

Abstract
Background and Purpose
Acetaminophen‐induced acute liver injury (AILI) is the most frequent cause of acute liver failure in developed countries. Given the significant limitations associated with N‐acetyl cysteine, the only antidote used to treat AILI, the development of novel therapeutic approaches that can offer a wide range of therapeutic time‐windows is clearly needed. Glycycoumarin (GCM), a natural coumarin purified from liquorice, has been previously demonstrated to possess potent hepatoprotective effects. In the present study, we aimed to investigate the therapeutic potential of GCM against AILI.
Experimental Approach
Acetaminophen (300 mg·kg−1) was administered to male C57BL/6 mice, with and without GCM. Serum transaminases, haematoxylin and eosin staining and Western blot were used to assess hepatic damage.
Key Results
GCM (50 mg·kg−1) was highly effective against acetaminophen‐induced hepatotoxicity. Moreover, GCM was superior to N‐acetyl cysteine, in terms of the dosage and the therapeutic time‐windows. Further mechanistic investigations revealed that the therapeutic action of GCM was not a result of inhibition of acetaminophen metabolic activation or associated with Nrf2. Instead, the protective effect of GCM appeared to be predominantly dependent on sustained activation of autophagy, which attenuated acetaminophen‐induced mitochondrial oxidative stress and JNK activation.
Conclusions and Implications
Collectively, our results indicate that GCM alleviated acetaminophen‐induced oxidative stress through activating autophagy, thereby protecting against AILI. Our findings suggest that GCM has potential as a novel therapeutic agent for treating AILI.
Abbreviations used
- 3‐NT
3‐nitrotyrosine
- AILI
acetaminophen‐induced liver injury
- ALF
acute liver failure
- ALT
alanine aminotransferase
- ER
endoplasmic reticulum
- GCM
glycycoumarin
- KO
knockout
- NAC
N‐acetyl cysteine
- NAPQI
N‐acetyl‐q‐benzoquinone imine
Introduction
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5239 is one of the most frequently used analgesic and antipyretic drugs. It is safe and effective when administered at recommended doses, whereas its overdose may lead to hepatotoxicity and acute liver failure (ALF) (Bernal et al., 2010). In fact, acetaminophen hepatotoxicity remains the leading cause of ALF in many countries and received ample attention in clinical situation (Lee, 2013). Mechanistically, acetaminophen overdose causes saturation of anti‐toxic phase II enzymes, and the excess acetaminophen is metabolized by cytochrome P450 enzymes into the highly reactive intermediate N‐acetyl‐q‐benzoquinone imine (NAPQI), which is detoxified by conjugating with GSH (Nelson, 1990). As such, NAPQI accumulates and depletes GSH, consequently leading to covalent binding with sulfhydryl groups in mitochondrial proteins (Qiu et al., 1998), which in turn results in oxidative stress and mitochondrial dysfunction (Jaeschke et al., 2012). This then triggers the phosphorylation of http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=518 and its subsequent mitochondrial translocation (Win et al., 2016). Although controversial, it is believed that sustained JNK activation promotes hepatocyte injury and death and has a detrimental effect on acetaminophen toxicity. Given the vital role of NAPQI in the initiation of acetaminophen hepatotoxicity, N‐acetyl cysteine (NAC) is used clinically as the only standard antidote for acetaminophen‐induced liver injury (AILI). It acts mainly by replenishing GSH to enhance the detoxification of NAPQI; however, due to its narrow therapeutic time window, it is not always effective in patients suffering from an acetaminophen overdose presenting at a late stage (Du et al., 2016). Hence, the development of new drugs that can extend the therapeutic time frame for AILI is clearly needed.
Multiple cellular processes have been identified to be involved in acetaminophen‐mediated hepatotoxicity, including the metabolic activation of acetaminophen, mitochondrial oxidative stress, autophagy, endoplasmic reticulum (ER) stress and liver regeneration, and these have provided new potential therapeutic targets for treating AILI. Earlier results demonstrated that autophagy is activated by acetaminophen to remove damaged mitochondria and acetaminophen‐induced protein adducts (Ni et al., 2012, 2016). Thus, autophagy seems to act as a cellular adaptation mechanism in response to acetaminophen‐induced hepatocyte damage. Considering the pivotal role of mitochondrial damage in the pathogenesis of AILI, we hypothesized that autophagy may participate in maintaining redox balance by eliminating ROS‐generating mitochondria, supporting the possibility of using the induction of autophagy as a therapeutic approach for patients with AILI.
Glycycoumarin (GCM) is a natural coumarin isolated from http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=413. Our previous studies showed that GCM was effective at protecting against alcohol‐induced hepatotoxicity and nonalcoholic steatohepatitis in vitro and in vivo (Song et al., 2015; Zhang et al., 2016). In the present work, we aimed to assess the therapeutic effect of GCM on acetaminophen‐induced hepatotoxicity and elucidate the molecular basis of this effect.
