

Abstract
To date, there are no biomarkers for major depressive disorder (MDD) treatment response in clinical use. Such biomarkers could allow for individualized treatment selection, reducing time spent on ineffective treatments and the burden of MDD. In search of such a biomarker, multisite pretreatment and early-treatment (1 week into treatment) structural magnetic resonance (MR) images were acquired from 184 patients with MDD randomized to an 8-week trial of the selective serotonin reuptake inhibitor (SSRI) sertraline or placebo. This study represents a large, multisite, placebo-controlled effort to examine the association between pretreatment differences or early-treatment changes in cortical thickness and treatment-specific outcomes. For standardization, a novel, robust site harmonization procedure was applied to structural measures in a priori regions (rostral and caudal anterior cingulate, lateral orbitofrontal, rostral middle frontal, and hippocampus), chosen based on previously published reports. Pretreatment cortical thickness or volume did not significantly associate with SSRI response. Thickening of the rostral anterior cingulate cortex in the first week of treatment was associated with better 8-week responses to SSRI (p = 0.010). These findings indicate that frontal lobe structural alterations in the first week of treatment may be associated with long-term treatment efficacy. While these associational findings may help to elucidate the specific neural targets of SSRIs, the predictive accuracy of pretreatment or early-treatment structural alterations in classifying treatment remitters from nonremitters was limited to 63.9%. Therefore, in this large sample of adults with MDD, structural MR imaging measures were not found to be clinically translatable biomarkers of treatment response to SSRI or placebo.
Introduction
Major depressive disorder (MDD) is a leading global cause of disability (World Health Organization) and first-step antidepressant therapy only yields remission in a third of cases [1]. Further, there is typically a delay in onset of the therapeutic effects of selective serotonin reuptake inhibitors (SSRIs) ranging from 2 to 4 weeks [2]. Biomarkers predicting effective treatment courses prospectively, or early in the course of treatment, during this period of delayed therapeutic onset [3] could afford individualized treatment planning, reducing time spent on ineffective treatments, shortening patient suffering, and reducing the overall cost burden of MDD [4].
Cortical thickness (CT) and volume in cortico-limbic circuits have been reported to be reduced in adults with MDD relative to healthy adults [5–19]. Postmortem data also shows reduced glial density and CT in orbitofrontal (OFC), anterior cingulate (ACC) and dorsolateral prefrontal (dlPFC) cortices in adults with MDD [20–23]. There are many hypothesized mechanisms to explain structural deficits in MDD including, but not limited to, stress-induced structural alterations via glucocorticoid overproduction [24] and genetic loading for MDD, such as familial history of mood disorders [25, 26].
Based on the evidenced structural deficits in MDD, pretreatment structural biomarkers for treatment response, such as hippocampal volume [27], have been suggested. However, a replicable, clinically translatable biomarker has yet to emerge, perhaps due to small, heterogeneous samples, disparate treatment courses, and small effect sizes in previous studies [13]. This study overcomes some of these limitations with a large sample size, specific treatment arms, and restriction to two analyzable MDD subtypes. Investigations into structural magnetic resonance imaging (MRI) biomarkers of MDD treatment response have further been limited to trials without placebo-control groups, preventing assessment of biomarker specificity [6, 7, 28–33]. To support causal inferences regarding a treatment's effect on the brain and to control for nonspecific treatment effects, placebo and/or active treatment control arms are critical [34]. Further, if a significant biological response to a medication can be observed early-in-treatment, this may provide additional information above and beyond pretreatment characteristics to aid in prediction. Short-term structural alterations during the period of delayed SSRI effectiveness have yet to be investigated as a means to predict eventual antidepressant treatment efficacy.
The clinical trial Establishing Moderators and Biosignatures of Antidepressant Response in Clinical Care (EMBARC) was designed to address critical gaps in the MDD literature with a large, multisite, randomized, placebo-controlled structure [35]. Unique to EMBARC’s study design is acquisition of both pretreatment and early-treatment (1 week into treatment) structural MRI scans, with the goal of investigating associations with the treatment’s eventual efficacy [35].
Prospective, mixed-treatment studies have found greater hippocampal and dlPFC volumes at baseline in remitters compared to nonremitters [30], and greater hippocampal volume to be associated with better clinical outcomes [31]. In fluoxetine trials, greater pretreatment hippocampal volume was found in female responders compared to nonresponders and was associated with better treatment outcomes across sexes [31], as well as just in males [32]. Greater pretreatment volume and grey matter density in the ACC were also found to be predictive of remission [28] and to be associated with faster improvement [33].
Longitudinal, mixed-treatment studies have found that OFC CT [7] and hippocampal volume [6] increased in patients with MDD over 6–36 months of treatment. Hippocampal volume, rostral middle frontal (RMF, largely overlapping with the dlPFC) CT, and OFC CT have been found to increase longitudinally in remitters and decrease in nonremitters [31]. These studies provide preliminary evidence that hippocampal, OFC, RMF, and ACC morphology may associate with MDD treatment response. However, large sample sizes and placebo-controlled treatment trials are critically needed to validate any of these regions as replicable, reproducible, and treatment-specific biomarkers of MDD treatment response.
Based on these pretreatment and longitudinal neuroimaging studies, the a priori measures of interest: rostral ACC, caudal ACC, OFC, and RMF cortical thickness and volume, and hippocampal volume, were selected in the hopes that with EMBARC’s large, placebo-controlled design, more conclusive evidence of their role in MDD treatment response may emerge. As these regions have been associated with executive functioning, cognition, memory formation, and emotional processing [29], these regions may be specifically implicated in the antidepressant effects of SSRIs. Our primary hypothesis was that individuals with a positive response to SSRI treatment, characterized by changes in a continuous measure of depression severity, would present with thicker/larger cortices pretreatment and would further display cortical thickening/enlargening in the acute phase of treatment, relative to those with a nonresponse to SSRI treatment. We sought to determine if the potential relationship between brain structure and treatment response was differentially detectable across the treatment arms. Lastly, we sought to determine if pretreatment and early-treatment changes in CT and volume could accurately predict eventual treatment response and response trajectories, and if there was an effect of chronic or recurrent MDD subtypes.
Materials and methods
Participants
The rationale and study design for EMBARC has been previously published [35]. EMBARC is a double-blind, randomized, placebo-controlled trial of the SSRI sertraline in patients with MDD. Basic inclusion criteria were being 18–65-years old, meeting criteria for nonpsychotic MDD (DSM-IV TR using the SCID-I/P [36]) and scoring ≥14 on the Quick Inventory of Depressive Symptoms (QIDS-SR; [37]). To reduce heterogeneity, only patients with early onset MDD (before age 30) and chronic (episode duration >2 years) or recurrent MDD (≥2 recurrences) were enrolled, as described [35].
Pretreatment, patients were assessed with the 17-item Hamilton Depression Rating Scale (HDRS-17; [38]) and randomized to placebo or sertraline (200 mg daily), with at least a 21-day medication free period prior to randomization. In this analysis, only patients with pretreatment HDRS-17 scores ≥15 were included to ensure treatment response was only assessed in patients experiencing moderate depressive symptomatology at the time of the pretreatment MRI scan [38]. Trivedi et al. [1] showed that the mean time to SSRI-induced remission was less than 7 weeks. Therefore, the treatment course for this trial was restricted to 8 weeks, wherein participants were readministered the HDRS-17. Remission was defined a priori within EMBARC as a posttreatment HDRS-17 score of ≤7 [35].
MRI acquisition
MRI scans were performed at pretreatment and 1 week into treatment. MRI scanning took place across 5 sites: University of Texas Southwestern Medical Center (TX—Philips Achieva, 8-channel head-coil), University of Michigan (UM—Philips Ingenia, 15-channel), Massachusetts General Hospital (MGH—Siemens TrioTim, 12-channel), Columbia University Medical Center (CU—GE Signa HDx, 8-channel), and Stony Brook Medical Center (SBU—Siemens TrioTim, 12-channel). T1-weighted image acquisition and test–retest reliability of processing have been published [39]. In short, MPRAGE sequences were acquired at TX, UM, MGH, and SBU, while an IR-FSPGR sequence was acquired at CU. Sequence parameters were as follows: TR/TE = 5.9–8.2/2.4–4.6 ms, 8–12° flip angle, 1 mm slice thickness, 4.4–5.5 min acquisition, and 1 mm isotropic voxel dimensions.
