader spectrum of glycoscientists beyond the current group of more specialized organic chemists.
Acknowledgments
DC thanks the NIH (GM62160 and GM125271) for support of his programs on glycosylation.
Biographies
Philip Ouma Adero obtained his bachelor’s degree in chemistry from University of Eastern Africa, Baraton, Kenya in 2009. He then moved to Youngstown State University, Ohio, USA, where he received his master’s degree in chemistry in 2012 under the supervision of Prof. Peter Norris, working on “Heterocycle Synthesis via Rhodium (II)-Catalyzed Azido Carbenoid Cyclization.” He continued his Ph.D. studies at Wayne State University under the guidance of Prof. David Crich. During this time, he worked on the development of cyclization reactions as clocks for the determination of the molecularity of glycosylation reactions. He was also involved in the development of selective hydrogenolytic cleavage of naphthylmethyl ethers in the presence of sulfides.
Harsha Amarasekara received his bachelor’s degree in chemistry from the University of Peradeniya, Sri Lanka in 2010, where he studied the mechanism of the degradation of carbofuran in the gas phase by computational chemistry. He joined Wayne State University in 2012 as a PhD candidate in organic chemistry and started working under the direction of Professor David Crich. His research focuses on the identification and characterization of intermediates in glycosylation reactions and on measuring the kinetics of those reactions. He is also very interested in the influence of side chain conformation on the reactivity and stereoselectivity of glycosylation reactions.
Peng Wen received his bachelor’s degree in bioengineering from Southwest Jiaotong University (SWJTU), China, in 2009. He continued his graduate studies at the same university under the supervision of Prof. Qun Lu and conducted research on the synthesis of Vitamin D analogs. In 2012, he received his master’s degree in biochemical engineering from SWJTU and subsequently moved to Wayne State University where he is currently a PhD candidate in organic chemistry under the guidance of Prof. David Crich. He has strong research interests in carbohydrate chemistry and has been involved in several projects including mechanistic studies of glycosylation reactions, methodology development for the construction of glycosydic linkages and the synthesis of natural oligosaccharide mimetics.
Luis Bohé is Chargé de Recherche at the CNRS Institute for Natural Products Chemistry in Gif-sur-Yvette, France and maintains a strong interest in synthetic methodology and reaction mechanisms. Since 2009, in collaboration with David Crich, he has directed his efforts toward the elucidation of glycosylation reactions employing kinetic isotope effect measurements made at natural abundance by NMR methods, and cation clock reactions.
David Crich is Schaap Professor of Organic Chemistry at Wayne State University where he and his coworkers conduct an extensive program of research in the fields of glycochemistry and the design and development of antibiotics for the treatment of multidrug resistant infectious diseases. He has received several awards for his work in carbohydrate chemistry including the Wolfram and Hudson Awards from the American Chemical Society Carbohydrate Division, the Emil Fischer Award from the European Carbohydrate Organization, and the Tate and Lyle and Haworth medals from the Royal Society. In 2018 he will receive the Whistler Award from the International Carbohydrate Organization.
References
- 1.For an excellent summary of the seminal works in the field and a exempliary study of nucleophilic substitution of benzhydryl systems see: Phan TB, Nolte C, Kobayashi S, Ofial AR, Mayr H. Can One Predict Changes from SN1 to SN2 Mechanisms? J Am Chem Soc. 2009;131:11392–11401. doi: 10.1021/ja903207b.
- 2.NB. In the absence of further substituents on the electrophile, the glycosylation reaction is nothing more than the protection of an alcohol as a tetrahydropyranyl ether.
- 3.Rhind-Tutt AJ, Vernon CA. Nucleophilic Substitution in 2,3,4,6-Tetra-O-Methyl Glycopyranosyl Chlorides. J Chem Soc. 1960:4637–4644. [Google Scholar]
- 4.Eby R, Schuerch C. The Use of 1-O-Tosyl-D-Glucopyranose Derivatives in α-D-Glucoside Synthesis. Carbohydr Res. 1974;34:79–90. [Google Scholar]
- 5.Lucas TJ, Schuerch C. Methanolysis as a Model Reaction for Oligosaccharide Synthesis of Some 6-Substituted 2,3,4-Tri-O-Benzyl-D-Galactopyranosyl Derivatives. Carbohydr Res. 1975;39:39–45. [Google Scholar]
- 6.Wallace JE, Schroeder LR. Koenigs-Knorr Reactions. Part II. A Mechanisitc Study of Mercury (II) Cyanide-Promoted Reactions of 2,3,4,6-Tetra-O-Methyl-α-D-Glucopyranosyl Bromide with Cyclohexanol in Benzene-Nitromethane. J Chem Soc, Perkin Trans 2. 1976:1632–1636. [Google Scholar]
- 7.Lemieux RU, Hendriks KB, Stick RV, James K. Halide Ion Catalyzed Glycosidation Reactions. Synthesis of α-Linked Disaccharides. J Am Chem Soc. 1975;97:4056–4062. [Google Scholar]
- 8.Horenstein NA. Mechanism for Nucleophilic Aliphatic Substitution at Glycosides. Adv Phys Org Chem. 2006;41:275–314. [Google Scholar]
- 9.Capon B. Mechanism in Carbohydrate Chemistry. Chem Rev. 1969;69:407–498. [Google Scholar]
- 10.Bochkov AF, Zaikov GE. Chemistry of the O-Glycosidic Bond. Pergamon; Oxford: 1979. [Google Scholar]
- 11.Schmidt RR. New Methods for the Synthesis of Glycosides and Oligosaccharides - Are There Alternatives to the Koenigs-Knorr Method? Angew Chem Int Ed. 1986;25:212–235. [Google Scholar]
- 12.Schmidt RR. Synthesis of Glycosides. In: Trost BM, Fleming I, editors. Comprehensive Organic Synthesis. Vol. 6. Pergamon Press; Oxford: 1991. pp. 33–64. [Google Scholar]
- 13.Sinnott ML. Carbohydrate Chemistry and Biochemistry. RSC Publishing; Cambridge: 2007. [Google Scholar]
- 14.Zhu X, Schmidt RR. New Principles for Glycoside-Bond Formation. Angew Chem Int Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- 15.Crich D. Mechanism of a Chemical Glycosylation. Acc Chem Res. 2010;43:1144–1153. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 16.Mydock LK, Demchenko AV. Mechanism of Chemical O-Glycosylation: From Early Studies to Recent Discoveries. Org Biomol Chem. 2010;8:497–510. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]
- 17.Bohé L, Crich D. A Propos of Glycosyl Cations and the Mechanism of Chemical Glycosylation. CR Chimie. 2011;14:3–16. doi: 10.1016/j.carres.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ranade SC, Demchenko AV. Mechanism of Chemical Glycosylation: Focus on the Mode of Activation and Departure of Anomeric Leaving Groups. J Carbohydr Chem. 2013;32:1–43. [Google Scholar]
- 19.Bohé L, Crich D. Comprehensive Organic Synthesis. 2. Vol. 6. Elsevier; Oxford: 2014. Synthesis of Glycosides; pp. 1–33. [Google Scholar]
- 20.Bohé L, Crich D. A Propos of Glycosyl Cations and the Mechanism of Chemical Glycosylation; the Current State of the Art. Carbohydr Res. 2015;403:48–59. doi: 10.1016/j.carres.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye X-S, Lu W. General Aspects in O-Glycosidic Bond Formation. In: Hung S-C, Zulueta MML, editors. Glycochemical Synthesis: Strategies and Applications. Wiley; Hoboken: 2016. pp. 69–95. [Google Scholar]
- 22.Sasaki K, Tohda K. Recent Topics in β-Selective Mannosylation. Tetrahedron Lett. 2018;59:496–503. [Google Scholar]
- 23.Fascione MA, Brabham R, Turnbull WB. Mechanistic Investigations into the Application of Sulfoxides in Carbohydrate Synthesis. Chem Eur J. 2016;22:3916–3928. doi: 10.1002/chem.201503504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagen B, van der Vorm S, Hansen T, van der Marel GA, Codée JDC. Stereoselective Glycosylations - Additions to Oxocarbenium Ions. In: Bennett CS, editor. Selective Glycosylations: Synthetic Methods and Catalysts. Wiley-VCH; Weinheim: 2017. pp. 3–28. [Google Scholar]
- 25.Capon B, Collins PM, Levy AA, Overend WG. Nucleophilic Displacement Reactions of Acetylglycosyl Halides. J Chem Soc. 1964:3242–3254. [Google Scholar]
- 26.Lemieux RU, Hayami JI. The Mechanism of the Anomerization of the Tetra-O-Acetyl-D-Glucopyranosyl Chlorides. Can J Chem. 1965;43:2162–2173. [Google Scholar]
- 27.Schroeder LR, Green JW, Johnson DC. Alcoholyses of 2,3,4,6-Tetra-O-Acetyl-α-D-Glucopyranosyl Bromide; Kinetics and Products at 25–40 °C. J Chem Soc (B) 1966:447–453. [Google Scholar]
- 28.Meerwein H, Hinz G, Hofmann P, Kroning E, Pfeil E. Uber Tertiare Oxoniumsalze. J Prakt Chem. 1937;147:257–285. [Google Scholar]
- 29.Meerwein H, Hederich V, Wunderlich K. Untersuchungen Mit Wasserfreiem Silberfluoroborat Und Kupfer(I)-Fluoroborat. Arch Pharm. 1958;291:541–554. [PubMed] [Google Scholar]
- 30.Rakhmankulov DL, Akhmatdinov RT, Kantor EA. Alkoxycarbenium Ions. Russ Chem Rev. 1984;53:888–899. [Google Scholar]
- 31.Parent JP, Deslongchamps P. Bent Bonds (τ) and the Antiperiplanar Hypothesis, and the Reactivity at the Anomeric Center in Pyranosides. Org Biomol Chem. 2016;14:11183–11198. doi: 10.1039/c6ob02263d. [DOI] [PubMed] [Google Scholar]
- 32.Keeffe JR, More O’Ferrall RA. The Protonation of Carbenes: Structural Effects on the α-Proton Acidity of Carbocations. ARKIVOC. 2008:183–204. [Google Scholar]
- 33.Kazem Abadi SSK, Tran M, Yadav AK, Adabala PJP, Chakladar S, Bennet AJ. New Class of Glycoside Hydrolase Mechanism-Based Covalent Inhibitors: Glycosylation Transition State Conformations. J Am Chem Soc. 2017;139:10625–10628. doi: 10.1021/jacs.7b05065. [DOI] [PubMed] [Google Scholar]
- 34.Danby PM, Withers SG. Glycosyl Cations Versus Allylic Cations in Spontaneous and Enzymatic Hydrolysis. J Am Chem Soc. 2017;139:10629–10632. doi: 10.1021/jacs.7b05628. [DOI] [PubMed] [Google Scholar]
- 35.Lustgarten RK, Brookhart M, Winstein S. The 7-Methoxynorbornadienyl Cation: Evidence for a Symmetrically Bridged Structure. Tetrahedron Lett. 1971;12:141–144. [Google Scholar]
- 36.Olah GA, Berrier Al, Prakash GKS. 17O NMR Spectroscopic Study of Oxonium and Carboxonium Ions. J Am Chem Soc. 1982;104:2373–2376. [Google Scholar]
- 37.Olah GA, White AM, O’Brien DH. Protonated Heteroaliphatic Compounds. Chem Rev. 1970;70:561–591. [Google Scholar]
- 38.Meyer WP, Martin JC. Substituent Effects of Alkoxy and Amino Groups Directly Bonded to Cationic Carbon in the Perpendicularly Twisted Geometry. 2-Oxa- and 2-Aza-1-Adamantanyl Tosylates. J Am Chem Soc. 1976;98:1231–1241. [Google Scholar]
- 39.Garcia Martinez A, Rios IE, Vilar ET. Herstellung Von Gem-Bis-Triflaten. Synthesis. 1979:382–383. [Google Scholar]
- 40.Lambert JB, Mark HW. Increased Electron Demand in the Solvolysis of Secondary 2-Norbornyl Tosylates. J Am Chem Soc. 1978;100:2501–2505. [Google Scholar]
- 41.Kirmse W, Mrotzeck U. Zerfall Von 2-Oxa-5- Und -6-Norbornandiazonium-Ionen. Chem Ber. 1988;121:485–492. [Google Scholar]
- 42.Crich D, Yang F. Phenylthiomethyl Glycosides: Convenient Synthons for the Formation of Azidomethyl and Glycosylmethyl Glycosides and Their Derivatives. Angew Chem Int Ed. 2009;48:8896–8899. doi: 10.1002/anie.200904168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chenault HK, Chafin LF. Hydrolysis of O-Isopropenyl Glucopyranosides Involves C-Protonation and Alkenyl Ether Cleavage and Exhibits a Kinetic Influence of Anomeric Configuration. J Org Chem. 1994;59:6167–6169. [Google Scholar]
- 44.Chenault HK, Chafin LF. Mechanism of Hydrolysis of Isopropenyl Glucopyranosides. Spectroscopic Evidence Concerning the Greater Reactivity of the α-Anomer. J Org Chem. 1998;63:833–840. doi: 10.1021/jo9721249. [DOI] [PubMed] [Google Scholar]
- 45.Chenault HK, Castro A, Chafin LF, Yang J. The Chemistry of Isopropenyl Glycopyranosides. Transglycosidations and Other Reactions. J Org Chem. 1996;61:5024–5031. [Google Scholar]
- 46.Overend WG, Rees CW, Sequeira JS. Reactions at Position 1 of Carbohydrates. The Acid-Catalyzed Hydrolysis of Glycosides. J Chem Soc. 1962:3429–3440. [Google Scholar]
- 47.Namchuk MN, McCarter JD, Becalski A, Andrews T, Withers SG. The Role of Sugar Substituents in Glycoside Hydrolysis. J Am Chem Soc. 2000;122:1270–1277. [Google Scholar]
- 48.A glycoside is considered to undergo spontaneous hydrolysis, that is, without acid or base catalysis, when the rate does not vary across a wide pH range Cocker D, Sinnott ML. Generation of Glycopyranosyl Cations in the Spontaneous Hydrolyses of 2,4-Dinitrophenyl Glycopyranosides. Evidence for the General Intermediacy of Glycopyranosyl Cations in the Acid-Catalysed Hydrolyses of Methyl Glycopyranosides. J Chem Soc, Perkin Trans 2. 1975:1391–1393.