Methods
Animals and treatments
All animal experiments were approved by the Institutional Animal Care and Use Committee, China Agricultural University. Animal studies are reported incompliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Male C57BL/6N mice (6 to 8 weeks old) were purchased from Charles River Laboratories (Beijing, China). Nrf2 knockout (KO) mice of C57BL/6J background and their wild‐type littermates were kindly provided by Professor Siwang Yu at Peking University School of Pharmaceutical Sciences. The animals weighed 20–22 g at the beginning of the corresponding experiments. All animals were allowed to acclimatize for at least 1 week under controlled conditions of temperature (23 ± 2°C), humidity (60 ± 5%) and 12 h light–dark cycle at the Department of Laboratory Animal Science, Peking University Health Science Centre. They were supplied with standard laboratory chow and water ad libitum and housed, six per cage, in plastic cages with corncob bedding.
The mice were divided into groups randomly. The acetaminophen hepatotoxicity model and the specific strain have been chosen in accordance with previously published studies (Jaeschke et al., 2013). Mice were fasted overnight and then received acetaminophen (300 mg·kg−1, i.p., prepared in 55°C warm saline). Some mice were pretreated with ML385 (40 mg·kg−1, i.p.) (Singh et al., 2016) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5535 (CQ, 60 mg·kg−1, i.p.) (Ni et al., 2012) before acetaminophen to suppress activation of Nrf2 and autophagy. GCM dissolved in 2% Tween 80 was injected i.p. 2 h after acetaminophen treatment. Comparative experiments on the therapeutic effects of GCM (100 mg·kg−1, i.p.) and NAC (300 mg·kg−1, i.p.) were carried out 4 h after acetaminophen injection. At the end of the experiment, body weights were recorded, the mice were anaesthetized with pentobarbital sodium (80 mg·kg−1, i.p.), and blood samples were obtained from the abdominal aorta. Finally, the animals were killed by cervical dislocation, and livers were quickly removed and weighed. Blinding was undertaken during the surgery and the subsequent data analysis.
ALT measurement
Serum was collected 3, 6, 12 and 24 h after acetaminophen treatment. Serum alanine aminotransferase (ALT) activities were measured using reagent kits from Nanjing Jiancheng (Nanjing, China).
Histology and immunohistochemistry
Liver tissue was collected 3, 6, 12 and 24 h after acetaminophen treatment. A portion of liver tissue was fixed in 10% neutral buffered formalin for histology and immunohistochemistry, and the rest of the sample was used for mitochondrial fractionation and immunoblotting. Formalin‐fixed, paraffin‐embedded liver tissues were cut into 5‐μm‐thick sections and stained with haematoxylin and eosin. For nitrotyrosine protein adducts, liver sections were immunostained with anti‐3‐nitrotyrosine antibody at a 1:250 dilution followed by detection using standard procedures, as described previously (Win et al., 2011). All experiments were performed in a blinded manner by Dr Fei Pei, a pathologist in the Department of Pathology at Peking University.
Fractionation of liver mitochondria
Mitochondria were isolated from murine livers using a tissue mitochondria isolation kit from Thermo Scientific (Waltham, MA, USA). Liver tissues were homogenized with a Dounce grinder and centrifuged as instructed in the kit. The final supernatant was collected as cytoplasm. The remaining pallet was washed with Wash Buffer and recentrifuged to yield mitochondria.
Western blotting
Liver tissues, cytoplasmic fractions and mitochondria were lysed with RIPA buffer. Forty micrograms of denatured protein were subjected to SDS‐PAGE and transferred onto a nitrocellulose membrane (0.2 μm). After incubation in blocking buffer (5% non‐fat milk in PBS), membranes were incubated with specific antibodies to perform immunoblot analyses.
Materials
GCM (purity >99%) was obtained from BioBioPha (Kunming, Yunnan, China). Acetaminophen was purchased from Cayman Chemical (Ann Arbor, MI, USA). Chloroquine diphosphate salt was purchased from Sigma‐Aldrich (St. Louis, MO, USA). ML385 was purchased from MedChem Express (Monmouth Junction, NJ, USA). Primary antibodies specific for p‐JNK (Cat# 9251S), JNK (Cat# 9258P), Drp1 (Cat# 8570S) and PHB1 (Cat# 2426S) were purchased from Cell Signalling Technology (Denvers, MA, USA). Primary‐antibody specific for β‐actin (Cat# AT0001) was obtained from CMCTAG (Milwaukee, WI, USA). 3‐Nitrotyrosine (3‐NT; ab53232) antibody for immunohistochemistry and immunoblotting was purchased from Abcam (Cambridge, MA, USA). Primary antibody specific for LC3 (Cat# M152–3), horseradish peroxidase‐conjugated goat anti‐rabbit immunoglobulin G (Cat# 485) and anti‐mouse immunoglobulin G (Cat# 330) secondary antibodies were purchased from MBL (Woburn, MA, USA).
