

Abstract
Cytoglobin is a widely expressed heme protein that binds oxygen, carbon monoxide and nitric oxide. Recent examination of cytoglobin in the vasculature indicates that it contributes to nitric oxide availability, which is central to normal blood vessel function through regulation of smooth muscle cell tone and physiological response. Given the potential implications of cytoglobin in vascular function, we examined how cytoglobin might be uniquely regulated in vascular smooth muscle cells. Our data demonstrate that endothelial cells can increase the expression of cytoglobin in vascular smooth muscle cells, and the induction of cytoglobin is cell contact-dependent. We show that Notch signaling is necessary for endothelial cell-induced cytoglobin expression and Notch2 and Notch3 are sufficient to drive its expression in aortic smooth muscle cells. We further reveal that in cytoglobin-depleted smooth muscle cells there is increased cellular nitric oxide. These data demonstrate that, in addition to being the main producer of vascular nitric oxide, endothelial cells facilitate the ability of smooth muscle cells to metabolize nitric oxide through upregulation of cytoglobin. Our results reveal a novel mechanism by which Notch signaling contributes to vascular function through regulation of a gene that controls nitric oxide levels.
Keywords: smooth muscle, cytoglobin, Notch signaling, nitric oxide, endothelial cell
Graphical Abstract
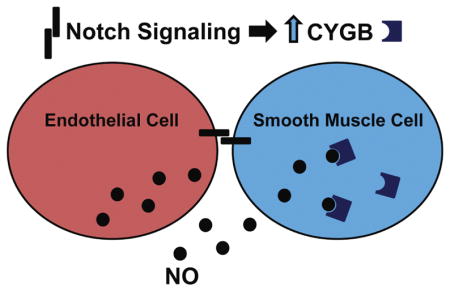
1.1 Introduction
Cytoglobin is a hexacoordinate hemoglobin, which is found in various organs and cell types (Kawada et al., 2001; Oleksiewicz et al., 2011). While its relationship to the globin family has made its ability to bind to gaseous ligands somewhat obvious, its direct role in cellular processes has remained elusive. Cytoglobin has been implicated in oxygen storage and transport, nitric oxide scavenging and protection against oxidative stress (Oleksiewicz et al., 2011; Rahaman and Straub, 2013). Several diseases have been associated with cytoglobin, including neurodegenerative disorders, fibrosis and cancer (Oleksiewicz et al., 2011; Rahaman and Straub, 2013). More recently, the role of cytoglobin has focused on its vascular function (Halligan et al., 2009; Jourd’heuil et al., 2017; Liu et al., 2017). Investigators have shown that cytoglobin is robustly expressed in vascular smooth muscle cells (VSMCs), with its expression concordant with differentiated contractile smooth muscle markers (Jourd’heuil et al., 2017; Liu et al., 2017; Rahaman and Straub, 2013; Thuy le et al., 2016). Data from cytoglobin-deficient mice revealed multi-organ abnormalities, some of which could be ameliorated by nitric oxide inhibition (Thuy le et al., 2016). Examination of the role cytoglobin in blood vessels demonstrated that it functions to regulate vascular tone and nitric oxide metabolism (Liu et al., 2017). Additionally, consistent with its expression in mature smooth muscle, cytoglobin was downregulated in dedifferentiated VSMCs, and contributed to the regulation of vascular remodeling in injury models through an anti-apoptotic mechanism (Jourd’heuil et al., 2017). The ability of cytoglobin to regulate nitric oxide consumption and metabolism indicate an important role in vascular homeostasis that could have clinical implications by serving as a therapeutic target.
Despite the fact that cytoglobin is widely expressed, evidence indicates that its expression is tightly regulated and responsive to environmental cues (Guo et al., 2006; Oleksiewicz et al., 2011; Shaw et al., 2009; Shivapurkar et al., 2008). Several reports have demonstrated that cytoglobin is transcriptionally regulated by hypoxia through hypoxia-inducible factor-1α (HIF1α) transactivation (Chakraborty et al., 2014; Guo et al., 2007; Oleksiewicz et al., 2013). In addition, other transcription factors associated with cellular stress response have been identified in the mammalian cytoglobin promoter, including NF-kB, and NFAT (Latina et al., 2016; Singh et al., 2009). Whereas, growth factors such as fibroblast growth factor (FGF) have also been shown to induce cytoglobin expression (Sato-Matsubara et al., 2017). Although cytoglobin expression in VSMCs is linked to a differentiated phenotype, the transcriptional mediators within this cell type have not been explored. Previous studies from our lab have focused on the role of endothelial cell-derived Notch signaling to mediate VSMC phenotypes (Lilly and Kennard, 2009; Liu et al., 2009; Pajaniappan et al., 2011; Zhao et al., 2012). Our data and others indicate that Notch signaling is activated in VSMCs via the Jagged-1 ligand on endothelial cells, which contributes to differentiation and maturation of blood vessels (High et al., 2008; Liu et al., 2009). The role of Notch signaling in vascular homeostasis and maintenance in adult vessels is less defined (Gridley, 2010; Siebel and Lendahl, 2017).
In this study, we examined the regulation and function of cytoglobin in vascular cells, specifically in relation to endothelial and smooth muscle cell interactions. We show that cytoglobin expression is induced in VSMCs by coculture with endothelial cells (ECs), and this induction is mediated through Notch signaling. We further demonstrate that deletion of VSMC cytoglobin causes an increase in nitric oxide. These data indicate that endothelial cells promote cytoglobin expression in smooth muscle cells as a means to handle nitric oxide, and suggest that it functions in the vasculature to facilitate vascular reactivity through regulation of nitric oxide. Our results imply that endothelial cell-derived Notch signaling acts to prime smooth muscle cells for vascular function by, not only governing contractile gene expression, but also through the regulation of mediators of vascular reactivity.