- 49.The three staggered conformation of the hexopyranose side chain are labelled according to the location of the side chain oxygen (O6) relative to the ring oxygen (O5) and C4 in the ring. They are therefore gauche,gauche (gg), gauche,trans (gt), and trans,gauche (tg). Bock K, Duus JO. A Conformational Study of Hydroxymethyl Groups in Carbohydrates Investigated by 1H NMR spectroscopy. J Carbohydr Chem. 1994;13:513–543.
- 50.Jensen HH, Nordstrom M, Bols M. The Disarming Effect of the 4,6-Acetal Group on Glycoside Reactivity: Torsional or Electronic. J Am Chem Soc. 2004;126:9205–9213. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- 51.Moumé-Pymbock M, Furukawa T, Mondal S, Crich D. Probing the Influence of a 4,6-O-Acetal on the Reactivity of Galactopyranosyl Donors: Verification of the Disarming Influence of the trans-gauche Conformation of C5–C6 Bonds. J Am Chem Soc. 2013;135:14249–14255. doi: 10.1021/ja405588x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andrews CW, Rodebaugh R, Fraser-Reid B. A Solvation Assisted Model for Estimating Anomeric Reactivity, Predicted Verus Observed Trends in Hydrolysis of N-Pentenyl Glycosides. J Org Chem. 1996;61:5280–5289. [Google Scholar]
- 53.Gylling Frihed T, Walvoort MTC, Codée JDC, van der Marel GA, Bols M, Pedersen CM. Influence of O6 in Mannosylations Using Benzylidene Protected Donors: Stereoelectronic or Conformational Effects? J Org Chem. 2013;78:2191–2205. doi: 10.1021/jo302455d. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. Programmable One-Pot Oligosaccharide Synthesis. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 55.Hsu CH, Hung SC, Wu CY, Wong CH. Toward Automated Oligosaccharide Synthesis. Angew Chem Int Ed. 2011;50:11872–11923. doi: 10.1002/anie.201100125. [DOI] [PubMed] [Google Scholar]
- 56.Douglas NL, Ley SV, Lucking U, Warriner SL. Tuning Glycoside Reactivity: New Tool for Efficient Oligosaccharide Synthesis. J Chem Soc, Perkin Trans 1. 1998:51–65. [Google Scholar]
- 57.Gomez AM. A Survey of Ley’s Reactivity Tuning in Oligosaccharide Synthesis. Top Curr Chem. 2011;301:31–68. doi: 10.1007/128_2010_112. [DOI] [PubMed] [Google Scholar]
- 58.Lahman M, Oscarson S. Investigation of the Reactivity Difference between Thioglycoside Donors with Variant Aglycon Parts. Can J Chem. 2002;80:889–893. [Google Scholar]
- 59.Li X, Huang L, Hu X, Huang X. Thio-Arylglycosides with Various Aglycon para-Substituents: A Probe for Studying Chemical Glycosylation Reactions. Org Biomol Chem. 2009;7:117–127. doi: 10.1039/b813048e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matsumoto K, Ueoka K, Suzuki S, Suga S, Yoshida J-i. Direct and Indirect Electrochemical Generation of Alkoxycarbenium Ion Pools from Thioacetals. Tetrahedron. 2009;65:10901–10907. [Google Scholar]
- 61.Saito K, Ueoka K, Matsumoto K, Suga S, Nokami T, Yoshida J-i. Indirect Cation-Flow Method: Flash Generation of Alkoxycarbenium Ions and Studies on the Stability of Glycosyl Cations. Angew Chem Int Ed. 2011;50:5153–5156. doi: 10.1002/anie.201100854. [DOI] [PubMed] [Google Scholar]
- 62.Nokami T, Hayashi R, Saigusa Y, Shimizu A, Liu CY, Mong KKT, Yoshida J-i. Automated Solution-Phase Synthesis of Oligosaccharides Via Iterative Electrochemical Assembly of Thioglycosides. Org Lett. 2013;15:4520–4523. doi: 10.1021/ol402034g. [DOI] [PubMed] [Google Scholar]
- 63.Akien GR, Subramaniam B. 245th ACS National Meeting & Exposition; New Orleans. 2013; p. CARB-105. [Google Scholar]
- 64.Akien GR, Subramaniam B. 247th ACS National Meeting & Exhibition; Dallas. 2014; p. CARB-96. [Google Scholar]
- 65.Martin A, Arda A, Désiré J, Martin-Mingot A, Probst N, Sinaÿ P, Jiménez-Barbero J, Thibaudeau S, Blériot Y. Catching Elusive Glycosyl Cations in a Condensed Phase with HF/SbF5 Superacid. Nat Chem. 2016;8:186–191. doi: 10.1038/nchem.2399. [DOI] [PubMed] [Google Scholar]
- 66.Bohé L, Crich D. Glycosyl Cations out on Parole. Nat Chem. 2016;8:99–100. doi: 10.1038/nchem.2436. [DOI] [PubMed] [Google Scholar]
- 67.Chamberland S, Ziller JW, Woerpel KA. Structural Evidence That Alkoxy Substituents Adopt Electronically Preferred Pseudoaxial Orientations in Six-Membered Ring Dioxocarbenium Ions. J Am Chem Soc. 2005;127:5322–5323. doi: 10.1021/ja050830i. [DOI] [PubMed] [Google Scholar]
- 68.Yang MT, Woerpel KA. The Effect of Electrostatic Interactions on Conformational Equilibria of Multiply Substituted Tetrahydropyran Oxocarbenium Ions. J Org Chem. 2009;74:545–553. doi: 10.1021/jo8017846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Woods RJ, Andrews CW, Bowen JP. Molecular Mechanical Investigations of the Properties of Oxocarbenium Ions. 1. Parameter Development. J Am Chem Soc. 1992;114:850–858. [Google Scholar]
- 70.Woods RJ, Andrews CW, Bowen JP. Molecular Mechanical Investigations of the Properties of Oxacarbenium Ions. Application to Glycoside Hydrolysis. J Am Chem Soc. 1992;114:859–864. [Google Scholar]
- 71.Miljkovic M, Yeagley D, Deslongchamps P, Dory YL. Experimental and Theoretical Evidence of through-Space Electrostatic Stabilization of the Incipient Oxocarbenium Ion by an Axially Oriented Electronegative Substituent During Glycopyranoside Acetolysis. J Org Chem. 1997;62:7597–7604. [Google Scholar]
- 72.Smith DM, Woerpel KA. Electrostatic Interactions in Cations and Their Importance in Biology and Chemistry. Org Biomol Chem. 2006;4:1195–1201. doi: 10.1039/b600056h. [DOI] [PubMed] [Google Scholar]
- 73.Jensen HH, Bols M. Stereoelectronic Substituent Effects. Acc Chem Res. 2006;39:259–265. doi: 10.1021/ar050189p. [DOI] [PubMed] [Google Scholar]
- 74.Heuckendorff M, Pedersen CM, Bols M. Quantifying Electronic Effects of Common Carbohydrate Protecting Groups in a Piperidine Model System. Chem Eur J. 2010;16:13982–13994. doi: 10.1002/chem.201002313. [DOI] [PubMed] [Google Scholar]
- 75.NB. The equilibrium constant for cyanohydrin formation from cyclopentanone is only 3.3 whereas that for the same reaction with cyclohexanone is 70. Prelog V, Kobelt M. The Dependence of the Dissociation Constants of the Ring-Homologous Cyclanone Cyanohydrins Upon the Size of the Ring. Helv Chim Acta. 1949;32:1187–1192.