Image processing
Raw structural images were passed through Freesurfer 5.3.0 (http://surfer.nmr.mgh.harvard.edu/) to extract CT (rACC, cACC, OFC, RMF) and volume (CT regions + hippocampus) from the Desikan-Killiany atlas [40]. Freesurfer surface models underwent systematic quality control, shown to improve statistical power, at a single site for optimal reliability [39].
Of 677 patients screened, 309 were randomized, and 305 underwent pretreatment MRI [35]. Of the 305, 270 (89%) had approved Freesurfer segmentations. Of the 270 approved, 216 patients completed 8 weeks of treatment and pretreatment and posttreatment HDRS-17 assessments (70% of randomization sample). Of these, 192 also had approved week 1 Freesurfer segmentations (62% of randomization sample). An additional 32 of the 216 pretreatment MRI scans (N = 184 remaining) and 27 of the 192 pretreatment + week 1 scans (N = 165 remaining) were excluded for pretreatment HDRS-17 scores <15.
Statistical analyses: site harmonization
An open-source site harmonization procedure, ComBat, previously validated on CT data from the EMBARC study, was used to remove variability associated with different scanners and sites. To generate site-corrected neuroimaging data, Empirical Bayes estimation linearly models the biological variables and site effects, where an error term accounts for site-specific scaling factors, a property unique to ComBat harmonization [41]. Fortin et al. [41] showed that ComBat corrected for mean site differences, and compared to residuals harmonization and adjusted residuals harmonization, ComBat was the only method to significantly correct for site-specific scaling effects. Age, age2, and sex were entered as biological variables to ComBat, along with raw week 0 and 1 CT and volume data and the site variable, to generate site harmonized CT and volume. Harmonization results were examined with percent differences (| harmonized – unharmonized |/unharmonized*100) and two-tailed, paired t -tests.
Site harmonization yielded average percent differences of 2.08 ± 1.78% and 2.10 ± 1.80% for week 0 and week 1 data, respectively. Importantly, in the unharmonized data, across the 4 sites (SBU scans (the fifth site) were excluded from the following t -tests across sites given that only 2 scans were performed there) and 9 a priori structural measures (volume + CT measurements from the cACC, rACC, RMF, and OFC, as well as hippocampal volume), there were nine cases in the week 0 data and eight cases in the week 1 data with Bonferonni-corrected significant site-wise differences in CT or volume (54 tests, Bonferonni significance threshold: p < 0.00093), indicating a substantial site-by-region effect. However, in the ComBat harmonized data, these site effects were removed, whereby across week 0 and 1 data, there were no significant differences between imaging sites. Therefore, site was not used as a model factor in subsequent testing.
Statistical analyses: a priori cortical thickness testing
To investigate whether pretreatment or early-treatment changes in cortical thickness were associated with SSRI or placebo clinical responses, linear mixed models were fit with the site harmonized pretreatment CT or the percent change in CT from week 0 to 1 (ΔCT = [week 1 – week 0]/week 0) as the dependent variable with percent change in HDRS-17 from week 0 to 8 (ΔHDRS-17 = [week 8 – week 0]/week 0) included as the measure of treatment outcome, along with treatment assignment, age, age2, sex, and region as additional model factors. To determine which regions were driving significant interaction terms, follow-up analyses of these effects were conducted.
Statistical analyses: a priori volumetric testing
In addition to investigating CT, the above linear mixed models were also examined with volume estimates. Site harmonized volume estimates were log transformed to fulfill normality assumptions. (Harmonized cortical thickness was normally distributed.)
Statistical analyses: machine learning classification models
Two methods for predictive modeling were investigated: (1) random forest (RF) and (2) penalized logistic regression (PLR). The harmonized neuroimaging data (pretreatment CT/volume and early-treatment change in CT/volume), psychometric data (pretreatment HDRS-17), and demographic data (age, age2, sex, and treatment assignment) were used to predict remission status. Ten repetitions of five-fold cross-validation were used to determine average model performance. The top 10 predictors determined using variable importance ranking for remission status were selected in the final models. Predictive analyses were performed in R 3.3.1 (R Core Team, 2016. Vienna, Austria; https://www.R-project.org/).
Statistical analyses: exploratory post hoc
Any significant regional post hoc CT or volume effects found were further examined in exploratory analyses to determine if the neuroimaging effects (1) predicted the trajectory of HDRS-17 scores during the 8-week trial and (2) were driven by patients of either the chronic or recurrent MDD subtypes.
Results
Participants
Of the total patient sample used in this analysis (N = 184), 34% of patients remitted (30% placebo and 39% sertraline remission rates). The difference in remission rates across treatments was not significant (p = 0.19). Study randomization resulted in treatment groups with similar ages, sex distributions, years of education, and ages of first MDE onsets. Pretreatment HDRS-17 was not significantly different across treatment or remission groups (Table 1).
Table 1.
Demographic and clinical characteristics for treatment and remission status stratification
| Treatment (n = 87) | Placebo (n = 97) | p-value (stat~testdf) | Remitter (n = 63) | Nonremitter (n = 121) | p-value (stat~ testdf) | |
|---|---|---|---|---|---|---|
| Age (years) | 37.18 ± 13.78 | 37.02 ± 12.92 | p = 0.93 | 34.59 ± 12.23 | 38.40 ± 13.69 | p = 0.06 |
| Sex | 58 female, 29 male | 57 female, 40 male | p = 0.27 | 39 female, 24 male | 76 female, 45 male | p = 0.91 |
| Race* | 15 African American, 4 Asian, 61 White, 7 Other | 15 African American, 8 Asian, 65 White, 8 Other, 1 American Indian/Alaskan | p = 0.83 | 9 African American, 7 Asian, 43 White, 3 Other, 1 American Indian/Alaskan | 21 African American, 5 Asian, 83 White, 12 Other | p= 0.14 |
| Education (years) | 15.11 ± 2.71 | 15.46 ± 2.43 | p = 0.38 | 15.23 ± 2.42 | 15.32 ± 2.65 | p = 0.81 |
| Pretreatment HDRS-17 | 19.95 ± 3.60 | 19.72 ± 3.56 | p = 0.66 | 19.27 ± 3.22 | 20.12 ± 3.72 | p = 0.12 |
| Posttreatment HDRS-17 | 10.98 ± 6.62 | 12.26 ± 7.52 | p = 0.22 | 3.95 ± 2.25 | 15.66 ± 5.23 | – |
| Age of first MDE onset | 15.62 ± 6.00 | 16.67 ± 6.00 | p = 0.24 | 15.55 ± 5.86 | 16.49 ± 6.07 | p = 0.32 |
| Lifetime comorbidity data* | ||||||
| Agoraphobia (no history of panic disorder) | 0% | 2.20% | p = 0.50 | 1.64% | 0.88% | p = 1.00 |
| Anorexia nervosa | 2.30% | 1.06% | p = 0.62 | 3.23% | 0.84% | p = 0.55 |
| Bulimia nervosa | 2.30% | 6.38% | p = 0.28 | 4.84% | 4.20% | p = 1.00 |
| Anxiety disorder due to general medical condition | 0% | 0% | – | 0% | 0% | – |
| Anxiety disorder not otherwise specified | 2.50% | 3.33% | p = 1.00 | 3.45% | 2.68% | p = 1.00 |
| Bipolar disorder (I, II, or other) | 0% | 0% | – | 0% | 0% | – |
| Obsessive compulsive disorder | 4.65% | 2.13% | p = 0.43 | 4.84% | 2.54% | p = 0.66 |
| Panic disorder | 17.24% | 17.58% | p = 1.00 | 17.74% | 17.24% | p = 1.00 |
| Posttraumatic stress disorder | 13.79% | 11.70% | p = 0.82 | 11.29% | 13.45% | p = 0.81 |
| Psychotic symptoms | 0% | 0% | – | 0% | 0% | – |
| Social phobia | 24.14% | 19.15% | p = 0.47 | 22.58% | 21.01% | p = 0.86 |
| Specific phobia | 3.45% | 9.57% | p = 0.14 | 4.84% | 7.56% | p = 0.55 |
| Substance-induced anxiety disorder | 0% | 0% | – | 0% | 0% | – |
CI confidence interval, HDRS-17 17-item Hamilton Depression Rating Scale, rACC rostral anterior cingulate cortex, RMF rostral middle frontal cortex. *Because of the small cell count, Monte Carlo simulated exact Chi-squared test was used to calculate p-values
Across the pretreatment and early-treatment CT and volume models tested, the structural metrics did not significantly associate with nonspecific treatment response (main effect of ΔHDRS-17 or interaction between ΔHDRS-17 and region p-values = 0.14–0.80; full model results for all tests in Supplementary Tables), indicating that the significant effects outlined below are treatment-specific.