1.2 Materials and Methods
Cell Culture
Primary cultures of human smooth muscle cells derived from aorta, coronary artery, pulmonary artery, and umbilical artery were purchased from Lonza and grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Mediatech, Inc.) supplemented with 5% fetal bovine serum (FBS) (Hyclone), 2mM glutamine, 1mM sodium pyruvate and 100U/ml penicillin-streptomycin. Human endothelial cells, from aorta, coronary artery, pulmonary artery, and umbilical artery were purchased from Lonza, and grown in EBM-2 supplemented with the bullet kit as recommended (Lonza). Cells between passages 6–9 were used for all experiments. For coculture, an equal number of cells were added to a culture well simultaneously and incubated for 24 to 48 hours prior to cell separation and processing. All coculture and alone control experiments were performed in media consisting of EBM-2 supplemented with the bullet kit, unless otherwise indicated. Notch inhibitor, N-[(3,5-Difluorophenyl)acetyl]-L-alanyl-2-phenyl]glycine-1,1-dimethylethyl ester (DAPT) (Calbiochem), Nitric oxide donor, DETA NONOate (153μM) (Cayman), inhibitor Carboxy-PTIO (25μM) (Cayman), and CoCl2 (100μM) (Sigma), were added to specified wells at the time of plating. To separate endothelial cells from smooth muscle cells, anti-PECAM1-conjugated Dynabeads (Invitrogen) were used according to manufacturer’s instructions (Lilly and Kennard, 2009; Liu et al., 2009). Transwell inserts (12-well type) (Corning Costar) with 0.4μm pores were coated with 50μg/ml rat-tail collagen I (BD Biosciences). 2×104 cells were plated in the bottom and top chambers as indicated with 0.3 ml media added to the insert and 1 ml of media added to the bottom well. For conditioned media experiments, endothelial cells and smooth muscle cells were cultured in EBM-2 media. The media was transferred to pre-plated smooth muscle cells at 24 and 48 hours. Following incubation for 48 hours, cells grown in the bottom chamber of the inserts were harvested and processed for qPCR.
RNA Interference/siRNA
Human aortic smooth muscle cells were plated in a 12 well plate at 5×104 cells/well. After 12 hours, the cells were transfected with siRNA using RNAiMAX (Invitrogen). Efficiency of knockdown was assessed using qPCR (Figure 4 and 6) and western blot (Figure 6). siRNAs against NOTCH3 were synthesized by IDT: 5′-AAC UGC GAA GUG AAC AUU G, and was used as previously described (Liu et al., 2009), NOTCH2 from Qiagen (Assay ID:GS4853), CYGB siRNA from ThermoFisher (Assay ID:s41571). All siRNAs were transfected at 100nM. Following transfection, cells were cocultured with endothelial cells for 48 hours, separated and collected for qPCR analysis and western blotting.
Figure 4. Cytoglobin expression is regulated by Notch signaling.

siRNA specific to (A) Notch2 and (B) Notch3 were transfected into human aortic smooth muscle cells then cocultured with endothelial cells for 48 hours followed by cell separation and qPCR to detect sufficiency of knockdown. Of note, Notch3 is known to be regulated by Notch signaling, and therefore is reduced by Notch2 as well as Notch3 siRNA. (C) Cytoglobin expression following Notch receptor-specific knockdown. (D) Lentiviral delivery of activated forms of Notch2 (NICD2) and Notch3 (NICD3), or control GFP into smooth muscle cells. Cells were cultured for 72 hours post-infection before RNA isolation and qPCR to detect Cytoglobin. n = 4, *P < 0.05, n.s.= not significant.
Figure 6. Inhibition of cytoglobin expression by siRNA.

(A) Detection of Cytoglobin transcripts by qPCR following transfection with control (siCon) or Cytoglobin (siCYGB) siRNA. (B) Western blot to detect Cytoglobin protein expression from control or Cytoglobin siRNA transfected cells. After transfection, smooth muscle cells were cultured alone or cocultured with endothelial cells for 48 hours, followed by cell separation and RNA or protein isolation. n = 4, *P < 0.05.
Lentivirus Expression
Human NOTCH2 intracellular domain (NICD2) and NOTCH3 intracellular domain (NICD3) were made as described previously (Liu et al., 2009; Zhao et al., 2012). The lentivirus plasmids were transfected into TN-293 cells using Lipofectamine 2000 (Invitrogen), and the viral particles were amplified and purified as described (Liu et al., 2009; Zhao et al., 2012). For aortic smooth muscle cell infection, equal volumes of viral particles were diluted in 10% FBS in DMEM and were incubated with cells for 24 hours. The efficiency of infection was evaluated using GFP expression and qPCR. Viral particles were titrated to achieve 90% to 100% infection. Expression of cDNAs were confirmed using qPCR and western blot analysis (not shown and (Liu et al., 2009; Zhao et al., 2012)).
Nitric oxide detection (DAR-4M) and Tracker Dye
Following transfection of human aortic smooth muscle cells with siRNA, cells were incubated in serum free Opti-MEM (Invitrogen) with 5μM CellTracker Green CMFDA (Invitrogen) dye for 30 minutes. Labeled smooth muscle cells were then plated with or without aortic endothelial cells for 24 hours. DETA NONOate (153μM), nitric oxide donor was added to the control alone cultures prior to processing. 10μM of DAR-4M (Millipore) was added to all cells in culture media for 30 minutes, followed by a buffered saline wash, formaldehyde fixation and DAPI staining (Wood et al., 2005). Images were captured at 100X magnification and quantified using ImageJ software. Pixel number for each image was determined using a set threshold for DAR-4M (red) and smooth muscle tracker dye (green), and the ratio of red/green was calculated to determine the number of DAR-4M positive smooth muscle cells.
