

Abstract
Purine radical cations (dA•+ and dG•+) are the primary hole carriers of DNA hole migration owing to their favorable oxidation potential. Much less is known about the reactivity of higher energy pyrimidine radical cations. The thymidine radical cation (T•+) was produced at a defined position in DNA from a photochemical precursor for the first time. T•+ initiates hole transfer to dGGG triplets in DNA. Hole localization in a dGGG sequence accounts for ~26 % of T•+ formed under aerobic conditions in 9. Reduction to yield thymidine is also quantified. In poly(dA-T) sequences, 5-formyl-2′-deoxyuridine, which results from deprotonation from the C5-methyl group of T•+, is formed in low yield. This is inconsistent with mechanistic proposals concerning product formation from electron transfer in poly(dA-T) sequences, following hole injection by a photoexcited anthraquinone. Instead, the strand damage patterns observed in those studies, in which thymidine is oxidized, are reproduced by independent generation of 2′-deoxyadenosin-N6-yl radical (dA•). The original mechanism was disproven using hole injection by a photoexcited anthraquinone in DNA. Tandem lesion formation by dA• provides the basis for an alternative mechanism for thymidine oxidation ascribed to hole migration in poly(dA-T) sequences. Overall, these experiments indicate that the final products formed following DNA hole transfer in poly(dA-T) sequences do not result from deprotonation or hydration of T•+, but rather from deprotonation of the more stable dA•+, to form dA•, which produces tandem lesions in which 5′-flanking thymidines are oxidized.
DNA hole migration is initiated by one electron oxidation of DNA, i.e. hole injection, and is a primary consequence of the direct effect of ionizing radiation.1–7 Hole injection by chemical and/or photochemical methods take advantage of the differences in redox potentials to selectively generate the radical cation of 2′-deoxyguanosine (dG) and/or dA.8–11 Short wavelength UV-irradiation (≤ 254 nm) and other forms of ionizing radiation (e.g. γ-radiolysis, photosensitization) are unselective and generate pyrimidine (e.g. T•+) and purine radical cations.12–17 Nucleotide radical cation formation is a hallmark of the direct effect of ionizing radiation. The role of dG•+ in hole migration and subsequent hole trapping by reacting with H2O/O2 is well documented.18,19 The reactivity of T•+ is less well understood than that of dG•+ and dA•+, due to the lack of methods to selectively produce the pyrimidine radical cation.20 Most reports on T•+ reactivity have focused on the nucleoside. However, in a series papers T•+ was invoked as a direct precursor to products formed in electron transfer studies within poly(dA-T) sequences.21–25 We wish to report on T•+ reactivity at a defined site in DNA for the first time via its independent generation from a photochemical precursor. In addition to providing insight into the competitive pathways for T•+ reactivity in DNA, these studies lead to an alternative mechanism for final products resulting from thymidine oxidation upon one electron oxidation in poly(dA-T).
Monomeric T•+ undergoes 4 competing reactions (Scheme 1).12,15–17 In DNA, these processes must compete with electron transfer. Hydration (1) occurs preferentially at C6, and when carried out in the presence of O2 and reducing agent, ultimately yields thymidine glycol (Tg). Deprotonation from the C5-methyl group (2) under aerobic conditions ultimately yields 5-formyl-2′-deoxyuridine (fdU). Direct reduction to restore thymidine has a large driving force and occurs directly, as well as via N-H deprotonation (3), followed by formal hydrogen transfer. The relative rates for these competing processes vary, particularly with respect to the contributions of the deprotonation and hydration pathways. Furthermore, it is often difficult to quantify the contribution of the pathways that yield thymidine since most methods generate T•+ from the nucleoside via one-electron oxidation. However, recently T•+ was produced from a photochemical precursor (4) other than thymidine.20 Irradiation (350 nm) of 4 produces an intermediate radical (S) that undergoes β-heterolysis to produce T•+ (Scheme 2).26–30 Generating monomeric T•+ from 4 revealed that thymidine is the major product under aerobic conditions, even in the absence of exogenous reducing agent. Products attributable to T•+ hydration are formed in ~1/3 the yield of thymidine. Hydration (NMe-Tg) competed more effectively with reduction (NMe-T) when the respective radical cation was generated from NMe-4 (via NMe-5), suggesting that N-H deprotonation contributed significantly to thymidine formation from T•+. However, the corresponding nucleosides attributable to C5-methyl deprotonation (fdU, NMe-fdU) were minor products from both precursors.
Scheme 1.
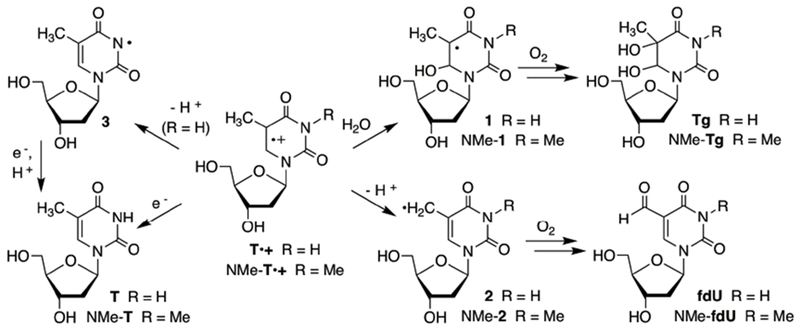
Thymidine Radical Cation Reactivity
Scheme 2.
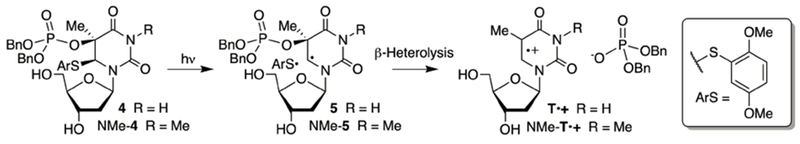
Independent Generation of the Thymidine Radical Cation
The low yield of (NMe-)fdU is contrary to some studies on monomeric T•+. It also is inconsistent with product studies upon electron transfer in poly(dA-T) sequences. Elegant studies by Schuster showed that when poly(dA-T) sequences are oxidized by a tethered photoexcited anthraquinone, the holes migrate through the duplex. The final sites of oxidation, detected via denaturing polyacrylamide gel electrophoresis (PAGE), following alkaline treatment, are at thymidine.21–23 This is surprising because dA•+ is ~3.5 - 6.5 kcal/mol more stable than T•+.31,32 Preferential damage at thymidine was rationalized by a kinetic preference for deprotonation from the C5-methyl of T•+, ultimately resulting in the formation of fdU and 5-hydroxymethyl-2′-deoxyuridine (hmU). Tg was detected in these experiments was attributed to T•+ hydration. The reactivity of independently generated T•+ within DNA described herein corroborates the observations reported concerning the monomeric radical cation. C5-Methyl deprotonation is a minor contributor to T•+ reactivity in DNA. An alternative mechanism involving formation of dA• via deprotonation of the more stable dA•+ is put forth and tested using the sequences similar to those employed in Schuster’s original studies.33 Finally, the alternative mechanism for product formation in one-electron oxidized poly(dA-T) is tested using the anthraquinone method for hole injection.
RESULTS AND DISCUSSION
Validation of 6 as a photochemical precursor for T•+ and its incorporation in oligonucleotides.
