

Summary
Cancer stem cell (CSC) identification relies on transplantation assays of cell subpopulations sorted from fresh tumor samples. Here, we attempt to bypass limitations of abundant tumor source and predetermined immune selection by in vivo propagating patient-derived xenografts (PDX) from human malignant rhabdoid tumor (MRT), a rare and lethal pediatric neoplasm, to an advanced state in which most cells behave as CSCs. Stemness is then probed by comparative transcriptomics of serial PDXs generating a gene signature of epithelial to mesenchymal transition, invasion/motility, metastasis, and self-renewal, pinpointing putative MRT CSC markers. The relevance of these putative CSC molecules is analyzed by sorting tumorigenic fractions from early-passaged PDX according to one such molecule, deciphering expression in archived primary tumors, and testing the effects of CSC molecule inhibition on MRT growth. Using this platform, we identify ALDH1 and lysyl oxidase (LOX) as relevant targets and provide a larger framework for target and drug discovery in rare pediatric cancers.
Keywords: stem cells, cancer stem cells, PDX, MRT, targeted therapy, ALDH1, LOX inhibition
Graphical Abstract
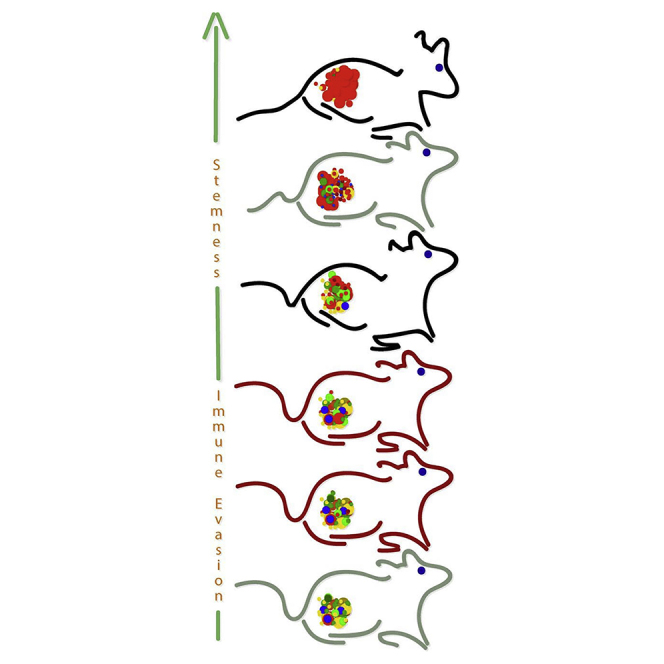
Highlights
-
•
Human malignant rhabdoid tumor (MRT) can be propagated in vivo as tumor xenografts
-
•
Long-term propagated PDX enrich for CSC frequency with no need for immune selection
-
•
Distinct gene signature in stem-like MRT xenografts reveals putative CSC biomarkers
-
•
Screening of putative CSC biomarkers allows identification of therapeutic targets
Golan et al. demonstrate that long-term propagation of human MRT xenografts robustly enriches for cancer stem cell frequency. This was exploited in turn for the identification of potential therapeutic targets in MRT such as lysyl oxidase and disclosed a platform to identify CSC targets in other rare pediatric tumors for which novel therapeutics are sought.
Introduction
Recent years have witnessed the exciting discoveries of cancer stem cells (CSCs)/tumor-initiating cells (TICs) in solid tumors. These are subpopulations of tumor cells that are thought to have crucial roles in treatment failure, cancer recurrence, and metastasis, due to their resistance to drug therapy, quiescent phenotype, migratory ability, and evasion of the immune system (Batlle and Clevers, 2017, Moore, 2011). Identification of CSC/TIC-specific markers is of great importance and can lead to the development of new targeted drugs that can be integrated into the current protocols and improve survival rates of aggressive and resistant disease.
Different methodologies are used to identify and isolate human CSCs/TICs. The most commonly used assays are prospective isolation based on surface marker expression followed by functional and tumorigenicity assays (Ailles and Weissman, 2007, Batlle and Clevers, 2017, Pode-Shakked et al., 2009). In cases for which surgical samples are less frequent, patient-derived xenografts (PDXs) have been utilized to first establish a renewable tissue source and then analyze for the presence of CSCs/TICs (Pleniceanu et al., 2017, Pode-Shakked et al., 2009, Pode-Shakked et al., 2013). Importantly, immune selection from both parental tumors and early-passage PDX for prospective isolation are usually done according to predetermined markers imported from the stem cell literature. We have previously established a collection of PDX from human Wilms tumors (WT), a pediatric renal malignancy, deciphered molecular events in WT tumorigenesis and linked them to an arrested renal stem/progenitor state (Dekel et al., 2006, Metsuyanim et al., 2009), and then went on to identify CSCs that propagate and sustain human WT in vivo (Pode-Shakked et al., 2009, Pode-Shakked et al., 2013, Shukrun et al., 2014). Importantly, when implementing WT-PDX for WT CSC discovery, we utilized early PDXs (up to passage 4; P4) for prospective isolation of a tumorigenic cell subset that could initiate multi-lineage WT in serial xenografts with as few as 200 cells (Pode-Shakked et al., 2013). Comparably, when analyzing the in vivo tumorigenicity of unsorted dissociated cells derived from the P4 WT-PDX as controls, we noted that 10,000 cells were required for tumor xenograft initiation and growth. Further comparison with P0/P1 WT-PDX, in which a significantly higher number of unsorted cells were required to initiate a Wilms tumor in the transplantation assay, indicated that some enrichment for CSC activity might be occurring in the PDX irrespective of cell sorting. Since enrichment was still limited when analyzing P4 WT-PDX and an immune selection step was required for further CSC enrichment, we reasoned that continued PDX propagation might gradually lead to a point in which most of the cells within the tumor act as CSCs leading to significant enrichment of CSC activity and disclosing new CSC targets. In order to study this hypothesis, we chose to model malignant rhabdoid tumor (MRT), a prototypical SMARCB1-deficient tumor that usually arises in the kidneys but also occurs in soft tissue and the brain (where it is referred to as atypical teratoid rhabdoid tumor or ATRT) and runs a lethal course in very young (Parham et al., 1994, Wick et al., 1995). Morphologically, most MRTs contain a population of "rhabdoid" cells, which are large cells with abundant cytoplasm and perinuclear spherical inclusions. Despite currently optimized available medical care, MRT maintains a very poor prognosis, generating aggressive and disseminated disease early on with overall survival approximating 25% (Bondareva et al., 2009, Olson et al., 1995, Versteege et al., 1998). Here, we propagated MRT PDX with characteristic "rhabdoid" morphology and functionally linked advanced PDX to highly enriched CSC/TIC activity. Late-passage stem-like MRT PDX afforded a screen for CSC-related molecules shown, in turn, to be relevant in initiation, propagation, and therapeutic targeting of aggressive MRT. Broadly, late-passage PDX may represent CSC/TIC hubs relevant for drug discovery of targets related to aggressive and disseminated disease.