Aortic Rings
Adult 6-week-old wild-type mice (C57BL/6 strain) were euthanized, and the descending thoracic aortas were isolated and cleaned of extraneous tissue. Aortas were cut into 1 mm rings with a scalpel and place into 96-well culture dish with DMEM supplemented with 2mM glutamine, 1mM sodium pyruvate, and 1%FBS, with 100μg/ml Primocin. 10μM DAPT or vehicle control was added to rings (using adjacent ring sections for paired experimental and control conditions) for 24 hours prior to harvesting RNA by TRIzol (Invitrogen) extraction using the TissueLyzer (Qiagen). The mouse studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol (AR12-00049) was approved by the Institutional Animal Care and Use Committee at Nationwide Children’s hospital.
Quantitative Real-Time PCR (qPCR)
Total RNA was isolated using TRIzol reagent following manufacturers’ instructions (Invitrogen), and reverse transcribed with M-MLV reverse transcriptase (Promega) to generate cDNA. Real-time PCR was performed using a StepOne PCR system (Applied Biosystems) with SYBR Green and 50ng cDNA template. The fold difference in various transcripts was calculated using ΔΔCT method with GAPDH or RPL13A as internal controls. Primer sequences were as follows: Human sequences: CYGB, 5-AAC ACT GTC GTG GAG AAC CTG CAT (forward) and 5-TGA AGT ACA CCG GTT CCA CCT TGT (reverse); SMOOTH MUSCLE α-ACTIN (ACTA2), 5′-CAA GTG ATC ACC ATC GGA AAT G (forward) and 5′-GAC TCC ATC CCG ATG AAG GA (reverse); CALPONIN (CNN1), 5′-TGA AGC CCC ACG ACA TTT TT (forward) and 5′-GGG TGG ACT GCA CCT GTG TA (reverse); NOTCH3, 5- GAG CCA ATG CCA ACT GAA GAG (forward) and 5-GGC AGA TCA GGT CGG AGA TG (reverse); NOTCH2, 5-ACA GTT GTG TCT GCT CAC CAG GAT (forward) and 5-GCG GAA ACC ATT CAC ACC GTT GAT (reverse); GAPDH, 5-ATG GAA ATC CCA TCA CCA TCT T (forward) and 5-CGC CCC ACT TGA TTT TGG (reverse); RPL1A, 5-CTT GTG AGT GGG GCA TCT G (forward) and 5-CCC TGT GTA CAA CAG CAA GC (reverse). Mouse sequences: Cygb 5-GGG TTC AGA GTA GGG TTC ATT C (forward) and 5-GGC CTT TGG GTA CTA TGT CTA TC (reverse); Notch3 5-TTG TCT GGA TGG AAG CCC ATG T (forward) and 5-ACT GAA CTC TGG CAA ACG CCT (reverse); Rpl1A 5-TCC CTG CTG CTC TCA AGG 5- GCC CCA GGT AAG CAA ACT T
Western Blot
Equal amounts of total cellular protein were separated on 10% SDS-PAGE gels, transferred to Amersham–Nitrocellulose membranes (GE healthcare), and subjected to incubation using primary antibodies to cytoglobin (Proteintech, 13317-1-AP) and β-Tubulin 1 (Sigma, T7816). Secondary antibodies conjugated to HRP (Amersham) or IRDye (LI-COR) were used for detection. Protein was detected by enhanced chemiluminescence (ECL) and with the LI-COR imaging system, and quantitated using the LI-COR Image Studio software by normalizing to β-Tubulin expression.
Statistical Analysis
Data analyses were performed using GraphPad Prism and comparisons between data sets were made using a Student’s t test, or ANOVA. Differences were considered significant if P < 0.05, and data are presented as mean ± standard deviation (SD) unless otherwise indicated. Data shown are representative of at least three independent experiments.
1.3 Results
Cytoglobin is induced in smooth muscle cells by cocultured endothelial cells
In an effort to examine the functional relationship between vascular smooth muscle cells and endothelial cells, our laboratory previously examined expression profiles of genes implicated in vascular structure and function (Lilly and Kennard, 2009; Liu et al., 2009; Pajaniappan et al., 2011; Zhao et al., 2015; Zhao et al., 2012). In this process, we measured the expression of cytoglobin (CYGB) in vascular smooth muscle cells cultured alone or in the presence of endothelial cells. Human aortic and coronary artery smooth muscle cells were cultured for 48 hours followed by cell separation using endothelial cell-specific Pecam-1 conjugated beads. We previously determined that the purity of smooth muscle cells following coculture and the cell separation procedure was >99% (Lilly and Kennard, 2009). Quantitative (q)PCR was performed to measure cytoglobin mRNA (Figure 1A). Transcript levels of cytoglobin were robustly increased (~10-fold) in human aortic smooth muscle cells that were cocultured with human aortic endothelial cells. Similarly, coronary artery smooth muscle cells showed a significant, albeit smaller increase of cytoglobin after coculture with coronary endothelial cells. To evaluate if cytoglobin exhibited a similar inductive trend in cells from other vascular beds, we examined cocultured cells derived from human pulmonary arteries and neonatal umbilical arteries. VSMCs from umbilical arteries showed a coculture-dependent induction of cytoglobin, however, we observed no endothelial cell-induced increase of cytoglobin expression in pulmonary artery smooth muscle cells. Previous studies from our lab revealed that cocultured endothelial cells promote expression of smooth muscle differentiation genes (Lin and Lilly, 2014a, b; Liu et al., 2009). Consistent with this, evaluation of smooth muscle α-actin (ACTA2) and calponin1 (CNN1) mRNA showed increased expression following coculture in the aortic, coronary and umbilical artery smooth muscle cells (Figure 1B, C), while pulmonary artery smooth muscle cells failed to show a similar response. Interestingly, we did observe ACTA2 and CNN1 induction in pulmonary artery smooth muscle cells in later cell passages (passage 8), however cytoglobin was not similarly increased (Supplementary Figure 1). We performed additional cocultures in which we swapped pulmonary and aortic endothelial cells with pulmonary and aortic smooth muscle cells. These experiments demonstrated that while pulmonary artery endothelial cells could induce cytoglobin expression in aortic smooth muscle cells, pulmonary artery smooth muscle cells failed to respond to either endothelial cell sub-type and increase cytoglobin (Supplementary Figure 1). These data suggest that cytoglobin shares some but not all regulatory pathways with classical smooth muscle differentiation markers, and possesses unique sub-type dependent regulation. Furthermore, its basal expression in the smooth muscle sub-types was 10-fold higher or more than that observed in endothelial cells (data not shown and Figure 3A), indicating smooth muscle enrichment. Given the robust induction of cytoglobin in aortic smooth muscle cells we focused on these cells for further analysis. Detection of cytoglobin protein by western blot in aortic smooth muscle cells showed a consistent pattern of endothelial cell-dependent induction. A ~3-fold increase in cytoglobin protein was observed after 24-hour coculture, which was increased to ~6-fold after 48 hours (Figure 2A, B). These data demonstrate that endothelial cells can induce expression of cytoglobin mRNA and protein in vascular smooth muscle cells, suggesting a novel means through which endothelial cells govern vascular function.