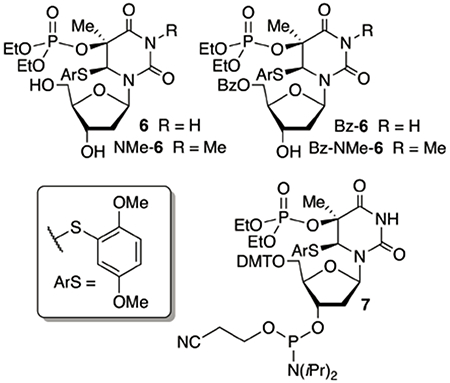
Photochemical generation of T•+ in aqueous solution proceeded cleanly.20 However, the dibenzyl phosphate triester present in 4 proved to be unstable to the standard alkaline conditions (e.g. concentrated aqueous ammonia) used to deprotect chemically synthesized oligonucleotides. Consequently, we synthesized chemically stable diethyl phosphate triesters 6 and NMe-6. We also synthesized the corresponding 5′-benzoates (Bz-6, Bz-NMe-6). Bz-6 and Bz-NMe-6 were used to validate that the radical cation is produced (Table S1, S2). The benzoyl group provides a useful chromophore for UV-absorbance detection and quantification of products. Photolyses of Bz-6 and Bz-NMe-6 provided very similar results as product studies from irradiation of 4 and NMe-4.34 Mass balances from Bz-6 photolyses ranged from 65-79%, depending upon solvent and pH. Thymidine was the major product formed from Bz-6 and the combined yields of hydration products increased significantly in photolysates of Bz-NMe-6 (28.3%), where N-H deprotonation is not possible, compared to those of Bz-6 (11.3%). In the absence of β-mercaptoethanol (BME, 10 mM), hydration products were formed in slightly higher yield than NMe-T from Bz-NMe-6, but the latter was favored by more than 3:1 when the photolysis was carried
out in the presence of BME. Importantly, products resulting from C5-methyl deprotonation (e.g. 5′-benzoylated fdU or 5′-benzoylated NMe-fdU) were minor products (<4%) under all reaction conditions.
The diethyl phosphate triester in 6 was stable to solid phase oligonucleotide synthesis and deprotection conditions, enabling the use of phosphoramidite 7 as a vehicle for introducing the radical cation precursor at defined sites in oligonucleotides. Oligonucleotides containing 6 were prepared using slight modifications of typical protocols. (Please note that for convenience the same numbering is used for molecules as monomeric nucleosides and in oligonucleotides.) Commercially available “fast deprotecting” phosphoramidites were used for introducing dA and dG. The phosphoramidite was coupled for 5 min and acetic anhydride was replaced as a capping agent by pivalic anhydride.35 The oligonucleotides were deprotected and cleaved from the solid phase support using concentrated aqueous ammonia at room temperature for 12 h. Following purification by denaturing polyacrylamide gel electrophoresis, the oligonucleotides were characterized by mass spectrometry.34
Thymidine Radical Cation (T•+) Reactivity in DNA.
A series of duplexes (8a,b - 10a,b) were prepared to examine T•+ generation and reactivity. Each of these duplexes contained at least one dGGG sequence, an often-used reporter group for hole transfer.36–38 This trinucleotide sequence has the lowest ionization potential of any trinucleotide sequence and serves as a hole trap in DNA.31 Hole trapping is typically detected as a strand break via denaturing PAGE following alkaline (piperidine) treatment or incubation with a base excision repair enzyme (e.g. formamidopyrimidine DNA glycosylase, Fpg). Furthermore, characteristic strand damage resulting from hole transfer is preferentially observed at the middle and 5′-dG of the trinucleotide sequence. The duplexes were designed to contain the dGGG sequence on the strand opposite that in which T•+ is produced. The efficiency for hole trapping has been estimated to be ~ 8%.36 Hence, the level of hole migration is actually ~12-fold higher than the strand cleavage reported below. Time course studies were carried out to establish the irradiation time (75 min) needed to reach maximum conversion of 6 (Figure S3). Strand damage levels reported were corrected for background cleavage in unphotolyzed samples, as well as for damage in substrates containing thymidine (8b - 10b) at the position where 6 is incorporated in otherwise identical duplexes.

Following UV-photolysis and piperidine treatment, strand damage (background subtracted)34 was approximately equal within dG11-13 (0.63 ± 0.14 %) and dG17-19 (0.70 ± 0.22 %) within 8a (Figure S4). In addition, the preference for strand damage within each triplet (dG12 > dG11 >> dG13, dG18 ≈ dG17 >> dG19) was consistent with the expectations of damage resulting from hole transfer (Figure 1A).34 Overall, the damage detected at the two trinucleotide reporter sequences corresponds to ~16% of the possible T•+ that can be produced.
Figure 1.
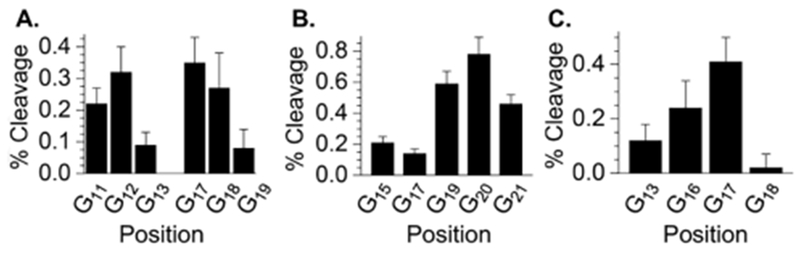
Strand damage due to hole migration from T•+. (A.) 8a (B.) 9a (C.) 10a. The data are the ave. ± std. dev. of 3 replicates.
Similarly, construct 9a was designed to probe hole transfer from T•+ to dGs located at the complementary strand. Strand damage in 9a was also consistent with the expectations for hole transfer (Figure 1B). Alkaline cleavage within dG19-21 was significantly greater than at dG15 or dG17 and although the preference for strand scission at dG20 and dG19 relative to dG21 was not as great as in 8a, the expected pattern was observed (Figure S5). The total hole transfer detected over the 5 dGs was ~26% of the possible radical cation produced.
Finally, substrate 10a was designed to most closely match that employed by Giese where he established hole trapping efficiency and enable estimating the fraction of T•+ resulting in hole transfer by incorporating 6 within a sequence such that a restriction site for MseI (5′-d(TTAA/AATT)) is created when thymidine is produced at that position.36 MseI does not cleave 10a prior to photolysis (Figures S6 and S7). In addition, although we cannot rule out the possibility that hmdU and fdU were also substrates for MseI, other experiments presented above and later in this manuscript indicate that these products are formed in minor amounts. Thymidine is produced from reduction of T•+, which occurs upon hole transfer but can also be produced via other pathways. MseI cleaves 36.4 ± 2.5 % of photolyzed 10a. The sum-total of piperidine-induced cleavage at dG13 and dG16-18 (0.79 ± 0.26 %), which when adjusted for 8% hole trapping efficiency accounts for 26.9% of the thymidine produced (Figure 1C).

In addition to alkali-labile strand scission at dG’s due to hole transfer and thymidine formation, piperidine-induced cleavage was observed at the original T•+ generation site (44.3 ± 0.4%) in 5′−32P-10a. Overall, we accounted for ~81% of the photolyzed 6 in the substrate.
The reactivity of T•+ was also examined in duplexes lacking dG (11, 12). Previous studies involving DNA hole transfer in poly(dA-T) sequences in which hole injection was induced via photoexcitation of anthraquinones led to the proposal that T•+ was the precursor to the observed alkali-labile lesions.21–25 Product formation from the higher energy pyrimidine radical cation than dA•+ was rationalized based upon kinetic over thermodynamic selectivity. In a dinucleotide step within poly(dA-T), alkali-labile lesion formation was highly dependent upon the flanking pyrimidine.22 For instance, alkaline-labile cleavage was observed predominantly at the 5′-thymidine in a TT step, but no damage was detected at 5′-d(TU) sequences. 5′-d(UT) sequences provided yet a different pattern, damage at the two nucleotides was approximately equal. LC-MS analysis of the nucleosides released upon enzyme digestion of the photolyzed oligonucleotides revealed fdU and Tg. (See Scheme 1 for structures.) The authors proposed a mechanism to explain the sequence dependence, as well as the formation of fdU and Tg from T•+ (Scheme 3). The absence of any alkaline-labile lesions at 5′-d(TU) steps was surprising. If T•+ is generated why would it undergo C5-methyl deprotonation to yield fdU, but not trapping by H2O and O2 to produce alkaline-labile Tg?
