

Abstract
Alterations in glutamate neurotransmission have been implicated in the pathophysiology of schizophrenia, as well as in symptom severity and cognitive deficits. The hippocampus, in particular, is a site of key functional and structural abnormalities in schizophrenia. Yet few studies have investigated hippocampal glutamate in antipsychotic-naïve first episode psychosis patients or in individuals at clinical high risk (CHR) of developing psychosis. Using proton magnetic resonance spectroscopy (1H-MRS), we investigated glutamate metabolite levels in the left hippocampus of 25 CHR (19 antipsychotic-naïve), 16 patients with first-episode psychosis (13 antipsychotic-naïve) and 31 healthy volunteers. We also explored associations between hippocampal glutamate metabolites and glial activation, as indexed by [18F]FEPPA positron emission tomography (PET); symptom severity; and cognitive function. Groups differed significantly in glutamate plus glutamine (Glx) levels (F(2, 69) = 6.39, p = 0.003). Post-hoc analysis revealed that CHR had significantly lower Glx levels than both healthy volunteers (p = 0.003) and first-episode psychosis patients (p = 0.050). No associations were found between glutamate metabolites and glial activation. Our findings suggest that glutamate metabolites are altered in CHR.
Introduction
Glutamate, the principal excitatory neurotransmitter in the brain, is implicated in the pathogenesis and pathophysiology of schizophrenia [1]. The glutamate hypothesis originated in the observation that whereas dopamine agonists could induce only the positive symptoms of schizophrenia in healthy volunteers (HV), N-Methyl-D-aspartate receptor (NMDA-R) antagonists (e.g., ketamine) could also induce the negative symptoms and cognitive deficits of schizophrenia [2, 3]. The clinical findings were linked to glutamatergic neurotransmission dysfunction [4], and subsequent studies supported a contribution of NMDA-R hypofunction to the pathogenesis of schizophrenia [5, 6]. Whereas early reports suggested NMDA-R hypofunction was a result of lower glutamate levels in schizophrenia [7], the current prevailing hypothesis implicates excess glutamate [8]. Over the last decades, additional evidence from neuroimaging studies have supported a role for glutamate dysregulation in symptom severity and cognitive dysfunction [9].
Hippocampal dysfunction is implicated in the pathophysiology of schizophrenia, particularly glutamatergic abnormalities of this region [10]. For instance, post-mortem studies have reported lower levels of hippocampal NMDA receptor gene expression in schizophrenia patients [11, 12]. These results are consistent with an earlier imaging study, which found a significant reduction in left hippocampal NMDA receptor expression in medication-free schizophrenia patients [13]. Moreover, studies have also shown functional and structural hippocampal abnormalities in schizophrenia [14], with researchers consistently reporting reduced hippocampal volume in these patients [15]. Similarly, a large study reported that individuals at clinical high risk (CHR) for psychosis (n = 61) had reduced left hippocampal volumes relative to HV (n = 25) [16]. The hippocampal atrophy may be driven by glutamate hypermetabolism [17], which has been shown to predict progression to psychosis in CHR [18].
Proton magnetic resonance spectroscopy (1H-MRS) of the hippocampus is challenging because of the region’s small size and vicinity to bony structures, resulting in small voxel sizes and large susceptibility changes that compromise the signal-to-noise ratio (SNR) and spectral resolution [19]. As a result of these challenges, few 1H-MRS studies of the hippocampus have been published.
To date, studies of hippocampal glutamatergic metabolites in CHR or those at genetic high risk for psychosis have reported no significant differences relative to HV [16, 20–23]. However, no studies have directly compared CHR with first-episode psychosis (FEP). Hippocampal 1H-MRS studies comparing FEP patients with HV have reported no group differences in glutamatergic metabolites [24–28], with the exception of one study that found higher glutamate plus glutamine (Glx) levels in the left medial temporal lobe in FEP [29]. However, most of these studies were done at magnetic field strengths at which glutamate and glutamine peaks significantly overlap (1.5 T), making glutamate quantification difficult [30].
Studies comparing chronic schizophrenia patients and HV have produced mixed results, with some reporting no group differences [31–33], some reporting higher glutamate metabolites in patients [34, 35] and others reporting lower glutamate markers in patients [12, 36]. One notable study on unmedicated chronic schizophrenia patients with a history of antipsychotic exposure observed elevated ratios of hippocampal Glx to creatine (Cr) in patients relative to HV (n = 27; [37]). Although noted, these studies are not directly comparable to the current study given that chronic schizophrenia patients are generally older, and have a longer duration of antipsychotic treatment and illness, than FEP patients.
Neuroinflammation, particularly microglial activation [38], may play a role in the aetiology of schizophrenia [39]. Recent preclinical and clinical studies reported that changes in glutamate metabolites are associated with excessive inflammatory responses [40]. However, no studies have investigated how alterations in glutamate metabolites relate to neuroinflammation in vivo. Because activated glia increase the expression of the 18-kDa translocator protein (TSPO), immune activation can be quantified by imaging TSPO using positron emission tomography (PET) [41]. Of the 12 studies that have investigated in vivo TSPO expression in psychosis, most studies have reported no significant differences in CHR or schizophrenia relative to HV [42–48], while one study reported a decrease in FEP [49], consistent with a recent meta-analysis [50]. Four studies observed an increase in schizophrenia [51–54]. However, three of the four latter studies [51–53] used the prototypical TSPO radiotracer [11C]PK11195 with known methodological limitations including low brain penetration, high nonspecific binding and high plasma protein binding [55]; the fourth study was limited by their use of distribution volume ratio [54], an alternative outcome measure which is used controversially with TSPO radiotracers [56].
Therefore, in the current study, we used 1H-MRS to investigate hippocampal glutamate and Glx levels in FEP, CHR and HV. We used multimodal imaging with 1H-MRS and [18F]FEPPA PET to explore, for the first time, the association between hippocampal glutamate metabolites and TSPO expression. Moreover, we explored whether alterations in glutamate metabolites were associated with symptom severity and cognitive function. Based on evidence of hyperactive glutamatergic activity in the hippocampus of chronic schizophrenia patients and preclinical models, we expect to observe elevated levels of Glu and Glx in the hippocampus of CHR and FEP, and that the metabolites would be positively associated with TSPO expression.