Figure 1. Cytoglobin mRNA is induced in smooth muscle cells by endothelial cells.
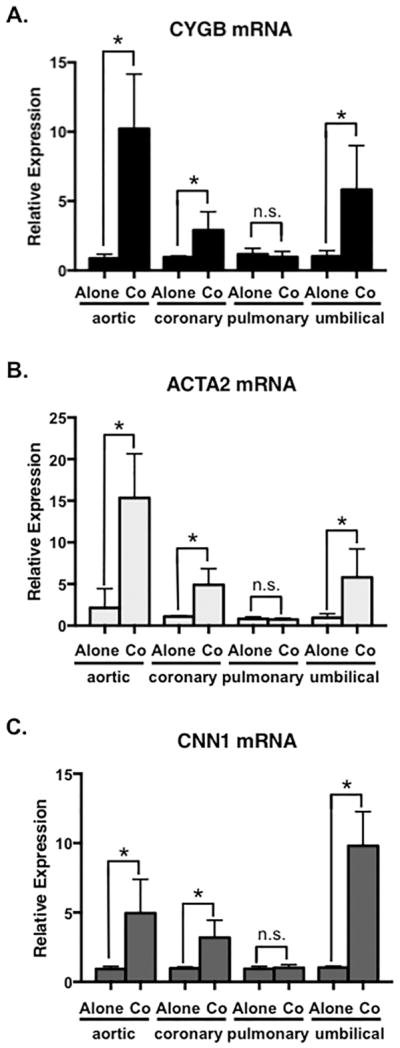
(A) qPCR to detect cytoglobin (CYGB) expression in cultured human aortic, coronary artery, pulmonary artery and umbilical artery smooth muscle cells. Smooth muscle cells were cultured alone or cocultured with appropriate human aortic or coronary/pulmonary/umbilical artery endothelial cells for 48 hours, followed by cell separation to remove endothelial cells. (B, C) qPCR to detect smooth muscle α-actin (ACTA2) and calponin (CNN1) in smooth muscle cells cultured alone or with endothelial cells. n ≥ 4, *P < 0.05.
Figure 3. Cytoglobin induction requires cell-cell contact and is blocked by Notch inhibition.

(A) Human aortic smooth muscle cells (SMC) and endothelial cells (EC) were cultured in the upper and lower chambers of 12-well transwell plates for 48 hours as indicated. RNA was isolated from the lower chambers and subjected to qPCR to detect cytoglobin mRNA expression. (B) Aortic smooth muscle cells were cultured alone or cocultured with endothelial cells for 48 hours in the presence of nitric oxide donor (DETA-NONOate) or inhibitor (CPITO), or the hypoxia mimic CoCl2. Cells were not separated prior to analysis for cytoglobin mRNA. (C, D) Smooth muscle cells were cultured alone or cocultured with endothelial cells for 48 hours in the presence of indicated amounts of Notch inhibitor, DAPT. Smooth muscle cells were separated from endothelial cells prior to RNA and protein isolation for qPCR (C) and western blotting (D), respectively. n ≥ 3, *P < 0.05, n.s.= not significant.
Figure 2. Cytoglobin protein is induced in aortic smooth muscle cells by endothelial cells.

(A) Western blot of aortic smooth muscle cells after 24 and 48-hour (h) coculture with endothelial cells followed by cell separation and detection of CYGB and β-tubulin (TUBB). (B) Quantification of western blots represented in (A) from 3 independent experiments to detect cytoglobin expression standardized to tubulin. *P < 0.05.
To begin to tease apart the mechanism in which cytoglobin is induced in smooth muscle cells by endothelial cells, we performed transwell coculture experiments to determine if the induction was mediated by an endothelial cell-derived secreted factor or required cell-cell contact. Smooth muscle cells cultured alone (bottom well) and in the presence of endothelial cells or control smooth muscle cells (top well) in transwells showed similar expression patterns of cytoglobin transcripts (Figure 3A). Utilization of endothelial cell-conditioned media added to smooth muscle cells showed similar results (Supplementary Figure 2), indicating that the induction of cytoglobin requires cell-cell contact. It is well established that cytoglobin binds oxygen and its expression is regulated by hypoxia (Chakraborty et al., 2014). Given that endothelial cells produce ample nitric oxide, we tested whether cytoglobin expression might be similarly regulated by nitric oxide in VSMCs. Consistent with the transwell experiments, neither a soluble nitric oxide donor (DETA-NONOate) nor inhibitor (CPITO) had a significant effect on cytoglobin expression compared to controls (Figure 3B). As expected, utilization of CoCl2 as a hypoxia mimic, showed significant cytoglobin mRNA induction in aortic smooth muscle cells cultured alone. Despite this, we observed no cooperative induction of cytoglobin in coculture and CoCl2 conditions, indicating distinct mechanisms of transcriptional regulation. Previously, our lab showed that endothelial cells activate Notch signaling in cocultured smooth muscle cells and this contributes to the induction of several genes, including the Hes/Hey family of transcriptional regulators (Lilly and Kennard, 2009; Liu et al., 2009). Incubation of smooth muscle cells in the presence of the Notch inhibitor, DAPT abolished the induction of cytoglobin mRNA and protein by cocultured endothelial cells (Figure 3C, D). These data indicate that endothelial-dependent cytoglobin expression in smooth muscle cells is regulated by Notch signaling.