Scheme 3.
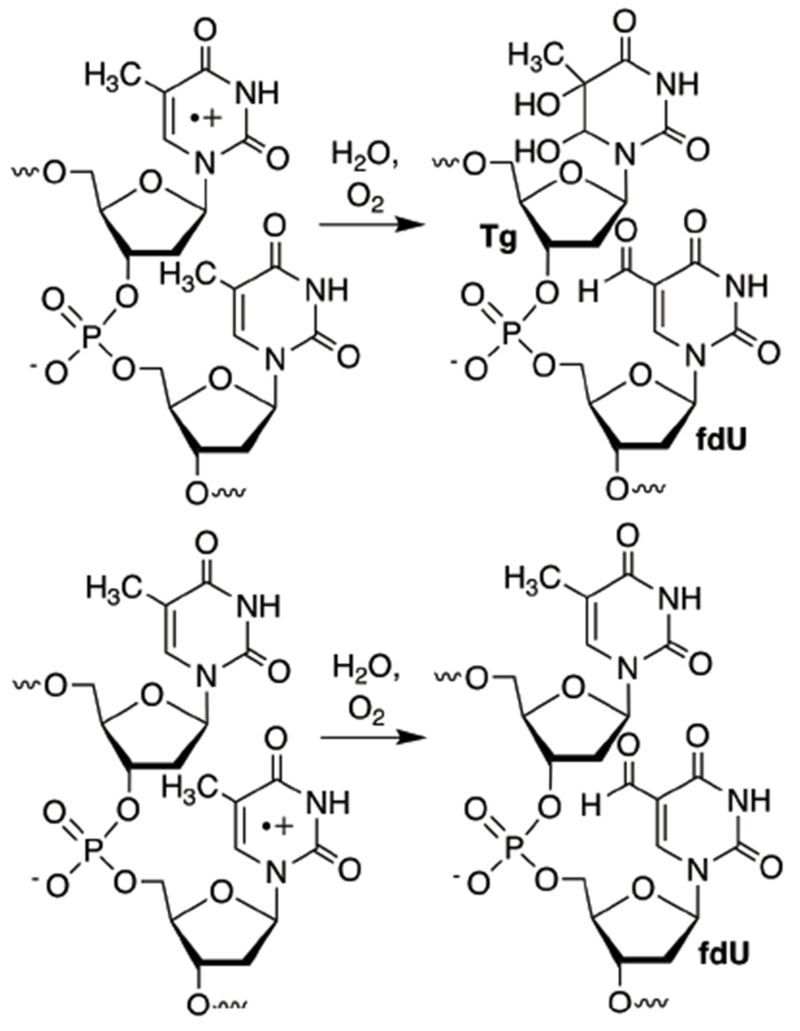
Previously proposed T•+ reactivity.
Denaturing PAGE analysis of 5′-32P-11 produced a piperidine-induced strand cleavage pattern that is qualitatively consistent with that previously reported (Figure S8). The major site of cleavage was at the nucleotide where T•+ was originally generated (49.3 ± 1.1 %), and approximately 10.4 ± 0.1 % strand damage was observed at T14. If the 5′-Tg-fdU tandem lesion (Scheme 3) was formed as proposed, one would expect strand scission at the 5′-nucleotide to predominate since it is closer to the 32P-label.22 Furthermore, fdU is inefficiently cleaved by piperidine.39 It is more difficult to reconcile the observation of comparable levels of strand damage at the radical cation site in 5′-32P-12 (46.6 ± 2.4 %) and no damage was at dU14. Formation of fdU as the major product (Scheme 3) is a foundation of the existing mechanism. Because fdU is inefficiently cleaved by piperidine, we utilized the glycosylase, hSMUG1, followed by NaOH to detect this modified nucleotide (Figure S8).39,40 fdU was not detected in the photolyzate of 11. This is also inconsistent with the previously proposed mechanism, but consistent with product studies on monomeric T•+, which showed that fdU formation is a minor product.20,22
We propose that the major site of alkaline lability is the position at which T•+ is independently generated. The major product(s) results from trapping by water regardless of the 5′-flanking (Scheme 1). Moreover, the lack of influence of a flanking dU on alkali-labile strand scission when T•+ is flanked by a 3′-pyrimidine is very different than the results obtained using anthraquinone containing duplexes.22 We believe that this difference is because T•+ is not produced upon photooxidation of DNA by anthraquinone.
An alternative mechanism for alkaline-labile lesion formation from hole transfer in poly(dA-T) sequences.
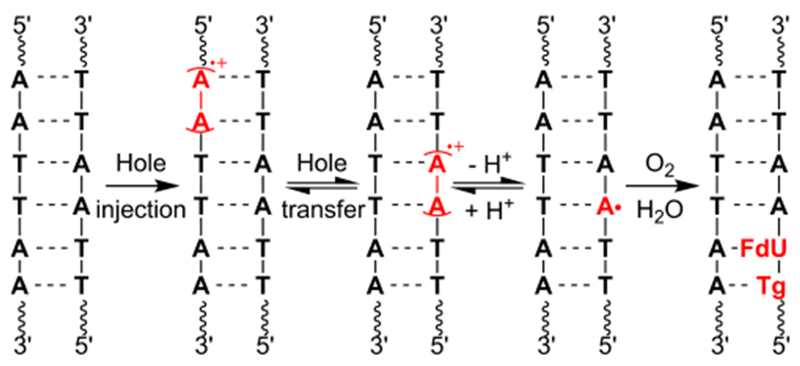
The failure of independently generated T•+ to reproduce the products observed when holes were injected into poly(dA-T) duplexes via photoexcited anthraquinone led us to consider an alternative mechanism.21–25 Since the preferential involvement of the pyrimidine radical cation versus the more stable dA•+ was initially a surprise, we considered whether the purine radical may provide an alternative explanation for the observed products. Recently, dA• and dA•+ were shown to equilibrate in duplex DNA in a sequence dependent manner (Scheme 4).33,41 As expected, the latter gives rise to hole transfer, which localize at dGGG sites. In the absence of such hole sinks, such as in poly(dA-T) substrates, one might expect that the migrating holes are carried by dA•+ ,which can deprotonate to form dA•. dA• has been shown to selectively abstract the C5-methyl hydrogen from the 5′-adjacent
Scheme 4.