Results
Establishment of the MRT PDX Model
Primary human MRT samples (MRT-01 and MRT-02) were obtained from patients' biopsies. Tumor grafts were formed by subcutaneous transplantations of 2–5 mm tumor pieces obtained from an MRT-01 sample into immunodeficient mice (Figure S1). Sequential propagation of MRT PDX was performed by single-cell suspension grafting utilizing a fixed number of 1 × 106 cells (Table S1 and Figure S1). Serial propagation allowed us to establish early- (<P5), intermediate- (P5–P10), and late-passage (P10–P16) PDX that were studied for MRT CSC phenotype characterization and elucidation of pathogenic pathways associated with MRT-initiating capacity (Table S2).
Xenotransplantation Assays of MRT Shows Increased CSC Frequency along Serial Propagation
Sequential propagation of MRT PDX correlated with shorter time to tumor engraftment (Figure 1A) and accelerated tumor growth (Table S2), indicating the promotion of tumor aggressiveness along passages. We next queried whether CSC capacity is functionally enhanced with MRT propagation. We performed limiting dilution (LD) xenotransplantation assays with MRT cells derived from early-, intermediate-, and late-passage PDX. This analysis shows significant positive selection for CSC frequency in late-passage PDX (CSC frequency of 1/3,930 in early PDX passages in comparison with 1/252 in late PDX passages, p < 0.001) (Table S3).
Figure 1.

Long-Term Propagation of MRT Is Associated with an Increase in CSC Frequency
(A) Serial PDX propagation correlated with shorter time to tumor engraftment (mean of 26 days in early PDX passages in comparison with 16 days in late PDX passages. Results are presented as the mean of pooled data from early, intermediate, and late passages. p values were generated using the Mann-Whitney test, p < 0.001), indicating change in tumor behavior toward a more aggressive phenotype.
(B) Representative images of H&E staining of primary MRT-01 and late-passage PDX (P14). PDX tumors cells maintain the basic rhabdoid-like cellular morphology with some morphological differences, including the acquisition of spindle-like cells, vast areas of necrosis, less apoptotic bodies, and more mitoses (white arrows indicate mitoses). Scale bar, 100 μm (top) and 50 μm (bottom).
(C) Representative images of IHC of primary MRT-01, early-passage PDX (P2), intermediate-passage PDX (P7), and late-passage PDX (P14) in serial sections for cytokeratin (AE1/AE3), epithelial membrane antigen (EMA), neurofilament protein (NFP), and vimentin. Late-passage PDX showed loss of differentiation markers (top three panels) while primary tumor and PDX tissues strongly express vimentin (bottom panel). Specifically, vimentin staining was 70% in primary tumor as well as in P14 while staining of cytokeratin decreased from 10% to 5%, EMA decreased from 30% to 0, and NFP decreased from 50% to 5% in primary tumor in comparison with P14, accordingly. Scale bar, 200 μm.
(D) qRT-PCR analysis comparing the expression levels of E-cadherin and vimentin between primary tumor, early-passage PDX (P3), and late-passage PDX (P17). The expression of the epithelial marker, E-cadherin, is downregulated throughout serial propagation while the expression of vimentin remains permanent. For qRT-PCR analyses, the values for primary tumor cells were used to normalize (therefore = 1), and all other values were calculated with respect to them. Results are presented as the mean ± SEM of triplicates on three separated experiments. p values were generated using a two-tailed unpaired t test. ∗p < 0.05.
(E) Representative flow cytometry analysis of PDX cells from early, intermediate and late MRT PDX (MRT-01) passages for the expression of several CSC markers including CD24, CD34, CD90, CD56, and ALDH1 antigens on single-cell suspensions. The results revealed that ALDH1 is a good candidate for MRT CSC marker, presenting a pattern of increased expression through PDX serial propagation. Results are presented as the mean of three separated experiments.
See also Figures S1 and Tables S1–S3.
MRT PDX Retains Rhabdoid Morphology in Advanced Generations while Adopting an Epithelial-Mesenchymal Transition Phenotype
Having observed that late-passage MRT PDX enriches for higher numbers of CSCs, we analyzed the histological and immunohistochemically changes that accompany the acquisition of the CSC phenotype along passages (Figures 1B and 1C). H&E staining revealed that PDX-derived tumors maintain the basic rhabdoid-like cellular morphology. Nevertheless, some morphological differences were observed in late-passage PDX, including the acquisition of spindle-like cell morphology, vast areas of necrosis, less apoptotic bodies, and more mitoses (Figure 1B). Immunohistochemistry (IHC) disclosed both primary tumor and PDX tissues to strongly express the mesenchymal marker vimentin (Figure 1C). However, late-passage PDX showed loss of differentiation markers, including cytokeratin, epithelial membrane antigen (EMA), and neurofilament (Figure 1C). To validate the IHC results, qRT-PCR and fluorescence-activated cell sorting (FACS) analysis were performed. The gene expression analysis demonstrated the maintenance of vimentin alongside the loss of the epithelial differentiation marker, E-cadherin (Figure 1D). FACS analysis showed decreased expression of CD34, CD24, and CD90 and increased expression of the CSC markers ALDH1 and NCAM1 along PDX serial propagation (Figure 1E). Thus, advanced MRT PDX harboring significantly enhanced CSC frequency retained the histopathologic rhabdoid features alongside signs of de-differentiation and a mesenchymal phenotype.
Distinct Gene Signature in Advanced Stem-like PDX Reveals Putative CSC Biomarkers
We next sought to characterize the global molecular profile of sequential MRT PDX accompanying the selection for the CSC phenotype. For this purpose, gene expression profiles were determined in primary, intermediate, and late MRT PDXs (from MRT-01) as well as in human embryonic stem cells (ESCs) and primary MRT of kidney (MRT-02) (Figure 2A). Unsupervised hierarchical clustering distinguished primary MRT tissues from PDX tissues. Closer similarity between all PDX samples and ESCs was demonstrated, suggesting selection for a stem cell phenotype during PDX propagation. Closer examination of primary MRT and sequential MRT PDX gene expression profiles identified 1,536 genes that were differentially expressed in a progressive manner throughout the serial propagation: 337 genes were upregulated and 1,199 genes were downregulated (Figure 2B and Table S4). To functionally characterize the differentially regulated genes, we performed gene ontology analysis using the Ingenuity Pathway Analysis software. The upregulated genes were found to be distributed among several categories, mostly developmental processes, cell-cycle regulation, and cancer (Figure 2C). Functional annotation of the downregulated genes demonstrated predominance of immune response processes as well as processes related to cell death and regulation of apoptosis (Figure 2D). Among the most overexpressed genes, we observed CXCL5, CXCL6, SEMA3C, GPM6A, ALDH1, FSTL5, LOX, TSPAN12, HGF/MET, which have been implicated in tumorigenicity, migration/invasion, EMT, proliferation, angiogenesis, and metastases (Figure 2E). Genes that were found to be excessively downregulated include immune recognition molecules such as HLA class I, II, and co-stimulatory molecules as well as an immune cluster of differentiation markers (Figure 2F). This phenomenon may be related to immune evasion properties of CSC-enriched PDX. Alternatively, immune cell subsets present in the primary tumor are likely lost with tumor progression, resulting in diminished immune cell markers. Overall, these results support the results obtained from the H&E and IHC staining that demonstrated decreased apoptosis, increased mitoses, and loss of differentiation in advanced MRT PDX compared with primary tumor. Moreover, gain of the CSC phenotype in progressive MRT was linked to a gene signature set.