Materials and methods
Participants
A total of 88 participants took part in this study, including 20 FEP, 33 CHR and 35 HV. Three FEP, 11 CHR and ten HV were also included in our previous [18F]FEPPA studies [44, 45]. To be eligible, CHR subjects had to fulfil the Criteria of Prodromal Syndromes [57]. FEP patients had to have a diagnosis of schizophrenia, schizophreniform disorder, delusional disorder, or psychosis not otherwise specified, as determined with the SCID (Supplementary Table 1) [58]. All but two patients were within 36 months of their initial diagnosis with a primary psychotic disorder. Subjects with bipolar disorder, or concurrent DSM IV axis I disorders that better explained their psychotic symptoms, were excluded. HV were excluded if they had a history of psychiatric illness, first-degree relative with a psychotic disorder, or history of substance abuse or dependence (except nicotine or cannabis). Subjects from all groups were excluded if they met any of the following criteria: pregnancy or current breastfeeding, concomitant or severe medical illness, metal implants precluding magnetic resonance imaging (MRI).
This study was approved by the Research Ethics Board at the Centre for Addiction and Mental Health (CAMH). All participants provided informed, written consent after receiving a description of all study procedures.
Clinical assessments
In CHR, symptoms were assessed with the Structured Interview for Psychosis-risk Syndromes (SIPS), which includes the Scale of Psychosis Risk Symptoms (SOPS) [57]. In FEP, symptom severity was assessed with the Positive and Negative Syndrome Scale (PANSS) [59]. Neurocognitive performance in all groups was assessed using the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS); RBANS subscales assess immediate memory, delayed memory, attention, language and visuospatial ability [60]. Assessments are described in detail in the supplementary materials.
PET acquisition and analysis
Details of PET data acquisition are described in detail elsewhere [44, 46]. Proton density-weighted brain MRI scans were obtained using a 3 T MR-750 scanner (General Electric Medical Systems). All [18F]FEPPA PET scans were acquired using a high-resolution neuro-PET camera system (HRRT, Siemens Molecular Imaging, Knoxville, TN, USA) for 125 min following an intravenous bolus injection of 188.15 ± 9.5 (mean ± SD) MBq of [18F]FEPPA. Arterial blood samples were collected using an automatic blood sampling system (Model PBS-101, Veenstra Instruments, Joure, Netherlands) for the first 22.5 min after the radiotracer injection. Samples were collected at a rate of 2.5 ml/min and manually at −5, 2.5, 7, 12, 15, 20, 30, 45, 60, 90 and 120 min to measure radioactivity in blood and determine the relative proportion of radiolabeled metabolites. Dispersion- and metabolite-corrected plasma input function was generated as previously described [61].
A validated in-house image analysis pipeline was used for processing images and extracting time activity-curves [62]. The hippocampus was delineated using Proton Density (PD) MRI [62]. Kinetic parameters of [18F]FEPPA were derived from the time-activity curves using the two-tissue compartment model (2TCM) and plasma input function to obtain the total distribution volume (VT), which has been validated for [18F]FEPPA quantification [61].
Subjects were classified as high-, mixed- or low-affinity binders based on the rs6971 polymorphism of the TSPO gene, which has been shown to affect [18F]FEPPA binding [63, 64]. Because the low-affinity binder genotype does not allow for accurate [18F]FEPPA PET quantification, subjects with this genotype were excluded from the [18F]FEPPA analyses. Genotyping details are described in more detail elsewhere [63, 64].
1H-MRS acquisition and analysis
1H-MRS scans were performed at the CAMH Research Imaging Centre (Toronto, Canada) using a 3 T General Electric Discovery MR750 scanner (Milwaukee, WI, USA) equipped with an 8-channel head coil. To minimise head motion, each subject was positioned at the centre of the head coil with tape strapped across the forehead and soft restraint padding around the head. T1-weighted fast spoiled gradient-echo 3-dimensional sagittal acquisition scans were acquired for each participant (FSPGR sequence, TE = 3.0 ms, TR = 6.7 ms, TI = 650 ms, flip angle = 8°, FOV = 28 cm, acquisition matrix 256 × 256 matrix, slice thickness = 0.9 mm).
Single voxel 1H-MRS spectra were obtained using the standard GE Proton Brain Examination (PROBE) sequence with point-resolved spectroscopy (PRESS sequence, TE = 35 ms, TR = 2000 ms, number of excitations = 8, bandwidth = 5000 Hz, 4096 data points used, 256 water-suppressed, and 16 water-unsuppressed averages). The voxel was oriented along the long axis of the head of the left hippocampus (30 × 14 × 10 mm; Fig. 1). The signal over the voxel was shimmed to achieve a linewidth of 12 Hz or less, measured from the unsuppressed water signal in the voxel.
Fig. 1.

a Voxel placement in the left hippocampus (30 × 14 × 10 mm3). b Typical spectrum of the left hippocampus LCModel output
1H-MRS data processing and analysis
The water-suppressed spectra were analysed with LCModel version 6.3-0E [65], using a standard basis set of metabolites, lipids and macromolecules listed in the supplementary material. Spectra were normalised to the unsuppressed water signal to quantify neurometabolites, which are expressed in institutional units. Metabolites with %SD values greater than 15% were excluded from analyses (Supplementary Table 2; [66]). Spectra with signal-to-noise ratios (SNR) below 10 or full-width-at-half-maximum (FWHM) above 0.10 ppm were excluded (Supplementary Table 3).
1H-MRS voxel tissue heterogeneity
T1-weighted MRI scans used for voxel localisation were segmented into grey matter (GM), white matter (WM) and cerebrospinal fluid (CSF) using FSL5.0 (FAST; FMRIB Analysis Group, Oxford University, UK). Voxel location, orientation and size information obtained from spectra file headers were used to generate binary masks in the same matrix as the T1-image (Gannet 2.1) [67]. Binary voxel masks were applied to the segmented T1 image in order to determine the percentage of GM, WM and CSF content within the 1H-MRS voxel [68]. CSF-corrected metabolite concentrations (institutional units) were calculated as follows: Ccorrected = Cuncorrected/(1-CSF), where CSF is the CSF fraction within the 1H-MRS voxel, and C is the metabolite concentration.
Statistical analysis
Analyses were performed using SPSS (version 22.0; IBM Corporation, Armonk, NY, USA), with p ≤ 0.05 two-tailed test considered significant. Demographic measures were examined for any group differences using analysis of variance (ANOVA; continuous variables) or chi-square tests (categorical variables). Glutamate metabolites between groups were compared using ANOVA, and Bonferroni correction was used to correct for multiple comparisons. Pearson correlations were used to test for associations between glutamate metabolites and clinical and cognitive measures (RBANS total score and sub-scales). Partial correlations controlling for TSPO genotype were used to test for associations between glutamate metabolites and [18F]FEPPA VT.