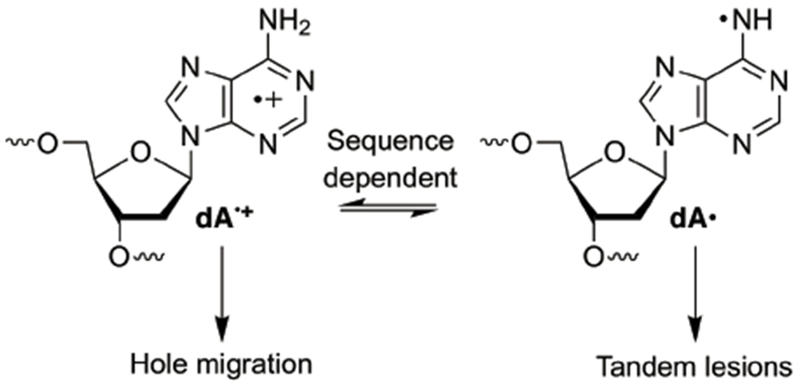
DNA damage from dA reactive intermediates.
thymidine which led to the formation of tandem lesion in 5′-d(GT) sequences.41
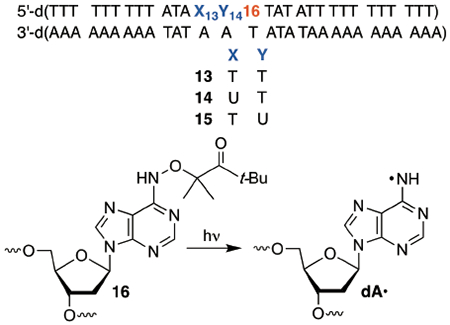
TT steps, such as those present in previously studied substrates, are well-positioned to react with 3′-dA•.22 Consequently, we tested the possibility that dA• is the species directly responsible for the products observed following hole injection in poly(dA-T) substrates. Substrates 13-15 were prepared containing one of the 3 aforementioned pyrimidine-pyrimidine steps flanked by a photochemical precursor (16) for dA•.42 The dA• precursor was not flanked by dA, so as to minimize dA»+ formation under the reaction conditions. Piperidine treatment of photolyzed 5′-32P-13-15 produced cleavage patterns that were fully consistent with those reported when duplexes containing the same dinucleotide steps and anthraquinone were irradiated (Figure 2). Specifically, duplex 13, containing a 5′-T13-T14-dA• sequence results in predominant cleavage at the 5′-thymidine of the dinucleotide sequence. Similarly, strand damage is detected in approximately equal amounts at dU13 and T14 in 5′-32P-14; whereas no damage is detected in the duplex (15) containing 5′-T-dU-dA•.
Figure 2.
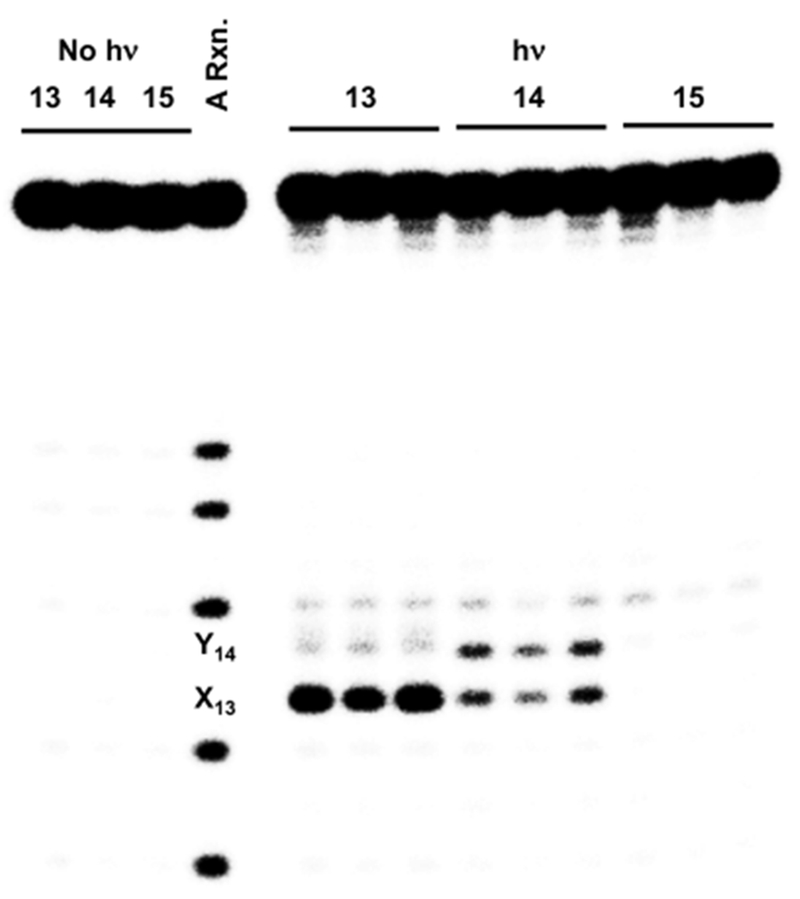
Strand damage in duplexes (5′-32P-13-15) containing 5′-pyr-pyr-dA• Three replicates are shown for each substrate.
A mechanism that is consistent with these observations and previously characterized dA• reactivity involves initial hydrogen atom abstraction from the C5-methyl group of the thymidine bonded to the 5′-phosphate of the neutral nitrogen radical (Scheme 5). Under the aerobic conditions the 5-(2′-deoxyuridinyl)-methyl peroxyl radical (Tp•) reacts with the 5′-adjacent pyrimidine by adding to the pyrimidine double bond at position 13. Addition at the C6-position of thymidine is shown. However, we cannot distinguish between this pathway and addition to the C5-position. Tp• addition to the 5′-adjacent pyrimidine may also explain why alkali-labile strand damage at the 5′-nucleotide in a 5′-d(UT) sequence is less favored than in a 5′-d(TT) sequence. The presence of a C5-proton in the former can give rise to products such as 5-hydroxy-2′-deoxyuridine, which do not yield strand scission upon alkali-treatment.43–45 Furthermore, previous investigations in which pyrimidine peroxyl radicals are independently generated in duplex DNA have shown that alkali-labile lesion formation at a 5′-dU is less efficient than when thymidine is present at this position.46 We also cannot rule out contributions pathways Tp• is ultimately converted to fdU, which is observed when photoexcited anthraquinone is the source of damage but not when T•+ is independently generated in DNA.22 Importantly, hydrogen atom abstraction from the thymidine C5-methyl group by dA• explains why strand damage at 5′-d(TU) sequences is not detected in experiments utilizing anthraquinone modified DNA.
Scheme 5.
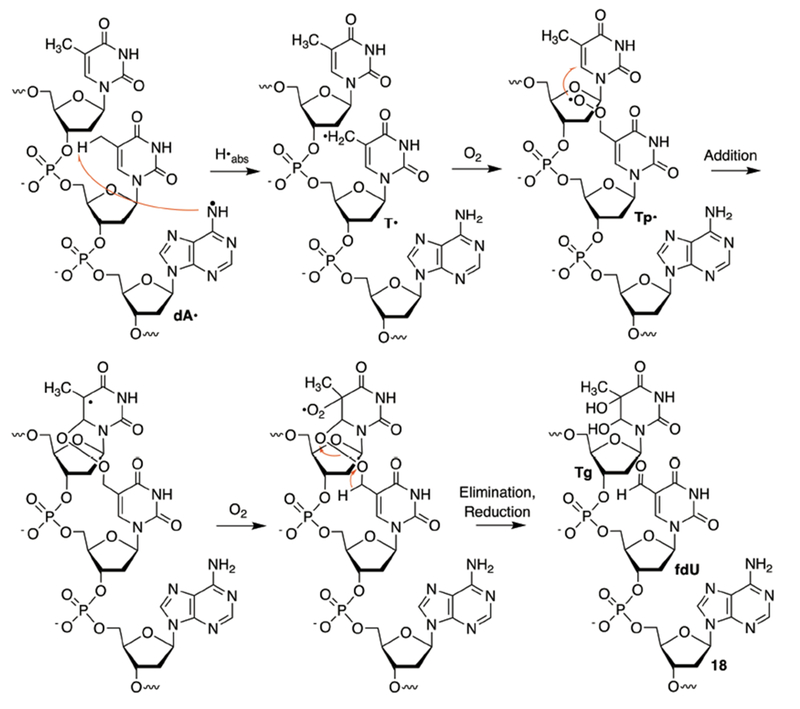
Tandem lesion formation from dA•.