Figure 2.

Global Gene Signature Associated with Enhanced Tumor-Initiating Activity Reveals Putative CSC Biomarkers and a New Therapeutic Target
(A) Microarray gene expression analysis comparing several different samples: primary MRTs (P, primary MRT that was propagated [MRT-01], and primary MRT of kidney [MRT-02]), Early MRT PDX passage (Xn2, Xn3), intermediate MRT PDX passage (Xn7), late MRT PDX passage (Xn17, Xn19), and human embryonic stem cells (ESCs). Unsupervised hierarchical clustering revealed great similarity between MRT PDXs and ESCs, emphasizing their undifferentiated nature.
(B) Comparison of the MRT tissues revealed two distinct gene expression patterns: genes that were upregulated (red) and genes that were downregulated (green) throughout the sequential propagation.
(C and D) Functional category grouping of the (C) upregulated and (D) downregulated differentially expressed genes during propagation.
(E and F) Gene heatmaps comparing the expression pattern of (E) several upregulated and (F) several downregulated genes related to various biological functions.
Validation of ALDH1 as MRT CSC Biomarker
ALDH1 has been previously suggested as a CSC marker (Ginestier et al., 2007, Pode-Shakked et al., 2013). Importantly, our molecular analysis independently pinpointed ALDH1 as a marker related to CSC phenotype gain in progressive MRT PDX, indicating initial validation of the platform. We therefore further examined its expression and functional roles. Protein analysis including FACS and IHC staining demonstrated the proportion of ALDH1 increased along tumor propagation (Figures 3A–3C) and that the ALDH1+ expressing cells were mainly the large rhabdoid cells (Figure 3D). At the gene expression level, qRT-PCR analysis revealed high ALDH1 expression in late-passage PDX, about 40 times higher in comparison with primary tumor (Figure 3E). To validate the relevance of our findings, we have stained other archived primary MRT tissues and found that indeed they all express ALDH1 in varying amounts similar to our findings (Figure 3F). CSCs have the functional ability to form clones in vitro and reform tumors upon transplantation into immunodeficient mice in vivo. We therefore performed colony-forming assays that showed significantly higher number of clones and larger colonies in ALDH1+ compared with ALDH1− MRT cells in accordance with the CSC phenotype (Figure 4A). To investigate the in vivo tumorigenicity of ALDH1+ cells, we used the LD xenotransplantation assay (Hu and Smyth, 2009) and injected decreasing amounts of FACS-sorted ALDH1+ and ALDH1− cells into the flanks of NOD/SCID mice (Table 1). Only mice injected with ALDH1+ cells from early PDX (P3) formed tumors compared with none of the mice injected with ALDH1− cells (p < 0.05), suggesting that indeed the ALDH1 molecule enriched for CSCs/TICs (Table 1). An additional sorting experiment according to ALDH1 expression performed on a small dissociated fragment from another primary MRT revealed again that only ALDH1+ cells were able to generate PDX in NOD/SCID mice (Table 1). All in all, a comparison of ALDH1+ with ALDH1− cells sorted from primary, early, and late MRT xenografts demonstrated a highly significant tumor-initiation potential (p < 0.0001; 19/21 injections of ALDH1+ cells formed tumors in comparison with 2/21 injections of ALDH1− cells). Importantly, tumors that arose from ALDH1+ cells could be further serially transplanted into secondary recipients demonstrating in vivo self-renewal capacity in addition to tumor-initiation capacity of ALDH1+ cells (Figure 4B, left panel). Moreover, tumors generated from ALDH + cells are heterogeneous and composed of ALDH1+ (24.9%) and ALDH1− cells (Figure 4B, right panel). In contrast, we show that the ALDH1− fraction cannot self-renew because once it is sorted from a xenograft generated from ALDH1+ cells (and differentiating to ALDH1− cells), it fails to initiate a tumor. Altogether, these data functionally confirmed that ALDH1 is indeed a marker for MRT CSC, validating the link we proposed between long-term PDX propagation and gain of cancer stemness to specific overexpressed molecules such as ALDH1.
Figure 3.

ALDH1 Is a Putative Marker for MRT CSC
(A) Serial propagation of human MRT PDX in NOD/SCID mice results in enrichment of ALDH1-expressing cells (scheme).
(B) Representative FACS analyses of different MRT PDX passages. As tumors progressed the proportion of ALDH1 expressing cells significantly increased, 4% in cells derived from Xn4 (left) and 25% in cells derived from Xn10 cells (right), indicating an enrichment in ALDH1+ expressing cells through PDX serial propagation.
(C) Representative images of IHC of primary MRT-01, intermediate-passage PDX (Xn7), and late-passage PDX (Xn13) for ALDH1 reveals increased expression with PDX serial propagation. Scale bar, 200 μm.
(D) Large magnification IHC of primary MRT for ALDH1 demonstrating that high ALDH1 expressing cells are mainly large rhabdoid cells. Scale bar, 200 μm.
(E) Validation via qRT-PCR revealed high ALDH1 expression in late passages, about 40 times higher in comparison to primary tumor. For qRT-PCR analyses the values for primary tumor cells were used to normalize (therefore = 1) and all other values were calculated accordingly. Results are presented as the mean ± SEM of triplicates on three separated experiments. p values were generated using a 2-tailed unpaired t test ∗p < 0.05; ∗∗p < 0.01.
(F) ALDH1 staining of other primary MRT tissues (03, 04, and 05) demonstrates that they all express ALDH1 in varying amounts. Scale bar, 100 μm.
Figure 4.

Functional Validation of ALDH1 as MRT CSC Biomarker
(A) Colony-forming ability was compared between ALDH1+ and ALDH1− MRT cells. Number of colonies formed by ALDH1+ cells was significantly higher in comparison with ALDH1− cells (left top bar graph; ∗∗p = 0.0083). Number of cells/colony was significantly higher in ALDH1+ compared with ALDH1− cells (left bottom bar graph; ∗∗p = 0.0024). Representative images of colonies formed from ALDH1+ and ALDH1− cells are presented on the right. Results are presented as the means ± SEM of triplicates in three separate experiments.