A priori cortical thickness testing
Pretreatment CT was significantly differentially associated with clinical response across the treatment arms (p = 0.040; Table 2). Follow-up revealed only a placebo-specific effect of thinner pretreatment RMF cortices associating with better responses to placebo (p = 0.025; Table 2; Fig. 1). The difference in the association between pretreatment RMF and response to either placebo or sertraline had a Cohen’s d effect size of 0.090.
Table 2.
Treatment-specific relationships of cortical morphology and treatment response results summary
| Continuous association model interaction | p-value (stat~testdf) | Post hoc effect | Unstandardized beta coefficient | 95% CI | p-alue (stat~testdf) | |
|---|---|---|---|---|---|---|
| Primary hypothesis testing: sertraline effects | ||||||
| Early-treatment harmonized CT change | ΔHDRS-17 by region by treatment assignment | 0.004 | rACC thickening associated with better clinical response to sertraline | −0.044 | [−0.078, −0.011] | 0.010 |
| Primary hypothesis testing: placebo effects | ||||||
| Pretreatment harmonized CT | ΔHDRS-17 by region by treatment assignment | 0.040 | Thicker pretreatment RMF associated with worse clinical response to placebo | 0.072 | [0.009, 0.134] | 0.025 |
| Early-treatment harmonized CT change | ΔHDRS-17 by region by treatment assignment | 0.004 | RMF thickening associated with better clinical response to placebo | −0.021 | [−0.039, −0.002] | 0.033 |
| Volumetric testing | ||||||
| Early-treatment harmonized volume change | ΔHDRS-17 by region by treatment assignment | 0.046 | RMF volume increase associated with better clinical response to placebo | −0.027 | [−0.050, −0.004] | 0.022 |
CI confidence interval, HDRS-17 17-item Hamilton Depression Rating Scale, rACC rostral anterior cingulate cortex, RMF rostral middle frontal cortex
Fig. 1.

Location of Freesurfer Desikan-Killiany defined rostral middle frontal (LEFT) and rostral anterior cingulate (RIGHT) found to be significant with harmonized morphology data
Across the whole sample, on average, cortical thickness increased in all regions during the first week of treatment with the largest changes seen in the rACC and RMF (percent differences—OFC: 0.05 ± 4.08%, cACC: 0.18 ± 4.87%, RMF: 0.25 ± 3.24%, rACC: 0.31 ± 5.48%). Early-treatment CT alterations were significantly differentially associated with clinical response across the treatment arms (p = 0.004; Table 2). Follow-up revealed that this effect was driven by rACC early-treatment changes in the sertraline group and RMF changes in the placebo group. Early-treatment rACC thickening significantly associated with better clinical response to sertraline (p = 0.010), while early-treatment RMF thickening significantly associated with better clinical response to placebo (p = 0.033) (Table 2; Fig. 1). The difference in the association between rACC and RMF early-treatment changes and response to either sertraline or placebo had effect sizes of d = 0.250 and 0.036, respectively.
A priori volumetric testing
Pretreatment volume did not significantly associate with treatment response differentially across the arms (p = 0.65). However, early-treatment volume alterations were significantly differentially associated with clinical response across the treatment arms (p = 0.046; Table 2). Post hoc analysis revealed that RMF enlargening in the first week of treatment associated with better responses to placebo (p = 0.022; Table 2; Fig. 1). The difference in the association between RMF early-treatment change and response to either placebo or sertraline had an effect size of d = 0.104.
Machine learning classification models
The RF and PLR models yielded similar results. Using the neuroimaging, psychometric, and demographic data, remission status was predicted more accurately with RF than PLR (Accuracy = 63.9 ± 1.7%, AUC = 58.6 ± 2.2%, sensitivity = 22.6 ± 5.0%, specificity = 85.8 ± 2.4% with RF and Accuracy = 60.6 ± 1.8%, AUC = 64.8 ± 2.4%, sensitivity = 12.8 ± 7.3%, specificity = 86.1 ± 5.5% with PLR). The receiver operating characteristic curve for the RF method is shown in Fig. 2.
Fig. 2.
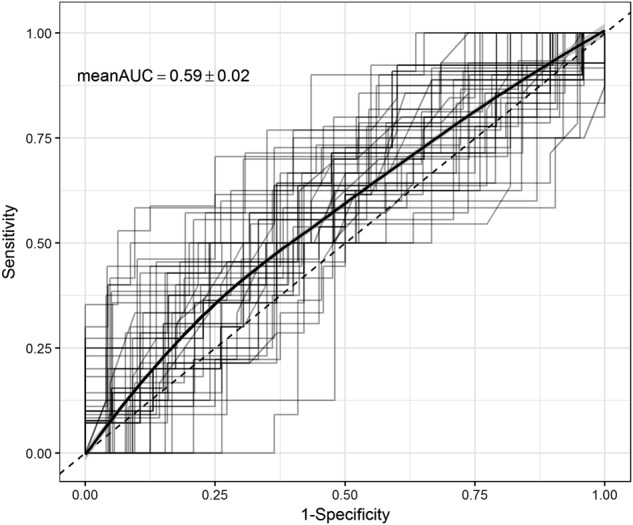
Receiver operating characteristic (ROC) curves for classifying posttreatment remission status generated from 10-repeated 5-fold cross-validation. Averaged ROC curve shown using threshold averaging based on the random forest model using the subset of the 10 most important predictors. Mean and standard deviation for the area under the curve (AUC) shown
Exploratory post hoc
To investigate whether brain structure predicts the trajectory of HDRS-17 throughout the trial, all HDRS-17 scores collected were analyzed (weeks 0–4, 6, and 8). Multivariable linear mixed models were fit with HDRS-17 score trajectory modeled as a linear function of time as the outcome to investigate the interaction between the significant predictors from primary testing (rACC CT change with sertraline, pretreatment RMF with placebo, and RMF CT and volume change with placebo) and treatment week (baseline to week 8). Results showed that the early-treatment change in rACC CT in the sertraline treatment arm significantly predicted the negatively sloped HDRS-17 trajectory over the course of treatment (p = 0.016, β = −5.373, df = 435, t = −2.41), bolstering the a priori, primary finding that rACC early-treatment thickening was associated with better responses to sertraline.
Within the set of n = 165 patients with pretreatment + week 1 scans, 73 exhibited chronic MDD (44.2%), 88 exhibited recurrent MDD (53.3%), and 4 had missing data. Remission rates across the MDD subtypes (chronic = 28.8% and recurrent = 38.6%) did not significantly differ (p = 0.19, df = 1, χ2 = 1.73). To investigate if the significant effects from primary testing were driven by differences between the MDD subtypes, the same linear mixed models were fit as performed in the a priori hypothesis testing section, but with MDD subtype added as a fixed factor. Only the significant effects from above were examined. The effect of thicker pretreatment RMF associating with better placebo response was driven by the chronic MDD subtype (p = 0.01, β = 0.12, df = 169, t = 2.59), whereas the early-treatment change in RMF CT and volume effects were driven by the recurrent MDD subtype (CT: p = 0.02, β = −0.03, df = 150, t = −2.37; volume: p = 0.0008, β = −0.05, df = 150, t = −3.41). Whereas for sertraline, the effect of early-treatment change in rACC CT remained marginally significant in the chronic subtype (p = 0.057, β = -0.05, df = 150, t = −1.92), but was nonsignificant in the recurrent subtype.