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). Results are presented as mean ± SD. Statistical comparisons were performed by one‐way ANOVA followed by Tukey's post hoc test using SPSS19.0. P < 0.05 was considered statistically significant. Post hoc tests were run only if F achieved P < 0.05 and there is no significant variance inhomogeneity. Graphs were drawn using GraphPad Prism (version 5.0 for Windows).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
GCM therapy attenuates acetaminophen‐induced acute liver injury
To identify the therapeutic effects of GCM on acetaminophen‐induced hepatotoxicity, C57BL/6N mice were treated with GCM (50 mg·kg−1) 0.5, 1 or 2 h after acetaminophen injection. In this therapeutic time frame, we observed a significant protection by all the GCM treatments at 24 h, as revealed by a 70–80% decrease in serum ALT levels (Figure 1A). We next performed a time course experiment to investigate the mechanism of the therapeutic effect of GCM on AILI, when GCM was administered 2 h following a toxic dose of acetaminophen. As illustrated by the increased ALT levels and histological analysis, acetaminophen treatment caused severe liver injury from 3 h, aggravated after 6 h and resulted in extensive hepatic necrosis at 24 h (Figure 1B, C). Post‐treatment with GCM attenuated acetaminophen toxicity at 6 h, which was sustained at 24 h after acetaminophen administration. Furthermore, AILI was also ameliorated by GCM post‐treatment in a dose‐dependent manner, as demonstrated by progressively reduced serum levels of ALT (data not shown).
Figure 1.
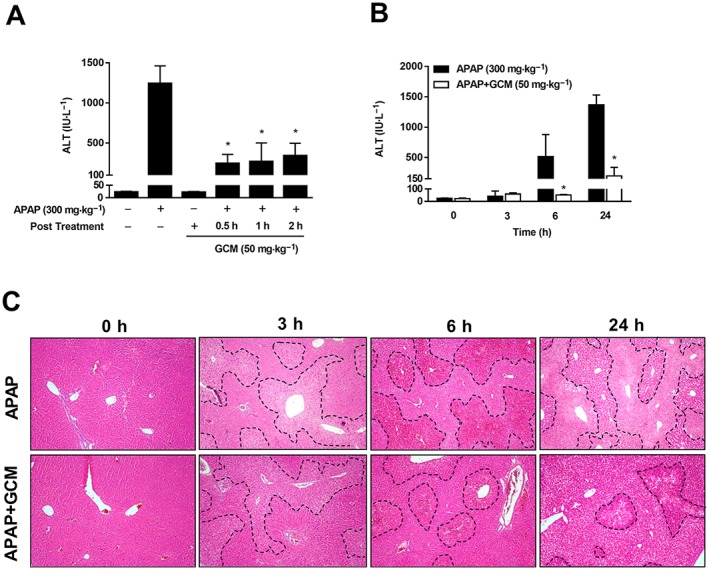
GCM protects against AILI. (A) C57BL/6N mice received 50 mg·kg−1 GCM at 0.5, 1 or 2 h after acetaminophen (APAP) treatment, and serum ALT was determined at 24 h. GCM 50 mg·kg−1 was administered 2 h after acetaminophen. Blood and liver tissue were collected at 3, 6 and 24 h. (B) Serum ALT levels were measured at various time points as indicated. (C) Representative images of haematoxylin and eosin‐stained liver sections at 100× magnification. Data are presented as mean ± SD for n = 6 mice per group. *P < 0.05 compared with the corresponding acetaminophen group.
GCM protects against acetaminophen hepatotoxicity through a JNK‐dependent pathway
Since JNK activation has been implicated in acetaminophen‐induced acute liver injury and actually acts as a pro‐death signal, we next analysed whether the JNK signalling pathway is involved in the protective effect of GCM against acetaminophen‐induced hepatotoxicity. As expected, GCM notably suppressed the JNK phosphorylation evoked by acetaminophen. Furthermore, the translocation of p‐JNK to mitochondria was also abolished by GCM treatment (Figure 2A–C). In further support for an inhibitory effect of GCM on p‐JNK, we assessed Drp1, which has been considered to act downstream of p‐JNK and account for mitochondrial division (Dara et al., 2015). Indeed, translocation of Drp1 from the cytoplasm to mitochondria was inhibited by GCM (Figure 2D), in concert with the effect on p‐JNK.
Figure 2.
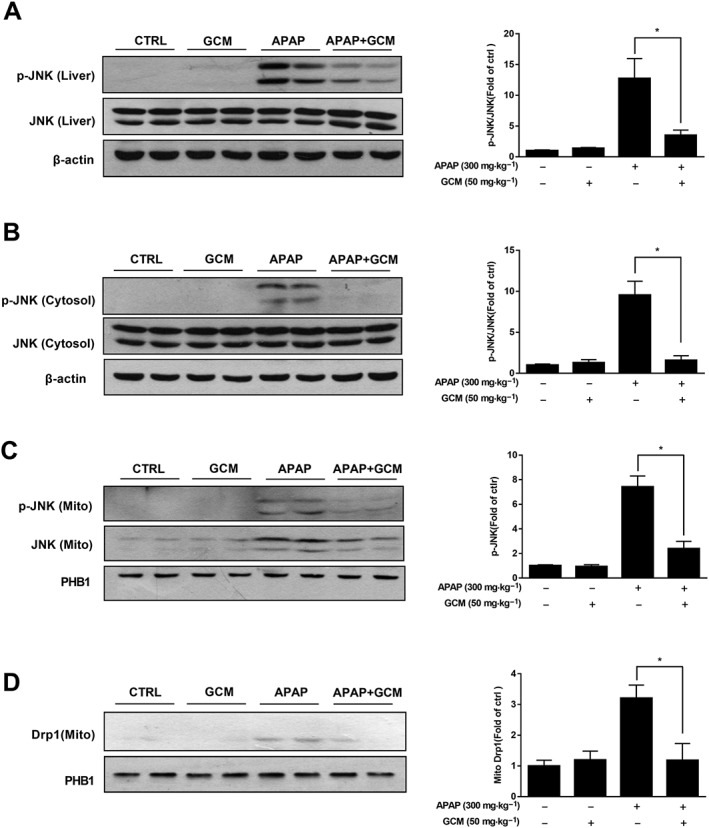
GCM inhibits JNK activation and mitochondria translocation induced by acetaminophen (APAP). C57BL/6N mice were injected with 300 mg·kg−1 acetaminophen, subsequently treated with GCM 50 mg·kg−1 at 2 h and killed at 6 h. Livers were collected to perform the isolation of hepatic mitochondria and cytoplasm. (A–C) Western blot of whole liver homogenate (A), cytoplasm (B) and mitochondria (C) for p‐JNK in mice with or without GCM (50 mg·kg−1). Bar graphs show densitometry of p‐JNK/JNK or p‐JNK/PHB1. (D) Western blot of mitochondrial fraction for Drp1 and densitometry. Quantitative data are presented as mean ± SD for n = 6 mice per group. *P < 0.05 compared with the corresponding acetaminophen group. PHB1, prohibitin 1.