(B) Tumors that arose from ALDH1+ cells could be further serially transplanted into secondary recipients, demonstrating in vivo self-renewal capacity, consistent with the presence of tumor-initiation capacity in this population (left panel). Moreover, tumors generated from ALDH+ cell were heterogeneously composed of ALDH+ cells (24.9%) and ALDH− cells (left panel) demonstrating differentiating capacity of the ALDH+ cells.
(C) GSEA of correlation between differentially expressed genes in P17 (versus primary MRT cells) and genes differentially expressed in ALDH1+ cells (versus ALDH1− cells). Enrichment score = 0.81, nominal p value = 0.001.
(D) RNA sequencing experiment demonstrates that among the most upregulated genes in the ALDH1+ cells in comparison with ALDH1− cells were those significantly overexpressed genes in late PDX passages in comparison with the primary tumor (e.g., ANXA1, GPM6A, HGF, and LOX).
Table 1.
Sorted ALDH1 Expressing Cells Show an Increased Capacity for MRT PDX Propagation
| Passage | Cell Type | No. of Injected Cells | Frequency of Engrafted Tumors | Total Frequency of Engrafted Tumors | CSC Frequencya (95% CI) |
|---|---|---|---|---|---|
| Primary (MRT-02)b | ALDH1+ | 1,000 | 4/4 | 4/4 | >1/1,000 (1,562, 1) |
| ALDH1− | 1,000 | 0/4 | 0/4 | – | |
| P3 (MRT-01)c | US | 2,000 | 1/1 | 1/5 | 1/3,915 (25,830, 593) |
| 1,000 | 0/2 | ||||
| 500 | 0/2 | ||||
| ALDH1+ | 2,000 | 2/2 | 4/6 | 1/745 (2,201, 252) | |
| 1,000 | 2/2 | ||||
| 500 | 0/2 | ||||
| ALDH1− | 2,000 | 0/2 | 0/6 | – | |
| 1,000 | 0/2 | ||||
| 500 | 0/2 | ||||
| P12 (MRT-01)c | US | 500 | 3/5 | 7/9 | 1/242 (61,495) |
| 100 | 4/4 | ||||
| ALDH1+ | 500 | 3/3 | 11/11 | >1/50 (58, 1) | |
| 100 | 4/4 | ||||
| 50 | 4/4 | ||||
| ALDH1− | 500 | 2/3 | 2/11 | 1/773 (2,976, 201) | |
| 100 | 0/4 | ||||
| 50 | 0/4 |
Sorted ALDH1-expressing cells show an increased capacity for MRT PDX propagation in primary, early (P3), and late (P12) passages.
In the comparison between ALDH− and ALDH+ PDX passages: p = 0.0285 in primary, p = 0.002 in P3, and p < 0.001 in P12; Fisher exact test.
Primary (MRT-02), cells obtained from primary MRT of kidney (RTK) of patient 2.
P3 and P12 (MRT-01), cells obtained from MRT PDX of patient 1.
Finally, we sought to better characterize sorted ALDH1+ CSCs at the molecular level and determine whether they enrich for the CSC molecules predicted by long-term PDX propagation. For this purpose, we performed RNA sequencing experiments on sorted ALDH1+ and ALDH1− cells. Gene set enrichment analysis (GSEA) was used to analyze the similarity between differentially expressed genes in the ALDH1+ cell fraction (in comparison with ALDH1− cells) versus overexpressed genes in unsorted PDX cells from passage 17 (in comparison with primary tumor cells). A strong positive correlation was demonstrated (ES = 0.81, nominal p value = 0.001), further supporting our results regarding enhancement of functional CSC capacity along MRT propagation (Figure 4C). Interestingly, the most upregulated genes alongside ALDH1 in late-passage MRT PDX, such as GPM6A, HGF, CXCL5, TSPAN12, and LOX, were also enriched in sorted ALDH1+ cells (Figure 4D). We chose LOX for further analysis and validated high LOX expression in ALDH1+ in comparison with ALDH1– cells using qRT-PCR (Figure S2A). The high expression of LOX in progressive MRT PDX and in the sorted ALDH1+ CSC fraction led us to examine whether LOX can serve as a therapeutic target.
Functional Validation of LOX Inhibitor as MRT CSC Therapeutic Agent
IHC staining analyzing the expression of LOX in the primary MRT tissue (MRT-01) demonstrated focal expression. Importantly, staining of other primary MRT tissues (MRT-03, MRT-04, MRT-05) for LOX was positive in all tissues analyzed (Figure 5A). IHC staining of LOX expression in early- (P2), intermediate- (P7), and late- (P14) passage PDX confirmed our gene profile studies demonstrating increased expression in late-passage PDX in comparison with early-passage PDX (Figure 5B). The LOX-expressing cells were mainly the large rhabdoid cells. Interestingly, in early-passage PDX cells, LOX was detected in the cytoplasm, while in late PDX cells, LOX was detected both in the cytoplasm and in the nucleus (Figure 5B, lower panel). Finally, to further confirm our results, we examined LOX expression in Xn obtained from G401 MRT cell line and demonstrated again positive LOX staining (Figure 5C).
Figure 5.

LOX Expression and Activity in MRT
(A) LOX staining of primary MRT tissues (MRT-01, MRT-03, MRT-04, and MRT-05) demonstrates that they all express LOX in varying amounts. Scale bar, 100 μm.
(B) LOX staining of MRT PDX passages: early (P2), intermediate (P7) and late (P14), revealed increasing expression along PDX serial propagation (upper panel, magnification ×20; scale bar, 100 μm). LOX was detected in the cytoplasm of cells from early-passage PDX while in late PDX cells, LOX was detected both in the cytoplasm and in the nucleus (black arrows, lower panel, magnification ×100; scale bar, 20 μm).
(C) Representative image of IHC staining of Xn obtained from the G401 cell line demonstrates abundant LOX expression.
(D) Treatment of MRT-01 PDX cells with 100 μM BAPN resulted in a significantly reduced proliferation. As a control treatment of WT, PDX with BAPN showed no effect on WT cell proliferation, indicating a specific effect of LOX inhibition on MRT cells. Results are presented as the means ± SEM of triplicates in three separate experiments. p values were generated using a two-tailed unpaired t test. ∗∗p < 0.01.
(E) Treatment of MRT G401 cell line with 100 μM BAPN resulted in a significantly reduced proliferation. Results are presented as the means ± SEM of triplicates in three separate experiments. p values were generated using a 2-tailed unpaired t test. ∗p < 0.05.
See also Figure S2.