Discussion
EMBARC provides a unique opportunity to examine pretreatment and short-term alterations in brain structure occurring within the first week of SSRI and placebo treatment. Cortical thickness alterations in the 1-week interval used in this study have been shown to be detectable via 3T structural MRI, for example, in learning new tasks [42] and following electroconvulsive shock therapy [43]. EMBARC’s double-blind, randomized, placebo-controlled structure provides a basis for understanding whether potential structural treatment response markers are specific to the SSRI, sertraline. While not significant, the remission rate for the SSRI arm in this trial was higher than the placebo arm (39% and 30%, respectively). These rates are marginally higher, but in agreement with, a pooled analysis of double-blind, randomized sertraline trials totaling over 4000 patients with MDD (35% and 24% for sertraline and placebo, respectively) [44]. We report evidence of early-treatment CT and volume alterations that differentially associate with clinical treatment response across placebo and sertraline treatment groups. Specifically, we found that bilaterally, rostral ACC CT alterations in the first week of treatment associate with the eventual change in symptom severity during a trial of sertraline, while RMF pretreatment CT, and CT and volume alterations in the first week of treatment, associate with the change in symptom severity during the placebo trial.
The magnitude of effect sizes between the treatment arms (placebo and sertraline) in this study are consistent with, and even larger than, structural findings distinguishing over 1700 patients with MDD from over 7000 healthy controls (compare effect sizes equaling −0.10 to −0.14 in two ENIGMA studies [12, 13] to effect sizes ranging from 0.036 to 0.250 in this study). Although consistent, or larger than, previous findings in MDD, it is critical to emphasize that the size of these effects indicate that the structural measures in this study are not likely effective biomarkers for MDD treatment response. Based on the large sample size used to determine these effects, this study is not underpowered and, as such, future work should instead focus on a combination of psychometric and structural and functional neuroimaging data to develop a combinatory biosignature capable of clinical translation.
The accuracy of pretreatment or early-treatment structural metrics to perform individualized classification of treatment remission was limited here to less than 65% across the classification methods tested. With a specificity of 86%, 107 of 109 nonremitters were correctly identified as such. However, with a sensitivity of approximately 13%, only 2 of 56 remitters were correctly identified. Therefore, if pretreatment and early-treatment structural data were presently translated to the clinic as a biomarker for treatment response, 97.6% of patients would be expected to not remit. Clearly, a treatment biomarker that classifies 2% of patients as remitters, when SSRI remission rates are typically in the range of 30–40% [44], is not at a stage to be of immediate clinical use. The fact that the expected, and experimentally determined, nonremission rate for sertraline is approximately 60–70% makes the threshold for accepting a treatment response predictor much higher than if response rates were higher. As shown in this analysis, although the algorithm classified patients with 64% accuracy, it identified essentially all patients as nonremitters. Therefore, prediction accuracy must be well above the nonremission rate, in this case, to be of any clinical use. Findings of this nature, where significant relationships emerge, but do not provide predictive power, can however, provide insights into SSRI and placebo mechanisms of action.
Our main finding that early-sertraline rACC thickening associates with efficacious SSRI treatment courses indicates that specific, short-term rACC CT alterations may play a role in the sertraline antidepressant response. Interestingly, we also found in exploratory analyses that changes in rACC CT in the first week of sertraline treatment predicted the trajectory of HDRS-17 scores across the trial, strengthening our finding that rACC thickening is associated with better responses to sertraline. The rACC is thought to be the “affective subdivision” of the ACC [45], supporting the regulation of stress response and assigning emotional valence to stimuli [46]. This is especially relevant given that a body of evidence suggests that antidepressants exert their effects through remediating affective biases early following administration [47]. Harmer et al. (2009) proposed the cognitive neuropsychological hypothesis of antidepressant drug action, where rapid shifts toward positive affective biases occur after pharmacological initiation and these shifts in emotional and introspective processing gradually change behavior and mood in order to alleviate depressive symptoms. Not only are short-term reversal of negative biases in recognition of facial expressions and emotional categorization seen in depressed patients acutely administered antidepressants [48], but this shift toward positive affective bias was also observed in healthy volunteers administered SSRIs [49]. This idea that affective, potentially introspective or self-referential, adaptions occur very early in treatment hinges on the notion that there are in fact, relevant pharmacological changes induced immediately, i.e., activation of second messenger systems and alterations to gene expression [47]. Short-term increases in rACC GABA concentration, acquired with MR spectroscopy, have been shown to significantly associate with SSRI clinical response [50], supporting the notion that pharmacoactive alterations can occur in this small time window. The exact biological mechanism underlying short-term thickening of this region is difficult to pinpoint. Downstream GABAergic signaling is one promising hypothesis. However, direct serotonergic input from the SSRI early-in-treatment could also lead to this effect.
It has been widely accepted that there is a 2–4 week delay in the onset of the therapeutic effects of SSRIs [2]. Despite this, it has been shown that SSRIs exert their serotonin transporter blocking effects within minutes [51]. Thus, hypotheses have emerged to explain the short- and long-term adaptations that might explain the delayed onset of antidepressant effects. Within the first 2 weeks of SSRI administration, serotonin reuptake is blocked at neurons originating in the dorsal raphe nucleus [52, 53]. This reuptake blockage increases endogenous serotonin at the presynaptic cell body, yielding an immediate inhibition of serotonergic firing, mediated by serotonin 5-HT1A autoreceptor negative feedback [51]. This short-term inhibition of serotonergic activity results in decreased serotonin availability cortically [52]. Decreased endogenous serotonin in the cortex would then cause increased expression of postsynaptic 5-HT1A receptors (up-regulation) in the short-term following SSRI administration [53].
With continued SSRI administration, however, serotonergic neurons become disinhibited [51], mediated by a down-regulation of desensitized 5-HT1A autoreceptors and the continual blockade of serotonin transporters [53]. This yields increased serotonin available postsynaptically [53]. Thus, these adaptations are thought to underlie the delay in the therapeutic effect of SSRIs [54]. This is bolstered by evidence that concomitant administration of SSRIs with the 5-HT1A autoreceptor antagonist pindolol accelerates the antidepressant response by simulating autoreceptor down-regulation at the initiation of treatment [55].
We posit that the associations between early-treatment rACC cortical thickening and sertraline treatment response may be a marker for a normative SSRI mechanism of action, whereby increased 5-HT1A receptor concentrations occur cortically in the acute phase of treatment. This hypothesis is bolstered by recent evidence suggesting a positive relationship between CT and 5-HT1A receptor concentration in a range of cortical regions [56]. Although speculative and unable to be tested within this study, the cortical thickness alterations may be a consequence of fluctuations in cortical 5-HT1A receptor concentrations. Individuals with short-term cortical thickening may either have a stronger 5-HT1A up-regulation response than those with less thickening (or even thinning) or may have the same rate of 5-HT1A up-regulation, but have more serotonergic neurons that can express the receptor. In either case, these conditions create the setting for a positive SSRI treatment response. While all a priori regions thickened during the first week of SSRI intervention, the hypothesized targeted role of the rACC in enhanced adaptive introspective processing or assigning affective bias may contribute to this localized relationship with eventual treatment response.