Results
1H-MRS analysis
Across all subjects, the mean (±SD) SNR and FWHM were 14.2 ± 2.4 p.p.m. and 0.060 ± 0.016 p.p.m., respectively. The SNR and FWHM did not differ among groups (Supplementary Table 3). With mean ( ± SD) CRLB values of 7.1 ± 1.1% and 6.2 ± 1.0% for Glu and Glx, respectively, the CRLB values did not differ across groups. Moreover, voxel tissue composition (GM, WM or CSF) did not differ among groups (Supplementary Table 4). Sixteen participants were excluded due to either 1H-MRS quality control criteria or other technical reasons (Supplementary Table 3).
Participant demographics
Demographics of the participants are presented in Table 1. Of the 16 FEP subjects, 13 were antipsychotic-naïve, and three were taking quetiapine (two with 100 mg and one with 400 mg, equivalent to 66 mg and 337 mg of chlorpromazine [69], respectively). CHR subjects were antipsychotic-naïve, with the exception of six subjects taking antipsychotic medications at low daily doses ( < 75 mg chlorpromazine equivalent: one with 20 mg quetiapine, one with 0.5 mg risperidone, two with 1 mg, and two with 2 mg aripiprazole).
Table 1.
Demographic characteristics of participants
| Demographics | HV (n = 31) | CHR (n = 25) | FEP (n = 16) | |
|---|---|---|---|---|
| Mean age ± SD (years) | 22.2 ± 3.1 | 21.0 ± 2.1 | 24.5 ± 5.8 | F(2,69) = 4.69, p = 0.012 |
| Sex (male/female) | 13/18 | 12/13 | 15/1 | X2(2,72) = 12.36, p = 0.02 |
| Cannabis use | 13 | 2 | 4 | |
| Antipsychotic use | 0 | 6 | 3 | |
| Antidepressant usea | 0 | 6 | 5 | |
| Benzodiazepine use | 0 | 3 | 1 | |
| Duration of illness (months) | – | – | 19.6 ± 26.3 | |
| RBANS total | – | 90.3 ± 13.7 | 84.5 ± 16.3 | |
| PANSS positive | – | – | 16.5 ± 4.7 | |
| PANSS negative | – | – | 12.4 ± 4.8 | |
| SOPS positive | – | 11.4 ± 3.5 | – | |
| SOPS negativeb | – | 11.2 ± 5.5 | – |
RBANS repeatable battery for the assessment of neuropsychological status, SOPS scale of psychosis-risk symptoms, PANSS positive and negative syndrome scale
aThe antidepressants were selective serotonin reuptake inhibitors (SSRI), selective norepinephrine reuptake inhibitors (SNRI) and bupropion
bSOPS negative score was unavailable for one CHR
Glutamate metabolite levels in FEP, CHR and HV
We found a significant effect of group on Glx (F(2, 69) = 6.39, p = 0.003). The Bonferroni-corrected post-hoc analysis revealed that CHR had significantly lower Glx levels than both HV (p = 0.003) and FEP (p = 0.050; Fig. 2). We also found a trend toward an effect of group on glutamate (F(2,69) = 3.03, p = 0.055). Excluding subjects using anti-psychotic medications did not change the outcome of the multivariate analysis (Glu: F(2,60) = 1.76, p = 0.18; Glx: F(2,60) = 4.56, p = 0.014; Supplementary Table 5). Exclusion of HV that screened positive for cannabis did not produce meaningful changes to the results (Supplementary Fig. 1). The outcome of the multivariate analysis was not altered by including age, sex or tobacco use as covariates (Supplementary Table 6).
Fig. 2.
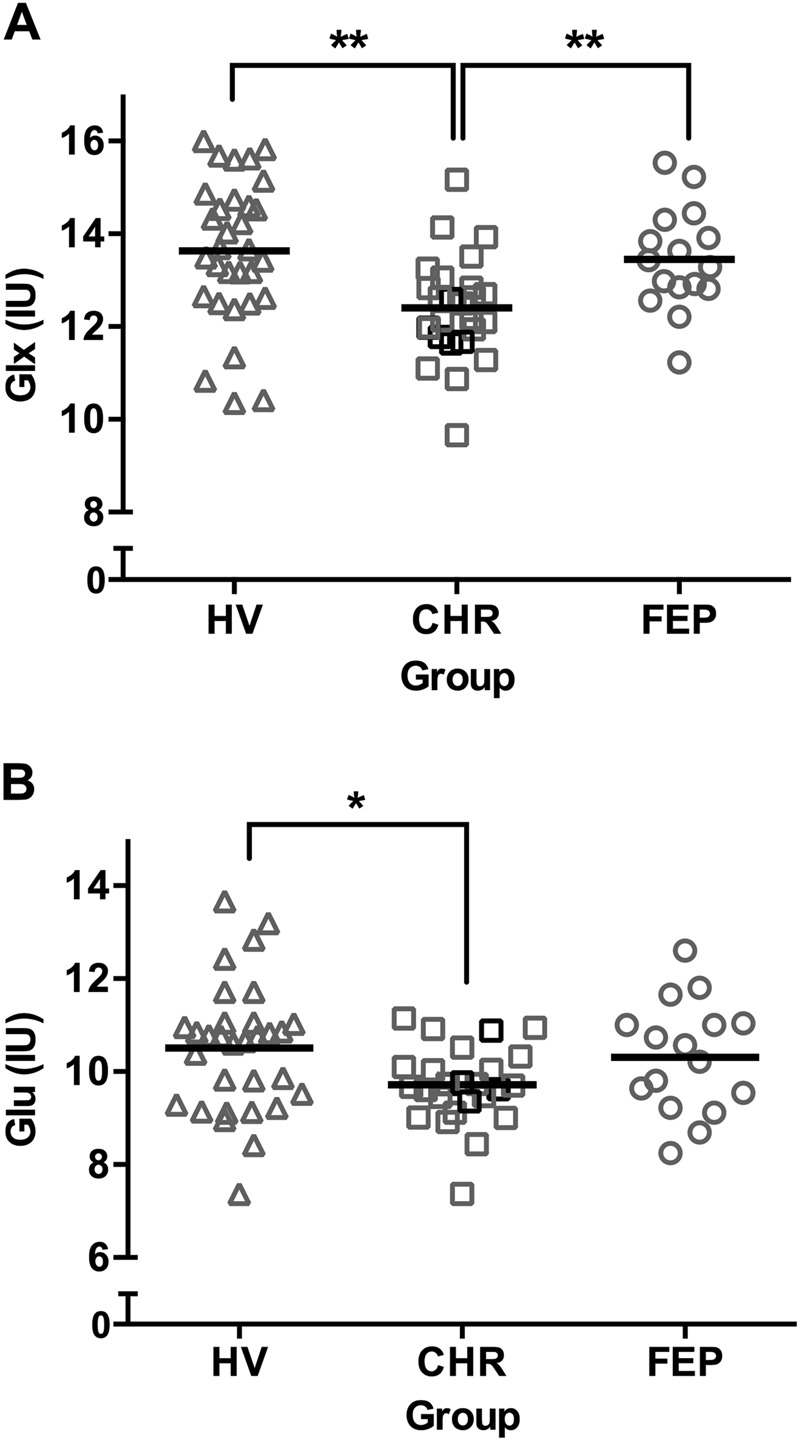
Scatter plots of a Glx (Glutamate + Glutamine) and b: Glu (Glutamate) in institutional units (IU) in the hippocampus of healthy volunteers (HV), clinical high risk (CHR) and first episode psychosis (FEP). CHR subjects that converted to psychosis after the study are shown as black squares. Group means are indicated by black bars. **Significance (p ≤ 0.05), *trend-level significance (0.05 < p < 0.1)
In our sample, four CHR subjects (16% of the sample) subsequently converted to psychosis after the study, and while small numbers preclude investigating associations with conversion to psychosis, we did not observe differences in Glx (F(1, 23) = 0.82, p = 0.37) or glutamate (F(1, 23) = 0.17, p = 0.68) between CHR converters (n = 4) and non-converters (n = 21).