Cytoglobin expression in smooth muscle cells is regulated by Notch signaling
While Notch signaling has been linked to regulating smooth muscle differentiation/contractile gene transcription (Gridley, 2010; Siebel and Lendahl, 2017), the role of Notch activity in controlling gene expression linked to vascular homeostasis is less defined. To delineate the contribution of Notch receptors in cytoglobin expression, we utilized siRNA to specifically knockdown Notch2 and Notch3 in human aortic smooth muscle cells (Figure 4A, B). Both Notch2 and Notch3 are predominantly expressed and contribute to vascular smooth muscle function (Baeten and Lilly, 2017; Gridley, 2010). Under coculture conditions, we observed that Notch2, but not Notch3 was required for endothelial cell-induced cytoglobin expression (Figure 4C). This receptor specificity may reflect that Notch3 is auto-regulated by Notch signaling (Liu et al., 2009)(Figure 4B), and thus is dispensable for expression of some Notch targets. To test the sufficiency of the Notch receptors to increase cytoglobin expression, we overexpressed the activated intracellular domains (ICD) of Notch2 and Notch3 via lentivirus delivery in smooth muscle cells. Interestingly, both activated forms of Notch2 (NICD2) and Notch3 (NICD3) were sufficient to induce cytoglobin expression in smooth muscle cells in the absence of endothelial cells (Figure 4D). These data support the notion that Notch signaling can regulate cytoglobin expression, and suggest that Notch2 may preferentially modulate its endothelial cell-dependent expression.
In intact blood vessels, Notch signaling is propagated and maintained by lateral induction between the smooth muscle cell layers of the vascular wall (Feng et al., 2010; Hoglund and Majesky, 2012; Manderfield et al., 2012). To determine if Notch signaling contributed to the expression of cytoglobin in intact blood vessels, we isolated and cultured aortic rings from wild-type mice and treated them with DAPT for 24 hours (Figure 5). Similar to cultured human smooth muscle cells, inhibition of Notch signaling by DAPT caused a decrease in cytoglobin mRNA expression. This decrease paralleled that of Notch3, which is a known auto-regulated target of the Notch pathway (Liu et al., 2009). Taken together, these results indicate that endothelial cells via Notch signaling induce cytoglobin, and the regulation of cytoglobin by the Notch pathway is conserved from mouse to human.
Figure 5. Cytoglobin expression is blunted by Notch inhibition in the mouse aorta.
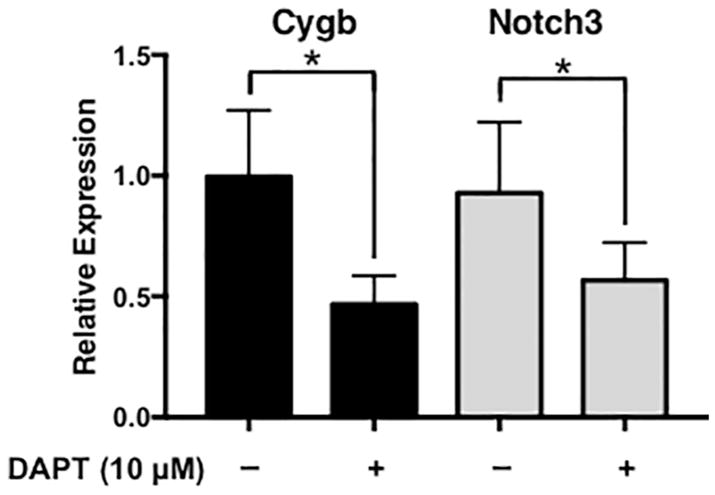
Analysis of cytoglobin (red bars) and Notch3 (yellow bars) expression by qPCR of mRNA isolated from aortic rings of wild-type mice cultured for 24 hours in the presence or absence of Notch inhibitor, DAPT. n = 4, *P < 0.05.
Induction of Cytoglobin by endothelial cells modulates nitric oxide
It is well established that within intact blood vessels endothelial cells produce nitric oxide that diffuses to neighboring smooth muscle cells and acts to control vascular functions, including proliferation and tone (Napoli and Ignarro, 2009). More recently, cytoglobin has been implicated in nitric oxide metabolism, and therefore endothelial cells might activate cytoglobin expression in smooth muscle cells to facilitate nitric oxide handling (Liu et al., 2017; Liu et al., 2012; Liu et al., 2013). To test if cytoglobin affected nitric oxide levels in smooth muscle cells, we knocked down cytoglobin expression by siRNA in aortic smooth muscle cells and cultured with or without endothelial cells. The efficacy of cytoglobin ablation at the transcript and protein level was verified as shown in Figure 6. Prior to coculture, smooth muscle cells were tagged with a green tracker dye to allow for identification in the presence of cocultured endothelial cells. Nitric oxide was directly measured using the DAR-4M reactive dye (Wood et al., 2005). The data indicate that in the absence of cytoglobin, there is increased nitric oxide present in smooth muscle cells either cultured alone or cocultured with endothelial cells (Figure 7A, B). In summary, these data demonstrate that cytoglobin is a contributing factor to nitric oxide levels in smooth muscle cells. Moreover, our results provide a mechanism through which Notch signaling regulates vascular homeostasis through regulation of cytoglobin and nitric oxide bioavailability.
Figure 7. Nitric oxide is increased in cytoglobin-deficient smooth muscle cells.