This mechanism was further validated via LC-MS/MS analysis of photolyzates of 17. A dodecameric substrate was employed to ensure detection of fragments via collision-induced dissociation (CID), which is crucial for product assignment.46–48 The complementary strand contained two additional thymidines to facilitate separation on the LC. Substrate 17 contained dG-dC base pairs to stabilize the duplex (Tm = 35.5 °C) against melting during the photolysis. If the products are due to the intermediacy of dA•, the presence of dG-dC base pairs should not influence the chemistry. The observation of an identical damage pattern in 20 and 13, despite the presence of multiple dG-dC base pairs, is consistent with this hypothesis (Figure S10).
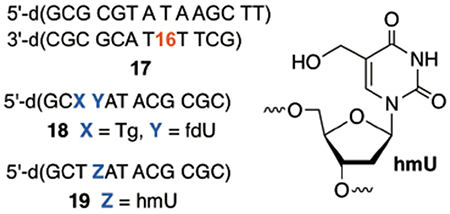
A product (18) with m/z =3666.6240 that is consistent with the expected mass and fragmentation pattern for the tandem lesion containing 5′-d(Tg-fdU) and dA at the position where dA• was generated was observed (Figure 3). Furthermore, the fragment [a3 – 143 Da], which is characteristic for Tg-containing DNA is detected by LC-MS/MS (Figure S14).49 Lastly, photolysis of 17 in the presence of β-mercaptoethanol (1 mM) eliminates formation of 18, and only 19 is observed. This is consistent with the intermediacy of Tp• in tandem lesion formation.
Figure 3.
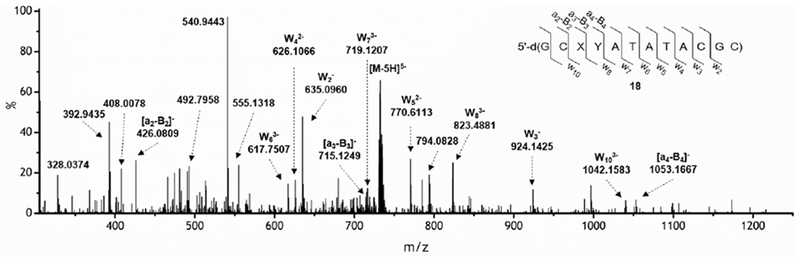
CID mass spectrum of the ion (m/z = 732.7, z= 5) of tandem lesion 18. X = Tg, Y = fdU.
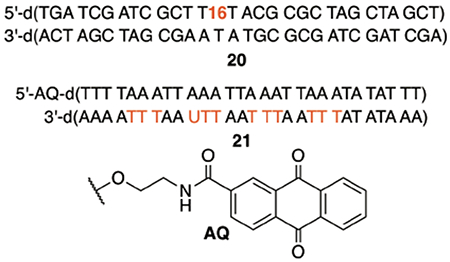
The above observations suggested an experiment to distinguish between the mechanism involving T•+ and that proposed herein involving dA•. We utilized the anthraquinone system to photochemically induce oxidative damage. It was previously shown that when photoexcited anthraquinone was used to introduce holes, 5′-d(TTT) sequences were preferentially damaged at the central thymidine, followed by the 3′-terminal T.21,22 One of the four 5′-d(TTT ) steps in a previously reported substrate was substituted by a 5′-d(TTU) sequence. If the mechanism involves T•+, the TT step in 5′-d(TTU) should give rise to tandem lesion since the hole transferred from the dA•+ on the opposite strand will not be affected by the presence of dU. If the alternative mechanism proposed above is responsible for the products, substituting a single dU for thymidine at the 3′-terminus of a 5′-d(TTT) sequence should eliminate strand damage at that location because the 3′-adjacent dA• radical responsible for initiating tandem lesion formation will not have a C5-methyl hydrogen atom to abstract. Indeed, photolysis of 5′-32P-21 generated the previously reported strand damage pattern in 5′-d(TTT) upon piperidine treatment (Figure 4). Moreover, no damage was detected at the 5′-d(TTU) sequence. Furthermore, piperidine treatment of photolyzed 5′-32P-22 and 5′-32P-23 reproduced the reactivity disparity between 5′-d(TTT) and 5′-d(TTU) (Figure S11). The alternative mechanism proposed (Scheme 5) is consistent with all of these observations
Figure 4.

Alkali-labile strand damage in 5′-32P-21 following photolysis for 8 h.
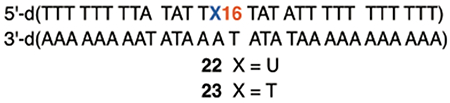
CONCLUSIONS
Radical cations (“holes”) are important intermediates in DNA damage imparted by the direct effect of ionizing radiation. Chemists have employed clever ways of producing holes in DNA but these usually involve producing lower energy purine radical cations, as opposed to pyrimidine radical cations. Using a photochemical precursor, a pyrimidine radical cation (T•+) was independently generated at a defined site within DNA for the first time. T•+ engages in hole transfer, but is also trapped by water to produce alkaline-labile lesions. T•+ does not yield detectable levels of fdU in DNA. This is consistent with studies on monomeric T•+ in which fdU was a minor product.42
T•+ reactivity in DNA composed of only dA and T was inconsistent with previous proposals concerning the involvement of this species when the photoexcited state of anthraquinone was used to introduce a hole.21–25 Combining observations made from independently generated T•+ with a photochemical method for producing dA• at defined sites in DNA led to an alternative proposal for explaining the products of hole transfer in poly(dA-T) DNA.
Rather than utilizing the higher energy T•+, we propose that dA•+ holes migrate throughout the poly(dA-T) sequence (Scheme 6). Holes are fixed at a particular location upon deprotonation of dA•+. The neutral-nitrogen radical (dA•) generates the observed products via tandem lesion formation. The tandem lesions are formed under aerobic conditions via initial hydrogen atom abstraction from the C5-methyl group of a 5′-adjacent thymidine, a process previously observed for dA• (Scheme 5).41 Independent generation of dA• reproduced the sequence specificity of tandem lesion formation when holes are injected by photoexcited anthraquinones.22 Finally, unlike the previously proposed mechanism, the alternative mechanism also predicted the lack of strand damage in a 5′-d(TTU) sequence using anthraquinone.
Scheme 6.
2′-Deoxyadenosin-N6-yl radical (dA•) was recently shown to be a traceless participant in tandem lesion formation in γ-radiolysis mediated DNA damage. The above experiments demonstrate that it plays a comparable role in DNA damage resulting from hole transfer within poly(dA-T) sequences.33,41
EXPERIMENTAL SECTION
Materials and methods.