Although not the focus of the current investigation, other standard 1H-MRS metabolites did not differ between groups (Supplementary Table 7).
Correlations between glutamate metabolites and [18F]FEPPA VT
Five subjects were excluded based on TSPO genotyping, because low-affinity binders cannot be quantified with [18F]FEPPA. Two subjects were excluded due to technical issues affecting [18F]FEPPA quantification, and nine did not complete a [18F]FEPPA scan. [18F]FEPPA total distribution volume (VT) did not differ significantly across groups (F(2, 52) = 2.1, p = 0.13). When all groups were combined (n = 56), we found no associations between left hippocampal [18F]FEPPA VT and glutamate metabolites (Glx: r(53) = 0.017, p = 0.90; Glu: r(53) = 0.061, p = 0.66), controlling for TSPO genotype. In FEP, we found a trend toward significance for the association between hippocampal [18F]FEPPA VT and Glu (r(6) = −0.64, p = 0.086), but not with Glx (r(6) = −0.22, p = 0.6). These correlations were not significant in CHR or HV.
Correlations between glutamate metabolites, symptom severity and cognitive function
In the sample as a whole (n = 70; RBANS scores were not available for two participants), cognitive function as measured by RBANS total score was not associated with glutamate metabolites (Glx: r(68) = −0.084, p = 0.49; Glu: r(68) = −0.052, p = 0.67). In CHR, RBANS visuospatial ability was significantly associated with Glx (r(23) = −0.48, p = 0.016; Supplementary Fig. 2). In FEP, PANSS positive and negative symptoms were not associated with glutamate metabolites. In CHR, SOPS positive and negative symptoms did not correlate with glutamate metabolites.
As correlations were exploratory, they were not corrected for multiple comparisons.
Discussion
Summary of results
In summary, the CHR group had significantly lower hippocampal Glx levels than both HV and FEP. Our results therefore suggest that hippocampal glutamate metabolites are altered in CHR. We also observed, in our exploratory correlations, an association in CHR between hippocampal Glx and RBANS visuospatial ability. We did not observe any significant associations between glutamate metabolites and [18F]FEPPA VT, suggesting that TSPO expression may not be related to changes in hippocampal glutamate metabolites, particularly in CHR.
Glutamate metabolite levels in FEP, CHR and HV
We found significant differences in hippocampal glutamate metabolites between CHR and HV. These findings are consistent with two slightly smaller studies, which reported trends toward lower glutamate levels in the left hippocampus of 24 CHR and 17 HV [20], and 22 CHR and 14 HV [22]. Three other studies investigated hippocampal glutamatergic metabolites and found no group differences [16, 21, 23]. The discrepancy in findings may be due to differences in methodology and quality control (e.g., CRLB limit 15 vs. 35% in [16]). Another discrepancy may pertain to the clinical populations. For example, one study investigated first-degree relatives of schizophrenia patients (not CHR), and used metabolic ratios instead of absolute metabolite concentrations [23].
Consistent with previous studies, we found no group differences in hippocampal glutamatergic metabolites between FEP and HV [24, 26–28, 70]. Notably, with the exception of our current study and one previous study [28], all other studies were conducted at low field strengths (1.5 T-2 T). Further strengths of the current study include a larger sample size than the only other ≥3 T study in FEP, stricter quality control criteria (CRLB < 15 vs. 35% in ref. [28]), and CSF corrections.
No previous studies have directly compared hippocampal glutamate metabolites between CHR and FEP. As the first study to compare the two groups, we report that hippocampal Glx levels were significantly lower in CHR relative to FEP.
Our findings of lower glutamate metabolites in CHR relative to HV and FEP suggest abnormal hippocampal neurochemistry early in the illness trajectory. Contrary to the observation of hippocampal hypermetabolism in high risk subjects and FEP patients [17], available 1H-MRS evidence does not provide clear support of elevated tissue glutamate metabolites in the hippocampus of individuals with CHR or FEP [8]. Indeed, studies are inconsistent, with glutamate levels varying depending on the subpopulation and methodology [9]. Our findings of lower glutamate metabolites (significantly lower Glx and trending toward lower Glu) in CHR relative to HV is in agreement with a recent 7 T glutamate imaging study, which observed significantly lower glutamate levels across cortical and subcortical regions in CHR relative to HV [71].