(A) Images of cultured cells costained with a tracker dye (green) to visualize aortic smooth muscle cells, and DAR4M (red) to detect nitric oxide. DAPI (blue) was used to highlight nuclei and can be seen in the unlabeled endothelial cells in images from cocultured cells. Aortic smooth muscle cells were transfected with control siRNA (siCon) or siRNA to target cytoglobin (siCYGB), then labeled with tracker dye prior to coculture. (B) Graph of DAR4M staining (nitric oxide quantification) of captured images. DAR4M (red pixels) normalized to SMC (green pixels). Data points represent individual images taken from three separate experiments. *P < 0.05.
1.4 Discussion
Cytoglobin is a cellular globin that directly functions by binding gaseous ligands, oxygen, carbon monoxide and nitric oxide (Oleksiewicz et al., 2011; Rahaman and Straub, 2013). The importance of nitric oxide in vascular function has been realized for over 30 years, but the mechanisms by which it is regulated remain elusive (Napoli and Ignarro, 2009; Vanhoutte et al., 2016). Here we show that cytoglobin expression is increased in vascular smooth muscle cells by cocultured endothelial cells. The induction of cytoglobin parallels that of smooth muscle-specific genes, ACTA2 and CNN1, which suggests it shares a regulatory network with these classical smooth muscle differentiation genes. Interestingly, while cytoglobin expression was increased by coculturing vascular cells from the aorta, coronary and umbilical arteries, this induction was not observed in cells derived from pulmonary arteries. Somewhat perplexing was that the cocultured pulmonary artery cells also failed to show an endothelial cell-dependent increase in smooth muscle marker genes in earlier passage cells (passage 5 and 6). However in later passages pulmonary artery smooth muscle cells showed an increase in these markers, but not cytoglobin, indicating unique subtype specific regulation of cytoglobin. Our data show that cytoglobin is regulated in smooth muscle cells by endothelial cells, the same cells that supply nitric oxide through the actions of endothelial nitric oxide synthase (NOS3). While it is not uncommon for signaling mediators to have feed-forward loops that regulate gene transcription and subsequent downstream activities, our data indicate that Notch signaling, not nitric oxide induces cytoglobin expression. Thus, endothelial cells have developed multiple strategies to regulate smooth muscle cell phenotype and function. A 1.5kb fragment of the cytoglobin promoter has been previously identified and shown to be responsive to hypoxia and calcium-dependent signaling (Guo et al., 2006, 2007). Consistent with this, conserved HIF1α and NFAT binding elements are present in the promoter along with sites for ETS-1 and SP1. We were unable to identify consensus CBF/RBPJk binding elements in this promoter region, which convey Notch activity. Furthermore, examination of the promoter’s responsiveness to Notch signaling via a luciferase reporter transfected into smooth muscle cells revealed the 1.5kb fragment was not sufficient to drive Notch-dependent expression, nor could it be activated by endothelial cells (data not shown). These results indicate that additional, yet to be identified, regulatory elements are required for endothelial cell and Notch-dependent expression of cytoglobin.
Previously, our laboratory demonstrated that when cocultured with endothelial cells, the gene expression profile of smooth muscle cells is significantly altered (Lin and Lilly, 2014b; Liu et al., 2009; Zhao et al., 2015; Zhao et al., 2012). Although there are likely several endothelial cell-dependent factors that contribute to this, the Notch signaling pathway is arguably a main contributor. Indeed, data have shown that endothelial cell-derived Notch activity regulates expression of Notch3, Sydecan-2, smooth muscle differentiation components, including smooth muscle α-actin (ACTA2), smooth muscle myosin heavy chain (MYH11) and miR-143/145 (Baeten et al., 2015; Boucher et al., 2012; Boucher et al., 2011; Gridley, 2010; High et al., 2008; Lin and Lilly, 2014b; Liu et al., 2009; Wang et al., 2012; Zhao et al., 2012). Similarly, we show that cytoglobin is also downstream of Notch signaling in smooth muscle cells and Notch activation is sufficient to induce cytoglobin expression in cultured vascular smooth muscle cells. Additionally, Notch inhibition in intact vessels shows reduced cytoglobin expression, indicating that it is regulated within smooth muscle cells by laterally induced Notch activity (Feng et al., 2010; Hoglund and Majesky, 2012; Manderfield et al., 2012). Upon selective knockdown of Notch2 and Notch3, we show that cytoglobin induction by endothelial cells is preferentially through the Notch2 receptor. The significance of this is not clear, and could simply be that Notch3 is autoregulated and dispensable for initial activation of some Notch-regulated genes. However, others and we have demonstrated overlapping, yet unique functions of these Notch receptors in smooth muscle cells that are linked to proliferation and cell survival (Baeten and Lilly, 2015; Boucher et al., 2013; Gridley, 2010). Cytoglobin has also been linked to governing proliferation and apoptosis (Halligan et al., 2009; Jourd’heuil et al., 2017; Liu et al., 2017; Thuy le et al., 2016). Whether the downstream actions of the Notch receptors to govern these processes is facilitated through cytoglobin remains to be determined.
Endothelial cells promote smooth muscle differentiation and quiescence, and in addition to governing genes that contribute to a contractile phenotype. The induction of cytoglobin suggests endothelial cells also facilitate the ability of smooth muscle cells to process nitric oxide. Previous studies have indicated that in cytoglobin-deficient mice, nitric oxide levels are higher, and that chemical inhibition of nitric oxide rescues some of their phenotypes (Liu et al., 2014; Thuy le et al., 2016). Our data are consistent with this and show that by direct measurement using DAR-4M reactive dye, nitric oxide is significantly increased in smooth muscle cells deficient in cytoglobin. Nitric oxide levels were significantly higher in smooth muscle cells cultured with or without endothelial cells, suggesting that smooth muscle cells may be the source of nitric oxide bound to cytoglobin. Alternatively, increased fluorescence might be due to increase oxidation in the absence of cytoglobin, or involve other metabolites such as nitrite. Despite this, endothelial cells appear to mediate smooth muscle cells ability to metabolize nitric oxide by increasing cytoglobin expression, which likely serves as feedback control to maintain vascular reactivity and homeostasis. Future studies are needed to determine if cytoglobin expression is perturbed in vascular disease in humans, and whether alterations of endothelial cell-mediated Notch-signaling contributes to alteration in smooth muscle function and disease progression.