Since LOX gene was upregulated in PDX during propagation and CSC phenotype selection as well as in sorted MRT CSCs, we sought to examine whether treatment with β-aminopropionitrile monofumarate (BAPN), an LOX inhibitor shown to harbor positive effects on various cancer cells (Bondareva et al., 2009), has an effect on MRT cells. First, we performed MTS analysis and demonstrated significantly reduced proliferation following treatment of MRT-01 PDX cells with 100 μM BAPN. As a control, we examined the effect of BAPN on WT-PDX cells and showed no effect on WT cell proliferation, indicating a specific effect of LOX inhibition on MRT cells (Figure 5D). The response to the LOX inhibitor BAPN was also demonstrated in the xenografts derived from the G401 MRT cell line, indicating that the observed effect can be generalized to other MRTs (Figure 5E). Next, we examined the morphology of MRT cells following BAPN treatment and showed that treatment induced morphological changes, including nuclear condensation and cell swelling (Figure 6A). In addition, we performed migration and invasion assays that revealed significant inhibition of migration and invasion capacity in BAPN-treated MRT cells compared with untreated cells (Figures 6B and 6C). Since all in vitro results were obtained using an intermediate-passage PDX (P10), we set to determine whether BAPN will have a similar effect on cells more resembling the primary tumor. Similar experiments were conducted on early-passage PDX (P2) cells and revealed identical results (Figures S2B and S2C). Following analysis of the in vitro effects of LOX inhibition on MRT, we assessed whether in vivo MRT initiation can be dampened via LOX modulation. Usually, MRT engraftment can be clearly visualized approximately 1 month following cell injection. Therefore, the growth of BAPN pretreated and untreated mCherry-expressing MRT PDX (P4) cells was monitored 1 month following inoculation into the right flanks of NOD/SCID mice. Results demonstrated that the mCherry signals of tumors formed from untreated cells were higher in comparison to the mCherry signals of tumors formed from pretreated cells (Figures 6D and 6E). These results suggest that targeting a novel MRT CSC-related molecule can abrogate in vivo tumor growth and warrants LOX as a possible MRT therapeutic target.
Figure 6.

Functional Validation of LOX Inhibitor as a Possible MRT Therapeutic Target
(A) BAPN treatment induced morphological changes on MRT PDX P9 cells, including nuclear condensation and cell swelling. Representative image: scale bar, 100 μm (top) and 50 μm (bottom).
(B) Migration assay revealed that 100 μM BAPN treatment for 48 hr significantly inhibited MRT cell's migration capacity compared with untreated cells. Scale bar, 1,000 μm.
(C) Representative image of 100 μM BAPN treatment for 48 hr demonstrating inhibition of tumor cell's invasion capacity.
(D) Matrigel invasion assay revealed 17% relative invasion of treated cells compared with 67% in untreated cells. The assay was performed in triplicate on two separate early PDX. Values are presented as means ± SEM. p values were generated using a two-tailed unpaired t test. ∗∗p < 0.01.
(E) In vitro treatment of mCherry-expressing P4 PDX cells, with 100 μM BAPN for 48 hr followed by inoculation of the pretreated cells into NOD/SCID mice (103 cells/mouse) demonstrated that untreated PDX signals 1 month following injection were higher in comparison with treated PDX. Results are shown as means ± SEM: treated (n = 6), untreated (n = 5). p values were generated using the Mann-Whitney test. ∗p < 0.05.
(F) Representative image of treated (right) and untreated (left) PDX demonstrating a higher enhancement of the untreated PDX.
See also Figure S2.
Discussion
Here, we have generated a progressive human MRT PDX in which most cells behave as CSCs/TICs. Long-term in vivo propagation results in vigorously proliferating cells so that practically each and every one can initiate MRT xenografts harboring the rhabdoid histopathology features with no need for immune selection. Accordingly, we divert from classical PDX studies (Petrillo et al., 2012), which utilize few passages, and try to show complete similarities to the primary tumor. Alternatively, by refining stemness in advanced late-passage MRT PDX, we probe the differences in comparison with the primary tumor, leading to the formation of a putative CSC-related gene signature set with the aim of deciphering CSC-related molecules.
Previous studies suggest that the hallmark rhabdoid cells of MRT may constitute a highly malignant tumor stem cell population that arises from transformation of an early embryonic multipotent progenitor cell with marked proliferative and invasive features (Deisch et al., 2011). Importantly, we observed a predominance of rhabdoid cellularity in progressive MRT PDX and possibly expansion of an early stem cell lineage contributing to the stemness phenotype. Accordingly, an early developmental gene module is elevated in advanced MRT PDX. These observations may parallel our previous findings with human WT in which undifferentiated blastema that preserves the embryonic renal progenitor state was expanded in WT-PDX models (Dekel et al., 2006, Pode-Shakked et al., 2017). Nevertheless, bearing in mind the plasticity of CSC (non-CSC transiting to CSC), the possibility that the experimental setting we have generated may just reflect serial selection of tumor cell populations that adapt to the mouse microenvironment (Batlle and Clevers, 2017) as well as the appearance of successive activation of tumor-initiating cell clones during serial xenotransplantation (Ball et al., 2017), we have designed a series of experiments to test the relevance of our platform. First, the putative CSC-related gene signature set disclosed molecules previously implicated in CSC such as ALDH1. ALDH1, a cytosolic detoxifying enzyme (Bertucci et al., 2013) has been recognized as an important CSC marker (Martinez-Cruzado et al., 2016, Pode-Shakked et al., 2013, Tomita et al., 2016) Importantly, analysis of ALDH1 protein levels during serial MRT PDX propagation showed distinct staining in primary tumors shifting toward widespread expression in advanced PDX validating gene data. Moreover, serial transplantation assays of sorted ALDH1+ cell populations derived from earlier MRT PDX as well as from an additional primary MRT, in which ALDH1 is confined to a relatively small population, functionally validated ALDH1 as a CSC marker in MRT. This indicated the relevance of the gene signature of late-passage stemness-enriched PDX in predicting a bona fide CSC marker. Yet, another level of validation was demonstrated via RNA sequencing of the sorted ALDH1+ CSCs, showing overlap and significant enrichment of the genes that were overexpressed in advanced MRT PDX. This suggested that the stem-like MRT PDX indeed harbor a CSC-related gene program and as such may inform on valuable therapeutic targets relevant to the human disease. LOX, overexpressed in advanced MRT PDX (gene and protein) and in the sorted ALDH1+ cell fraction was further shown to localize to subpopulations of cells in archived primary MRT samples and to MRT G401 cell line. Importantly, the increased nuclear LOX expression in advanced PDX is consistent with emerging evidence showing that high nuclear LOX expression is in correlation with tumor aggressiveness and poor prognosis (De Donato et al., 2017, Liu et al., 2017). Inhibition of LOX showed effects on in vitro proliferation, migration, and invasiveness of MRT cells derived from progressive PDX, G401 cell line, and G401 xenografts. In addition, blocking LOX activity in an MRT cell suspension prior to cell inoculation into mice significantly abrogated MRT-initiating capacity and xenograft growth. Interestingly LOX, an extracellular matrix (ECM)-remodeling enzyme has been shown to be overexpressed in several aggressive malignancies (Bondareva et al., 2009, Melstrom et al., 2008), and to be associated with tumor progression, metastases, and increased mortality (Barker et al., 2012, Cox et al., 2015). Moreover, LOX has been shown to have significant roles in mesenchymal stem cell-driven breast cancer malignancy, regulating EMT and potentially controlling the CSC phenotype (Boufraqech et al., 2016, Thomas and Karnoub, 2013) Intriguingly, the recent demonstration that LOX family members were overexpressed in a mouse model of MRT (Vitte et al., 2017) and recent roles clarified for LOX as a histone-modifying enzyme and in chromatin modifications and remodeling (Iturbide et al., 2015, Mello et al., 1995, Mello et al., 2011, Millanes-Romero et al., 2013, Mizikova et al., 2017) may indicate a more specific role in MRT in which the SWI/SNF chromatin-remodeling complex is altered due to lack of SMARCB1(Kordes et al., 2003, Versteege et al., 1998, Wick et al., 1995).