The fact that no significant relationships emerged between these structural metrics and nonspecific treatment response argues for the investigation of placebo-specific biomarkers. While ethical considerations prevent these markers from being clinically useful in prescribing placebo, meaningful conclusions may be drawn from them regarding the placebo mechanism of action. The idea that placebo treatment induces distinct alterations in the brain has been previously theorized [57, 58]. Leuchter et al. (2002) [59] showed an increase in prefrontal cortex activity early in placebo treatment that was absent in the SSRI treatment condition. The novel short-term RMF cortex effects we find to be specific to positive placebo responses may be linked to the opioid system, which has been heavily implicated in the distinct antidepressant role of placebo treatment [60, 61]. In fact, Petrovic et al. [62] found increased functional MRI activation in a portion of the ventrolateral prefrontal cortex overlapping with the RMF used in this study, in analgesia and anxiolytic placebo treatments relative to specific treatments. Because these prefrontal placebo activations also covaried with rACC activity in the placebo condition, the authors hypothesized that placebo treatment follows a top-down influence over the opioid system, where portions of the prefrontal cortex, including the RMF, are thought to be responsible for processing the expectation for treatment and initiate cascades of mu-opioid activation that act on the rACC [62]. This expectation for treatment pathway appears to only play a significant role in the placebo antidepressant response, as Leuchter et al. (2014) showed that expecting an efficacious medication only predicted placebo response and was not significantly related to response to antidepressant medication [63].
The expectation for treatment, mediated opioid system cascade may initiate through activation of local enkephalinergic inter-neurons within the RMF [62], yielding a local increase in thickness and volume as observed in this study that may be mediated by spine and synapse turnover or dendritic branching. Future studies with structural MRI 2 or 3 weeks into treatment would be critical in testing whether the potential top-down regulation of opioid transmission in the ACC via the RMF also modulates ACC structure during treatment. We hypothesize that the observed effect of thicker pretreatment RMF cortices associating with worse responses to placebo represents a ceiling effect of the RMF mediated top-down influence on the opioid system. In the placebo group, post hoc analysis showed a negative relationship between pretreatment RMF CT and the early-treatment change in RMF CT (Pearson’s r = −0.21, p = 0.047), where individuals presenting with thicker RMF cortices exhibited less cortical thickening and even thinning, in the first week of placebo treatment. This indicates that initiating placebo treatment with a thicker cortex may prevent further thickening, meaning the neural plasticity necessary for treatment expectation to confer a top-down modulation of the opioid system would be absent or reduced.
This neuroimaging analysis of a large, placebo-controlled SSRI trial has some limitations. EMBARC was focused to early-onset, chronic/recurrent patients with MDD to reduce heterogeneity since varying ages of onset, chronicity, and recurrency have all shown to be associated with treatment response [35, 64]. Because there are no current clinical biomarkers predicting treatment outcome in MDD as a whole and given the heterogeneity of MDD, focusing on specific subtypes of MDD may be a necessary approach for biomarker identification. Our exploratory analyses showed that the chronic MDD subtype in the sertraline arm and the recurrent MDD subtype in the placebo arm drove the association between response and early-treatment alterations. This data highlights a specificity not only across treatment arms, but also between subtypes and provides a foundation for future studies aimed at subtype-specific biomarker discovery.
While EMBARC contains a placebo arm, active-control arms can also provide valuable information about the treatment’s biological specificity [65]. EMBARC does, however, contain a second stage where nonresponders were switched to buproprion (for sertraline nonresponders) or to sertraline (for placebo nonresponders) [35]. Future analyses using this data could build upon this work. Further, as with any treatment, there is also a placebo effect present (e.g., the patient expecting an effective treatment), so although the rACC results were specific to the sertraline arm relative to the placebo arm, this effect is potentially entangled with a placebo effect. Further, the specificity of findings to the rACC and RMF cortices were discovered in follow-up analyses (post hoc) testing of the linear mixed model interaction terms. These interaction terms showed that there was a region-specific effect of brain structure and treatment response that differed across the treatment arms. Post hoc testing was subsequently performed to determine, which regions drove the effect. However, the post hoc nature of these follow-up analyses should be considered. Also, the duration between medication initiation and the week 1 scan was not controlled for in this analysis. It may be that due to the short duration between scans in the EMBARC study and the relatively small magnitude of morphologic changes observed, that these measurements may be particularly sensitive to deviations from an exact 7-day duration study design. Further work might investigate this effect and consider this factor as a potential covariate. Although MR scanners across the sites were of 3T strength, scanner differences necessitated a statistical site harmonization method to be employed. Though site differences can be a challenge, inclusion of a representative sample without geographical or scanner restrictions that can be postprocessed to remove potential confounding biases is also a study strength.
To conclude, we used noninvasive structural estimates to determine if short-term alterations associate with long-term, treatment-specific responses. We found that early-treatment rACC thickening associates with efficacious sertraline responses, while early-treatment RMF thickening and enlargement correlates with efficacious placebo responses. Moreover, thicker RMF cortices at baseline associated with worse placebo responses. The differential early-treatment morphologic alterations accompanying sertraline and placebo responses provides evidence of short-term, antidepressant-specific neuroplasticity associated with amelioration of MDD symptomatology. It is likely that brain structure could play a role in multimodal neuroimaging studies aimed at developing clinically translatable biomarkers of MDD treatment response. Further, combined PET/MRI studies are necessary to test our hypothesis that specific serotonergic activity gives rise to cortical thickness alterations early-in SSRI treatment.
Electronic supplementary material
Acknowledgments
We would like to thank the Center for Understanding Biology using Imaging Technology (CUBIT) image analysts at Stony Brook University for their work in MRI importing, processing, and quality control. We acknowledge the biostatistical consultation and support from the Biostatistical Consulting Core at the School of Medicine, Stony Brook University. We would also like to thank the EMBARC National Coordinating Center at UT Southwestern Medical Center and the Data Center at Columbia University.
Funding
The EMBARC study was supported by the National Institute of Mental Health of the National Institutes of Health under Award numbers U01MH092221 (Trivedi, M.H.) and U01MH092250 (McGrath, P.J., Parsey, R.V., and Weissman, M.M.) The study was also supported in part by Valeant Pharmaceuticals who donated Wellbutrin X.L. used in the second phase of this study.