Correlations between glutamate metabolites and cognition and symptoms
Few studies have investigated the relation between hippocampal glutamate metabolites and cognitive dysfunction, with most studies reporting no relation [9]. However, a study comparing recurrent and first-episode psychotic patients found that increased hippocampal glutamate was associated with poorer cognitive function, as measured by the Wisconsin Card Sorting Test [72]. The association may be related to hippocampal hypermetabolism leading to atrophy and perhaps cognitive dysfunction [17]. The NMDA receptor, which exists in high density in the hippocampus, and particularly in the cornu ammonus 1 (CA1) region, is highly sensitive to excitotoxicity [73, 74]; as a result, CA1 cells are highly vulnerable to NMDA-induced apoptosis triggered by excess glutamate. Notably, in the present study, CHR participants with the highest Glx levels exhibited the poorest visuospatial performance. This preliminary result is consistent with the role of the hippocampus in tasks involving visuospatial function [75]. However, because this association is exploratory, the observations need to be replicated by studies designed and powered to test the role of hippocampal glutamate metabolites in cognitive function.
We found no association between hippocampal glutamate metabolites and clinical symptom severity in FEP or CHR. Although few 1H-MRS studies have explored these associations in FEP, our preliminary results are consistent with a previous study in FEP (n = 9) [27], which found no significant correlations between hippocampal glutamate levels and negative clinical symptoms. To our knowledge, no studies have investigated associations between in vivo hippocampal glutamate levels and prodromal symptoms.
Correlations between glutamate metabolites and [18F]FEPPA VT
We report the first preliminary investigation of the relation between hippocampal glutamate metabolites and TSPO expression in early psychosis. Glutamate metabolites did not correlate with hippocampal [18F]FEPPA binding, suggesting that TSPO expression and glutamate metabolites were not associated in our cohorts.
Limitations
We acknowledge several limitations inherent to 1H-MRS and PET studies. First, 1H-MRS measures total tissue content of metabolites, and is not specific to synaptic content. Although glutamine may be contributing to the glutamate 1H-MRS signal due to the difficulty in discriminating between their partially overlapping spectra [76], it is generally accepted that glutamate can be well quantified at 3 T [30]. Secondly, HV that used cannabis were included in this study to increase the sample size. Nevertheless, glutamate metabolite levels did not differ between users and non-users (Supplementary Table 8), and the conclusions of the main analysis do not change after controlling for cannabis use (Supplementary Table 9) or removing cannabis users (Supplementary Fig. 1); although these measures reduce power, the direction of the effect remains similar. Thirdly, the FEP group differed significantly from HV and CHR groups with respect to its smaller sample size, older mean age (~two years difference) and greater number of males. The outcome of the analysis did not change after including age or sex as covariates (Supplementary Table 6), and our results in FEP are consistent with previously reported larger studies in this population. The trend level association between hippocampal [18F]FEPPA binding and Glx in FEP remains to be confirmed in larger studies. Finally, although [18F]FEPPA binding is mainly attributed to microglial activation, TSPO is also expressed in astrocytes and vascular endothelial cells [77].
The importance of understanding the status of glutamate signalling before psychosis onset is suggested by evidence that in rodents, elevating synaptic glutamate release recapitulated the hypermetabolism and atrophy observed in the NMDA-receptor rich hippocampal CA1 region in prodromal psychosis [73, 74]. Beyond glutamate metabolites alone, a role for excitatory-inhibitory balance in psychosis is increasingly supported by evidence from clinical and pre-clinical studies [78]. Future studies measuring both glutamate and GABA in the same individuals, as previously done in schizophrenia and CHR in other brain regions [79–81], may lead to a better understanding of the hippocampal dysfunction observed in psychosis. The present study is the first to test differences in glutamate metabolite levels between HV, CHR and FEP within a single cohort and thus serves as a starting point for future investigations in the hippocampus, as previously tested in the striatum [68].
Conclusion
Multiple factors have been implicated in the aetiology of schizophrenia, supporting the need for multimodal imaging studies. The current study suggests that glutamate metabolites differ across groups in the psychosis spectrum. No associations were found between glutamate metabolites and TSPO expression. Future studies should investigate how glutamate metabolites are related to cognitive function, hippocampal volume and other neurochemicals.
Electronic supplementary material
Acknowledgements
The authors would like to thank the excellent staff of the CAMH Research Imaging Centre and the FYPP clinic. This research was supported by an operating grant from Canadian Institutes of Health Research (CIHR) and CAMH Foundation, as well as the National Institutes of Health (NIH) R01 grant MH100043 to Dr. Mizrahi. Work by the first author was partially supported by the Mach-Gaensslen Foundation of Canada and the CREMS Program at the University of Toronto.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Shima Shakory, Jeremy J. Watts.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41386-018-0163-0).
References
- 1.Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37:4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Arch Gen Psychiatry. 1994;51:199. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 3.Luby ED, Cohen BD, Rosenbaum G, Gottlieb JS, Kelley R. Study of a new schizophrenomimetic drug—sernyl. Arch Neurol Psychiatry. 1959;81:363. doi: 10.1001/archneurpsyc.1959.02340150095011. [DOI] [PubMed] [Google Scholar]
- 4.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–8. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 5.Coyle JT, Tsai G, Goff D. Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann N Y Acad Sci. 2003;1003:318–27. doi: 10.1196/annals.1300.020. [DOI] [PubMed] [Google Scholar]