GCM attenuates acetaminophen‐induced mitochondrial oxidative stress
In the acetaminophen‐induced model of hepatotoxicity, phosphorylated JNK binds to mitochondria and subsequently amplifies mitochondrial ROS and forms a self‐sustaining activation loop (Win et al., 2016). Oxidative stress products then cause mitochondrial dysfunction, which has been suggested as the predominant injury mechanism of acetaminophen hepatotoxicity. Hence, we assessed mitochondrial 3‐nitrotyrosine (3‐NT) levels to determine whether GCM can attenuate acetaminophen‐induced mitochondrial oxidative stress. In line with previous studies, there was a significant elevation of 3‐NT in mitochondria at 6 h after acetaminophen as demonstrated by Western blotting (Figure 3A). Immunohistochemistry also showed a massive accumulation of nitrotyrosine protein adducts in the centrilobular areas (Figure 3B). By contrast, GCM treatment substantially reduced the elevated levels of this major biochemical marker of mitochondrial oxidative stress induced by acetaminophen, further illustrating the therapeutic effects of GCM on AILI.
Figure 3.
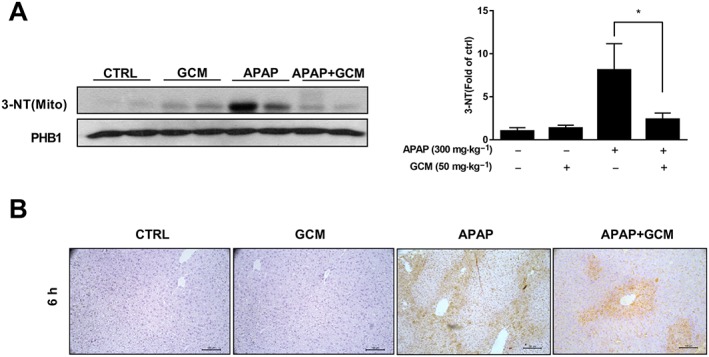
GCM reduced acetaminophen (APAP)‐induced 3‐NT generation in mice. Animal experiments were performed as described in the legend of Figure 2. (A) Western blot of mitochondria 3‐NT and densitometry; (B) immunohistochemistry for nitrotyrosine protein adducts in liver tissue after acetaminophen treatment with or without GCM. Quantitative data are presented as mean ± SD for n = 6 mice per group. *P < 0.05 compared with the corresponding acetaminophen group.
Protection by GCM against acetaminophen‐induced hepatotoxicity is independent of Nrf2
To investigate which signalling pathway was involved in this attenuation of acetaminophen‐induced mitochondrial oxidative stress, we tested the effect of GCM in Nrf2 KO mice, to determine whether the Nrf2 antioxidant system is involved in it protection against acetaminophen hepatotoxicity. However, KO of Nrf2 did not abolish the therapeutic effects of GCM on acetaminophen‐induced acute liver injury, as revealed by serum ALT levels (Figure 4A). Accordingly, histological analysis showed that areas of acetaminophen‐induced necrosis in Nrf2 KO mice were increased compared with that observed in WT controls, but were still overtly reduced after GCM treatment (Figure 4B). Therefore, we concluded that Nrf2 activation did not participate in mediating the protective effect of GCM against acetaminophen hepatotoxicity, which means that other signalling pathways are responsible for its therapeutic action.
Figure 4.
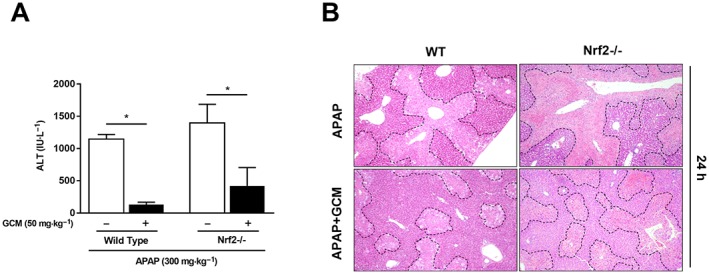
Protection by GCM against acetaminophen (APAP)‐induced hepatotoxicity is independent of Nrf2. Eight‐week‐old male C57BL/6J background Nrf2 KO mice and their wild‐type littermates were treated with GCM (50 mg·kg−1, i.p.) 2 h post‐acetaminophen (300 mg·kg−1, i.p.) administration; blood and liver tissues were collected at 24 h after acetaminophen injection. (A) Serum ALT levels were measured at 24 h post‐acetaminophen treatment. (B) Representative images of haematoxylin and eosin‐stained liver sections at 100× magnification. Data are presented as mean ± SD for n = 6 mice per group. *P < 0.05 compared with the corresponding acetaminophen group.