Conflict of interest
Ms. Bartlett, Dr. Ogden, Ms. Malchow, and Dr. Petkova report no conflicting interests. Dr. Yang, Dr. Sharma, and Ms. Zhang report no conflicting interests relating to this paper. Dr. DeLorenzo reports no financial disclosures. Dr. Cooper and Dr. Adams report no competing interests. Dr. McInnis has no conflicts of interest with respect to this paper. Dr. Trombello owns stock in Merck and Gilead Sciences. Within the past 36 months he has also owned stock in Johnson & Johnson. Dr. Trivedi, is or has been an advisor/consultant and received fee from (lifetime disclosure): Abbott Laboratories, Inc., Abdi Ibrahim, Akzo (Organon Pharmaceuticals Inc.), Alkermes, AstraZeneca, Axon Advisors, Bristol-Myers Squibb Company, Cephalon, Inc., Cerecor, CME Institute of Physicians, Concert Pharmaceuticals, Inc., Eli Lilly & Company, Evotec, Fabre Kramer Pharmaceuticals, Inc., Forest Pharmaceuticals, GlaxoSmithKline, Janssen Global Services, LLC, Janssen Pharmaceutica Products, LP, Johnson & Johnson PRD, Libby, Lundbeck, Meade Johnson, MedAvante, Medtronic, Merck, Mitsubishi Tanabe Pharma Development America, Inc., Naurex, Neuronetics, Otsuka Pharmaceuticals, Pamlab, Parke-Davis Pharmaceuticals, Inc., Pfizer Inc., PgxHealth, Phoenix Marketing Solutions, Rexahn Pharmaceuticals, Ridge Diagnostics, Roche Products Ltd., Sepracor, SHIRE Development, Sierra, SK Life and Science, Sunovion, Takeda, Tal Medical/Puretech Venture, Targacept, Transcept, VantagePoint, Vivus, and Wyeth-Ayerst Laboratories. In addition, he has received grants/research support from: Agency for Healthcare Research and Quality (AHRQ), Cyberonics, Inc., National Alliance for Research in Schizophrenia and Depression, National Institute of Mental Health and National Institute on Drug Abuse. In the past two years, Dr. Weissman received funding from the National Institute of Mental Health (NIMH), the National Alliance for Research on Schizophrenia and Depression (NARSAD), the Sackler Foundation, the Templeton Foundation; and receives royalties from the Oxford University Press, Perseus Press, the American Psychiatric Association Press, and MultiHealth Systems. Dr. McGrath has received funding from the National Institute of Mental Health, New York State Department of Mental Hygiene, Research Foundation for Mental Hygiene (New York State), Forest Research Laboratories, Sunovion Pharmaceuticals, and Naurex Pharmaceuticals (now Allergan). Dr. Fava has received research support from Abbot Laboratories; Alkermes, Inc.; American Cyanamid; Aspect Medical Systems; AstraZeneca; Avanir Pharmaceuticals; BioResearch; BrainCells Inc.; Bristol-Myers Squibb; CeNeRx BioPharma; Cephalon; Clintara, LLC; Cerecor; Covance; Covidien; Eli Lilly and Company; EnVivo Pharmaceuticals, Inc.; Euthymics Bioscience, Inc.; Forest Pharmaceuticals, Inc.; Ganeden Biotech, Inc.; GlaxoSmithKline; Harvard Clinical Research Institute; Hoffman-LaRoche; Icon Clinical Research; i3 Innovus/Ingenix; Janssen R&D, LLC; Jed Foundation; Johnson & Johnson Pharmaceutical Research & Development; Lichtwer Pharma GmbH; Lorex Pharmaceuticals; Lundbeck Inc.; MedAvante; Methylation Sciences Inc.; National Alliance for Research on Schizophrenia & Depression (NARSAD); National Center for Complementary and Alternative Medicine (NCCAM); National Institute of Drug Abuse (NIDA); National Institute of Mental Health (NIMH); Neuralstem, Inc.; Novartis AG; Organon Pharmaceuticals; PamLab, LLC.; Pfizer Inc.; Pharmacia-Upjohn; Pharmaceutical Research Associates., Inc.; Pharmavite® LLC; PharmoRx Therapeutics; Photothera; Reckitt Benckiser; Roche Pharmaceuticals; RCT Logic, LLC (formerly Clinical Trials Solutions, LLC); Sanofi-Aventis US LLC; Shire; Solvay Pharmaceuticals, Inc.; Stanley Medical Research Institute (SMRI); Synthelabo; Tal Medical; Wyeth-Ayerst Laboratories; he has served as advisor or consultant to Abbott Laboratories; Acadia; Affectis Pharmaceuticals AG; Alkermes, Inc.; Amarin Pharma Inc.; Aspect Medical Systems; AstraZeneca; Auspex Pharmaceuticals; Avanir Pharmaceuticals; AXSOME Therapeutics; Bayer AG; Best Practice Project Management, Inc.; Biogen; BioMarin Pharmaceuticals, Inc.; Biovail Corporation; BrainCells Inc; Bristol-Myers Squibb; CeNeRx BioPharma; Cephalon, Inc.; Cerecor; CNS Response, Inc.; Compellis Pharmaceuticals; Cypress Pharmaceutical, Inc.; DiagnoSearch Life Sciences (P) Ltd.; Dinippon Sumitomo Pharma Co. Inc.; Dov Pharmaceuticals, Inc.; Edgemont Pharmaceuticals, Inc.; Eisai Inc.; Eli Lilly and Company; EnVivo Pharmaceuticals, Inc.; ePharmaSolutions; EPIX Pharmaceuticals, Inc.; Euthymics Bioscience, Inc.; Fabre-Kramer Pharmaceuticals, Inc.; Forest Pharmaceuticals, Inc.; Forum Pharmaceuticals; GenOmind, LLC; GlaxoSmithKline; Grunenthal GmbH; i3 Innovus/Ingenis; Intracellular; Janssen Pharmaceutica; Jazz Pharmaceuticals, Inc.; Johnson & Johnson Pharmaceutical Research & Development, LLC; Knoll Pharmaceuticals Corp.; Labopharm Inc.; Lorex Pharmaceuticals; Lundbeck Inc.; MedAvante, Inc.; Merck & Co., Inc.; MSI Methylation Sciences, Inc.; Naurex, Inc.; Nestle Health Sciences; Neuralstem, Inc.; Neuronetics, Inc.; NextWave Pharmaceuticals; Novartis AG; Nutrition 21; Orexigen Therapeutics, Inc.; Organon Pharmaceuticals; Osmotica; Otsuka Pharmaceuticals; Pamlab, LLC.; Pfizer Inc.; PharmaStar; Pharmavite® LLC.; PharmoRx Therapeutics; Precision Human Biolaboratory; Prexa Pharmaceuticals, Inc.; Puretech Ventures; PsychoGenics; Psylin Neurosciences, Inc.; RCT Logic, LLC Formerly Clinical Trials Solutions, LLC; Rexahn Pharmaceuticals, Inc.; Ridge Diagnostics, Inc.; Roche; Sanofi-Aventis US LLC.; Sepracor Inc.; Servier Laboratories; Schering-Plough Corporation; Solvay Pharmaceuticals, Inc.; Somaxon Pharmaceuticals, Inc.; Somerset Pharmaceuticals, Inc.; Sunovion Pharmaceuticals; Supernus Pharmaceuticals, Inc.; Synthelabo; Taisho Pharmaceutical; Takeda Pharmaceutical Company Limited; Tal Medical, Inc.; Tetragenex Pharmaceuticals, Inc.; TransForm Pharmaceuticals, Inc.; Transcept Pharmaceuticals, Inc.; Vanda Pharmaceuticals, Inc.; VistaGen; he has received speaking or publishing fees from Adamed, Co; Advanced Meeting Partners; American Psychiatric Association; American Society of Clinical Psychopharmacology; AstraZeneca; Belvoir Media Group; Boehringer Ingelheim GmbH; Bristol-Myers Squibb; Cephalon, Inc.; CME Institute/Physicians Postgraduate Press, Inc.; Eli Lilly and Company; Forest Pharmaceuticals, Inc.; GlaxoSmithKline; Imedex, LLC; MGH Psychiatry Academy/Primedia; MGH Psychiatry Academy/Reed Elsevier; Novartis AG; Organon Pharmaceuticals; Pfizer Inc.; PharmaStar; United BioSource, Corp.; Wyeth-Ayerst Laboratories; he has equity holdings in Compellis and PsyBrain, Inc.; he has a patent for Sequential Parallel Comparison Design (SPCD), which are licensed by MGH to Pharmaceutical Product Development, LLC (PPD); and patent application for a combination of Ketamine plus Scopolamine in Major Depressive Disorder (MDD), licensed by MGH to Biohaven; and he receives copyright royalties for the MGH Cognitive & Physical Functioning Questionnaire (CPFQ), Sexual Functioning Inventory (SFI), Antidepressant Treatment Response Questionnaire (ATRQ), Discontinuation-Emergent Signs & Symptoms (DESS), Symptoms of Depression Questionnaire (SDQ), and SAFER; Lippincott, Williams & Wilkins; Wolkers Kluwer; World Scientific Publishing Co. Pte.Ltd. Dr. Kurian has received research grant support from the following organizations: Targacept, Inc., Pfizer, Inc., Johnson & Johnson, Evotec, Rexahn, Naurex, Forest Pharmaceuticals and the National Institute of Mental Health (NIMH). Mary L. Phillips has received funding from NIMH and the Emmerling-Pittsburgh Foundation. Dr. Oquendo receives royalties for use of the Columbia Suicide Severity Rating Scale. Her family owns stock in Bristol Myers Squibb. Over the past 3 years, Dr. Pizzagalli has received honoraria/consulting fees from Akili Interactive Labs, BlackThorn Therapeutics, Pfizer, and Posit Science for activities unrelated to this project. Dr. Parsey reports no relevant or material financial interests that relate to the research described in this paper.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41386-018-0122-9).