- 76.De Meo C, Wallace CE, Geringer SA. Synthesis and Elimination of C-3-Labeled Thiosialosides. Org Lett. 2014;16:2676–2679. doi: 10.1021/ol500917k. [DOI] [PubMed] [Google Scholar]
- 77.Amyes TL, Jencks WP. Lifetimes of Oxocarbenium Ions in Aqueous Solution from Common Ion Inhibition of the Solvolysis of α-Azido Ethers by Added Azide Ion. J Am Chem Soc. 1989;111:7888–7900. [Google Scholar]
- 78.Huang X, Surry C, Hiebert T, Bennet AJ. Hydrolysis of (2-Deoxy-β-D-Glucopyranosyl)Pyridinium Salts. J Am Chem Soc. 1995;117:10614–10621. [Google Scholar]
- 79.Zhu J, Bennet AJ. Hydrolysis of (2-Deoxy-α-D-Glucopyranosylpyridinium) Salts: The 2-Deoxyglucosyl Oxacarbenium Is Not Solvent-Equilibrated in Water. J Am Chem Soc. 1998;120:3887–3893. [Google Scholar]
- 80.Zhu J, Bennet AJ. Solvolyses of 2-Deoxy-α- and β-D-Glucopyranosyl 4′-Bromoisoquinolinium Tetrafluoroborates. J Org Chem. 2000;65:4423–4430. doi: 10.1021/jo0004106. [DOI] [PubMed] [Google Scholar]
- 81.Horenstein BA, Bruner M. The N-Acetyl Neuraminyl Oxecarbenium Ion Is an Intermediate in the Presence of Anionic Nucleophiles. J Am Chem Soc. 1998;120:1357–1362. [Google Scholar]
- 82.Horenstein B. Quantum Mechanical Analysis of an α-Carboxylate-Substituted Oxocarbenium Ion. Isotope Effects for Formation of the Sialyl Cation and the Origin of an Unusually Large Secondary 14C Isotope Effect. J Am Chem Soc. 1997;119:1101–1107. [Google Scholar]
- 83.Huang M, Retailleau P, Bohé L, Crich D. Cation Clock Permits Distinction between the Mechanisms of α- and β-O- and β-C-Glycosylation in the Mannopyranose Series; Evidence for the Existence of a Mannopyranosyl Oxocarbenium Ion. J Am Chem Soc. 2012;134:14746–14749. doi: 10.1021/ja307266n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adero PO, Furukawa T, Huang M, Mukherjee D, Retailleau P, Bohé L, Crich D. Cation Clock Reactions for the Determination of Relative Reaction Kinetics in Glycosylation Reactions: Applications to Gluco- and Mannopyranosyl Sulfoxide and Trichloroacetimidate Type Donors. J Am Chem Soc. 2015;137:10336–10345. doi: 10.1021/jacs.5b06126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang M, Furukawa T, Retailleau P, Crich D, Bohé L. Further Studies on Cation Clock Reactions in Glycosylation: Observation of a Configuration Specific Intramolecular Sulfenyl Transfer and Isolation and Characterization of a Tricyclic Acetal. Carbohydr Res. 2016;427:21–28. doi: 10.1016/j.carres.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Amarasekara H, Crich D. Synthesis and Intramolecular Glycosylation of Sialyl Mono-Esters of o-Xylylene Glycol. The Importance of Donor Configuration and Nitrogen Protecting Groups on Cyclization Yield and Selectivity; Isolation and Characterization of a N-Sialyl Acetamide Indicative of Participation by Acetonitrile. Carbohydrate Res. 2016;435:113–120. doi: 10.1016/j.carres.2016.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Denekamp C, Sandlers Y. Anomeric Distinction and Oxonium Ion Formation in Acetylated Glycosides. J Mass Spectrom. 2005;40:765–771. doi: 10.1002/jms.848. [DOI] [PubMed] [Google Scholar]
- 88.Denekamp C, Sandlers Y. Formation and Stability of Oxocarbenium Ions from Glycosides. J Mass Spectrom. 2005;40:1055–1063. doi: 10.1002/jms.880. [DOI] [PubMed] [Google Scholar]
- 89.Okada Y, Nagata O, Taira M, Yamada H. Highly β-Selective and Direct Formation of 2-O-Glycosylated Glucosides by Ring Restriction into Twist Boat. Org Lett. 2007;9:2755–2758. doi: 10.1021/ol070720b. [DOI] [PubMed] [Google Scholar]
- 90.Bols M, Pedersen CM. Silyl-Protective Groups Influencing the Reactivity and Selectivity in Glycosylations. Beil J Org Chem. 2017;13:93–105. doi: 10.3762/bjoc.13.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kancharla PK, Navuluri C, Crich D. Dissecting the Influence of Oxazolidinones and Cyclic Carbonates in Sialic Acid Chemistry. Angew Chem Int Ed. 2012;51:11105–11109. doi: 10.1002/anie.201204400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chizhov AO, Podvalnyy NM, Zinin AI, Malysheva NN, Kononov LO. Electrospray Ionization Mass Spectra of Derivatives of 2-Phenylthio-N-Trifluoroacetylneuraminic Acid. J Anal Chem. 2015;70:1664–1670. [Google Scholar]
- 93.Chizhov AO, Shpirt AM, Savel’eva NY, Kononov LO. A Comparison of Electrospray Tandem Mass Spectra of Some Sialic Acid Derivatives: Ion Trap and High Resolution QqTOF Mass Spectrometers. J Anal Chem. 2016;71:1392–1396. [Google Scholar]
- 94.Vasella A. Glycosylidene Carbenes. In: Hecht SM, editor. Bioorganic Chemistry: Carbohydrates. Oxford University Press; New York: 1999. pp. 56–88. [Google Scholar]
- 95.Vasella A, Bernet B, Weber M, Wenger W. Glycosylidene Diazirines. In: Ernst B, Hart GW, Sinaÿ P, editors. Carbohydrates in Chemistry and Biology: Chemistry of Saccharides. Vol. 1. Wiley-VCH; Weinheim: 2000. pp. 155–175. [Google Scholar]
- 96.Briner K, Vasella A. Glycosylidene Carbenes. A New Approach to Glycoside Synthesis. Preparation of Glycosylidene-Derived Diaziridines and Diazirines. Helv Chim Acta. 1989;72:1371–1382. [Google Scholar]
- 97.Briner K, Vasella A. Glycosylidene Carbenes. Synthesis of Alkyl and Fluoroalkyl Glycosides. Helv Chim Acta. 1992;75:621–637. [Google Scholar]
- 98.Takahashi Y, Vasella A. Glycosylidene Carbenes. Neighboring Group Participation by the 2-Benzyloxy Group in the Glycosidation of Strongly Acidic Hydroxy Compounds. Helv Chim Acta. 1992;75:1563–1571. [Google Scholar]
- 99.Krumper JR, Salamant WA, Woerpel KA. Correlations between Nucleophilicities and Selectivities in the Substitutions of Tetrahydropyran Acetals. J Org Chem. 2009;74:8039–8050. doi: 10.1021/jo901639b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wu CY, Wong CH. Programmable One-Pot Glycosylation. Top Curr Chem. 2011;301:223–252. doi: 10.1007/128_2010_109. [DOI] [PubMed] [Google Scholar]
- 101.Mootoo DR, Konradsson P, Udodong U, Fraser-Reid B. “Armed” and “Disarmed” N-Pentenyl Glycosides in Saccharide Couplings Leading to Oligosaccharides. J Am Chem Soc. 1988;110:5583–5584. [Google Scholar]
- 102.Fraser-Reid B, Lopez C. Armed–Disarmed Effects in Carbohydrate Chemistry: History, Synthetic and Mechanistic Studies. Top Curr Chem. 2011;301:1–29. doi: 10.1007/128_2010_105. [DOI] [PubMed] [Google Scholar]
- 103.Paulsen H, Richter A, Sinnwell V, Stenzel W. Darstellung Selektiv Blockierter 2-Azido-2-Desoxy-D-Gluco und Galactopyranosylhalogenide: Reaktivitat und 13C-Nmr-Spektren. Carbohydr Res. 1978;64:339–362. [Google Scholar]
- 104.Glaudemans CPJ, Fletcher HG. Long Range Effects of Acyl Groups in the Solvolysis of Glycofuranosyl Halides. The Synthesis of 2,3-Di-O-Benzyl-5-O-p-Nitrobenzoyl-α-D-Arabinofuranosyl Chloride and of 2-O-Benzyl-3,5-Di-O-p-Nitrobenzoyl-α-D-Arabinofuranosyl Chloride. J Am Chem Soc. 1965;87:4636–4641. [Google Scholar]
- 105.Pedersen CM, Olsen J, Brka AB, Bols M. Quantifying the Electronic Effects of Carbohydrate Hydroxy Groups by Using Aminosugar Models. Chem Eur J. 2011;17:7080–7086. doi: 10.1002/chem.201100020. [DOI] [PubMed] [Google Scholar]
- 106.Pedersen CM, Bols M. On the Nature of the Electronic Effect of Multiple Hydroxyl Groups in the 6-Membered Ring – the Effects Are Additive but Steric Hindrance Plays a Role Too. Org Biomol Chem. 2017;15:1164–1173. doi: 10.1039/c6ob02427k. [DOI] [PubMed] [Google Scholar]
- 107.Pedersen CM, Nordstrom LU, Bols M. “Super Armed” Glycosyl Donors: Conformational Arming of Thioglycosides by Silylation. J Am Chem Soc. 2007;129:9222–9235. doi: 10.1021/ja071955l. [DOI] [PubMed] [Google Scholar]
- 108.Pedersen CM, Marinescu LG, Bols M. Glycosyl Donors in Unusual Conformations - Influence on Selectivity and Reactivity. CR Chimie. 2011;14:17–43. [Google Scholar]
- 109.Okada Y, Mukae T, Okajimia K, Taira M, Fujita M, Yamada H. Highly β-Selective O-Glucosidation Due to the Restricted Twist-Boat Conformation. Org Lett. 2007;9:1573–1576. doi: 10.1021/ol070427b. [DOI] [PubMed] [Google Scholar]
- 110.Marzabadi CH, EAJ, Gonzalez-Outeirino J, Gaffney PJR, White CGH, Tocher DA, Todaro LJ. Why Are Silyl Ethers Conformationally Different from Alkyl Ethers? Chair-Chair Conformational Equilibria in Siloxycyclohexanes and Their Dependence on the Substituents on Silicon. The Wider Roles of Eclipsing, of 1,3-Repulsive Steric Interactions, and of Attractive Steric Interactions. J Am Chem Soc. 2003;125:15163–15173. doi: 10.1021/ja035936x. [DOI] [PubMed] [Google Scholar]
- 111.Komarova BS, Gerbst AG, Finogenova AM, Dmitrenok AS, Tsvetkov YE, Nifantiev NE. 1,3-syn-Diaxial Repulsion of Typical Protecting Groups Used in Carbohydrate Chemistry in 3-O-Substituted Derivatives of Isopropyl D-Idopyranosides. J Org Chem. 2017;82:8897–8908. doi: 10.1021/acs.joc.7b01167. [DOI] [PubMed] [Google Scholar]
- 112.Okajima K, Mukae T, Imagawa H, Kawamura Y, Nishizawa M, Yamada H. Achievement of Ring Inversion of myo-Inositol Derivatives Due to Silyloxy/Silyloxy Repulsion Enhanced by the trans-Substituents on Both Sides. Tetrahedron. 2005;61:3497–3506. [Google Scholar]
- 113.Yamada H, Okajima K, Imagawa H, Mukae T, Kawamura Y, Nishizawa M. Stable Diaxial Chair Cyclohexanes Due to the Adjacent and Bulky trans-Siloxy Groups. Bull Chem Soc Jpn. 2007;80:583–585. [Google Scholar]
- 114.Lewis MD, Cha JK, Kishi Y. Highly Stereoselective Approaches to α- and β-C-Glycopyranosides. J Am Chem Soc. 1982;104:4976–4978. [Google Scholar]
- 115.Mayr H, Kempf B, Ofial AR. π-Nucleophilicity in Carbon–Carbon Bond-Forming Reactions. Acc Chem Res. 2003;36:66–77. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]
- 116.Deslongchamps P. Stereoelectronic Effects in Organic Chemistry. Pergamon; Oxford: 1983. [Google Scholar]
- 117.Kirby AJ. The Anomeric Effect and Related Stereoelectronic Effects at Oxygen. Springer-Verlag; Berlin: 1983. [Google Scholar]
- 118.Sinnott ML. The Principle of Least Nuclear Motion and the Theory of Stereoelectronic Control. Adv Phys Org Chem. 1988;24:113–204. [Google Scholar]
- 119.Cumpstey I. On a So-Called “Kinetic Anomeric Effect” in Chemical Glycosylation. Org Biomol Chem. 2012;10:2503–2508. doi: 10.1039/c2ob06696c. [DOI] [PubMed] [Google Scholar]
- 120.NB. In the absence of rigorous assignment of the H6 proR and proS hydrogens, the coupling constant data are also consistent with the gt conformation.