Sustained autophagy induced by GCM contributes to its protective effect against acetaminophen hepatotoxicity
Given that GCM is able to stimulate autophagy (Song et al., 2015) and autophagy has been established to have protective effects against acetaminophen hepatotoxicity (Ni et al., 2012, 2016), we next investigated whether GCM induces autophagy in our model, which in turn contributes to its therapeutic effects on AILI. The time course experiments showed a rapid increase in LC3II levels at 3 h post‐acetaminophen treatment, indicating the activation of autophagy in response to cellular stress caused by acetaminophen (Figure 5A). However, we then observed a gradual decline in LC3II levels at 6 and 12 h after acetaminophen administration, suggesting autophagy induced by acetaminophen acted as a cellular adaptation mechanism and was activated only transiently. Treatment with GCM further increased LC3II levels at each time point, compared with the corresponding acetaminophen group. Notably, an elevation of LC3II levels by GCM was still very prominent even at 12 h after acetaminophen treatment, a time when they were almost back to normal in the acetaminophen alone‐treated alone (Figure 5A). Importantly, in the presence of the lysosomal inhibitor chloroquine, the increase in LC3II induced by GCM was further elevated compared to either acetaminophen or chloroquine treatment alone, indicating that GCM induced an autophagic flux rather than blocked it in this model (Figure 5B). These results demonstrated that GCM therapy enhanced adaptive autophagy induced by acetaminophen, converting it from a transient response to persistent activation. To confirm the protective role of autophagy induced by GCM treatment against acetaminophen hepatotoxicity, the therapeutic effects of GCM on AILI were assessed by using the Nrf2 inhibitor ML385 and autophagic flux blocker chloroquine in combination (Since inhibition of autophagy flux by chloroquine may lead to an accumulation of p62, which subsequently activates Nrf2, interfering with the results, we first injected the Nrf2 inhibitor ML385 to preclude the involvement of Nrf2 and then utilized chloroquine to suppress autophagy). As expected, no statistical difference was observed in ALT levels and necrosis areas between groups with or without GCM (Figure 5C, D), suggesting that the therapeutic effect of GCM on AILI was nearly abolished under the simultaneous inhibition of autophagy and Nrf2.
Figure 5.
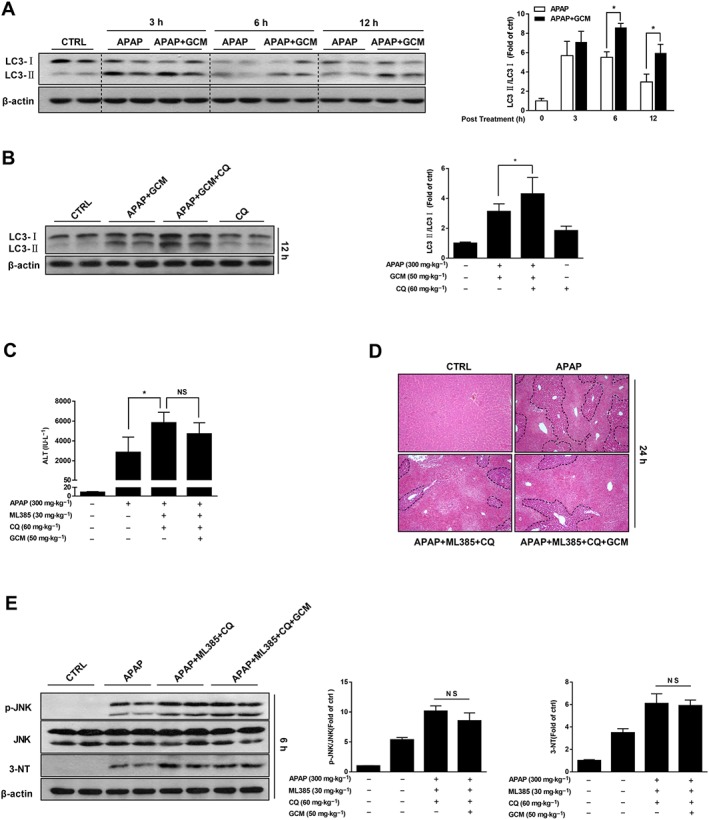
Sustained autophagy stimulated by GCM contributes to its protective effect against acetaminophen (APAP) hepatotoxicity. (A) C57BL/6N mice received 50 mg·kg−1 GCM at 2 h post‐acetaminophen challenge, and liver tissue were collected at 3, 6 and 12 h. LC3II proteins were measured by Western blot at various time points as indicated. Bar graphs show densitometry quantification of LC3II/LC3I. (B–E) Wild‐type C57BL/6N mice were injected (i.p.) with chloroquine (CQ; 60 mg·kg−1) and ML385 (40 mg·kg−1) 2 h before acetaminophen (300 mg·kg−1). Mice were then further treated with or without GCM at 2 h post‐acetaminophen administration. Blood and liver tissue were collected at 6, 12 and 24 h. (B) Western blot of whole liver homogenate for LC3 in mice with or without chloroquine (60 mg·kg−1) at 12 h. Bar graphs show densitometry of LC3II/LC3I. (C) Serum ALT levels were quantified at 24 h. (D) Representative images of haematoxylin and eosin staining are presented. (E) Western blot of whole liver homogenate for p‐JNK and 3‐NT in mice with or without chloroquine (60 mg·kg−1) at 6 h. Bar graphs show densitometry of p‐JNK/JNK or 3‐NT/β‐actin. Data are presented as mean ± SD for n = 6 mice per group. *P < 0.05 compared with the corresponding acetaminophen group.