- 6.Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- 7.Kim JS, Kornhuber HH, Schmid-Burgk W, Holzmüller B. Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci Lett. 1980;20:379–82. doi: 10.1016/0304-3940(80)90178-0. [DOI] [PubMed] [Google Scholar]
- 8.Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK, JW O, et al. Nature of glutamate alterations in schizophrenia. JAMA Psychiatry. 2016;52:998–1007. doi: 10.1001/jamapsychiatry.2016.0442. [DOI] [PubMed] [Google Scholar]
- 9.Merritt K, McGuire P, Egerton A. Relationship between glutamate dysfunction and symptoms and cognitive function in psychosis. Front Psychiatry. 2013;4:151. doi: 10.3389/fpsyt.2013.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci. 2011;32:507–13. doi: 10.1016/j.tips.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Law AJ, Deakin JF. Asymmetrical reductions of hippocampal NMDAR1 glutamate receptor mRNA in the psychoses. Neuroreport. 2001;12:2971–4. doi: 10.1097/00001756-200109170-00043. [DOI] [PubMed] [Google Scholar]
- 12.Stan AD, Ghose S, Zhao C, Hulsey K, Mihalakos P, Yanagi M, et al. Magnetic resonance spectroscopy and tissue protein concentrations together suggest lower glutamate signaling in dentate gyrus in schizophrenia. Mol Psychiatry. 2015;20:433–9. doi: 10.1038/mp.2014.54. [DOI] [PubMed] [Google Scholar]
- 13.Pilowsky LS, Bressan RA, Stone JM, Erlandsson K, Mulligan RS, Krystal JH, et al. First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol Psychiatry. 2006;11:118–9. doi: 10.1038/sj.mp.4001751. [DOI] [PubMed] [Google Scholar]
- 14.Heckers S, Konradi C. Hippocampal pathology in schizophrenia. Curr Top Behav Neurosci. 2010;4:529–53. doi: 10.1007/7854_2010_43. [DOI] [PubMed] [Google Scholar]
- 15.Haijma SV, Van Haren N, Cahn W, Koolschijn PCMP, Hulshoff Pol HE, Kahn RS. Brain volumes in schizophrenia: a meta-analysis in over 18,000 subjects. Schizophr Bull. 2013;39:1129–38. doi: 10.1093/schbul/sbs118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wood SJ, Kennedy D, Phillips LJ, Seal ML, Yücel M, Nelson B, et al. Hippocampal pathology in individuals at ultra-high risk for psychosis: a multi-modal magnetic resonance study. Neuroimage. 2010;52:62–8. doi: 10.1016/j.neuroimage.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 17.Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78:81–93. doi: 10.1016/j.neuron.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, et al. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66:938–46. doi: 10.1001/archgenpsychiatry.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allaïli N, Valabrègue R, Auerbach EJ, Guillemot V, Yahia-Cherif L, Bardinet E, et al. Single-voxel (1)H spectroscopy in the human hippocampus at 3 T using the LASER sequence: characterization of neurochemical profile and reproducibility. NMR Biomed. 2015;28:1209–17. doi: 10.1002/nbm.3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fusar-Poli P, Stone JM, Broome MR, Valli I, Mechelli A, McLean MA, et al. Thalamic glutamate levels as a predictor of cortical response during executive functioning in subjects at high risk for psychosis. Arch Gen Psychiatry. 2011;68:881. doi: 10.1001/archgenpsychiatry.2011.46. [DOI] [PubMed] [Google Scholar]
- 21.Stone JM, Day F, Tsagaraki H, Valli I, McLean MA, Lythgoe DJ, et al. Glutamate dysfunction in people with prodromal symptoms of psychosis: relationship to gray matter volume. Biol Psychiatry. 2009;66:533–9. doi: 10.1016/j.biopsych.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 22.Valli I, Stone J, Mechelli A, Bhattacharyya S, Raffin M, Allen P, et al. Altered medial temporal activation related to local glutamate levels in subjects with prodromal signs of psychosis. Biol Psychiatry. 2011;69:97–99. [DOI] [PubMed]
- 23.Capizzano AA, Nicoll Toscano JL, Ho BC. Magnetic resonance spectroscopy of limbic structures displays metabolite differences in young unaffected relatives of schizophrenia probands. Schizophr Res. 2011;131:4–10. doi: 10.1016/j.schres.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galińska B, Szulc A, Tarasów E, Kubas B, Dzienis W, Czernikiewicz A, et al. Duration of untreated psychosis and proton magnetic resonance spectroscopy (1H-MRS) findings in first-episode schizophrenia. Med Sci Monit. 2009;15:CR82–8. [PubMed] [Google Scholar]
- 25.Hasan A, Wobrock T, Falkai P, Schneider-Axmann T, Guse B, Backens M, et al. Hippocampal integrity and neurocognition in first-episode schizophrenia: a multidimensional study. World J Biol Psychiatry. 2014;15:188–99. doi: 10.3109/15622975.2011.620002. [DOI] [PubMed] [Google Scholar]
- 26.Bartha R, Al-Semaan YM, Williamson PC, Drost DJ, Malla AK, Carr TJ, et al. A short echo proton magnetic resonance spectroscopy study of the left mesial-temporal lobe in first-onset schizophrenic patients. Biol Psychiatry. 1999;45:1403–11. doi: 10.1016/S0006-3223(99)00007-4. [DOI] [PubMed] [Google Scholar]
- 27.Olbrich HM, Valerius G, Rüsch N, Büchert M, Thiel T, Hennig J, et al. Frontolimbic glutamate alterations in first episode schizophrenia: Evidence from a magnetic resonance spectroscopy study. World J Biol Psychiatry. 2008;9:59–63. doi: 10.1080/15622970701227811. [DOI] [PubMed] [Google Scholar]
- 28.Wood S, Berger G, Wellard R, Proffit T, Mcconchie M, Velakoulis D, et al. A 1H-MRS investigation of the medial temporal lobe in antipsychotic-naïve and early-treated first episode psychosis. Schizophr Res. 2008;102:163–70. doi: 10.1016/j.schres.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 29.Szulc A, Galinska B, Czernikiewicz A, Tarasow E, Kubas B, Dzienis W, et al. Glutamatergic system dysfunction in schizophrenia A proton magnetic resonance spectroscopy (1H MRS) study. Pol Prz Radiol. 2004;69:33–6. [Google Scholar]
- 30.Snyder J, Wilman A. Field strength dependence of PRESS timings for simultaneous detection of glutamate and glutamine from 1.5 to 7T. J Magn Reson. 2010;203:66–72. doi: 10.1016/j.jmr.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Hutcheson NL, Reid MA, White DM, Kraguljac NV, Avsar KB, Bolding MS, et al. Multimodal analysis of the hippocampus in schizophrenia using proton magnetic resonance spectroscopy and functional magnetic resonance imaging. Schizophr Res. 2012;140:136–42. doi: 10.1016/j.schres.2012.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kraguljac NV, Reid MA, White DM, den Hollander J, Lahti AC. Regional decoupling of N-acetyl-aspartate and glutamate in schizophrenia. Neuropsychopharmacology. 2012;37:2635–42. doi: 10.1038/npp.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lutkenhoff ES, Erp TG, van, Thomas MA, Therman S, Manninen M, Huttunen MO, et al. Proton MRS in twin pairs discordant for schizophrenia. Mol Psychiatry. 2010;15:308–18. doi: 10.1038/mp.2008.87. [DOI] [PubMed] [Google Scholar]