Finally, stemness-related molecules upregulated in late-passage PDX included targets that may be of clinical importance and relevant to therapeutic strategies; for instance, DYPD encodes dihydropyrimidine dehydrogenase, an enzyme that catalyzes the rate-limiting step in fluorouracil metabolism (Edwards et al., 2016). Accordingly, high levels of DYPD would preclude a response to 5-fluorouracil and inadvertently lead to CSC enrichment. In addition, among the most upregulated genes were CXCL6 and CXCL5, which are NFkB reactive oxygen species-related tumor targets. Thus, blockage with superoxide inhibitors might prevent CSC activity (Laidlaw et al., 2016). In sum, this study lays the foundation for targeting stemness and aggressiveness determinants in MRT. While we concentrated our efforts on LOX for proof-of-principle experiments, other molecules that synergize with LOX to control ECM proteolysis such as matrix metalloproteinase (Fouani et al., 2017) were also overexpressed in advanced MRT PDX, indicating that combined targeting of the CSC niche generated in aggressive and disseminated disease might be of importance. In addition, other pathways, including HGF/Met or novel CSC-related surface markers such as the GPM6A, may serve as relevant therapeutic targets. Importantly, our study provides a template for long-term PDX propagation and generation of a CSC phenotype to identify other CSC targets in a variety of tumors.
Experimental Procedures
Tissue Samples and Cell Lines
The primary MRT samples (MRT-01 from soft tissue of the neck and MRT-02 from primary MRT of kidney) were obtained from the patients within 1 hr of surgery. Archived formalin-fixed paraffin-embedded MRT samples (MRT-03, MRT-04, MRT-05) were obtained from the Department of Pathology, Sheba Medical Center. One MRT cell line (Garvin et al., 1993) was used in our study. Informed consent was given by the legal guardians of the patients involved according to the declaration of Helsinki.
In Vivo Xenograft Formation
The animal experiments were performed in accordance with the Guidelines for Animal Experiments of Sheba Medical Center. All animals in the individual experiments were 5- to 8-week-old, female, non-obese diabetic severe combined immunodeficiency (NOD/SCID). Time to engraftment (when a tumor was first palpable), time to resection, weight, and volume for each engrafted PDX were recorded. Tumor growth was followed by measuring perpendicular diameters of the tumors with a Vernier caliper. Tumor volume was calculated using the equation 1/2(length × width2). In addition, tumors were excised postmortem and weighed. Only tumors that could be excised completely without additional invaded tissue were used for weight measurements.
Estimation of the Relative Frequency of Tumor-Propagating Cells
To evaluate and compare the tumorigenic activity of PDX cells from early and late PDX passage and of sorted ALDH1+ and ALDH1− cells, serial dilutions (1×106 to 50 cells) of cells suspended in 100 μL of PBS and 100 μL of Matrigel were injected subcutaneously into the flank of NOD/SCID mice. Estimation of the relative frequency of cancer-propagating cells was calculated online using the ELDA software (http://bioinf.wehi.edu.au/software/elda/).
Gene Set Enrichment Analysis
GSEA was performed to determine whether the differentially expressed upregulated genes in passage 17 sample (in comparison with the primary tumor) are enriched within the differentially expressed genes in the ALDH1+ sample (in comparison with the ALDH1− sample). GSEA was performed to analyze the enrichment of the gene sets following the developer's protocol (Subramanian et al., 2005) (http://www.broad.mit.edu/gsea/).
MTS Cell Viability Assay
A total of 5 × 103 cells were plated in triplicate and grown in 96-well plates overnight. The following day, the medium was changed and supplemented with various concentrations of BAPN (Sigma, St Louis, MO, USA, cat. no. A3134-5G) and the cells were further incubated for 48 hr. Cell proliferation was measured using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega) according to the manufacturer's instructions. Three independent experiments were carried out.
Cell Migration Assay
Cells were grown overnight, after which the medium was changed and supplemented with 100 μM BAPN. After 48 hr, a scrape was made through the confluent monolayers with a plastic 1 mL pipette tip. Treated and control cells were then photographed at identical time points (24 and 48 hr).
Invasion Assay
The effect of BAPN on the tumor cells' invasion capacity was determined by Matrigel invasion assay using a 24-well transwell (pore size 8 μm; Corning Incorporated). Cells were grown in Iscove's modified Dulbecco's medium (IMDM) ± 100 μM BAPN. After 48 hr of incubation including 12 hr of starvation conditions (serum-free IMDM), the treated and untreated serum-starved cells were seeded at 10 × 104 cells/well in 350 μL of serum-free IMDM into the upper chamber of each insert coated with Matrigel (15 μL). The lower chamber contained 150 μL of IMDM supplemented with 10% fetal bovine serum serving as a chemoattractant. After 24 hr of incubation at 37°C and 5% CO2, the medium was aspirated, remaining cells and medium from the top of the membrane were removed using a cotton-tipped applicator, and the invading cells were stained with Giemsa stain. The invading cells were counted under light microscopy (11 random fields ×20 magnification per well). The assay was performed in triplicate with two sets of independent experiments, on two different PDX passages (P3, P5). Data are presented as relative invaded cells. The fraction of invaded cells counted per area was compared with the number of expected invaded cells per area, which was calculated based on the number of cells initially seeded.
In Vivo BAPN Treatment
PDX (P4)-expressing mCherry cells, were treated in vitro with 100 μM BAPN or saline for the control group. Forty-eight hours after treatment, MRT cells (103 cells/mouse) were subcutaneously inoculated into the right flanks of NOD/SCID mice. Tumors volume were analyzed during 1 month, using a CRi Maestro II in vivo imaging system (Caliper Life Sciences, Hopkinton, MA). Fluorescence images were obtained with an excitation wavelength of 465 nm and emission wavelength range of 500–800 nm.
Exclusion of Data
All data from animals that died or had to be killed prior to the scheduled termination of the experiment were excluded.