- 121.Kancharla PK, Crich D. Influence of Side Chain Conformation and Configuration on Glycosyl Donor Reactivity and Selectivity as Illustrated by Sialic Acid Donors Epimeric at the 7-Position. J Am Chem Soc. 2013;135:18999–19007. doi: 10.1021/ja410683y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dharuman S, Crich D. Determination of the Influence of Side Chain Conformation on Glycosylation Selectivity Using Conformationally Restricted Donors. Chem Eur J. 2016;22:4535–4542. doi: 10.1002/chem.201505019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Balmond EI, Benito-Alifonso D, Coe DM, Alder RW, McGarrigle EM, Galan MC. A 3,4-trans-Fused Cyclic Protecting Group Facilitates α-Selective Catalytic Synthesis of 2-Deoxyglycosides. Angew Chem Int Ed. 2014;53:8190–8194. doi: 10.1002/anie.201403543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Larsen CH, Ridgway BH, Shaw JT, Woerpel KA. A Stereoelectronic Model to Explain the Highly Stereoselective Reaction of Nucleophiles with Five-Membered-Ring Oxocarbenium Ions. J Am Chem Soc. 1999;121:11208–12209. [Google Scholar]
- 125.Larsen CH, Ridgway BH, Shaw JT, Smith DM, Woerpel KA. Stereoselective C-Glycosylation Reactions of Ribose Derivatives: Electronic Effects of Five-Membered Oxocarbenium Ions. J Am Chem Soc. 2005;127:10879–10884. doi: 10.1021/ja0524043. [DOI] [PubMed] [Google Scholar]
- 126.Tran VT, Woerpel KA. Nucleophilic Addition to Silyl-Protected Five-Membered Ring Oxocarbenium Ions Governed by Stereoelectronic Effects. J Org Chem. 2013;78:6609–6621. doi: 10.1021/jo400945j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lavinda O, Tran VT, Woerpel KA. Effect of Conformational Rigidity on the Stereoselectivity of Nucleophilic Additions to Five-Membered Ring Bicyclic Oxocarbenium Ion Intermediates. Org Biomol Chem. 2014;12:7083–7091. doi: 10.1039/c4ob01251h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hou D, Taha HA, Lowary TL. 2,3-Anhydrosugars in Glycoside Bond Synthesis: Mechanism of 2-Deoxy-2-Thioaryl Glycoside Formation. J Am Chem Soc. 2009;131:12937–12948. doi: 10.1021/ja9029945. [DOI] [PubMed] [Google Scholar]
- 129.van Rijssel ER, van Delft P, Lodder G, Overkleeft HS, van der Marel GA, Filippov DV, Codée JDC. Furanosyl Oxocarbenium Ion Stability and Stereoselectivity. Angew Chem Int Ed. 2014;53:10381–10385. doi: 10.1002/anie.201405477. [DOI] [PubMed] [Google Scholar]
- 130.Beaver MG, Buscagan TM, Lavinda O, Woerpel KA. Stereoelectronic Model to Explain Highly Stereoselective Reactions of Seven-Membered-Ring Oxocarbenium-Ion Intermediates. Angew Chem Int Ed. 2016;55:1816–1819. doi: 10.1002/anie.201507806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Igarashi K, Honma T, Irisawa J. Reaction of Glycosyl Chlorides with Silver Perchlorate. Carbohydr Res. 1970;15:329–337. [Google Scholar]
- 132.Igarashi K, Honma T, Irisawa J. Reaction of Glycosyl Chlorides with Silver Tetrafluoroborate. Carbohydr Res. 1970;13:49–55. [Google Scholar]
- 133.Nielsen MM, Stougaard BA, Bols M, Glibstrup E, Pedersen CM. Glycosyl Fluorides as Intermediates in BF3·OEt2-Promoted Glycosylation with Trichloroacetimidates. Eur J Org Chem. 2017:1281–1284. [Google Scholar]
- 134.Marousek V, Lucas TJ, Wheat PE, Schuerch C. The Influence of Reactant Structure and Solvent on Galactoside Synthesis from Galactosyl Sulfonates. Carbohydr Res. 1978;60:85–96. [Google Scholar]
- 135.Srivastava VK, Schuerch C. A Synthesis of β-D-Mannopyranosides by Glycosidation at C-1. Carbohydr Res. 1980;79:C13–C16. [Google Scholar]
- 136.Srivastava VK, Schuerch C. Synthesis of β-D-Mannopyranosides and β-L-Rhamnopyranosides by Glycosidation at C-1. J Org Chem. 1981;46:1121–1126. [Google Scholar]
- 137.Kronzer FJ, Schuerch C. The Methanolysis of Some Derivatives of 2,3,4-Tri-O-Benzyl-α-D-Glucopyranosyl Bromide in the Presence and Absence of Silver Salts. Carbohydr Res. 1973;27:379–390. [Google Scholar]
- 138.Crich D, Sun S. Are Glycosyl Triflates Intermediates in the Sulfoxide Glycosylation Method? A Chemical and 1H, 13C, and 19F-NMR Spectroscopic Investigation. J Am Chem Soc. 1997;119:11217–11223. [Google Scholar]
- 139.Crich D. Chemistry of Glycosyl Triflates: Synthesis of β-Mannopyranosides. In: Wang PG, Bertozzi CR, editors. Glycochemistry: Principles, Synthesis, and Applications. Dekker; New York: 2001. pp. 53–75. [Google Scholar]
- 140.Crich D. Chemistry of Glycosyl Triflates: Synthesis of β-Mannopyranosides. J Carbohydr Chem. 2002;21:667–690. [Google Scholar]
- 141.Crich D, Bowers AA. Sulfoxides, Sulfimides, and Sulfones. In: Demchenko AV, editor. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance. Wiley; Weinheim: 2008. pp. 303–329. [Google Scholar]
- 142.Aubry S, Sasaki K, Sharma I, Crich D. Influence of Protecting Groups on the Reactivity and Selectivity of Glycosylation: Chemistry of the 4,6-O-Benzylidene Protected Mannopyranosyl Donors and Related Species. Top Curr Chem. 2011;301:141–188. doi: 10.1007/128_2010_102. [DOI] [PubMed] [Google Scholar]
- 143.Crich D. Methodology Development and Physical Organic Chemistry: A Powerful Combination for the Advancement of Glycochemistry. J Org Chem. 2011;76:9193–9209. doi: 10.1021/jo2017026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bohé L, Crich D. Glycosylation with Glycosyl Sulfonates. In: Bennett SC, editor. Selective Glycosylations – Synthetic Methods and Catalysts. Wiley; Hoboken: 2017. pp. 115–133. [Google Scholar]
- 145.Crich D, Lim LBL. Glycosylation with Sulfoxides and Sulfinates as Donors or Promoters. Org React. 2004;64:115–251. [Google Scholar]
- 146.Walvoort MTC, van der Marel GA, Overkleeft HS, Codée JDC. On the Reactivity and Selectivity of Donor Glycosides in Glycochemistry and Glycobiology: Trapped Covalent Intermediates. Chem Sci. 2013;4:897–906. [Google Scholar]
- 147.Frihed TG, Bols M, Pedersen CM. Mechanisms of Glycosylation Reactions Studied by Low-Temperature Nuclear Magnetic Resonance. Chem Rev. 2015;115:4963–5013. doi: 10.1021/cr500434x. [DOI] [PubMed] [Google Scholar]
- 148.Hagen B, Ali S, Overkleeft HS, van der Marel GA, Codée JDC. Mapping the Reactivity and Selectivity of 2-Azidofucosyl Donors for the Assembly of N-Acetylfucosamine-Containing Bacterial Oligosaccharides. J Org Chem. 2017;82:848–868. doi: 10.1021/acs.joc.6b02593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hagen B, van Dijk JHM, Zhang Q, Overkleeft HS, van der Marel GA, Codée JDC. Synthesis of the Staphylococcus aureus Strain M Capsular Polysaccharide Repeating Unit. Org Lett. 2017;19:2514–2517. doi: 10.1021/acs.orglett.7b00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.van der Vorm S, Overkleeft HS, van der Marel GA, Codée JDC. Stereoselectivity of Conformationally Restricted Glucosazide Donors. J Org Chem. 2017;82:4793–4811. doi: 10.1021/acs.joc.7b00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Qiao Y, Ge W, Jia L, Hou X, Wang Y, Pedersen CM. Glycosylation Intermediates Studied Using Low Temperature 1H- and 19F-DOSY NMR: New Insight into the Activation of Trichloroacetimidates. Chem Commun. 2016;52:11418–11421. doi: 10.1039/c6cc05272j. [DOI] [PubMed] [Google Scholar]
- 152.Singh GP, Watson AJA, Fairbanks AJ. Achiral 2-Hydroxy Protecting Group for the Stereocontrolled Synthesis of 1,2-cis-α-Glycosides by Six-Ring Neighboring Group Participation. Org Lett. 2015;17:4376–4379. doi: 10.1021/acs.orglett.5b02226. [DOI] [PubMed] [Google Scholar]
- 153.Issa JP, Bennett CS. A Reagent-Controlled SN2-Glycosylation for the Direct Synthesis of β-Linked 2-Deoxy Sugars. J Am Chem Soc. 2014;136:5740–5744. doi: 10.1021/ja500410c. [DOI] [PubMed] [Google Scholar]
- 154.Kitowski A, Jiménez-Moreno E, Salvadó M, Mestre J, Castillón S, Jiménez-Osés G, Boutureira O, Bernardes GJL. Oxidative Activation of C–S Bonds with an Electropositive Nitrogen Promoter Enables Orthogonal Glycosylation of Alkyl over Phenyl Thioglycosides. Org Lett. 2017;19:5490–5493. doi: 10.1021/acs.orglett.7b02886. [DOI] [PubMed] [Google Scholar]
- 155.D’Angelo KA, Taylor MS. Borinic Acid Catalyzed Stereo- and Regioselective Couplings of Glycosyl Methanesulfonates. J Am Chem Soc. 2016;138:11058–11067. doi: 10.1021/jacs.6b06943. [DOI] [PubMed] [Google Scholar]
- 156.Watson AJA, Alexander SR, Cox DJ, Fairbanks AJ. Protecting Group Dependence of Stereochemical Outcome of Glycosylation of 2-O-(Thiophen-2-yl)methyl Ether Protected Glycosyl Donors. Eur J Org Chem. 2016:1520–1532. [Google Scholar]
- 157.Mensink RA, Elferink H, White PB, Pers N, Rutjes FPJT, Boltje TJ. A Study on Stereoselective Glycosylations via Sulfonium Ion Intermediates. Eur J Org Chem. 2016:4656–4667. [Google Scholar]
- 158.Dyapa R, Dockery LT, Walczak MA. Dehydrative Glycosylation with Cyclic Phosphonium Anhydrides. Org Biomol Chem. 2017;15:51–55. doi: 10.1039/c6ob01812b. [DOI] [PubMed] [Google Scholar]
- 159.Khomutnyk YY, Argüelles AJ, Winschel GA, Sun Z, Zimmerman PM, Nagorny P. Studies of the Mechanism and Origins of Enantioselectivity for the Chiral Phosphoric Acid-Catalyzed Stereoselective Spiroketalization Reactions. J Am Chem Soc. 2016;138:444–456. doi: 10.1021/jacs.5b12528. [DOI] [PubMed] [Google Scholar]
- 160.Brabham R, Fascione MA. Chiral Auxiliaries in Stereoselective Glycosylation Reactions. In: Bennett CS, editor. Selective Glyocsylations: Synthetic Methods and Catalysts. Wiley-VCH; Weinheim: 2017. pp. 97–113. [Google Scholar]
- 161.Nokami T. Sulfonium Ions as Reactive Glycosylation Intermediates. Trends in Glycosci Glycobiol. 2012;24:203–214. [Google Scholar]
- 162.West AC, Schuerch C. The Reverse Anomeric Effect and the Synthesis of α-Glycosides. J Am Chem Soc. 1973;95:1333–1335. [Google Scholar]
- 163.Fang T, Gu Y, Huang W, Boons GJ. Mechanism of Glycosylation of Anomeric Sulfonium Ions. J Am Chem Soc. 2016;138:3002–3011. doi: 10.1021/jacs.5b08436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cox DJ, Singh GP, Watson AJA, Fairbanks AJ. Neighbouring Group Participation During Glycosylation: Do 2-Substituted Ethyl Ethers Participate? Eur J Org Chem. 2014:4624–4642. [Google Scholar]