- 34.Chang L, Friedman J, Ernst T, Zhong K, Tsopelas ND, Davis K. Brain metabolite abnormalities in the white matter of elderly schizophrenic subjects: implication for glial dysfunction. Biol Psychiatry. 2007;62:1396–404. doi: 10.1016/j.biopsych.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Elst LT, Valerius G, Büchert M, Thiel T, Rüsch N, Bubl E, et al. Increased prefrontal and hippocampal glutamate concentration in schizophrenia: evidence from a magnetic resonance spectroscopy study. Biol Psychiatry. 2005;58:724–30. doi: 10.1016/j.biopsych.2005.04.041. [DOI] [PubMed] [Google Scholar]
- 36.Singh S, Khushu S, Kumar P, Goyal S, Bhatia T, Deshpande SN. Evidence for regional hippocampal damage in patients with schizophrenia. Neuroradiology. 2018;60:199–205. doi: 10.1007/s00234-017-1954-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kraguljac NV, White DM, Reid MA, Lahti AC, PF R, LW C. Increased hippocampal glutamate and volumetric deficits in unmedicated patients with schizophrenia. JAMA Psychiatry. 2013;70:1294. doi: 10.1001/jamapsychiatry.2013.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monji A, Kato T, Kanba S. Cytokines and schizophrenia: microglia hypothesis of schizophrenia. Psychiatry Clin Neurosci. 2009;63:257–65. doi: 10.1111/j.1440-1819.2009.01945.x. [DOI] [PubMed] [Google Scholar]
- 39.Müller N, Weidinger E, Leitner B, Schwarz MJ. The role of inflammation in schizophrenia. Front Neurosci. 2015;9:372. doi: 10.3389/fnins.2015.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haroon E, Miller AH, Sanacora G. Inflammation, glutamate, and glia: a trio of trouble in mood disorders. Neuropsychopharmacology. 2017;42:193–215. doi: 10.1038/npp.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venneti S, Wiley CA, Kofler J. Imaging microglial activation during neuroinflammation and Alzheimer’s disease. J Neuroimmune Pharmacol. 2009;4:227–43. doi: 10.1007/s11481-008-9142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coughlin JM, Wang Y, Ambinder EB, Ward RE, Minn I, Vranesic M, et al. In vivo markers of inflammatory response in recent-onset schizophrenia: a combined study using [11C]DPA-713 PET and analysis of CSF and plasma. Transl Psychiatry. 2016;6:e777. doi: 10.1038/tp.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doef TF, van der, Witte LD, de, Sutterland AL, Jobse E, Yaqub M, Boellaard R, et al. In vivo (R)-[(11)C]PK11195 PET imaging of 18kDa translocator protein in recent onset psychosis. NPJ Schizophr. 2016;2:16031. doi: 10.1038/npjschz.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hafizi S, Silva T Da, Gerritsen C, Kiang M, Bagby RM, Prce I, et al. Imaging microglial activation in individuals at clinical high risk for psychosis: an in vivo PET study with [18F]FEPPA. Neuropsychopharmacology. 2017a. 10.1038/npp.2017.111. [DOI] [PMC free article] [PubMed]
- 45.Hafizi S, Tseng HH, Rao N, Selvanathan T, Kenk M, Bazinet RP, et al. Imaging microglial activation in untreated first-episode psychosis: a PET study with [18 F]FEPPA. Am J Psychiatry. 2017;174:118–24. doi: 10.1176/appi.ajp.2016.16020171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kenk M, Selvanathan T, Rao N, Suridjan I, Rusjan P, Remington G, et al. Imaging neuroinflammation in gray and white matter in schizophrenia: an in-vivo PET study with [18F]-FEPPA. Schizophr Bull. 2015;41:85–93. doi: 10.1093/schbul/sbu157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Biase MA, Zalesky A, O’keefe G, Laskaris L, Baune BT, Weickert CS, et al. PET imaging of putative microglial activation in individuals at ultra-high risk for psychosis, recently diagnosed and chronically ill with schizophrenia. Transl Psychiatry. 2017;7:e1225. doi: 10.1038/tp.2017.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takano A, Arakawa R, Ito H, Tateno A, Takahashi H, Matsumoto R, et al. Peripheral benzodiazepine receptors in patients with chronic schizophrenia: a PET study with [11C]DAA1106. Int J Neuropsychopharmacol. 2010;13:943–50. doi: 10.1017/S1461145710000313. [DOI] [PubMed] [Google Scholar]
- 49.Collste K, Plavén-Sigray P, Fatouros-Bergman H, Victorsson P, Schain M, Forsberg A, et al. Lower levels of the glial cell marker TSPO in drug-naive first-episode psychosis patients as measured using PET and [11C]PBR28. Mol Psychiatry. 2017;22:850–6. doi: 10.1038/mp.2016.247. [DOI] [PubMed] [Google Scholar]
- 50.Plavén-Sigray P, Matheson GJ, Collste K, Ashok AH, Coughlin JM, Howes OD et al. Positron emission tomography studies of the glial cell marker translocator protein in patients with psychosis: a meta-analysis using individual participant data. Biol Psychiatry. 2018. 10.1016/j.biopsych.2018.02.1171. [DOI] [PMC free article] [PubMed]
- 51.Doorduin J, Vries EFJ, de, Willemsen ATM, Groot JC, de, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med. 2009;50:1801–7. doi: 10.2967/jnumed.109.066647. [DOI] [PubMed] [Google Scholar]
- 52.Holmes SE, Hinz R, Drake RJ, Gregory CJ, Conen S, Matthews JC, et al. In vivo imaging of brain microglial activity in antipsychotic-free and medicated schizophrenia: a [11C](R)-PK11195 positron emission tomography study. Mol Psychiatry. 2016;21:1672–9. doi: 10.1038/mp.2016.180. [DOI] [PubMed] [Google Scholar]
- 53.van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64:820–2. doi: 10.1016/j.biopsych.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 54.Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR, et al. Microglial activity in people at ultra high risk of psychosis and in schizophrenia: an [11C]PBR28 PET brain imaging study. Am J Psychiatry. 2016;173:44–52. doi: 10.1176/appi.ajp.2015.14101358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson AA, Garcia A, Parkes J, McCormick P, Stephenson KA, Houle S, et al. Radiosynthesis and initial evaluation of [18F]-FEPPA for PET imaging of peripheral benzodiazepine receptors. Nucl Med Biol. 2008;35:305–14. doi: 10.1016/j.nucmedbio.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 56.Narendran R, Frankle WG. Comment on analyses and conclusions of “microglial activity in people at ultra high risk of psychosis and in schizophrenia: an [11 C]PBR28 PET brain imaging study. Am J Psychiatry. 2016;173:536–7. doi: 10.1176/appi.ajp.2016.15111417. [DOI] [PubMed] [Google Scholar]