Statistical Analysis
Results are expressed as the means ± SEM unless otherwise indicated. For animal studies, sample size was estimated to be at least four mice per group to ensure power with statistical confidence. Statistical differences in gene expression between MRT PDX cell populations were evaluated using Student's t test. Statistical differences in the in vivo experiments were calculated using the Mann-Whitney test/chi-squared test or Fisher's exact test as indicated in the figure legends. For all statistical analysis, the level of significance was set as p < 0.05 unless otherwise indicated.
Author Contributions
B.D., A. Toren, H.G., and R.S. designed the experiments. H.G., D.D.B.-L., R.C., M.M.D., S.P.-C., and R.S. performed the in vivo experiments. H.G., R.S., N.P.-S., O.P., J.J.-H., G.S., D.D.B.-L., M.M.D., E.V., R.C., O.H.-S., and S.G. performed in vitro assays. B.D., H.G., J.J.-H., R.S., R.B., A. Trink, I.K., and T.K. analyzed the data. B.D., A. Toren, R.S., and H.G. wrote the manuscript.
Acknowledgments
We thank Itamar Goldstein for his assistance with the FACS experiments and Peleg Hasson for provision of BAPN. This work was supported by the Zeiring Foundation, the Israel Cancer Association (grant no. 20150916) and the Israel Cancer Research Fund (ICRF) (grant no. PG-14-112 to B.D.). A patent application on isolation and characterization of cancer stem cells has been filed by Sheba Medical Center (THM). This work is part of the requirements toward a PhD degree, Sackler School of Medicine, Tel Aviv University (R.S. and H.G.).
Published: August 16, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, two figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.07.010.
Accession Numbers
The accession number for the microarray data reported in this paper is GEO: GSE7269. The accession number for the RNA sequencing data reported in this paper is GEO: GSE114471.
Supplemental Information
References
- Ailles L.E., Weissman I.L. Cancer stem cells in solid tumors. Curr. Opin. Biotechnol. 2007;18:460–466. doi: 10.1016/j.copbio.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Ball C.R., Oppel F., Ehrenberg K.R., Dubash T.D., Dieter S.M., Hoffmann C.M., Abel U., Herbst F., Koch M., Werner J. Succession of transiently active tumor-initiating cell clones in human pancreatic cancer xenografts. EMBO Mol. Med. 2017;9:918–932. doi: 10.15252/emmm.201607354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker H.E., Cox T.R., Erler J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. 2012;12:540–552. doi: 10.1038/nrc3319. [DOI] [PubMed] [Google Scholar]
- Batlle E., Clevers H. Cancer stem cells revisited. Nat. Med. 2017;23:1124–1134. doi: 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- Bertucci F., Bouvier-Labit C., Finetti P., Metellus P., Adelaide J., Mokhtari K., Figarella-Branger D., Decouvelaere A.V., Miquel C., Coindre J.M. Gene expression profiling of solitary fibrous tumors. PLoS One. 2013;8:e64497. doi: 10.1371/journal.pone.0064497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondareva A., Downey C.M., Ayres F., Liu W., Boyd S.K., Hallgrimsson B., Jirik F.R. The lysyl oxidase inhibitor, beta-aminopropionitrile, diminishes the metastatic colonization potential of circulating breast cancer cells. PLoS One. 2009;4:e5620. doi: 10.1371/journal.pone.0005620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boufraqech M., Zhang L., Nilubol N., Sadowski S.M., Kotian S., Quezado M., Kebebew E. Lysyl oxidase (LOX) transcriptionally regulates SNAI2 expression and TIMP4 secretion in human cancers. Clin. Cancer Res. 2016;22:4491–4504. doi: 10.1158/1078-0432.CCR-15-2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox T.R., Rumney R.M.H., Schoof E.M., Perryman L., Hoye A.M., Agrawal A., Bird D., Latif N.A., Forrest H., Evans H.R. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature. 2015;522:106–110. doi: 10.1038/nature14492. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- De Donato M., Petrillo M., Martinelli E., Filippetti F., Zannoni G.F., Scambia G., Gallo D. Uncovering the role of nuclear lysyl oxidase (LOX) in advanced high grade serous ovarian cancer. Gynecol. Oncol. 2017;146:170–178. doi: 10.1016/j.ygyno.2017.05.001. [DOI] [PubMed] [Google Scholar]
- Deisch J., Raisanen J., Rakheja D. Immunohistochemical expression of embryonic stem cell markers in malignant rhabdoid tumors. Pediatr. Dev. Pathol. 2011;14:353–359. doi: 10.2350/10-09-0902-OA.1. [DOI] [PubMed] [Google Scholar]
- Dekel B., Metsuyanim S., Schmidt-Ott K.M., Fridman E., Jacob-Hirsch J., Simon A., Pinthus J., Mor Y., Barasch J., Amariglio N. Multiple imprinted and stemness genes provide a link between normal and tumor progenitor cells of the developing human kidney. Cancer Res. 2006;66:6040–6049. doi: 10.1158/0008-5472.CAN-05-4528. [DOI] [PubMed] [Google Scholar]
- Edwards L., Gupta R., Filipp F.V. Hypermutation of DPYD deregulates pyrimidine metabolism and promotes malignant progression. Mol. Cancer Res. 2016;14:196–206. doi: 10.1158/1541-7786.MCR-15-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouani L., Menezes S.V., Paulson M., Richardson D.R., Kovacevic Z. Metals and metastasis: exploiting the role of metals in cancer metastasis to develop novel anti-metastatic agents. Pharmacol. Res. 2017;115:275–287. doi: 10.1016/j.phrs.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Garvin A.J., Re G.G., Tarnowski B.I., Hazen-Martin D.J., Sens D.A. The G401 cell line, utilized for studies of chromosomal changes in Wilms' tumor, is derived from a rhabdoid tumor of the kidney. Am. J. Pathol. 1993;142:375–380. [PMC free article] [PubMed] [Google Scholar]
- Ginestier C., Hur M.H., Charafe-Jauffret E., Monville F., Dutcher J., Brown M., Jacquemier J., Viens P., Kleer C.G., Liu S. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Smyth G.K. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Iturbide A., Garcia de Herreros A., Peiro S. A new role for LOX and LOXL2 proteins in transcription regulation. FEBS J. 2015;282:1768–1773. doi: 10.1111/febs.12961. [DOI] [PubMed] [Google Scholar]
- Kordes U., Gesk S., Fruhwald M.C., Graf N., Leuschner I., Hasselblatt M., Jeibmann A., Oyen F., Peters O., Pietsch T. Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant rhabdoid tumor. Genes Chromosomes Cancer. 2003;49:176–181. doi: 10.1002/gcc.20729. [DOI] [PubMed] [Google Scholar]