- 165.Smoot JT, Demchenko AV. How the Arming Participating Moieties Can Broaden the Scope of Chemoselective Oligosaccharide Synthesis by Allowing the Inverse Armed-Disarmed Approach. J Org Chem. 2008;73:8838–8850. doi: 10.1021/jo801551r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fascione MA, Kilner CA, Leach AG, Turnbull WB. Do Glycosyl Sulfonium Ions Engage in Neighbouring-Group Participation? A Study of Oxathiane Glycosyl Donors and the Basis for Their Stereoselectivity. Chem Eur J. 2012;18:321–333. doi: 10.1002/chem.201101889. [DOI] [PubMed] [Google Scholar]
- 167.Nokami T, Shibuya A, Manabe S, Ito Y, Yoshida J-i. α- and β-Glycosyl Sulfonium Ions: Generation and Reactivity. Chem Eur J. 2009;15:2252–2255. doi: 10.1002/chem.200802293. [DOI] [PubMed] [Google Scholar]
- 168.Elferink H, Mensink RA, White PB, Boltje TJ. Stereoselective β-Mannosylation by Neighboring-Group Participation. Angew Chem Int Ed. 2016;55:11217–11220. doi: 10.1002/anie.201604358. [DOI] [PubMed] [Google Scholar]
- 169.Hanessian S, Banoub J. Chemistry of the Glycosidic Linkage. An Efficient Synthesis of 1,2-trans-Disaccharides. Carbohydr Res. 1977;53:C13–C16. [Google Scholar]
- 170.Bock K, Guzmán JFB, Refn S. Synthesis and Properties of 1,1,3,3-Tetramethyl-2-(2,3,4,6-Tetra-O-Acetyl-α-D-Glucopyranosyl)Uronium Triflate. Carbohydr Res. 1992;232:353–357. [Google Scholar]
- 171.Koto S, Morishima N, Owa M, Zen S. A Stereoselective α-Glucosylation by Use of a Mixture of 4-Nitrobenzenesulfonyl Chloride, Silver Trifluoromethanesulfonate, N,N-Dimethylacetamide and Triethylamine. Carbohydr Res. 1984;130:73–83. [Google Scholar]
- 172.Lu SR, Lai YH, Chen JH, Liu CY, Mong KKT. Dimethylformamide: An Unusual Glycosylation Modulator. Angew Chem Int Ed. 2011;50:7315–7320. doi: 10.1002/anie.201100076. [DOI] [PubMed] [Google Scholar]
- 173.Lin YH, Ghosh B, Mong KKT. In Situ Formation of β-Glycosyl Imidinium Triflate from Participating Thioglycosyl Donors: Elaboration to Disarmed–Armed Iterative Glycosylation. Chem Commun. 2012;48:10910–10912. doi: 10.1039/c2cc35032g. [DOI] [PubMed] [Google Scholar]
- 174.Ingle AB, Chao CS, Hung W-c, Mong KKT. Tuning Reactivity of Glycosyl Imidinium Intermediate for 2-Azido-2-Deoxyglycosyl Donors in α-Glycosidic Bond Formation. Org Lett. 2013;15:5290–5293. doi: 10.1021/ol402519c. [DOI] [PubMed] [Google Scholar]
- 175.Hu JC, Feng AFW, Chang BY, Lin CH, Mong KKT. A Flexible 1,2-cis α-Glycosylation Strategy Based on in situ Adduct Transformation. Org Biomol Chem. 2017;15:5345–5356. doi: 10.1039/c7ob00839b. [DOI] [PubMed] [Google Scholar]
- 176.Garcia BA, Gin DY. Dehydrative Glycosylation with Activated Diphenyl Sulfonium Reagents. Scope, Mode of C(1)-Hemiacetal Activation, and Detection of Reactive Glycosyl Intermediates. J Am Chem Soc. 2000;122:4269–4279. [Google Scholar]
- 177.Codée JDC, van den Bos LJ, Litjens REJN, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA. Chemoselective Glycosylations Using Sulfonium Triflate Activator Systems. Tetrahedron. 2004;60:1057–1064. [Google Scholar]
- 178.Crich D, Li W. Efficient Glycosidation of a Phenyl Thiosialoside Donor with Diphenyl Sulfoxide and Triflic Anhydride in Dichloromethane. Org Lett. 2006;8:959–962. doi: 10.1021/ol060030s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mulani SK, Hung WC, Ingle AB, Shiau KS, Mong KKT. Modulating Glycosylation with Exogenous Nucleophiles: An Overview. Org Biomol Chem. 2014;12:1184–1197. doi: 10.1039/c3ob42129e. [DOI] [PubMed] [Google Scholar]
- 180.Kaeothip S, Yasomanee JP, Demchenko AV. Glycosidation of Thioglycosides in the Presence of Bromine: Mechanism, Reactivity, and Stereoselectivity. J Org Chem. 2012;77:291–299. doi: 10.1021/jo2019174. [DOI] [PubMed] [Google Scholar]
- 181.Prévost M, St-Jean O, Guindon Y. Synthesis of 1′,2′-cis-Nucleoside Analogues: Evidence of Stereoelectronic Control for SN2 Reactions at the Anomeric Center of Furanosides. J Am Chem Soc. 2010;132:12433–12439. doi: 10.1021/ja104429y. [DOI] [PubMed] [Google Scholar]
- 182.Goswami M, Ashley DC, Baik MH, Pohl NLB. Mechanistic Studies of Bismuth(V)-Mediated Thioglycoside Activation Reveal Differential Reactivity of Anomers. J Org Chem. 2016;81:5949–5962. doi: 10.1021/acs.joc.6b00860. [DOI] [PubMed] [Google Scholar]
- 183.Smith RS, Müller-Bunz H, Zhu X. Investigation of α-Thioglycoside Donors: Reactivity Studies toward Configuration-Controlled Orthogonal Activation in One-Pot Systems. Org Lett. 2016;18:3578–3581. doi: 10.1021/acs.orglett.6b01572. [DOI] [PubMed] [Google Scholar]
- 184.Crich D, Li M. Revisiting the Armed-Disarmed Concept: The Importance of Anomeric Configuration in the Activation of S-Benzoxazolyl Glycosides. Org Lett. 2007;9:4115–4118. doi: 10.1021/ol701466u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Heuckendorff M, Pedersen CM, Bols M. The Influence of Neighboring Group Participation on the Hydrolysis of 2-O-Substituted Methyl Glucopyranosides. Org Lett. 2011;13:5956–5959. doi: 10.1021/ol202308y. [DOI] [PubMed] [Google Scholar]
- 186.Pougny JR, Sinaÿ P. Reaction d’Imidates de Glucopyranosyle avec l’Acetonitrile. Applications Synthetiques. Tetrahedron Lett. 1976;17:4073–4076. [Google Scholar]
- 187.Ratcliffe AJ, Fraser-Reid B. Generation of α-D-Glucopyranosylacetonitrilium Ions. Concerning the Reverse Anomeric Effect. J Chem Soc, Perkin Trans, 1. 1990:747–750. [Google Scholar]
- 188.Crich D, Patel M. On the Nitrile Effect in L-Rhamnopyranosylation. Carbohydr Res. 2006;341:1467–1475. doi: 10.1016/j.carres.2006.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Schmidt RR, Behrendt M, Toepfer A. Nitriles as Solvents in Glycosylation Reactions: Highly Selective β-Glycoside Synthesis. Synlett. 1990:694–696. [Google Scholar]
- 190.Elferink H, Pedersen CM. L-Rhamnosylation: The Solvent Is the Solution. Eur J Org Chem. 2017:53–59. [Google Scholar]
- 191.Lemieux RU, Ratcliffe RM. The Azidonitration of Tri-O-Acetyl-D-Galactal. Can J Chem. 1979;57:1244–1251. [Google Scholar]
- 192.Pavia AA, Ung-Chhun SN, Durand JL. Synthesis of N-Glycosides. Formation of Glucosylamine by Reaction of 2,3,4,6-Tetra-O-Benzyl-D-Glucopyranose with Acetonitrile in the Presence of Trifluoromethanesulfonic Anhydride. J Org Chem. 1981;46:3158–3160. [Google Scholar]
- 193.Klemer A, Kohla M. Eine Einfache Synthese von N-Acyl-Glyosylaminen. J Carbohydr Chem. 1988;7:785–797. [Google Scholar]
- 194.Gordon DM, Danishefsky SJ. Ritter-Like Reactions of 1,2-Anhydropyranose Derivatives. J Org Chem. 1991;56:3713–3715. [Google Scholar]
- 195.Schweizer F, Lohse A, Otter A, Hindsgaul O. One Pot Conversion of Ketoses into Sugar β-Peptides Via a Ritter Reaction. Synlett. 2001:1434–1436. [Google Scholar]
- 196.Meijer A, Ellervik U. Interhalogens (ICl/IBr) and AgOTf in Thioglycoside Activation: Synthesis of Bislactam Analogues of Ganglioside GD3. J Org Chem. 2004;69:6249–6256. doi: 10.1021/jo049184g. [DOI] [PubMed] [Google Scholar]
- 197.Tsuda T, Nakamura S, Hashimoto S. A Highly Stereoselective Construction of 1,2-trans-β-Glycosidic Linkages Capitalizing on 2-Azido-2-Deoxy-D-Glycosyl Diphenyl Phosphates as Glycosyl Donors. Tetrahedron. 2004;60:10711–10737. [Google Scholar]
- 198.Hoang KLM, Liu XW. The Intriguing Dual-Directing Effect of 2-Cyanobenzyl Ether for a Highly Stereospecific Glycosylation Reaction. Nat Commun. 2014;5:5051s. doi: 10.1038/ncomms6051. [DOI] [PubMed] [Google Scholar]
- 199.Braccini J, Derouet C, Esnault J, de Penhoat CH, Mallet JM, Michon V, Sinaÿ P. Conformational Analysis of Nitrilium Intermediates in Glycosylation Reactions. Carbohydr Res. 1993;246:23–41. [Google Scholar]
- 200.Stalford SA, Kilner CA, Leach AG, Turnbull WB. Neighbouring Group Participation vs. Addition to Oxacarbenium Ions: Studies on the Synthesis of Mycobacterial Oligosaccharides. Org Biomol Chem. 2009;7:4842–4852. doi: 10.1039/b914417j. [DOI] [PubMed] [Google Scholar]
- 201.Kanie O, Kiso M, Hasegawa A. Glycosylation Using Methylthioglycosides of N-Acetylneuraminic Acid and Dimethyl(Methylthio)Sulfonium Triflate. J Carbohydr Chem. 1988;7:501–506. [Google Scholar]
- 202.Hasegawa A, Kiso M. Synthesis of Sialyl Glycosides. In: Hanessian S, editor. Preparative Carbohydrate Chemistry. Marcel Dekker; New York: 1997. pp. 357–379. [Google Scholar]
- 203.Navuluri C, Crich D. Recent Advances in the Stereocontrolled Synthesis of Sialic Acid Glycosides. In: Hung S-C, Zulueta MML, editors. Glycochemical Synthesis: Strategies and Applications. Wiley; New York: 2016. pp. 131–154. [Google Scholar]
- 204.Martichonok V, Whitesides GM. Stereoselective α-Sialylation with Sialyl Xanthate and Phenylsulfenyl Triflate. J Org Chem. 1996;61:1702–1706. doi: 10.1021/jo951711w. [DOI] [PubMed] [Google Scholar]
- 205.Whitfield DM. Computational Studies of the Role of Glycopyranosyl Oxacarbenium Ions in Glycobiology and Glycochemistry. Adv Carbohydr Chem Biochem. 2009;62:83–159. doi: 10.1016/S0065-2318(09)00004-3. [DOI] [PubMed] [Google Scholar]
- 206.Crich D, Sun S. Formation of β-Mannopyranosides of Primary Alcohols Using the Sulfoxide Method. J Org Chem. 1996;61:4506–4507. doi: 10.1021/jo9606517. [DOI] [PubMed] [Google Scholar]
- 207.Crich D, Sun S. Direct Synthesis of β-Mannopyranosides by the Sulfoxide Method. J Org Chem. 1997;62:1198–1199. [Google Scholar]
- 208.Crich D, Sun S. Direct Chemical Synthesis of β-Mannopyranosides and Other Glycosides Via Glycosyl Triflates. Tetrahedron. 1998;54:8321–8348. [Google Scholar]
- 209.Cai F, Wu B, Crich D. Stereocontrolled Synthesis of Mannans and Rhamnans. Adv Carbohydr Chem Biochem. 2009;62:251–309. doi: 10.1016/S0065-2318(09)00006-7. [DOI] [PubMed] [Google Scholar]
- 210.Guinchard X, Picard S, Crich D. Synthetic Approaches to Bioactive Carbohydrates. In: Cossy J, Arseniyadis S, editors. Modern Tools for the Synthesis of Complex Bioactive Molecules. Wiley; Hoboken: 2012. pp. 395–432. [Google Scholar]
- 211.Yun M, Shin Y, Chun KH, Jen S. Direct Syntheses of β-Mannopyranosyl Disaccharides from 4,6-O-Benzylidene Derivatives of Ethylthio-α-D-Mannopyranoside Donors. Bull Korean Chem Soc. 2000;21:562–566. [Google Scholar]
- 212.Weingart R, Schmidt RR. Can Preferential β-Mannopyranoside Formation with 4,6-O-Benzylidene Protected Mannopyranosyl Sulfoxides Be Reached with Trichloracetimidates. Tetrahedron Lett. 2000;41:8753–8758. [Google Scholar]