Supplementary Material
Acknowledgments
Grants
This work was supported by the National Institutes of Health (R01HL132801 and R01HL135657 to BL; R00HL116769 and S10OD023438 to AJT) and Nationwide Children’s Hospital (to BL and AJT).
Disclosures
The authors have declared no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baeten JT, Jackson AR, McHugh KM, Lilly B. Loss of Notch2 and Notch3 in vascular smooth muscle causes patent ductus arteriosus. Genesis (New York, NY: 2000) 2015;53:738–748. doi: 10.1002/dvg.22904. [DOI] [PubMed] [Google Scholar]
- Baeten JT, Lilly B. Differential Regulation of NOTCH2 and NOTCH3 Contribute to Their Unique Functions in Vascular Smooth Muscle Cells. The Journal of biological chemistry. 2015;290:16226–16237. doi: 10.1074/jbc.M115.655548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeten JT, Lilly B. Notch Signaling in Vascular Smooth Muscle Cells. Advances in pharmacology (San Diego, Calif) 2017;78:351–382. doi: 10.1016/bs.apha.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher J, Gridley T, Liaw L. Molecular pathways of notch signaling in vascular smooth muscle cells. Frontiers in physiology. 2012;3:81. doi: 10.3389/fphys.2012.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher JM, Harrington A, Rostama B, Lindner V, Liaw L. A receptor-specific function for Notch2 in mediating vascular smooth muscle cell growth arrest through cyclin-dependent kinase inhibitor 1B. Circulation research. 2013;113:975–985. doi: 10.1161/CIRCRESAHA.113.301272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher JM, Peterson SM, Urs S, Zhang C, Liaw L. The miR-143/145 cluster is a novel transcriptional target of Jagged-1/Notch signaling in vascular smooth muscle cells. The Journal of biological chemistry. 2011;286:28312–28321. doi: 10.1074/jbc.M111.221945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, John R, Nag A. Cytoglobin in tumor hypoxia: novel insights into cancer suppression. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:6207–6219. doi: 10.1007/s13277-014-1992-z. [DOI] [PubMed] [Google Scholar]
- Feng X, Krebs LT, Gridley T. Patent ductus arteriosus in mice with smooth muscle-specific Jag1 deletion. Development (Cambridge, England) 2010;137:4191–4199. doi: 10.1242/dev.052043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gridley T. Notch signaling in the vasculature. Current topics in developmental biology. 2010;92:277–309. doi: 10.1016/S0070-2153(10)92009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Philipsen S, Tan-Un KC. Characterization of human cytoglobin gene promoter region. Biochimica et biophysica acta. 2006;1759:208–215. doi: 10.1016/j.bbaexp.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Guo X, Philipsen S, Tan-Un KC. Study of the hypoxia-dependent regulation of human CYGB gene. Biochemical and biophysical research communications. 2007;364:145–150. doi: 10.1016/j.bbrc.2007.09.108. [DOI] [PubMed] [Google Scholar]
- Halligan KE, Jourd’heuil FL, Jourd’heuil D. Cytoglobin is expressed in the vasculature and regulates cell respiration and proliferation via nitric oxide dioxygenation. The Journal of biological chemistry. 2009;284:8539–8547. doi: 10.1074/jbc.M808231200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:1955–1959. doi: 10.1073/pnas.0709663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglund VJ, Majesky MW. Patterning the artery wall by lateral induction of Notch signaling. Circulation. 2012;125:212–215. doi: 10.1161/CIRCULATIONAHA.111.075937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourd’heuil FL, Xu H, Reilly T, McKellar K, El Alaoui C, Steppich J, Liu YF, Zhao W, Ginnan R, Conti D, Lopez-Soler R, Asif A, Keller RK, Schwarz JJ, Thanh Thuy LT, Kawada N, Long X, Singer HA, Jourd’heuil D. The Hemoglobin Homolog Cytoglobin in Smooth Muscle Inhibits Apoptosis and Regulates Vascular Remodeling. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:1944–1955. doi: 10.1161/ATVBAHA.117.309410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada N, Kristensen DB, Asahina K, Nakatani K, Minamiyama Y, Seki S, Yoshizato K. Characterization of a stellate cell activation-associated protein (STAP) with peroxidase activity found in rat hepatic stellate cells. The Journal of biological chemistry. 2001;276:25318–25323. doi: 10.1074/jbc.M102630200. [DOI] [PubMed] [Google Scholar]
- Latina A, Viticchie G, Lena AM, Piro MC, Annicchiarico-Petruzzelli M, Melino G, Candi E. DeltaNp63 targets cytoglobin to inhibit oxidative stress-induced apoptosis in keratinocytes and lung cancer. Oncogene. 2016;35:1493–1503. doi: 10.1038/onc.2015.222. [DOI] [PubMed] [Google Scholar]
- Lilly B, Kennard S. Differential gene expression in a coculture model of angiogenesis reveals modulation of select pathways and a role for Notch signaling. Physiological genomics. 2009;36:69–78. doi: 10.1152/physiolgenomics.90318.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Lilly B. Endothelial cells direct mesenchymal stem cells toward a smooth muscle cell fate. Stem cells and development. 2014a;23:2581–2590. doi: 10.1089/scd.2014.0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Lilly B. Notch signaling governs phenotypic modulation of smooth muscle cells. Vascular pharmacology. 2014b;63:88–96. doi: 10.1016/j.vph.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Liu H, Kennard S, Lilly B. NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circulation research. 2009;104:466–475. doi: 10.1161/CIRCRESAHA.108.184846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Schroeder HJ, Barcelo L, Bragg SL, Terry MH, Wilson SM, Power GG, Blood AB. Role of blood and vascular smooth muscle in the vasoactivity of nitrite. American journal of physiology Heart and circulatory physiology. 2014;307:H976–986. doi: 10.1152/ajpheart.00138.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, El-Mahdy MA, Boslett J, Varadharaj S, Hemann C, Abdelghany TM, Ismail RS, Little SC, Zhou D, Thuy LT, Kawada N, Zweier JL. Cytoglobin regulates blood pressure and vascular tone through nitric oxide metabolism in the vascular wall. Nature communications. 2017;8:14807. doi: 10.1038/ncomms14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Follmer D, Zweier JR, Huang X, Hemann C, Liu K, Druhan LJ, Zweier JL. Characterization of the function of cytoglobin as an oxygen-dependent regulator of nitric oxide concentration. Biochemistry. 2012;51:5072–5082. doi: 10.1021/bi300291h. [DOI] [PubMed] [Google Scholar]