- 57.Miller TJ, McGlashan TH, Rosen JL, Somjee L, Markovich PJ, Stein K, et al. Prospective diagnosis of the initial prodrome for schizophrenia based on the structured interview for prodromal syndromes: preliminary evidence of interrater reliability and predictive validity. Am J Psychiatry. 2002;159:863–5. doi: 10.1176/appi.ajp.159.5.863. [DOI] [PubMed] [Google Scholar]
- 58.First M, Spitzer R, Gibbon M, Williams J. Structured clinical interview for DSM-IV Axis I Disorders: Patient Edition (SCIDI/P) New York: Biometric Research New York State Psychiatric Institute; 1995. [Google Scholar]
- 59.Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13:261–76. doi: 10.1093/schbul/13.2.261. [DOI] [PubMed] [Google Scholar]
- 60.Randolph C, Tierney MC, Mohr E, Chase TN. The repeatable battery for the assessment of neuropsychological status (RBANS): preliminary clinical validity. J Clin Exp Neuropsychol. 1998;20:310–9. doi: 10.1076/jcen.20.3.310.823. [DOI] [PubMed] [Google Scholar]
- 61.Rusjan PM, Wilson AA, Bloomfield PM, Vitcu I, Meyer JH, Houle S, et al. Quantitation of translocator protein binding in human brain with the novel radioligand [18 F]-FEPPA and positron emission tomography. J Cereb Blood Flow Metab. 2011;31:1807–16. doi: 10.1038/jcbfm.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rusjan P, Mamo D, Ginovart N, Hussey D, Vitcu I, Yasuno F, et al. An automated method for the extraction of regional data from PET images. Psychiatry Res Neuroimaging. 2006;147:79–89. doi: 10.1016/j.pscychresns.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 63.Mizrahi R, Rusjan PM, Kennedy J, Pollock B, Mulsant B, Suridjan I, et al. Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [18 F]-FEPPA. J Cereb Blood Flow Metab. 2012;32:968–72. doi: 10.1038/jcbfm.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32:1–5. doi: 10.1038/jcbfm.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Provencher SW. Automatic quantitation of localizedin vivo1H spectra with LCModel. NMR Biomed. 2001;14:260–4. doi: 10.1002/nbm.698. [DOI] [PubMed] [Google Scholar]
- 66.Provencher S (2016). LCModel and LCMgui User’s Manual. http://lcmodel.ca.
- 67.Edden RAE, Puts NAJ, Harris AD, Barker PB, Evans CJ. Gannet: a batch-processing tool for the quantitative analysis of gamma-aminobutyric acid-edited MR spectroscopy spectra. J Magn Reson Imaging. 2014;40:1445–52. doi: 10.1002/jmri.24478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de la Fuente-Sandoval C, León-Ortiz P, Favila R, Stephano S, Mamo D, Ramírez-Bermúdez J, et al. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first-episode psychosis. Neuropsychopharmacology. 2011;36:1781–91. doi: 10.1038/npp.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Andreasen NC, Pressler M, Nopoulos P, Miller D, Ho BC. Antipsychotic dose equivalents and dose-years: a standardized method for comparing exposure to different drugs. Biol Psychiatry. 2010;67:255–62. doi: 10.1016/j.biopsych.2009.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hasan A, Wobrock T, Falkai P, Schneider-Axmann T, Guse B, Backens M, et al. Hippocampal integrity and neurocognition in first-episode schizophrenia: a multidimensional study. World J Biol Psychiatry. 2014;15:188–99. doi: 10.3109/15622975.2011.620002. [DOI] [PubMed] [Google Scholar]
- 71.Roalf DR, Nanga RPR, Rupert PE, Hariharan H, Quarmley M, Calkins ME, et al. Glutamate imaging (GluCEST) reveals lower brain GluCEST contrast in patients on the psychosis spectrum. Mol Psychiatry. 2017;22:1298–305. doi: 10.1038/mp.2016.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rüsch N, Tebartz van Elst L, Valerius G, Büchert M, Thiel T, Ebert D, et al. Neurochemical and structural correlates of executive dysfunction in schizophrenia. Schizophr Res. 2008;99:155–63. doi: 10.1016/j.schres.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 73.Lieberman JA, Girgis RR, Brucato G, Moore H, Provenzano F, Kegeles L, et al. Hippocampal dysfunction in the pathophysiology of schizophrenia: a selective review and hypothesis for early detection and intervention. Mol Psychiatry. 2018. 10.1038/mp.2017.249. [DOI] [PMC free article] [PubMed]
- 74.Butler TR, Self RL, Smith KJ, Sharrett-Field LJ, Berry JN, Littleton JM, et al. Selective vulnerability of hippocampal cornu ammonis 1 pyramidal cells to excitotoxic insult is associated with the expression of polyamine-sensitive N-methyl-D-asparate-type glutamate receptors. Neuroscience. 2010;165:525–34. doi: 10.1016/j.neuroscience.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baddeley A, Jarrold C, Vargha-Khadem F. Working memory and the hippocampus. J Cogn Neurosci. 2011;23:3855–61. doi: 10.1162/jocn_a_00066. [DOI] [PubMed] [Google Scholar]
- 76.Hancu I. Optimized glutamate detection at 3T. J Magn Reson Imaging. 2009;30:1155–62. doi: 10.1002/jmri.21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Notter T, Coughlin JM, Gschwind T, Weber-Stadlbauer U, Wang Y, Kassiou M, et al. Translational evaluation of translocator protein as a marker of neuroinflammation in schizophrenia. Mol Psychiatry. 2017. 10.1038/mp.2016.248. [DOI] [PubMed]
- 78.Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kegeles LS, Mao X, Stanford AD, Girgis R, Ojeil N, Xu X, et al. Elevated prefrontal cortex γ-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 2012;69:449. doi: 10.1001/archgenpsychiatry.2011.1519. [DOI] [PubMed] [Google Scholar]
- 80.de la Fuente-Sandoval C, Reyes-Madrigal F, Mao X, León-Ortiz P, Rodríguez-Mayoral O, Jung-Cook H, et al. Prefrontal and striatal gamma-aminobutyric acid levels and the effect of antipsychotic treatment in first-episode psychosis patients. Biol Psychiatry. 2018;83:475–83. doi: 10.1016/j.biopsych.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.la Fuente-Sandoval C, de, Reyes-Madrigal F, Mao X, León-Ortiz P, Rodríguez-Mayoral O, Solís-Vivanco R, et al. Cortico-striatal GABAergic and glutamatergic dysregulations in subjects at ultra-high risk for psychosis investigated with proton magnetic resonance spectroscopy. Int J Neuropsychopharmacol. 2016;19:pyv105. doi: 10.1093/ijnp/pyv105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.