- 213.Tsuda T, Sato S, Nakamura S, Hashimoto S. Direct and Stereoselective Construction of β-Mannosidic Linkages Capitalizing on 4,6-O-Benzylidene-Protected D-Mannopyranosyl Diethylphosphate. Heterocycles. 2003;59:509–515. [Google Scholar]
- 214.Codée JDC, Kröck L, Castagner B, Seeberger PH. Automated Solid-Phase Synthesis of Protected Oligosaccharides Containing β-Mannosidic Linkages. Chem Eur J. 2008;14:3987–3994. doi: 10.1002/chem.200701864. [DOI] [PubMed] [Google Scholar]
- 215.Tanaka K, Mori Y, Fukase K. Practical Synthesis of a Man(1–4)GlcnTroc Fragment via Microfluidic β-Mannosylation. J Carbohydr Chem. 2009;28:1–11. [Google Scholar]
- 216.Tanifum CT, Chang CWT. Sonication-Assisted Oligomannoside Synthesis. J Org Chem. 2009;74:634–644. doi: 10.1021/jo8019835. [DOI] [PubMed] [Google Scholar]
- 217.Wang S, Lafont D, Rahkila J, Picod B, Leino R, Vidal S. Glycosylation of ‘Basic’ Alcohols: Methyl 6-(Hydroxymethyl)Picolinate as a Case Study. Carbohydr Res. 2013;372:35–43. doi: 10.1016/j.carres.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 218.Heuckendorff M, Bendix J, Pedersen CM, Bols M. β-Selective Mannosylation with a 4,6-Silylene Tethered Thiomannosyl Donor. Org Lett. 2014;16:1116–1119. doi: 10.1021/ol403722f. [DOI] [PubMed] [Google Scholar]
- 219.Heuckendorff M, Bols PS, Barry C, Frihed TG, Pedersen CM, Bols M. β-Mannosylation with 4,6-Benzylidene Protected Mannosyl Donors without Preactivation. Chem Commun. 2015:13283–13285. doi: 10.1039/c5cc04716a. [DOI] [PubMed] [Google Scholar]
- 220.Tanaka S-i, Takashima M, Tokimoto H, Fujimoto Y, Tanaka K, Fukase K. Highly β-Selective Mannosylation Towards Manβ–1–4-GlcNAc Synthesis: TMSB(C6F5)4 as a Lewis Acid/Cation Trap Catalyst. Synlett. 2005:2325–2328. [Google Scholar]
- 221.Moumé-Pymbock M, Crich D. Stereoselective C-Glycoside Formation with 2-O-Benzyl-4,6-O-Benzylidene Protected 3-Deoxy Gluco- and Mannopyranoside Donors: Comparison with O-Glycoside Formation. J Org Chem. 2012;77:8905–8912. doi: 10.1021/jo3011655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Huang M, Garrett GE, Birlirakis N, Bohé L, Pratt DA, Crich D. Dissecting the Mechanisms of a Class of Chemical Glycosylation Using Primary 13C Kinetic Isotope Effects. Nat Chem. 2012;4:663–667. doi: 10.1038/nchem.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Nigudkar SS, Wang T, Pistorio SG, Yasomanee JP, Stine KJ, Demchenko AV. OFox Imidates as Versatile Glycosyl Donors for Chemical Glycosylation. Org Biomol Chem. 2017;15:348–359. doi: 10.1039/c6ob02230h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zefirov NS, Koz’min AS. Competititve Binding of Nucleofugal Anions in Carbocationic-Like Processes. Acc Chem Res. 1985;18:154–158. [Google Scholar]
- 225.Dhakal B, Bohé L, Crich D. Trifluoromethanesulfonate Anion as Nucleophile in Organic Chemistry. J Org Chem. 2017;82:9263–9269. doi: 10.1021/acs.joc.7b01850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zhu Y, Yu B. Highly Stereoselective β-Mannopyranosylation Via the 1-α-Glycosyloxy-Isochromenylium-4-Gold(I) Intermediates. Chem Eur J. 2015;21:8771–8780. doi: 10.1002/chem.201500648. [DOI] [PubMed] [Google Scholar]
- 227.Kowalska K, Pedersen CM. Catalytic Stereospecific O-Glycosylation. Chem Commun. 2017;53:2040–2043. doi: 10.1039/c6cc10076g. [DOI] [PubMed] [Google Scholar]
- 228.Hashimoto Y, Tanikawa S, Saito R, Sasaki K. β-Stereoselective Mannosylation Using 2,6-Lactones. J Am Chem Soc. 2016;138:14840–14843. doi: 10.1021/jacs.6b08874. [DOI] [PubMed] [Google Scholar]
- 229.Hasty SJ, Ranade SC, Demchenko AV. A Study of Silver (I) Perchlorate as an Effective Promoter for Chemical Glycosylation with Thioimidates and Thioglycosides. Reports Org Chem. 2014;4:1–10. [Google Scholar]
- 230.Sasaki K, Matsumura S, Toshima K. A Novel Glycosidation of Glycosyl Fluorides Using a Designed Ionic Liquid and Its Effect on Stereoselectivity. Tetrahedron Lett. 2004;45:7043–7047. [Google Scholar]
- 231.Poletti L, Rencurosi A, Lay L, Russo G. Trichloroacetimidates as Glycosyl Donors in Recyclable Ionic Liquids. Synlett. 2003:2297–2300. [Google Scholar]
- 232.Rencurosi A, Lay L, Russo G, Caneva E, Poletti L. Glycosylation with Trichloroacetimidates in Ionic Liquids: Influence of the Reaction Medium on the Stereochemical Outcome. J Org Chem. 2005;70:7765–7768. doi: 10.1021/jo050704x. [DOI] [PubMed] [Google Scholar]
- 233.Worm-Leonhard K, Larsen K, Jensen KJ. 4,6-O-Benzylidene Directed β-Mannosylation without Intermediate Triflate Formation? Comparison of Trichloroacetimidate and Disal Donors in Microwave- Promoted Glycosylations under Neutral Conditions. J Carbohydr Chem. 2007;26:349–368. [Google Scholar]
- 234.Nigudkar SS, Stine KJ, Demchenko AV. Regenerative Glycosylation under Nucleophilic Catalysis. J Am Chem Soc. 2014;136:921–923. doi: 10.1021/ja411746a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kiyooka S-i, Kaneno D, Fujiyama R. Intrinsic Reactivity Index as a Single Scale Directed toward Both Electrophilicity and Nucleophilicity Using Frontier Molecular Orbitals. Tetrahedron. 2013;69:4247–4258. [Google Scholar]
- 236.Mayr H. Reactivity Scales for Quantifying Polar Organic Reactivity: The Benzhydrylium Methodology. Tetrahedron. 2015;71:5095–5111. [Google Scholar]
- 237.Mayr H, Ofial AR. Philicities, Fugalities, and Equilibrium Constants. Acc Chem Res. 2016;49:952–965. doi: 10.1021/acs.accounts.6b00071. [DOI] [PubMed] [Google Scholar]
- 238.Byrne PA, Kobayashi S, Würthwein EU, Ammer J, Mayr H. Why Are Vinyl Cations Sluggish Electrophiles? J Am Chem Soc. 2017;139:1499–1511. doi: 10.1021/jacs.6b10889. [DOI] [PubMed] [Google Scholar]
- 239.Nguyen HM, Poole JL, Gin DY. Chemoselective Iterative Dehydrative Glycosylation. Angew Chem Int Ed. 2001;40:414–417. doi: 10.1002/1521-3773(20010119)40:2<414::AID-ANIE414>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 240.Balmond EI, Galan MC, McGarrigle EM. Recent Developments in the Application of Organocatalysis to Glycosylations. Synlett. 2013;24:2335–2339. [Google Scholar]
- 241.Williams R, Galan MC. Recent Advances in Organocatalytic Glycosylations. Eur J Org Chem. 2017:6247–6264. [Google Scholar]
- 242.Cox DJ, Smith MD, Fairbanks AJ. Glycosylation Catalyzed by a Chiral Brønsted Acid. Org Lett. 2010;12:1452–1455. doi: 10.1021/ol1001895. [DOI] [PubMed] [Google Scholar]
- 243.Kimura T, Sekine M, Takahashi D, Toshima K. Chiral Brønsted Acid Mediated Glycosylation with Recognition of Alcohol Chirality. Angew Chem Int Ed. 2013;52:12131–12134. doi: 10.1002/anie.201304830. [DOI] [PubMed] [Google Scholar]
- 244.Liu D, Sarrafpour S, Guo W, Goulart B, Bennett CS. Matched/Mismatched Interactions in Chiral Bronsted Acid-Catalyzed Glycosylation Reactions with 2-Deoxy-Sugar Trichloroacetimidate Donors. J Carbohydr Chem. 2014;33:423–434. [Google Scholar]
- 245.Tay JH, Argüelles AJ, DeMars MD, Zimmerman PM, Sherman DH, Nagorny P. Regiodivergent Glycosylations of 6-Deoxy-Erythronolide B and Oleandomycin-Derived Macrolactones Enabled by Chiral Acid Catalysis. J Am Chem Soc. 2017;139:8570–8578. doi: 10.1021/jacs.7b03198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lee J, Borovika A, Khomutnyka Y, Nagorny P. Chiral Phosphoric Acid-Catalyzed Desymmetrizative Glycosylation of 2-Deoxystreptamine and Its Application to Aminoglycoside Synthesis. Chem Commun. 2017;53:8976–8979. doi: 10.1039/c7cc05052f. [DOI] [PubMed] [Google Scholar]
- 247.Schmidt RR, Stumpp M. Glycosylphosphate Aus Glycosyl(Trichloroacetimidaten) Liebigs Ann Chem. 1984:680–691. [Google Scholar]
- 248.Gould ND, Allen CL, Nam BC, Schepartz A, Miller SJ. Combined Lewis Acid and Brønsted Acid-Mediated Reactivity of Glycosyl Trichloroacetimidate Donors. Carbohydr Res. 2013;382:36–42. doi: 10.1016/j.carres.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 249.Peng P, Schmidt RR. Acid–Base Catalysis in Glycosidations: A Nature Derived Alternative to the Generally Employed Methodology. Acc Chem Res. 2017;50:1171–1183. doi: 10.1021/acs.accounts.6b00518. [DOI] [PubMed] [Google Scholar]
- 250.Geng Y, Kumar A, Faidallah HM, Albar HA, Mhkalid IA, Schmidt RR. Cooperative Catalysis in Glycosidation Reactions with O-Glycosyl Trichloroacetimidates as Glycosyl Donors. Angew Chem Int Ed. 2013;52:10089–10092. doi: 10.1002/anie.201302158. [DOI] [PubMed] [Google Scholar]
- 251.Palo-Nieto C, Sau A, Williams R, Galan MC. Cooperative Brønsted Acid-Type Organocatalysis for the Stereoselective Synthesis of Deoxyglycosides. J Org Chem. 2017;82:407–414. doi: 10.1021/acs.joc.6b02498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Park Y, Harper KC, Kuhl N, Kwan EE, Liu RY, Jacobsen EN. Macrocyclic Bis-Thioureas Catalyze Stereospecific Glycosylation Reactions. Science. 2017;355:162–166. doi: 10.1126/science.aal1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Kwan EE, Park Y, Besser HA, Anderson TL, Jacobsen EN. Sensitive and Accurate 13C Kinetic Isotope Effect Measurements Enabled by Polarization Transfer. J Am Chem Soc. 2017;139:43–46. doi: 10.1021/jacs.6b10621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Peng P, Schmidt RR. An Alternative Reaction Course in O-Glycosidation with O-Glycosyl Trichloroacetimidates as Glycosyl Donors and Lewis Acidic Metal Salts as Catalyst: Acid–Base Catalysis with Gold Chloride-Glycosyl Acceptor Adducts. J Am Chem Soc. 2015;137:12653–12659. doi: 10.1021/jacs.5b07895. [DOI] [PubMed] [Google Scholar]
- 255.Paulsen H. Selectivity and Reactivity in Oligosaccharide Synthesis. In: Bartmann W, Trost BM, editors. Selectivity a Goal for Synthetic Efficiency. Verlag Chemie; Weinheim: 1984. pp. 169–190. [Google Scholar]
- 256.Barresi F, Hindsgaul O. Chemically Synthesized Oligosaccharides, 1994. A Searchable Table of Glycosidic Linkages. J Carbohydr Chem. 1995;14:1043–1087. [Google Scholar]
- 257.Spijker NM, van Boeckel CAA. Double Stereodifferentiation in Carbohydrate Coupling Reactions: The Mismatched Interaction of Donor and Acceptor as an Unprecedented Factor Governing the α/β-Ratio of Glycoside Formation. Angew Chem Int Ed. 1991;30:180–183. [Google Scholar]
- 258.Horeau A, Kagan H-B, Vigneron JP. Synthèses Asymetriques Par Double Induction. Bull Soc Chim Fr. 1968:3795–3797. [Google Scholar]