References
- 1.Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163:28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 2.Quitkin FM, Rabkin JD, Markowitz JM, Stewart JW, McGrath PJ, Harrison W. Use of pattern analysis to identify true drug response. A replication. Arch Gen Psychiatry. 1987;44:259–64. doi: 10.1001/archpsyc.1987.01800150071009. [DOI] [PubMed] [Google Scholar]
- 3.Kraemer HC, Wilson GT, Fairburn CG, Agras WS. Mediators and moderators of treatment effects in randomized clinical trials. Arch Gen Psychiatry. 2002;59:877–83. doi: 10.1001/archpsyc.59.10.877. [DOI] [PubMed] [Google Scholar]
- 4.McGrath CL, Kelley ME, Holtzheimer PE, Dunlop BW, Craighead WE, Franco AR, et al. Toward a neuroimaging treatment selection biomarker for major depressive disorder. JAMA Psychiatry. 2013;70:821–9. doi: 10.1001/jamapsychiatry.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colloby SJ, Firbank MJ, Vasudev A, Parry SW, Thomas AJ, O’Brien JT. Cortical thickness and VBM-DARTEL in late-life depression. J Affect Disord. 2011;133:158–64. doi: 10.1016/j.jad.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 6.Frodl T, Jager M, Smajstrlova I, Born C, Bottlender R, Palladino T, et al. Effect of hippocampal and amygdala volumes on clinical outcomes in major depression: a 3-year prospective magnetic resonance imaging study. J Psychiatry Neurosci. 2008;33:423–30. [PMC free article] [PubMed] [Google Scholar]
- 7.Jarnum H, Eskildsen SF, Steffensen EG, Lundbye-Christensen S, Simonsen CW, Thomsen IS, et al. Longitudinal MRI study of cortical thickness, perfusion, and metabolite levels in major depressive disorder. Acta Psychiatr Scand. 2011;124:435–46. doi: 10.1111/j.1600-0447.2011.01766.x. [DOI] [PubMed] [Google Scholar]
- 8.Koolschijn PC, van Haren NE, Schnack HG, Janssen J, Hulshoff Pol HE, Kahn RS. Cortical thickness and voxel-based morphometry in depressed elderly. Eur Neuropsychopharmacol. 2010;20:398–404. doi: 10.1016/j.euroneuro.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 9.Liu CH, Jing B, Ma X, Xu PF, Zhang Y, Li F, et al. Voxel-based morphometry study of the insular cortex in female patients with current and remitted depression. Neuroscience. 2014;262:190–9. doi: 10.1016/j.neuroscience.2013.12.058. [DOI] [PubMed] [Google Scholar]
- 10.Malykhin NV, Carter R, Seres P, Coupland NJ. Structural changes in the hippocampus in major depressive disorder: contributions of disease and treatment. J Psychiatry Neurosci. 2010;35:337–43. doi: 10.1503/jpn.100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKinnon MC, Yucel K, Nazarov A, MacQueen GM. A meta-analysis examining clinical predictors of hippocampal volume in patients with major depressive disorder. J Psychiatry Neurosci. 2009;34:41–54. [PMC free article] [PubMed] [Google Scholar]
- 12.Schmaal L, Hibar DP, Samann PG, Hall GB, Baune BT, Jahanshad N, et al. Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Mol Psychiatry. 2016;22:900–909. doi: 10.1038/mp.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmaal L, Veltman DJ, van Erp TG, Samann PG, Frodl T, Jahanshad N, et al. Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry. 2016;21:806–12. doi: 10.1038/mp.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahashi T, Yucel M, Lorenzetti V, Tanino R, Whittle S, Suzuki M, et al. Volumetric MRI study of the insular cortex in individuals with current and past major depression. J Affect Disord. 2010;121:231–8. doi: 10.1016/j.jad.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Truong W, Minuzzi L, Soares CN, Frey BN, Evans AC, MacQueen GM, et al. Changes in cortical thickness across the lifespan in major depressive disorder. Psychiatry Res. 2013;214:204–11. doi: 10.1016/j.pscychresns.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Tu PC, Chen LF, Hsieh JC, Bai YM, Li CT, Su TP. Regional cortical thinning in patients with major depressive disorder: a surface-based morphometry study. Psychiatry Res. 2012;202:206–13. doi: 10.1016/j.pscychresns.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 17.van Eijndhoven P, van Wingen G, Katzenbauer M, Groen W, Tepest R, Fernandez G, et al. Paralimbic cortical thickness in first-episode depression: evidence for trait-related differences in mood regulation. Am J Psychiatry. 2013;170:1477–86. doi: 10.1176/appi.ajp.2013.12121504. [DOI] [PubMed] [Google Scholar]
- 18.Wagner G, Schultz CC, Koch K, Schachtzabel C, Sauer H, Schlosser RG. Prefrontal cortical thickness in depressed patients with high-risk for suicidal behavior. J Psychiatr Res. 2012;46:1449–55. doi: 10.1016/j.jpsychires.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 19.Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS. Hippocampal volume reduction in major depression. Am J Psychiatry. 2000;157:115–8. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- 20.Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb Cortex. 2002;12:386–94. doi: 10.1093/cercor/12.4.386. [DOI] [PubMed] [Google Scholar]
- 21.Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry. 2001;58:545–53. doi: 10.1001/archpsyc.58.6.545. [DOI] [PubMed] [Google Scholar]
- 22.Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA. 1998;95:13290–5. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–98. doi: 10.1016/S0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 24.Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry. 2000;57:925–35. doi: 10.1001/archpsyc.57.10.925. [DOI] [PubMed] [Google Scholar]
- 25.Papmeyer M, Giles S, Sussmann JE, Kielty S, Stewart T, Lawrie SM, et al. Cortical thickness in individuals at high familial risk of mood disorders as they develop major depressive disorder. Biol Psychiatry. 2015;78:58–66. doi: 10.1016/j.biopsych.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 26.Peterson BS, Warner V, Bansal R, Zhu H, Hao X, Liu J, et al. Cortical thinning in persons at increased familial risk for major depression. Proc Natl Acad Sci USA. 2009;106:6273–8. doi: 10.1073/pnas.0805311106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161:1957–66. doi: 10.1176/appi.ajp.161.11.1957. [DOI] [PubMed] [Google Scholar]
- 28.Costafreda SG, Chu C, Ashburner J, Fu CH. Prognostic and diagnostic potential of the structural neuroanatomy of depression. PLoS One. 2009;4:e6353. doi: 10.1371/journal.pone.0006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fonseka TM, MacQueen GM, Kennedy SH. Neuroimaging biomarkers as predictors of treatment outcome in Major Depressive Disorder. J Affect Disord. 2017;223:21–35. [DOI] [PubMed]
- 30.MacQueen GM, Yucel K, Taylor VH, Macdonald K, Joffe R. Posterior hippocampal volumes are associated with remission rates in patients with major depressive disorder. Biol Psychiatry. 2008;64:880–3. doi: 10.1016/j.biopsych.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 31.Phillips JL, Batten LA, Tremblay P, Aldosary F, Blier P. A prospective, longitudinal study of the effect of remission on cortical thickness and hippocampal volume in patients with treatment-resistant depression. international J Neuropsychopharmacol. 2015;18:pyv037. [DOI] [PMC free article] [PubMed]
- 32.Vakili K, Pillay SS, Lafer B, Fava M, Renshaw PF, Bonello-Cintron CM, et al. Hippocampal volume in primary unipolar major depression: a magnetic resonance imaging study. Biol Psychiatry. 2000;47:1087–90. doi: 10.1016/S0006-3223(99)00296-6. [DOI] [PubMed] [Google Scholar]