- Liu X, Tong J, Zweier JR, Follmer D, Hemann C, Ismail RS, Zweier JL. Differences in oxygen-dependent nitric oxide metabolism by cytoglobin and myoglobin account for their differing functional roles. The FEBS journal. 2013;280:3621–3631. doi: 10.1111/febs.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderfield LJ, High FA, Engleka KA, Liu F, Li L, Rentschler S, Epstein JA. Notch activation of Jagged1 contributes to the assembly of the arterial wall. Circulation. 2012;125:314–323. doi: 10.1161/CIRCULATIONAHA.111.047159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli C, Ignarro LJ. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Archives of pharmacal research. 2009;32:1103–1108. doi: 10.1007/s12272-009-1801-1. [DOI] [PubMed] [Google Scholar]
- Oleksiewicz U, Liloglou T, Field JK, Xinarianos G. Cytoglobin: biochemical, functional and clinical perspective of the newest member of the globin family. Cellular and molecular life sciences: CMLS. 2011;68:3869–3883. doi: 10.1007/s00018-011-0764-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiewicz U, Liloglou T, Tasopoulou KM, Daskoulidou N, Bryan J, Gosney JR, Field JK, Xinarianos G. Cytoglobin has bimodal: tumour suppressor and oncogene functions in lung cancer cell lines. Human molecular genetics. 2013;22:3207–3217. doi: 10.1093/hmg/ddt174. [DOI] [PubMed] [Google Scholar]
- Pajaniappan M, Glober NK, Kennard S, Liu H, Zhao N, Lilly B. Endothelial cells downregulate apolipoprotein D expression in mural cells through paracrine secretion and Notch signaling. American journal of physiology Heart and circulatory physiology. 2011;301:H784–793. doi: 10.1152/ajpheart.00116.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahaman MM, Straub AC. The emerging roles of somatic globins in cardiovascular redox biology and beyond. Redox biology. 2013;1:405–410. doi: 10.1016/j.redox.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato-Matsubara M, Matsubara T, Daikoku A, Okina Y, Longato L, Rombouts K, Thuy LTT, Adachi J, Tomonaga T, Ikeda K, Yoshizato K, Pinzani M, Kawada N. Fibroblast growth factor 2 (FGF2) regulates cytoglobin expression and activation of human hepatic stellate cells via JNK signaling. The Journal of biological chemistry. 2017 doi: 10.1074/jbc.M117.793794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Omar MM, Rokadiya S, Kogera FA, Lowe D, Hall GL, Woolgar JA, Homer J, Liloglou T, Field JK, Risk JM. Cytoglobin is upregulated by tumour hypoxia and silenced by promoter hypermethylation in head and neck cancer. British journal of cancer. 2009;101:139–144. doi: 10.1038/sj.bjc.6605121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivapurkar N, Stastny V, Okumura N, Girard L, Xie Y, Prinsen C, Thunnissen FB, Wistuba II, Czerniak B, Frenkel E, Roth JA, Liloglou T, Xinarianos G, Field JK, Minna JD, Gazdar AF. Cytoglobin, the newest member of the globin family, functions as a tumor suppressor gene. Cancer research. 2008;68:7448–7456. doi: 10.1158/0008-5472.CAN-08-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebel C, Lendahl U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol Rev. 2017;97:1235–1294. doi: 10.1152/physrev.00005.2017. [DOI] [PubMed] [Google Scholar]
- Singh S, Manda SM, Sikder D, Birrer MJ, Rothermel BA, Garry DJ, Mammen PP. Calcineurin activates cytoglobin transcription in hypoxic myocytes. The Journal of biological chemistry. 2009;284:10409–10421. doi: 10.1074/jbc.M809572200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuy le TT, Van Thuy TT, Matsumoto Y, Hai H, Ikura Y, Yoshizato K, Kawada N. Absence of cytoglobin promotes multiple organ abnormalities in aged mice. Scientific reports. 2016;6:24990. doi: 10.1038/srep24990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte PM, Zhao Y, Xu A, Leung SW. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circulation research. 2016;119:375–396. doi: 10.1161/CIRCRESAHA.116.306531. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhao N, Kennard S, Lilly B. Notch2 and Notch3 function together to regulate vascular smooth muscle development. PloS one. 2012;7:e37365. doi: 10.1371/journal.pone.0037365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MW, Hastings RC, Sygowski LA. A homogeneous fluorescent cell-based assay for detection of heterologously expressed nitric oxide synthase activity. J Biomol Screen. 2005;10:849–855. doi: 10.1177/1087057105280640. [DOI] [PubMed] [Google Scholar]
- Zhao N, Koenig SN, Trask AJ, Lin CH, Hans CP, Garg V, Lilly B. MicroRNA miR145 regulates TGFBR2 expression and matrix synthesis in vascular smooth muscle cells. Circulation research. 2015;116:23–34. doi: 10.1161/CIRCRESAHA.115.303970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Liu H, Lilly B. Reciprocal regulation of syndecan-2 and Notch signaling in vascular smooth muscle cells. The Journal of biological chemistry. 2012;287:16111–16120. doi: 10.1074/jbc.M111.322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.