- 259.Masamune S, Choy W, Peterson JS, Sita LR. Double Asymmetric Synthesis and a New Strategy for Stereochemical Control in Organic Synthesis. Angew Chem Int Ed. 1985;24:1–30. [Google Scholar]
- 260.Bohé L, Crich D. Double Diastereoselection, Regioselectivity, and the Importance of Donor-Acceptor Complementarity in the Stereoselectivity of Glycosylation Reactions. Trends Glycosci Glycotech. 2010;22:1–15. [Google Scholar]
- 261.Sylla B, Descroix K, Pain C, Gervaise C, Jamois F, Yvin JC, Legentil L, Nugier-Chauvin C, Daniellou R, Ferrieres V. Double Diastereoselection Explains Limitations in Synthesizing Mannose-Containing β-(1,3)-Glucans. Carbohydr Res. 2010;345:1366–1370. doi: 10.1016/j.carres.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 262.Guillemineau M, Auzanneau FI. Matched and Mismatched Acceptor/Donor Pairs in the Glycosylation of a Trisaccharide Diol Free at O-3 of Two N-Acylated Glucosamine Residues. Carbohydr Res. 2012;357:132–138. doi: 10.1016/j.carres.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 263.Abronina P, Fedina K, Podvalnyy N, Nikita M, Zinin A, Chizhov A, Kondakov N, Torgov V, Kononov L. The Use of O-Trifluoroacetyl Protection and Profound Influence of the Nature of Glycosyl Acceptor in Benzyl-Free Arabinofuranosylation. Carbohydr Res. 2014;396:25–36. doi: 10.1016/j.carres.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 264.Mancini RS, McClary CA, Anthonipillai S, Taylor MS. Organoboron-Promoted Regioselective Glycosylations in the Synthesis of a Saponin-Derived Pentasaccharide from Spergularia Ramosa. J Org Chem. 2015;80:8501–8510. doi: 10.1021/acs.joc.5b00950. [DOI] [PubMed] [Google Scholar]
- 265.Forman A, Auzanneau FI. Orthoester Formation Leading to Mismatched Helferich Glycosylations at O-3 of N-Trichloroacetylated Glucosamine Residues. Carbohydr Res. 2016;425:10–21. doi: 10.1016/j.carres.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 266.Fraser-Reid B, López JC, Gómez AM, Uriel C. Reciprocal Donor Acceptor Selectivity (RDAS) and Paulsen’s Concept of “Match” in Saccharide Coupling. Eur J Org Chem. 2004:1387–1395. [Google Scholar]
- 267.Islam M, Gayatri G, Hotha S. Influence of Steric Crowding on Diastereoselective Arabinofuranosylations. J Org Chem. 2015;80:7939–7945. doi: 10.1021/acs.joc.5b00964. [DOI] [PubMed] [Google Scholar]
- 268.Beaver MG, Woerpel KA. Erosion of Stereochemical Control with Increasing Nucleophilicity: O-Glycosylation at the Diffusion Limit. J Org Chem. 2010;75:1107–1118. doi: 10.1021/jo902222a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Shenoy SR, Smith DM, Woerpel KA. Nucleophilic Additions of Trimethylsilyl Cyanide to Cyclic Oxocarbenium Ions: Evidence for the Loss of Stereoselectivity at the Limits of Diffusion Control. J Am Chem Soc. 2006;128:8671–8677. doi: 10.1021/ja061110u. [DOI] [PubMed] [Google Scholar]
- 270.van der Vorm S, Hansen T, Overkleeft HS, van der Marel GA, Codée JDC. The Influence of Acceptor Nucleophilicity on the Glycosylation Reaction Mechanism. Chem Sci. 2017;8:1867–1875. doi: 10.1039/c6sc04638j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Sharma I, Bohé L, Crich D. Influence of Protecting Groups on the Anomeric Equilibrium; Case of the 4,6-O-Benzylidene Acetal in the Mannopyranose Series. Carbohydr Res. 2012;357:126–131. doi: 10.1016/j.carres.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kancharla PK, Kato T, Crich D. Probing the Influence of Protecting Groups on the Anomeric Equilibrium in Sialic Acid Glycosides with the Persistent Radical Effect. J Am Chem Soc. 2014;136:5472–5480. doi: 10.1021/ja501276r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Gouliaras C, Lee D, Chan L, Taylor MS. Regioselective Activation of Glycosyl Acceptors by a Diarylborinic Acid-Derived Catalyst. J Am Chem Soc. 2011;133:13926–13929. doi: 10.1021/ja2062715. [DOI] [PubMed] [Google Scholar]
- 274.Nakagawa A, Tanaka M, Hanamura S, Takahashi D, Toshima K. Regioselective and 1,2-cis-α-Stereoselective Glycosylation Utilizing Glycosyl-Acceptor-Derived Boronic Ester Catalyst. Angew Chem Int Ed. 2015;54:10935–10939. doi: 10.1002/anie.201504182. [DOI] [PubMed] [Google Scholar]
- 275.Tanaka M, Nashida J, Takahashi D, Toshima K. Glycosyl-Acceptor-Derived Borinic Ester-Promoted Direct and β-Stereoselective Mannosylation with a 1,2-Anhydromannose Donor. Org Lett. 2016;18:2288–2291. doi: 10.1021/acs.orglett.6b00926. [DOI] [PubMed] [Google Scholar]
- 276.Tanaka M, Nakagawa A, Nishi N, Iijima K, Sawa R, Takahashi D, Toshima K. Boronic-Acid-Catalyzed Regioselective and 1,2-cis-Stereoselective Glycosylation of Unprotected Sugar Acceptors via SNi-Type Mechanism. J Am Chem Soc. 2018;140:3644–3651. doi: 10.1021/jacs.7b12108. [DOI] [PubMed] [Google Scholar]
- 277.Uhlmann P, Vasella A. Regioselective Glycosylation of Diols and Triols: Intra- and Intermolecular Hydrogen Bonds. Helv Chim Acta. 1992;75:1979–1994. [Google Scholar]
- 278.Bozó E, Vasella A. Regioselective Glycosidation of 4,6-O-Benzylidene-D-Altropyranosides. Helv Chim Acta. 1992;75:2613–2633. [Google Scholar]
- 279.Muddasani PR, Bozó E, Bernet B, Vasella A. Glycosidation of Partially Protected Galactopyranose-, Glucopyranose-, and Mannopyranose-Derived Vicinal Diols. Helv Chim Acta. 1994;77:257–290. [Google Scholar]
- 280.Peng P, Linseis M, Winter RF, Schmidt RR. Regioselective Acylation of Diols and Triols: The Cyanide Effect. J Am Chem Soc. 2016;138:6002–6009. doi: 10.1021/jacs.6b02454. [DOI] [PubMed] [Google Scholar]
- 281.Mong KKT, Nokami T, Tran NTT, Nhi PB. Solvent Effect on Glycosylation. In: Bennett CS, editor. Selective Glycosylations: Synthetic Methods and Catalysts. Wiley-VCH; Weinheim: 2017. pp. 59–77. [Google Scholar]
- 282.De Meo C, Farris M, Ginder N, Gulley B, Priyadarshani U, Woods M. Solvent Effect in the Synthesis of Sialosyl (2–6) Galactosides: Is Acetonitrile the Only Choice? Eur J Org Chem. 2008:3673–3677. [Google Scholar]
- 283.Crich D, Dudkin V. Efficient, Diastereoselective Chemical Synthesis of a β-Mannopyranosyl Phosphoisoprenoid. Org Lett. 2000;2:3941–3943. doi: 10.1021/ol006725p. [DOI] [PubMed] [Google Scholar]
- 284.Kendale JC, Valentín EM, Woerpel KA. Solvent Effects in the Nucleophilic Substitutions of Tetrahydropyran Acetals Promoted by Trimethylsilyl Trifluoromethanesulfonate: Trichloroethylene as Solvent for Stereoselective C- and O-Glycosylations. Org Lett. 2014;16:3684–3687. doi: 10.1021/ol501471c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Cardona A, Boutureira O, Castillón S, Díaz Y, Matheu MI. Metal-Free and VOC-Free O-Glycosylation in Supercritical CO2. Green Chem. 2017;19:2687–2694. [Google Scholar]
- 286.Kononov LO, Malysheva NN, Kononova EG, Orlova AV. Intermolecular Hydrogen-Bonding Pattern of a Glycosyl Donor: The Key to Understanding the Outcome of Sialylation. Eur J Org Chem. 2008:3251–3254. [Google Scholar]
- 287.Kononov LO, Malysheva NN, Orlova AV. Stereoselectivity of Glycosylation May Change During the Reaction Course: Highly α-Stereoselective Sialylation Achieved by Supramer Approach. Eur J Org Chem. 2009:611–616. [Google Scholar]
- 288.Kononov LO. Modulation of Stereoselectivity of Glycosylation: A Supramer Approach. Adv Chem Res. 2013;18:143–177. [Google Scholar]
- 289.Kononov LO. Chemical Reactivity and Solution Structure: On the Way to a Paradigm Shift? RSC Adv. 2015;5:46718–46734. [Google Scholar]
- 290.Podvalnyy NM, Malysheva NN, Panova MV, Zinin AI, Chizhov AO, Orlova AV, Kononov LO. Stereoselective Sialylation with O-Trifluoroacetylated Thiosialosides: Hydrogen Bonding Involved? Carbohydr Res. 2017;451:12–28. doi: 10.1016/j.carres.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 291.Kononov LO, Fedina KG, Orlova AV, Kondakov NN, Abronina PI, Podvalnyy NM, Chizhov AO. Bimodal Concentration-Dependent Reactivity Pattern of a Glycosyl Donor: Is the Solution Structure Involved? Carbohydr Res. 2017;437:28–35. doi: 10.1016/j.carres.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 292.Kuznik G, Hörsch B, Kretzschmar G, Unverzagt C. Chemical and Enzymatic Synthesis of High-Affinity Selectin Ligands. Bioorg Med Chem Lett. 1997;7:577–580. [Google Scholar]
- 293.Yu CS, Niikura K, Lin CC, Wong CH. The Thioglycoside and Glycosyl Phosphite of 5-Azido Sialic Acid: Excellent Donors for the α-Glycosylation of Primary Hydroxy Groups. Angew Chem Int Ed. 2001;40:2900–2903. doi: 10.1002/1521-3773(20010803)40:15<2900::AID-ANIE2900>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 294.Gong J, Liu H, Nicholls JM, Li X. Studies on the Sialylation of Galactoses with Different C-5 Modified Sialyl Donors. Carbohydr Res. 2012;361:91–99. doi: 10.1016/j.carres.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 295.Lu KC, Tseng SY, Lin CC. 5-Azido Neuraminic Acid Thioglycoside as Sialylation Donor. Carbohydr Res. 2002;337:755–760. doi: 10.1016/s0008-6215(02)00057-5. [DOI] [PubMed] [Google Scholar]
- 296.Mukaiyama T, Mandai H, Jona H. A Catalytic and α-Selective Sialylation Using Novel 5-Azido Sialyl Fluoride. Chem Lett. 2002;31:1182–1183. [Google Scholar]
- 297.De Meo C, Priyadarshani U. C-5 Modifications in N-Acetyl-Neuraminic Acid: Scope and Limitations. Carbohydr Res. 2008;343:1540–1552. doi: 10.1016/j.carres.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 298.Dhakal B, Buda S, Crich D. Stereoselective Synthesis of 5-epi-α-Sialosides Related to the Pseudaminic Acid Glycosides. Reassessment of the Stereoselectivity of the 5-Azido-5-Deacetamidosialyl Thioglycosides and Use of Triflate as Nucleophile in the Zbiral Deamination of Sialic Acids. J Org Chem. 2016;81:10617–10630. doi: 10.1021/acs.joc.6b02221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Callam CS, Gadikota RR, Krein DM, Lowary TL. 2,3-Anhydrosugars in Glycoside Bond Synthesis. NMR and Computational Investigations into the Mechanism of Glycosylations with 2,3-Anhydrofuranosyl Glycosyl Sulfoxides. J Am Chem Soc. 2003;125:13112–13119. doi: 10.1021/ja0349610. [DOI] [PubMed] [Google Scholar]