THF was distilled over Na/benzophenone. DCM, TEA, DIPEA, DMF and pyridine were dried over CaH2. All other reagents were purchased from commercial sources and were directly used without further purification unless noted otherwise. T4 polynucleotide kinase (T4 PNK), human single-strand-selective monofunctional uracil-DNA Glycosylase (hSMUG1) and restriction enzyme MseI were obtained from New England Biolabs. γ-32P-ATP was purchased from Perkin Elmer. C18-Sep-Pak cartridges were obtained from Waters. Diethyl N, N-diethyl phosphoramidite was synthesized as described in the literature.50 All reactions were carried out under argon atmosphere and monitored by TLC on Silica Gel G-25 UV254 (0.25 mm). Flash column chromatography was performed with Silicycle grade 70–230 mesh, 60–200 μm, 60 Å silica. The ratio between silica gel and crude product ranged from 100:1 to 20:1 (w/w). Samples in a mixture of acetonitrile and water/buffer (phosphate buffer (20 mM) or Chelex-treated pH =5 citrate buffer (40 mM)) at a ratio of 1:1 (v/v), typically contain 100 μM precursor as well as 20 μM the internal standard 5′-benzoyl-2′-deoxyuridine (BzdU). Photolyses were performed in Pyrex tubes in a Rayonet photoreactor fitted with 16 lamps with maximum output at 350 nm. Samples containing Bz-6 were photolyzed for 30 min, while samples containing Bz-NMe-6 were photolyzed for 45 min. Photolyzed samples of Bz-6 were incubated at 37 °C in the presence of MeONH2•HCl (10 mM) and NaOAc (10 mM) for 1 h. The samples were neutralized using pH 8.0 PBS buffer (100 mM, 12% volume of samples) prior to UPLC analysis. Photolyzed samples were analyzed using UPLC with an Acquity 1.8 μm C18 UPLC HSS column (100 × 2.1 mm). Detection was carried out at 230 nm, following separation using water (solvent A) and acetonitrile (solvent B) with the following linear gradient (0.2 mL/min): (time (min), % B) 0, 5; 30, 40; 35, 70; 40, 97; 42, 5; 45, 5. Response factors (Rf) for each compound (X) versus 5′-benzoyl-2′-deoxyuridine (BzdU) was calculated using the following formula: ([X]/[BzdU]) = Rf(A(X)/A(BzdU)), where [X] is the concentration of compound X and [BzdU] is the concentration of BzdU. A(X) and A(BzdU) are the areas under the peaks corresponding to X versus BzdU. ESI-MS was carried out on a Thermoquest LCQDeca. UPLC-MS analyses were carried out on Waters Acquity/Xevo-G2 UPLC-MS system equipped with an Oligonucleotide BEH C18 Column (130Å, 1.7 μm, 2.1 mm × 100 mm). Oligonucleotide masses were obtained via deconvolution using MassLynx 4.1 software. CID was processed using Microsoft Excel. MALDI-TOF analyses were carried out on a Bruker AutoFlex III MALDI-TOF. Quantification of radiolabeled oligonucleotides was carried out using a Molecular Dynamics Phosphorimager 860 equipped with ImageQuant Version TL software.
Oligonucleotide synthesis.
Oligonucleotides were prepared on an Applied Biosystems Incorporated 394 oligonucleotide synthesizer. Cyanoethyl phosphoramidites with phenoxyacetyl protecting groups on the exocyclic amines of dA and dG were used for the synthesis of unnatural oligonucleotides. For the synthesis of oligonucleotides containing 6, the capping A reagent is the mixture of lutidine : trimethyl acetic anhydride : THF = 1:1:8.35 The coupling time for modified phosphoramidite was increased to 5 min. All functionalized oligonucleotides were deprotected and cleaved from solid support with 28% aq. NH3 at room temperature. The deprotection time for oligonucleotides containing 6 and 16 are 12 h and 16 h, repectively. The oligonucleotide containing anthraquinone 21 is synthesized according to reported procedure.51 The oligonucleotides are purified via 20% denaturing polyacrylamide gel electrophoresis (PAGE).
Photolysis of oligonucleotides.
The strand (1-2 μM) was labeled at the 5′ end with γ-32P-ATP using T4 PNK in T4 PNK buffer (70 mM Tris-HCl, pH 7.6, 10 mM MgCl2, 5 mM DTT, at least 90 min, 37 °C). The labeled strand was hybridized to the complementary strand (1.5 eq.) in PBS by heating at 90°C for 1 min and slowly cooling to room temperature. The hybridized duplexes were diluted to 0.1 μM in PBS before photolysis. All photolyses were carried out in Pyrex tubes using a Rayonet photoreactor equipped with 16 lamps having a maximum output at 350 nm. Oligonucleotides containing 6 and 16 are photolyzed for 75 min and 8 h, respectively.
Post-photolysis treatments.
Aliquots from photolyzed solutions or unphotolyzed controls were treated with piperidine (1 M, 30 min, 90 °C), MseI (5 Units, 50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 100 μg/ml BSA (pH 7.9), 1 h, 37 °C), hSMUG1 (5 Units, 50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 100 μg/ml BSA (pH 7.9), 1 h, 37 °C) or NaOH (0.1 M, 30 min, 37 °C). NaOH treated samples were neutralized with HCl (1 equiv.). All samples treated with enzymes were precipitated (0.3 M NaOAc, pH 5.2, 0.1 mg/mL calf thymus DNA) with ethanol. Piperidine treated samples were evaporated to dryness under vacuum, and washed with 2 × 10 μL water, which was also removed under vacuum. Samples were analyzed by dissolving in formamide loading buffer prior to analyzing by 20% denaturing PAGE. Piperidine solution was prepared freshly.
UPLC-MS/MS analysis of oligonucleotides.
Photolyzates (8 μL) containing 5 μM of duplex dodecamer were analyzed by UPLC-MS using the Oligonucleotide BEH C18 Column (A, 100 mM HFIP and 8.6 mM TEA; B, Methanol; 5% B from t = 0 to t = 5 min; 5-13% B linearly over 15 min; 13% B from t = 20 to t = 15 min; 9-30% B linearly over 5 min; 30% B from t = 25 to t = 30 min; 30-5% B linearly over 5 min; 5% B from t = 35 to t = 40 min; flow rate, 0.2 mL/min.). The column temperature was 60 °C. The collision energy was set to ramp from 10 to 45 V.
(5R, 6S)-Bis-TBDMS-5,6-dihydro-5-diethyl phosphate-6-(2,5-dimethoxythiophenyl)-thymidine (TBS-6).
Diethyl N, N-diethyl phosphoramidite (46.4 mg, 0.24 mmol) was added to a solution of (5R, 6S)-bis-TBDMS-5,6-dihydro-5-hydroxy-6-(2,5-dimethoxythiophenyl)-thymidine (131 mg, 0.20 mmol) in CH2Cl2 (2 mL) under argon, followed by ethyl-thiotetrazole (0.25 M in acetonitrile, 0.96 mL, 0.24 mmol). The solution was stirred at room temperature overnight and then cooled to 0 °C. t-BuOOH (5-6 M in decane, 0.3 mL) was added, and the mixture was stirred at 0 °C for 1 h. After warming to room temperature, the mixture was directly subjected to flash column chromatography on silica gel (EtOAc:CH2Cl2 = 1:3) to provide the product (82 mg, 51.8%) as a white foam. 1H NMR (400 MHz, CDCl3) δ 7.22 (s, 1H), 7.08 (d, J = 3.1 Hz, 1H), 6.88 (dd, J = 9.0, 3.1 Hz, 1H), 6.77 (d, J = 9.0 Hz, 1H), 6.15 (dd, J = 9.4, 5.3 Hz, 1H), 5.30 (s, 1H), 4.24 – 4.12 (m, 5H), 3.77 (s, 3H), 3.75 (s, 3H), 3.70 – 3.66 (m, 1H), 3.54 – 3.49 (m, 1H), 3.14 – 3.01 (m, 1H), 2.45 – 2.38 (m, 1H), 1.85 (s, 3H), 1.68 - 1.63 (m, 1H), 1.32 - 1.29 (m, 6H), 0.90 (s, 9H), 0.88 (s, 9H), 0.08 (s, 6H), 0.06 (s, 3H), 0.05 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.6, 167.5, 155.1, 153.2, 150.2, 123.3, 117.7, 117.1, 111.7, 86.3, 84.6, 79.2, 79.1, 72.7, 64.5, 64.2, 63.8, 63.5, 56.0, 55.8, 36.1, 30.0, 26.0, 25.8, 22.7, 18.4, 18.0, 16.1, 16.0, −4.7, −4.8, −5.4. 31P NMR (162 MHz, CDCl3) δ −5.89. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C34H62N2O11PSSi2 793.3350; Found 793.3344.