- Laidlaw K.M., Berhan S., Liu S., Silvestri G., Holyoake T.L., Frank D.A., Aggarwal B., Bonner M.Y., Perrotti D., Jorgensen H.G. Cooperation of imipramine blue and tyrosine kinase blockade demonstrates activity against chronic myeloid leukemia. Oncotarget. 2016;7:51651–51664. doi: 10.18632/oncotarget.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Cox T.R., Cui W., Adell G., Holmlund B., Ping J., Jarlsfelt I., Erler J.T., Sun X.F. Nuclear expression of lysyl oxidase enzyme is an independent prognostic factor in rectal cancer patients. Oncotarget. 2017;8:60015–60024. doi: 10.18632/oncotarget.9623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Cruzado L., Tornin J., Santos L., Rodriguez A., Garcia-Castro J., Moris F., Rodriguez R. Aldh1 expression and activity increase during tumor evolution in sarcoma cancer stem cell populations. Sci. Rep. 2016;6:27878. doi: 10.1038/srep27878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello M.L., Alvarenga E.M., Vidal Bde C., Di Donato A. Chromatin supraorganization, mitotic abnormalities and proliferation in cells with increased or down-regulated lox expression: indirect evidence of a LOX-histone H1 interaction in vivo. Micron. 2011;42:8–16. doi: 10.1016/j.micron.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Mello M.L., Contente S., Vidal B.C., Planding W., Schenck U. Modulation of ras transformation affecting chromatin supraorganization as assessed by image analysis. Exp. Cell Res. 1995;220:374–382. doi: 10.1006/excr.1995.1328. [DOI] [PubMed] [Google Scholar]
- Melstrom L.G., Bentrem D.J., Salabat M.R., Kennedy T.J., Ding X.Z., Strouch M., Rao S.M., Witt R.C., Ternent C.A., Talamonti M.S. Overexpression of 5-lipoxygenase in colon polyps and cancer and the effect of 5-LOX inhibitors in vitro and in a murine model. Clin. Cancer Res. 2008;14:6525–6530. doi: 10.1158/1078-0432.CCR-07-4631. [DOI] [PubMed] [Google Scholar]
- Metsuyanim S., Harari-Steinberg O., Buzhor E., Omer D., Pode-Shakked N., Ben-Hur H., Halperin R., Schneider D., Dekel B. Expression of stem cell markers in the human fetal kidney. PLoS One. 2009;4:e6709. doi: 10.1371/journal.pone.0006709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millanes-Romero A., Herranz N., Perrera V., Iturbide A., Loubat-Casanovas J., Gil J., Jenuwein T., Garcia de Herreros A., Peiro S. Regulation of heterochromatin transcription by Snail1/LOXL2 during epithelial-to-mesenchymal transition. Mol. Cell. 2013;52:746–757. doi: 10.1016/j.molcel.2013.10.015. [DOI] [PubMed] [Google Scholar]
- Mizikova I., Palumbo F., Tabi T., Herold S., Vadasz I., Mayer K., Seeger W., Morty R.E. Perturbations to lysyl oxidase expression broadly influence the transcriptome of lung fibroblasts. Physiol. Genomics. 2017;49:416–429. doi: 10.1152/physiolgenomics.00026.2017. [DOI] [PubMed] [Google Scholar]
- Moore L.S. Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J. Oncol. 2011;2011 doi: 10.1155/2011/396076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson T.A., Bayar E., Kosnik E., Hamoudi A.B., Klopfenstein K.J., Pieters R.S., Ruymann F.B. Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J. Pediatr. Hematol. Oncol. 1995;17:71–75. doi: 10.1097/00043426-199502000-00013. [DOI] [PubMed] [Google Scholar]
- Parham D.M., Weeks D.A., Beckwith J.B. The clinicopathologic spectrum of putative extrarenal rhabdoid tumors. An analysis of 42 cases studied with immunohistochemistry or electron microscopy. Am. J. Surg. Pathol. 1994;18:1010–1029. doi: 10.1097/00000478-199410000-00005. [DOI] [PubMed] [Google Scholar]
- Petrillo L.A., Wolf D.M., Kapoun A.M., Wang N.J., Barczak A., Xiao Y., Korkaya H., Baehner F., Lewicki J., Wicha M. Xenografts faithfully recapitulate breast cancer-specific gene expression patterns of parent primary breast tumors. Breast Cancer Res. Treat. 2012;135:913–922. doi: 10.1007/s10549-012-2226-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleniceanu O., Shukrun R., Omer D., Vax E., Kanter I., Dziedzic K., Pode-Shakked N., Mark-Daniei M., Pri-Chen S., Gnatek Y. Peroxisome proliferator-activated receptor gamma (PPARgamma) is central to the initiation and propagation of human angiomyolipoma, suggesting its potential as a therapeutic target. EMBO Mol. Med. 2017;9:1763. doi: 10.15252/emmm.201708507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pode-Shakked N., Gershon R., Tam G., Omer D., Gnatek Y., Kanter I., Oriel S., Katz G., Harari-Steinberg O., Kalisky T. Evidence of in vitro preservation of human nephrogenesis at the single-cell level. Stem Cell Rep. 2017;9:279–291. doi: 10.1016/j.stemcr.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pode-Shakked N., Metsuyanim S., Rom-Gross E., Mor Y., Fridman E., Goldstein I., Amariglio N., Rechavi G., Keshet G., Dekel B. Developmental tumourigenesis: NCAM as a putative marker for the malignant renal stem/progenitor cell population. J. Cell. Mol. Med. 2009;13:1792–1808. doi: 10.1111/j.1582-4934.2008.00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pode-Shakked N., Shukrun R., Mark-Danieli M., Tsvetkov P., Bahar S., Pri-Chen S., Goldstein R.S., Rom-Gross E., Mor Y., Fridman E. The isolation and characterization of renal cancer initiating cells from human Wilms' tumour xenografts unveils new therapeutic targets. EMBO Mol. Med. 2013;5:18–37. doi: 10.1002/emmm.201201516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukrun R., Pode-Shakked N., Pleniceanu O., Omer D., Vax E., Peer E., Pri-Chen S., Jacob J., Hu Q., Harari-Steinberg O. Wilms' tumor blastemal stem cells dedifferentiate to propagate the tumor bulk. Stem Cell Rep. 2014;3:24–33. doi: 10.1016/j.stemcr.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C., Karnoub A.E. Lysyl oxidase at the crossroads of mesenchymal stem cells and epithelial-mesenchymal transition. Oncotarget. 2013;4:376–377. doi: 10.18632/oncotarget.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita H., Tanaka K., Tanaka T., Hara A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget. 2016;7:11018–11032. doi: 10.18632/oncotarget.6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteege I., Sevenet N., Lange J., Rousseau-Merck M.F., Ambros P., Handgretinger R., Aurias A., Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- Vitte J., Gao F., Coppola G., Judkins A.R., Giovannini M. Timing of Smarcb1 and Nf2 inactivation determines schwannoma versus rhabdoid tumor development. Nat. Commun. 2017;8:300. doi: 10.1038/s41467-017-00346-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick M.R., Ritter J.H., Dehner L.P. Malignant rhabdoid tumors: a clinicopathologic review and conceptual discussion. Semin. Diagn. Pathol. 1995;12:233–248. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.