- 300.Crich D, Sharma I. Influence of the O3 Protecting Group on Stereoselectivity in the Preparation of C-Mannopyranosides with 4,6-O-Benzylidene Protected Donors. J Org Chem. 2010;75:8383–8391. doi: 10.1021/jo101453y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Nukada T, Berces A, Whitfield DM. Can the Stereochemical Outcome of Glycosylation Reactions Be Controlled by the Conformational Preferences of the Glycosyl Donor? Carbohydr Res. 2002;337:765–774. doi: 10.1016/s0008-6215(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 302.Crich D, Sharma I. Is Donor-Acceptor Hydrogen Bonding Necessary for 4,6-O-Benzylidene-Directed β-Mannopyranosylation? Stereoselective Synthesis of β-C-Mannopyranosides and α-C-Glucopyranosides. Org Lett. 2008;10:4731–4734. doi: 10.1021/ol8017038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Whitfield DM. Plausible Transition States for Glycosylation Reactions. Carbohydr Res. 2012;356:180–190. doi: 10.1016/j.carres.2012.03.040. [DOI] [PubMed] [Google Scholar]
- 304.Whitfield DM. Complications of Modeling Glycosylation Reactions: Can the Anomeric Conformation of a Donor Determine the Glycopyranosyl Oxacarbenium Ring Conformation? Carbohydr Res. 2012;356:191–195. doi: 10.1016/j.carres.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 305.Whitfield DM. In a Glycosylation Reaction How Does a Hydroxylic Nucleophile Find the Activated Anomeric Carbon? Carbohydr Res. 2015;403:69–89. doi: 10.1016/j.carres.2014.05.021. [DOI] [PubMed] [Google Scholar]
- 306.Whitfield DM, Guo J. Proton Transfer and Hydrogen Bonding in Glycosylation Reactions. J Carbohydr Chem. 2017;36:59–99. [Google Scholar]
- 307.Yasomanee JP, Demchenko AV. Effect of Remote Picolinyl and Picoloyl Substituents on the Stereoselectivity of Chemical Glycosylation. J Am Chem Soc. 2012;134:20097–20102. doi: 10.1021/ja307355n. [DOI] [PubMed] [Google Scholar]
- 308.Yasomanee JP, Demchenko AV. Hydrogen Bond Mediated Aglycone Delivery: Synthesis of Linear and Branched α-Glucans. Angew Chem Int Ed. 2014;53:10453–10456. doi: 10.1002/anie.201405084. [DOI] [PubMed] [Google Scholar]
- 309.Pistorio SG, Yasomanee JP, Demchenko AV. Hydrogen-Bond-Mediated Aglycone Delivery: Focus on β-Mannosylation. Org Lett. 2014;16:716–719. doi: 10.1021/ol403396j. [DOI] [PubMed] [Google Scholar]
- 310.Nigudkar SS, Demchenko AV. Stereocontrolled 1,2-cis-Glycosylation as the Driving Force of Progress in Synthetic Carbohydrate Chemistry. Chem Sci. 2015;6:2687–2704. doi: 10.1039/c5sc00280j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Liu QW, Bin HC, Yang JS. β-Arabinofuranosylation Using 5-O-(2-Quinolinecarbonyl) Substituted Ethyl Thioglycoside Donors. Org Lett. 2013;15:3974–3977. doi: 10.1021/ol401755e. [DOI] [PubMed] [Google Scholar]
- 312.Escopy S, Geringer SA, De Meo C. Combined Effect of the Picoloyl Protecting Group and Triflic Acid in Sialylation. Org Lett. 2017;19:2638–2641. doi: 10.1021/acs.orglett.7b00976. [DOI] [PubMed] [Google Scholar]
- 313.Crich D, Cai W, Dai Z. Highly Diastereoselctive α-Mannosylation in the Absence of Participating Protecting Groups. J Org Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]
- 314.Ustyuzhanina N, Komarova B, Zlotina N, Krylov V, Gerbst A, Tsvetkov Y, Nifantiev N. Stereoselective α-Glycosylation with 3-O-Acetylated D-Gluco Donors. Synlett. 2006:921–923. [Google Scholar]
- 315.Komarova BS, Ustyuzhanina NE, Tsvetkov YE, Nifantiev NE. Stereocontrol of 1,2-cis-Glycosylation by Remote O-Acyl Protecting Groups. In: Werz DB, Vidal S, editors. Modern Synthetic Methods in Carbohydrate Chemistry; from Monosaccharides to Complex Glycoconjugates. Wiley; Weinheim: 2014. pp. 125–160. [Google Scholar]
- 316.Komarova BS, Tsvetkov YE, Nifantiev NE. Design of α-Selective Glycopyranosyl Donors Relying on Remote Anchimeric Assistance. Chemical Record. 2016;16:488–506. doi: 10.1002/tcr.201500245. [DOI] [PubMed] [Google Scholar]
- 317.Wulff G, Roehle G. Kinetic Investigations on the Mechanisms of the Koenigs-Knorr Reaction. Chem Ber. 1972;105:1122–1132. doi: 10.1002/cber.19721050405. [DOI] [PubMed] [Google Scholar]
- 318.Banait NS, Jencks WP. Reactions of Anionic Nucleophiles with α-D-Glucopyranosyl Fluoride in Aqueous Solution through a Concerted, ANDN (SN2) Mechanism. J Am Chem Soc. 1991;113:7951–7958. [Google Scholar]
- 319.Wurst JM, Liu G, Tan DS. Hydrogen-Bonding Catalysis and Inhibition by Simple Solvents in the Stereoselective Kinetic Epoxide-Opening Spirocyclization of Glycal Epoxides to Form Spiroketals. J Am Chem Soc. 2011;133:7916–7925. doi: 10.1021/ja201249c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Taylor MS. Catalysis Based on Reversible Covalent Interactions of Organoboron Compounds. Acc Chem Res. 2015;48:295–305. doi: 10.1021/ar500371z. [DOI] [PubMed] [Google Scholar]
- 321.Lee D, Williamson CL, Chan L, Taylor MS. Regioselective, Borinic Acid-Catalyzed Monoacylation, Sulfonylation and Alkylation of Diols and Carbohydrates: Expansion of Substrate Scope and Mechanistic Studies. J Am Chem Soc. 2012;134:8260–8267. doi: 10.1021/ja302549c. [DOI] [PubMed] [Google Scholar]
- 322.Singleton DA, Thomas AA. High Precision Simultaneous Determination of Multiple Small Kinetic Isotope Effects at Natural Abundance. J Am Chem Soc. 1995;117:9357–9358. [Google Scholar]
- 323.Crich D, Chandrasekera NS. Mechanism of 4,6-O-Benzylidene Directed β-Mannosylation as Determined by α-Deuterium Kinetic Isotope Effects. Angew Chem Int Ed. 2004;43:5386–5389. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- 324.Berti PJ, Tanaka KSE. Transition State Analysis Using Multiple Kinetic Isotope Effects: Mechanisms of Enzymatic and Non-Enzymatic Glycoside Hydrolysis and Transfer. Adv Phys Org Chem. 2002;37:239–314. [Google Scholar]
- 325.E-Badri MH, Willenbring D, Tantillo DJ, Gervay-Hague J. Mechanistic Studies on the Stereoselective Formation of β-Mannosides from Mannosyl Iodides Using α-Deuterium Kinetic Isotope Effects. J Org Chem. 2007;72:4663–4672. doi: 10.1021/jo070229y. [DOI] [PubMed] [Google Scholar]
- 326.Chan J, Tang A, Bennet AJ. A Stepwise Solvent-Promoted SNi Reaction of α-D-Glucopyranosyl Fluoride: Mechanistic Implications for Retaining Glycosyltransferases. J Am Chem Soc. 2012;134:1212–1220. doi: 10.1021/ja209339j. [DOI] [PubMed] [Google Scholar]
- 327.Bennet AJ, Sinnott ML. Complete Kinetic Isotope Effect Description of Transition States for Acid-Catalyzed Hydrolyses of Methyl α- and β-Glucopyranosides. J Am Chem Soc. 1986;108:7287–7294. [Google Scholar]
- 328.Chan J, Sannikova N, Tang A, Bennet AJ. Transition-State Structure for the Quintessential SN2 Reaction of a Carbohydrate: Reaction of α-Glucopyranosyl Fluoride with Azide Ion in Water. J Am Chem Soc. 2014;136:12225–12228. doi: 10.1021/ja506092h. [DOI] [PubMed] [Google Scholar]
- 329.Pelletier G, Zwicker A, Allen CL, Schepartz A, Miller SJ. Aqueous Glycosylation of Unprotected Sucrose Employing Glycosyl Fluorides in the Presence of Calcium Ion and Trimethylamine. J Am Chem Soc. 2016;138:3175–3182. doi: 10.1021/jacs.5b13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Smith DM, Tran MB, Woerpel KA. Nucleophilic Additions to Fused Bicyclic Five-Membered Ring Oxocarbenium Ions: Evidence for Preferential Attack on the Inside Face. J Am Chem Soc. 2003;125:14149–14152. doi: 10.1021/ja0375176. [DOI] [PubMed] [Google Scholar]
- 331.Ingold KU, Griller D. Radical Clock Reactions. Acc Chem Res. 1980;13:317–323. [Google Scholar]
- 332.Zhang X-t, Gu Z-y, Xing G-w. Comparative Studies on the O-Sialylation with Four Different α/β-Oriented (N-Acetyl)-5-N,4-O-Carbonyl-Protected p-Toluenethiosialosides as Donors. Carbohydr Res. 2014;388:1–7. doi: 10.1016/j.carres.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 333.Ahiadorme DA, Podvalnyy NM, Orlova AV, Chizhov AO, Kononov LO. Glycosylation of Dibutyl Phosphate Anion with Arabinofuranosyl Bromide: Unusual Influence of Concentration of the Reagents on the Ratio of Anomeric Glycosyl Phosphates Formed. Russ Chem Bull. 2016;65:2776–2778. [Google Scholar]
- 334.Satoh H, Hansen HS, Manabe S, van Gunsteren WF, Hunenberger PH. Theoretical Investigation of Solvent Effects on Glycosylation Reactions: Stereoselectivity Controlled by Preferential Conformations of the Intermediate Oxacarbenium-Counterion Complex. J Chem Theor Comput. 2010;6:1783–1797. doi: 10.1021/ct1001347. [DOI] [PubMed] [Google Scholar]
- 335.Li Z. Computational Study of the Influence of Cyclic Protecting Groups in Stereoselectivity of Glycosylation Reactions. Carbohydr Res. 2010;345:1952–1957. doi: 10.1016/j.carres.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 336.Hosoya T, Takano T, Kosma P, Rosenau T. Theoretical Foundation for the Presence of Oxacarbenium Ions in Chemical Glycoside Synthesis. J Org Chem. 2014;79:7889–7894. doi: 10.1021/jo501012s. [DOI] [PubMed] [Google Scholar]
- 337.Hosoya T, Kosma P, Rosenau T. Contact Ion Pairs and Solvent-Separated Ion Pairs from D-Mannopyranosyl and D-Glucopyranosyl Triflates. Carbohydr Res. 2015;401:127–131. doi: 10.1016/j.carres.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 338.Hosoya T, Kosma P, Rosenau T. Theoretical Study on the Effects of a 4,6-O-Diacetal Protecting Group on the Stability of Ion Pairs from D-Mannopyranosyl and D-Glucopyranosyl Triflates. Carbohydr Res. 2015;411:64–69. doi: 10.1016/j.carres.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 339.Lee JK, Bain AD, Berti PJ. Probing the Transition States of Four Glucoside Hydrolyses with 13C Kinetic Isotope Effects Measured at Natural Abundance by NMR Spectroscopy. J Am Chem Soc. 2004;126:3769–3776. doi: 10.1021/ja0394028. [DOI] [PubMed] [Google Scholar]
- 340.Ionescu AR, Whitfield DM, Zgierski MZ. O-2 Substituted Pyranosyl Oxacarbenium Ions Are C2-O2 Rotors With a Strong syn Preference. Carbohydr Res. 2007;342:2793–2800. doi: 10.1016/j.carres.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 341.Kumar R, Whitfield DM. Could Diastereoselectivity in the Presence of O-2 Chiral Nonparticipating Groups Be an Indicator of Glycopyranosyl Oxacarbenium Ions in Glycosylation Reactions? J Org Chem. 2012;77:3724–3739. doi: 10.1021/jo202563f. [DOI] [PubMed] [Google Scholar]
- 342.Zhou J, Lv S, Zhang D, Xia F, Hu W. Deactivating Influence of 3-O-Glycosyl Substituent on Anomeric Reactivity of Thiomannoside Observed in Oligomannoside Synthesis. J Org Chem. 2017;82:2599–2621. doi: 10.1021/acs.joc.6b03017. [DOI] [PubMed] [Google Scholar]