In order to further establish the molecular mechanisms of autophagy in GCM‐induced protective effect against acetaminophen hepatotoxicity, p‐JNK and 3‐NT were assessed by Western blot. GCM administration to autophagy‐retarded mice did not reduce 3‐NT accumulation or inhibit the increase in p‐JNK induced by acetaminophen treatment (Figure 5E). These results suggest that autophagy, by attenuating the oxidative stress induced by acetaminophen, may be responsible for the protective effect of GCM.
GCM exerts better therapeutic effects on acetaminophen hepatotoxicity compared with NAC
To explore its possible clinical implication in AILI patients, we compared the therapeutic effects of GCM against acetaminophen hepatotoxicity with that of NAC, the only therapeutic option for acetaminophen overdose patients approved by the United States Food and Drug Administration (Polson et al., 2005). We found that NAC 100 mg·kg−1 as a 2 h post‐treatment only slightly lowered ALT levels elevated by acetaminophen, in contrast to an 80% of decrease induced by GCM (50 mg·kg−1) treatment (Figure 6A). It should be noted that NAC was still very effective if we increased the dose to 200 and 300 mg·kg−1 (Figure 6A). However, the delayed treatment with NAC 300 mg·kg−1 gradually lost its protective effect with an increase in time, until, at 4 h after acetaminophen intoxication, no protection was observed in the ALT levels (Figure 6B). However, in this situation, 100 mg·kg−1 GCM was still sufficient to rescue mice from AILI, as revealed by a 50% of reduction in ALT levels and approximately 60% decrease in necrosis around the centrilobular veins (Figure 6C, D). Together, these results suggest that GCM exerts superior therapeutic effects on acetaminophen hepatotoxicity compared to NAC both as an acute and a delayed treatment.
Figure 6.
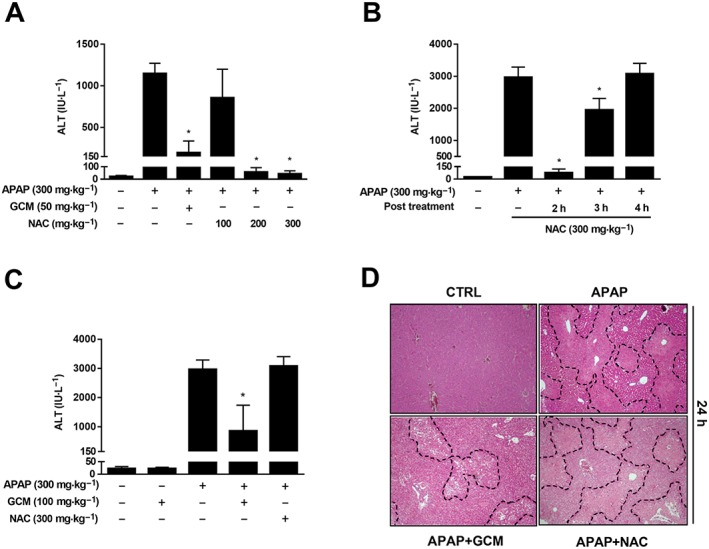
GCM has an extended therapeutic window for AILI compared with NAC. (A) C57BL/6N mice received GCM (50 mg·kg−1, i.p.) or NAC (100, 200 or 300 mg·kg−1, i.p.) at 2 h after acetaminophen (APAP) treatment, and serum ALT levels were determined at 24 h post‐acetaminophen. (B) Mice received NAC (300 mg·kg−1) or PBS at 2, 3 or 4 h after acetaminophen (300 mg·kg−1, i.p.) administration, and serum ALT levels were measured at 24 h post‐acetaminophen. (C) C57BL/6N mice received GCM (100 mg·kg−1, i.p.) or NAC (300 mg·kg−1, i.p.) at 4 h after acetaminophen treatment, and serum ALT levels were determined at 24 h post‐acetaminophen. (D) Representative images of haematoxylin and eosin‐stained liver sections at 100× magnification. Data are presented as mean ± SD for n = 6 mice per group. *P < 0.05 compared with the corresponding acetaminophen group.