(5R, 6S)-5,6-dihydro-5-diethyl phosphate-6-(2,5-dimethoxythiophenyl)-thymidine (6).
Et3N.3HF (290 mg, 1.8 mmol) was added to a solution of (5S, 6S)-3′5′-Bis-TBDMS-3-methyl-5,6-dihydro-5-diethyl phosphate-6-(2,5-dimethoxythiophenyl)-thymidine (79.2 mg, 0.1 mmol) in THF (3.0 mL). The mixture was stirred at room temperature overnight, at which time the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel (methanol: CH2Cl2 = 1:9) to afford the product 6 (47 mg, 83%) as a white foam. 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 1H), 7.06 (d J = 3.1 Hz, 1H), 6.90 (dd, J = 9.0, 3.1 Hz, 1H), 6.77 (d, J = 9.0 Hz, 1H), 6.17 (dd, J = 9.0, 5.8 Hz, 1H), 5.96 (s, 1H), 4.37 (ddd, J = 11.1, 10.2, 8.8 Hz, 3H), 4.27 – 4.11 (m, 3H), 3.83 (d, J = 12.5 Hz, 2H), 3.76 (s, 3H), 3.74 (s, 3H), 3.68 (d, J = 11.3 Hz, 1H), 2.40 (dd, J = 8.6, 5.3 Hz, 2H), 1.85 (s, 3H), 1.66 – 1.61 (m, 1H), 1.39 – 1.34 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 167.8, 167.7, 155.7, 153.0, 150.6, 124.0, 117.8, 116.9, 111.7, 86.9, 85.6, 78.9, 72.8, 65.2, 64.8, 62.7, 62.3, 55.9, 36.9, 22.9, 16.1, 16.0. 31P NMR (162 MHz, CDCl3) δ −6.20. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C22H34N2O11PS 565.1621; Found 565.1631.
(5R, 6S)-5′-DMT-5,6-dihydro-5-diethyl phosphate-6-(2,5-dimethoxythiophenyl)-thymidine (DMT-6).
Nucleoside 6 (141.1 mg, 0.25 mmol) was azeotropically co-evaporated with anhydrous pyridine (3 × 2 mL) and then re-dissolved in anhydrous pyridine (2 mL). 4,4’-Dimethoxytritylchloride (127.1 mg, 0.375 mmol) and DMAP (6.11 mg, 0.05 mmol) were added at 0 °C under argon. The solution was allowed to warm to room temperature overnight. The reaction was quenched with MeOH (0.5 mL). The solvent was removed under reduced pressure and then diluted with ethyl acetate (40 mL). The mixture was washed with water (30 mL), brine (30 mL), dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography on silica gel (EtOAc:Hexane = 1:1) to yield the product as a white foam (100 mg, 47.7%). 1H NMR (400 MHz, CDCl3) δ 7.68 (s, 1H), 7.47 - 7.37 (m, 2H), 7.37 - 7.24 (m, 6H), 7.23 - 7.15 (m, 1H), 7.06 (d, J = 3.1 Hz, 1H), 6.93 - 6.78 (m, 5H), 6.72 (d, J = 9.1 Hz, 1H), 6.12 (dd, J = 7.8, 6.5 Hz, 1H), 5.25 (s, 1H), 4.26 (d, J = 3.2 Hz, 1H), 4.22 - 3.96 (m, 4H), 3.82-3.72 (m, 7H), 3.71 (s, 3H), 3.66 (s, 3H), 3.40 (dd, J = 9.8, 5.2 Hz, 1H), 3.24 (dd, J = 9.8, 6.3 Hz, 1H), 2.71 (m, 1H), 2.37 (d, J = 2.5 Hz, 1H), 1.89 (m, 1H), 1.79 (s, 3H), 1.28 (m, 3H), 1.20 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 171.2, 167.7, 167.6, 158.5, 155.1, 153.1, 150.3, 144.7, 135.9, 130.1, 128.1, 127.9, 126.9, 123.2, 117.7, 117.2,113.2, 111.7, 86.5, 84.0, 83.4, 79.4, 79.3, 72.7, 64.5, 64.2, 64.1, 63.6, 60.4, 55.9, 55.8, 55.2, 35.8, 22.7, 21.1, 16.1, 14.2. 31P NMR (162 MHz, CDCl3) δ −6.05. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C43H51N2NaO13PS 889.2747; Found 889.2740.
Phosphoramidite (7).
Diisopropylethylamine (93.1 mg, 0.72 mmol) and cyanoethyl diisopropylphosphoramidic chloride (51.1 mg, 0.216 mmol) were added to a solution of DMT-6 (150.9 mg, 0.18 mmol) in CH2Cl2 (3 mL) at 0 °C under argon. The mixture was stirred at 0 °C for 2 h. The solution was diluted with ethyl acetate (40 mL), and then washed with saturated NaHCO3 (40 mL) and brine (30 mL). The organic phase was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel (CH2Cl2:ethyl acetate = 1:1, containing 1% Et3N) to yield 7 (170 mg, 88.6%) as a white foam. 1H NMR (400 MHz, CDCl3) δ 7.44 (dd, J = 7.2, 4.4 Hz, 2H), 7.37-7.27 (m, 6H), 7.20 (dd, J = 7.3, 5.6 Hz, 2H), 7.07 (d, J = 3.1 Hz, 1H), 6.83 (ddd, J = 9.0, 5.9, 1.2 Hz, 5H), 6.78 – 6.69 (m, 1H), 6.24 – 6.06 (m, 1H), 5.28 (d, J = 7.1 Hz, 1H), 4.35 (dd, J = 6.6, 3.2 Hz, 1H), 4.13 (dd, J = 15.7, 13.7 Hz, 2H), 4.01 (d, J = 7.1 Hz, 3H), 3.78 (d, J = 3.4 Hz, 6H), 3.72 (d, J = 3.0 Hz, 3H), 3.66 (d, J = 7.4 Hz, 3H), 3.60 (d, J = 2.0 Hz, 3H), 3.39-3.17 (m, 2H), 2.65 (ddd, J = 21.6, 14.4, 6.6 Hz, 2H), 2.42 (t, J = 6.7 Hz, 1H), 2.04-1.88 (m, 1H), 1.85 (d, J = 2.5 Hz, 3H), 1.28 (ddd, J = 11.7, 8.9, 4.0 Hz, 4H), 1.20-1.10 (m, 12H), 1.06 (d, J = 6.8 Hz, 3H). 31P NMR (162 MHz, CDCl3) δ 148.73, 148.30, −5.93, −5.99. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C52H69N4O14P2S 1067.4006; Found 1067.3971.
Supplementary Material
ACKNOWLEDGMENT
We are grateful for support from the National Institute of General Medicine (GM-054996). We thank Professor Jacqueline Barton and Dr. Phil Bartels for providing helpful suggestions concerning the manuscript.