- 33.Chen CH, Ridler K, Suckling J, Williams S, Fu CH, Merlo-Pich E, et al. Brain imaging correlates of depressive symptom severity and predictors of symptom improvement after antidepressant treatment. Biol Psychiatry. 2007;62:407–14. doi: 10.1016/j.biopsych.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 34.Bansal R, Hellerstein DJ, Peterson BS. Evidence for neuroplastic compensation in the cerebral cortex of persons with depressive illness. Mol Psychiatry. 2017;34:375–83. [DOI] [PMC free article] [PubMed]
- 35.Trivedi MH, McGrath PJ, Fava M, Parsey RV, Kurian BT, Phillips ML, et al. Establishing moderators and biosignatures of antidepressant response in clinical care (EMBARC): rationale and design. J Psychiatr Res. 2016;78:11–23. doi: 10.1016/j.jpsychires.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Association AP, Association AP. DSM-IV-TR: Diagnostic and statistical manual of mental disorders, text revision. Washington, DC: American Psychiatric Association; 2000. pp. 78–85. [Google Scholar]
- 37.Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry. 2003;54:573–83. doi: 10.1016/S0006-3223(02)01866-8. [DOI] [PubMed] [Google Scholar]
- 38.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iscan Z, Jin TB, Kendrick A, Szeglin B, Lu H, Trivedi M, et al. Test–retest reliability of freesurfer measurements within and between sites: effects of visual approval process. Hum Brain Mapp. 2015;36:3472–85. doi: 10.1002/hbm.22856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–80. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 41.Fortin J-P, Cullen N, Sheline YI, Taylor WD, Aselcioglu I, Adams P, et al. Harmonization of cortical thickness measurements across scanners and sites. Neuroimage. 2018;167:104–120. [DOI] [PMC free article] [PubMed]
- 42.Driemeyer J, Boyke J, Gaser C, Büchel C, May A. Changes in gray matter induced by learning—revisited. PLoS One. 2008;3:e2669. doi: 10.1371/journal.pone.0002669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pirnia T, Joshi S, Leaver A, Vasavada M, Njau S, Woods R, et al. Electroconvulsive therapy and structural neuroplasticity in neocortical, limbic and paralimbic cortex. Transl Psychiatry. 2016;6:e832. doi: 10.1038/tp.2016.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nemeroff CC, Entsuah AR, Willard L, Demitrack M, Thase M. Venlafaxine and SSRIs: pooled remission analysis. Eur Neuropsychopharmacol. 2003;13(Suppl 4):S255. doi: 10.1016/S0924-977X(03)91899-2. [DOI] [Google Scholar]
- 45.Devinsky O, Morrell MJ, Vogt BA. Contributions of anterior cingulate cortex to behaviour. Brain. 1995;118(Pt 1):279–306. doi: 10.1093/brain/118.1.279. [DOI] [PubMed] [Google Scholar]
- 46.Pizzagalli DA. Frontocingulate dysfunction in depression: toward biomarkers of treatment response. Neuropsychopharmacol. 2011;36:183–206. doi: 10.1038/npp.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harmer CJ, Goodwin GM, Cowen PJ. Why do antidepressants take so long to work? A cognitive neuropsychological model of antidepressant drug action. Br J Psychiatry. 2009;195:102–8. doi: 10.1192/bjp.bp.108.051193. [DOI] [PubMed] [Google Scholar]
- 48.Harmer CJ, O’Sullivan U, Favaron E, Massey-Chase R, Ayres R, Reinecke A, et al. Effect of acute antidepressant administration on negative affective bias in depressed patients. Am J Psychiatry. 2009;166:1178–84. doi: 10.1176/appi.ajp.2009.09020149. [DOI] [PubMed] [Google Scholar]
- 49.Harmer CJ, Shelley NC, Cowen PJ, Goodwin GM. Increased positive versus negative affective perception and memory in healthy volunteers following selective serotonin and norepinephrine reuptake inhibition. Am J Psychiatry. 2004;161:1256–63. doi: 10.1176/appi.ajp.161.7.1256. [DOI] [PubMed] [Google Scholar]
- 50.Brennan BP, Admon R, Perriello C, LaFlamme EM, Athey AJ, Pizzagalli DA, et al. Acute change in anterior cingulate cortex GABA, but not glutamine/glutamate, mediates antidepressant response to citalopram. Psychiatry Res: Neuroimaging. 2017;269:9–16. [DOI] [PMC free article] [PubMed]
- 51.Blier P, de Montigny C. Current advances and trends in the treatment of depression. Trends Pharmacol Sci. 1994;15:220–6. doi: 10.1016/0165-6147(94)90315-8. [DOI] [PubMed] [Google Scholar]
- 52.Artigas F, Romero L, de Montigny C, Blier P. Acceleration of the effect of selected antidepressant drugs in major depression by 5-HT1A antagonists. Trends Neurosci. 1996;19:378–83. doi: 10.1016/S0166-2236(96)10037-0. [DOI] [PubMed] [Google Scholar]
- 53.Norman TR. The new antidepressants-mechanisms of action. Issues. 1999;22:106–8.
- 54.Blier P, Pineyro G, el Mansari M, Bergeron R, de Montigny C. Role of somatodendritic 5-HT autoreceptors in modulating 5-HT neurotransmission. Ann N Y Acad Sci. 1998;861:204–16. doi: 10.1111/j.1749-6632.1998.tb10192.x. [DOI] [PubMed] [Google Scholar]
- 55.Artigas F, Perez V, Alvarez E. Pindolol induces a rapid improvement of depressed patients treated with serotonin reuptake inhibitors. Arch Gen Psychiatry. 1994;51:248–51. doi: 10.1001/archpsyc.1994.03950030084009. [DOI] [PubMed] [Google Scholar]
- 56.Pillai RL, Malhotra A, Rupert DD, Weschler B, Williams JC, Zhang M, et al. Relations between cortical thickness, serotonin 1A receptor binding, and structural connectivity: a multimodal imaging study. Hum Brain Mapp. 2018;39:1043–55. [DOI] [PMC free article] [PubMed]
- 57.Mayberg HS, Silva JA, Brannan SK, Tekell JL, Mahurin RK, McGinnis S, et al. The functional neuroanatomy of the placebo effect. Am J Psychiatry. 2002;159:728–37. doi: 10.1176/appi.ajp.159.5.728. [DOI] [PubMed] [Google Scholar]
- 58.Sikora M, Heffernan J, Avery ET, Mickey BJ, Zubieta JK, Peciña M. Salience network functional connectivity predicts placebo effects in major depression. Biol Psychiatry. 2016;1:68–76. doi: 10.1016/j.bpsc.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leuchter AF, Cook IA, Witte EA, Morgan M, Abrams M. Changes in brain function of depressed subjects during treatment with placebo. Am J Psychiatry. 2002;159:122–9. doi: 10.1176/appi.ajp.159.1.122. [DOI] [PubMed] [Google Scholar]
- 60.Levine JD, Gordon NC, Fields HL. The mechanism of placebo analgesia. Lancet. 1978;2:654–7. doi: 10.1016/S0140-6736(78)92762-9. [DOI] [PubMed] [Google Scholar]
- 61.Pecina M, Bohnert A, Sikora M, Avery E, Langenecker S, Mickey B, et al. Placebo-activated neural systems are linked to antidepressant responses. JAMA Psychiatry. 2015;72:1087–94. [DOI] [PMC free article] [PubMed]
- 62.Petrovic P, Kalso E, Petersson KM, Andersson J, Fransson P, Ingvar M. A prefrontal non-opioid mechanism in placebo analgesia. Pain. 2010;150:59–65. doi: 10.1016/j.pain.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 63.Leuchter AF, Hunter AM, Tartter M, Cook IA. Role of pill-taking, expectation and therapeutic alliance in the placebo response in clinical trials for major depression. Br J Psychiatry. 2014;205:443–9. doi: 10.1192/bjp.bp.113.140343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stewart JW, Quitkin M. Chronicity of antidepressant efficacy in atypical depression. Arch Gen Psychiatry. 1989;46:1080–7. doi: 10.1001/archpsyc.1989.01810120022005. [DOI] [PubMed] [Google Scholar]
- 65.Temple R, Ellenberg SS. Placebo-controlled trials and active-control trials in the evaluation of new treatments. Part 1: ethical and scientific issues. Ann Intern Med. 2000;133:455–63. doi: 10.7326/0003-4819-133-6-200009190-00014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.