Discussion
A hepatoprotective effect of liquorice has been manifested in multiple clinical and animal studies; this encouraged us to search for promising bioactive compounds that contribute to the hepatoprotective effect of this medicinal and edible plant. In our previous studies, we isolated and identified an active ingredient GCM from liquorice, and this coumarin compound proved to possess potent protective effects against palmitate‐induced lipoapoptosis and alcohol‐induced hepatotoxicity (Song et al., 2015; Zhang et al., 2016). Based on these in vitro and in vivo studies, we speculated that GCM could have a protective effect in other liver injury models. The findings of the present study indeed provide evidence that GCM is highly effective against acetaminophen‐induced hepatotoxicity. Moreover, GCM is superior to NAC in terms of the dosage and the therapeutic time windows.
Acetaminophen‐induced hepatotoxicity is initiated by the formation of an electrophilic metabolite via the cytochrome P450 enzymes (Nelson, 1990). Hence, any interference with this metabolic activation process might have a pronounced impact on the subsequent liver injury. Moreover, patients often present late in the course of AILI, and the treatment is often delayed. Taken together, therapeutic approaches rather than a pretreatment regimen are more clinically relevant. Based on this mechanistic insight, we chose a therapy model to perform our study. When GCM was administered 2 h after a toxic dose of acetaminophen (300 mg·kg−1), the mice showed reduced hepatotoxicity, as revealed by decreased serum ALT levels and areas of hepatic necrosis. It has been demonstrated that the conversion of acetaminophen into the highly reactive intermediate metabolite NAPQI is nearly complete at 2 h after acetaminophen injection (McGill et al., 2013). Therefore, it is unlikely that the production of NAPQI was affected by GCM when it was given at 2 h post‐acetaminophen challenge, ruling out inhibition of acetaminophen metabolic activation as a key mechanism contributing to the hepatoprotective effect of GCM.
As previously reported, the generation of ROS induced by acetaminophen leads to sustained activation of JNK, which then amplifies mitochondrial ROS and forms a self‐sustaining activation loop (Win et al., 2016). Hence, interference with any elements in this sustained JNK activation loop would limit ROS generation and JNK phosphorylation and finally provide protection against acetaminophen hepatotoxicity. Regarding the mechanisms underlying the protective effect of GCM against AILI, our data demonstrated that GCM inhibited JNK activation and its mitochondria translocation, as well as the induction of 3‐NT, suggesting a positive role of GCM in the alleviation of oxidative stress.
It is believed that NAPQI generation and the subsequent GSH depletion lead to an imbalance in the redox system of hepatocytes (Jaeschke et al., 2012). Some cell defence signalling pathways are therefore activated in response to this oxidative stress such as Nrf2. Mechanistically, changes in redox status of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2757 induced by NAPQI leads to Nrf2 translocation to the nucleus and transcriptional activation of antioxidant enzymes, which represents a compensatory mechanism to protect against acetaminophen hepatotoxicity. Hence, Nrf2 KO mice are much more susceptible to AILI than wild‐type mice (Goldring et al., 2004). Due to the potent ability of GCM to stimulate Nrf2, we examined whether the attenuation of oxidative stress induced by GCM in this model was dependent on an Nrf2‐mediated antioxidant defence pathway. Unexpectedly, in the Nrf2 KO mice, we still observed the hepatoprotective effect of GCM, precluding a role for the Nrf2 signalling pathway in the protective effect of GCM against acetaminophen hepatotoxicity. The aforementioned results raise a question –what is the mechanism of the GCM‐mediated protective effect in wild type and Nrf2 KO animals following acetaminophen administration? Apart from Nrf2, GCM has been reported to exhibit a potent ability to activate autophagy, which has been proved to be protective in acetaminophen‐induced hepatotoxicity. Autophagy is activated by acetaminophen‐induced cellular stress and serves as an adaptive, protective mechanism through the removal of acetaminophen‐protein adducts and damaged mitochondria (Ni et al., 2012, 2016). Indeed, our results showed that autophagy was activated rapidly at 3 h following the administration of a toxic dose of acetaminophen but then gradually got back to normal levels. By contrast, GCM treatment enhanced this adaptive cellular response at each time point, suggesting the involvement of autophagy in the therapeutic effect of GCM. Additionally, inhibition of autophagy by chloroquine abolished the down‐regulation of p‐JNK and 3‐NT induced by GCM treatment. Based on these data, we conclude that sustained autophagy stimulated by GCM protects against acetaminophen‐induced hepatotoxicity by alleviating oxidative stress.
NAC is the only recommended clinical antidote for treating acetaminophen‐induced acute liver injury (Polson et al., 2005). The mechanism of action of NAC is to provide a synthetic precursor for synthesis of GSH in hepatocytes, which can directly detoxify NAPQI (Lauterburg et al., 1983). As such, the therapeutic window for NAC in mice is limited to within the acetaminophen metabolism phase, and the efficacy of this drug gradually diminishes and ultimately disappears by 3–4 h (James et al., 2003). Unfortunately, many acetaminophen overdose patients often present beyond the metabolism phase when the liver injury is already evident (Larson et al., 2005). Hence, medications that still have therapeutic potential beyond the early stage would be of great help. Although it was generally thought that the protective role of autophagy in acetaminophen toxicity was limited to the oxidative damage phase and primarily targeted at damaged mitochondria (Ni et al., 2012, 2016), our result revealed a sustained activation of autophagy after GCM treatment, which was even observed 12 h post‐acetaminophen challenge, after oxidative stress had already developed. This suggests that a late stage mechanism, such as suppression of ER stress, might be involved in the autophagy‐mediated hepataprotective effect of GCM (Yan et al., 2018). Although the precise mechanism by which GCM exerts its therapeutic effect against acetaminophen hepatotoxicity in the late phase remains unclear, we observed a better therapeutic effect of GCM against acetaminophen hepatotoxicity compared with NAC, as revealed by the lower effective dose and wider therapeutic window. Nonetheless, it should be noted that the toxicity of GCM is as yet unclear, and this clearly needs further investigation.