Footnotes
Supporting Information
Experimental procedures for the synthesis of NMe-compounds and Bz-6. Representative autoradiograms, expanded LC-MS/MS data analysis, MS characterization of oligonucleotides containing modified nucleotides. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).von Sonntag C Free-Radical-Induced DNA Damage and Its Repair, Springer-Verlag: Berlin, 2006. [Google Scholar]
- (2).Purkayastha S; Milligan JR; Bernhard WA J. Rhys. Chem. B 2005,109,16967–16973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Yokoya A; Cunniffe SMT; O’Neill PJ Am. Chem. Soc 2002, 124,8859–8866. [DOI] [PubMed] [Google Scholar]
- (4).Banyasz A; Martínez-Fernández L; Balty C; Perron M; Douki T; Improta R; Markovitsi D J. Am. Chem. Soc 2017, 139, 10561–10568. [DOI] [PubMed] [Google Scholar]
- (5).Ming X; Matter B; Song M; Veliath E; Shanley R; Jones R; Tretyakova N J. Am. Chem. Soc 2014, 136,4223–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kanvah S; Joseph J; Schuster GB; Barnett RN; Cleveland CL; Landman U Acc. Chem. Res 2010, 43, 280–287. [DOI] [PubMed] [Google Scholar]
- (7).Ding H; Greenberg MM J. Am. Chem. Soc 2007, 129, 772–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Genereux JC; Barton JK Chem. Rev 2010, 110, 1642–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Genereux JC; Wuerth SM; Barton JK J. Am. Chem. Soc 2011, 133, 3863–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Harris MA; Mishra AK; Young RM; Brown KE; Wasielewski MR; Lewis FD J. Am. Chem. Soc 2016, 138, 5491–5494. [DOI] [PubMed] [Google Scholar]
- (11).Fujitsuka M; Majima T Chem. Sci 2017, 8, 1752–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Deeble DJ; Schuchmann MN; Steenken S; von Sonntag CJ Phys. Chem 1990, 94, 8186–8192. [Google Scholar]
- (13).Geimer J; Beckert DJ Phys. Chem. A 1999, 103, 3991–3998. [Google Scholar]
- (14).Wagner JR; van Lier JE; Johnston LJ Photochem. and Photobiol 1990, 52, 333–343. [DOI] [PubMed] [Google Scholar]
- (15).Malone ME; Symons MCR; Parker AW J. Chem. Soc. Perkin Trans 2 1993, 2067–2075. [Google Scholar]
- (16).Shaw AA; Voituriez L; Cadet J; Gregoli S; Symons MCR J. Chem. Soc. Perkin Trans 2 1988, 1303–1307. [Google Scholar]
- (17).Adhikary A; Kumar A; Heizer AN; Palmer BJ; Pottiboyina V; Liang Y; Wnuk SF; Sevilla MD J. Am. Chem. Soc 2013, 135, 3121–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Rokhlenko Y; Cadet J; Geacintov NE; Shafirovich VJ Am. Chem. Soc 2014, 136, 5956–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rokhlenko Y; Geacintov NE; Shafirovich VJ Am. Chem. Soc 2012, 134, 4955–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sun H; Taverna Porro ML; Greenberg MM J. Org. Chem 2017, 82, 11072–11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Joy A; Ghosh AK; Schuster GB J. Am. Chem. Soc 2006, 128, 5346–5347. [DOI] [PubMed] [Google Scholar]
- (22).Ghosh A; Joy A; Schuster GB; Douki T; Cadet J Org. Biomol. Chem 2008, 6, 916–928. [DOI] [PubMed] [Google Scholar]
- (23).Joseph J; Schuster GB Chem. Comm 2010, 46, 7872–7878. [DOI] [PubMed] [Google Scholar]
- (24).Barnett RN; Joseph J; Landman U; Schuster GB J. Am. Chem. Soc 2013, 135, 3904–3914. [DOI] [PubMed] [Google Scholar]
- (25).Kanvah S; Schuster GB Org. Biomol. Chem 2010, 8, 1340–1343. [DOI] [PubMed] [Google Scholar]
- (26).Horner JH; Lal M; Newcomb M Org. Lett 2006, 8, 5497–5500. [DOI] [PubMed] [Google Scholar]
- (27).Horner JH; Bagnol L; Newcomb MJ Am. Chem. Soc 2004, 126, 14979–14987. [DOI] [PubMed] [Google Scholar]
- (28).Horner JH; Taxil E; Newcomb MJ Am. Chem. Soc 2002, 124, 5402–5410. [DOI] [PubMed] [Google Scholar]
- (29).Newcomb M; Miranda N; Sannigrahi M; Huang X; Crich DJ Am. Chem. Soc 2001, 123, 6445–6446. [DOI] [PubMed] [Google Scholar]
- (30).Horner JH; Newcomb M J. Am. Chem. Soc 2001, 123, 4364–4365. [DOI] [PubMed] [Google Scholar]
- (31).Steenken S; Jovanovic SV J. Am. Chem. Soc 1997, 119, 617–618. [Google Scholar]
- (32).Psciuk BT; Lord RL; Munk BH; Schlegel HB J. Chem. Theory Computation 2012, 8, 5107–5123. [DOI] [PubMed] [Google Scholar]
- (33).Zheng L; Greenberg MM J. Am. Chem. Soc 2017, 139, 17751–17754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Information., S. S.
- (35).Zhu Q; Delaney MO; Greenberg MM Bioorg. & Med. Chem. Lett 2001, 11, 1105–1108. [DOI] [PubMed] [Google Scholar]
- (36).Meggers E; Michel-Beyerle ME; Giese BJ Am. Chem. Soc 1998, 120, 12950–12955. [Google Scholar]
- (37).Saito I; Nakanura T; Nakatani K; Yoshioka Y; Yamaguchi K; Sugiyama H J. Am. Chem. Soc 1998, 120, 12686–12687. [Google Scholar]
- (38).Hall DB; Holmlin RE; Barton JK Nature 1996, 382, 731–735. [DOI] [PubMed] [Google Scholar]
- (39).Rogstad DK; Heo J; Vaidehi N; Goddard WA; Burdzy A; Sowers LC Biochemistry 2004, 43, 5688–5697. [DOI] [PubMed] [Google Scholar]
- (40).Masaoka A; Matsubara M; Hasegawa R; Tanaka T; Kurisu S; Terato H; Ohyama Y; Karino N; Matsuda A; Ide H Biochemistry 2003, 42, 5003–5012. [DOI] [PubMed] [Google Scholar]
- (41).Zheng L; Greenberg MM J. Am. Chem. Soc 2018, 140, 6400–6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zheng L; Griesser M; Pratt DA; Greenberg MM J. Org. Chem 2017, 82, 3571–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Fujimoto J; Tran L; Sowers LC Chem. Res. Toxicol 1997, 10, 1254–1258. [DOI] [PubMed] [Google Scholar]
- (44).Morningstar ML; Kreutzer DA; Essigmann JM Chem. Res. Toxicol 1997, 10, 1345–1350. [DOI] [PubMed] [Google Scholar]
- (45).Cadet J; Douki T; Frelon S; Sauvaigo S; Pouget JP; Ravanat JL Free Rad. Biol. Med 2002, 33, 441–449. [DOI] [PubMed] [Google Scholar]
- (46).Ni J; Pomerantz SC; Rozenski J; Zhang Y; McCloskey JA Anal. Chem 1996, 68, 1989–1999. [DOI] [PubMed] [Google Scholar]
- (47).Chowdhury G; Guengerich FP Angew. Chem. Int. Ed 2008, 47, 381–384. [DOI] [PubMed] [Google Scholar]
- (48).Chowdhury G; Guengerich FP Chem. Res. Toxicol 2009, 22, 1310–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wang Y Chem. Res. Toxicol 2002, 15, 671–676. [DOI] [PubMed] [Google Scholar]
- (50).Chen SB; Li YM; Luo SZ; Zhao G; Tan B; Zhao YF Phosphorus Sulfur Silicon Relat Elem 2000, 164, 277–291. [Google Scholar]
- (51).Gasper SM; Schuster GB J. Am. Chem. Soc 1997, 119, 12762–12771. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.