In summary, the therapeutic effect of GCM against acetaminophen‐induced hepatotoxicity predominantly stems from its sustained activation of autophagy, which could inhibit acetaminophen overdose‐evoked oxidative stress, thereby protecting mice from liver damage. Given the remarkable hepatoprotective effect of GCM, this coumarin compound is worthy of further exploration as a novel potential therapeutic agent for treating patients who have overdosed on acetaminophen.
Author contributions
M.Y. designed and performed experiments and wrote the manuscript. L.Y. performed the isolation and purification of GCM. S.Y., X.L., X.L., S.L., J.C. and L.F. helped with the animal experiments. N.K. and H.H. supervised the study and revised the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Yan, M. , Ye, L. , Yin, S. , Lu, X. , Liu, X. , Lu, S. , Cui, J. , Fan, L. , Kaplowitz, N. , and Hu, H. (2018) Glycycoumarin protects mice against acetaminophen‐induced liver injury predominantly via activating sustained autophagy. British Journal of Pharmacology, 175: 3747–3757. 10.1111/bph.14444.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal W, Auzinger G, Dhawan A, Wendon J (2010). Acute liver failure. Lancet 376: 190–201. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D et al (2015). Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 62: 1847–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Ramachandran A, Jaeschke H (2016). Oxidative stress during acetaminophen hepatotoxicity: sources, pathophysiological role and therapeutic potential. Redox Biol 10: 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldring CE, Kitteringham NR, Elsby R, Randle LE, Clement YN, Williams DP et al (2004). Activation of hepatic Nrf2 in vivo by acetaminophen in CD‐1 mice. Hepatology 39: 1267–1276. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, McGill MR, Ramachandran A (2012). Oxidant stress, mitochondria, and cell death mechanisms in drug‐induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 44: 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Williams CD, McGill MR, Xie Y, Ramachandran A (2013). Models of drug‐induced liver injury for evaluation of phytotherapeutics and other natural products. Food Chem Toxicol 55: 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James LP, McCullough SS, Lamps LW, Hinson JA (2003). Effect of N‐acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol Sci 75: 458–467. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. J Pharmacol Pharmacother 1: 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS et al (2005). Acetaminophen‐induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42: 1364–1372. [DOI] [PubMed] [Google Scholar]
- Lauterburg BH, Corcoran GB, Mitchell JR (1983). Mechanism of action of N‐acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 71: 980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WM (2013). Drug‐induced acute liver failure. Clin Liver Dis 17: 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Lebofsky M, Norris HR, Slawson MH, Bajt ML, Xie Y et al (2013). Plasma and liver acetaminophen‐protein adduct levels in mice after acetaminophen treatment: dose‐response, mechanisms, and clinical implications. Toxicol Appl Pharmacol 269: 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SD (1990). Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin Liver Dis 10: 267–278. [DOI] [PubMed] [Google Scholar]
- Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX (2012). Activation of autophagy protects against acetaminophen‐induced hepatotoxicity. Hepatology 55: 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, McGill MR, Chao X, Du K, Williams JA, Xie Y et al (2016). Removal of acetaminophen protein adducts by autophagy protects against acetaminophen‐induced liver injury in mice. J Hepatol 65: 354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polson J, Lee WM, American Association for the Study of Liver Disease (2005). AASLD position paper: the management of acute liver failure. Hepatology 41: 1179–1197. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Benet LZ, Burlingame AL (1998). Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two‐dimensional gel electrophoresis and mass spectrometry. J Biol Chem 273: 17940–17953. [DOI] [PubMed] [Google Scholar]
- Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L et al (2016). Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1‐deficient NSCLC tumors. ACS Chem Biol 11: 3214–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Yin S, Huo Y, Liang M, Fan L, Ye M et al (2015). Glycycoumarin ameliorates alcohol‐induced hepatotoxicity via activation of Nrf2 and autophagy. Free Radic Biol Med 89: 135–146. [DOI] [PubMed] [Google Scholar]
- Win S, Than TA, Han D, Petrovic LM, Kaplowitz N (2011). cJun N‐terminal kinase (JNK)‐dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem 286: 35071–35078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win S, Than TA, Min RW, Aghajan M, Kaplowitz N (2016). c‐Jun N‐terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)‐dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 63: 1987–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Huo Y, Yin S, Hu H (2018). Mechanisms of acetaminophen‐induced liver injury and its implications for therapeutic interventions. Redox Biol 17: 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang E, Yin S, Song X, Fan L, Hu H (2016). Glycycoumarin inhibits hepatocyte lipoapoptosis through activation of autophagy and inhibition of ER stress/GSK‐3‐mediated mitochondrial pathway. Sci Rep 6: 38138. [DOI] [PMC free article] [PubMed] [Google Scholar]