

Abstract
Background and Purpose
The endocannabinoid system and PPARγ are important targets for the development of novel compounds against fibrotic diseases such as systemic sclerosis (SSc), also called scleroderma. The aim of this study was to characterize VCE‐004.3, a novel cannabidiol derivative, and study its anti‐inflammatory and anti‐fibrotic activities.
Experimental Approach
The binding of VCE‐004.3 to CB1 and CB2 receptors and PPARγ and its effect on their functional activities were studied in vitro and in silico. Anti‐fibrotic effects of VCE‐004.3 were investigated in NIH‐3T3 fibroblasts and human dermal fibroblasts. To assess its anti‐inflammatory and anti‐fibrotic efficacy in vivo, we used two complementary models of bleomycin‐induced fibrosis. Its effect on ERK1/2 phosphorylation induced by IgG from SSc patients and PDGF was also investigated.
Key Results
VCE‐004.3 bound to and activated PPARγ and CB2 receptors and antagonized CB1 receptors. VCE‐004.3 bound to an alternative site at the PPARγ ligand binding pocket. VCE‐004.3 inhibited collagen gene transcription and synthesis and prevented TGFβ‐induced fibroblast migration and differentiation to myofibroblasts. It prevented skin fibrosis, myofibroblast differentiation and ERK1/2 phosphorylation in bleomycin‐induced skin fibrosis. Furthermore, it reduced mast cell degranulation, macrophage activation, T‐lymphocyte infiltration, and the expression of inflammatory and profibrotic factors. Topical application of VCE‐004.3 also alleviated skin fibrosis. Finally, VCE‐004.3 inhibited PDGF‐BB‐ and SSc IgG‐induced ERK1/2 activation in fibroblasts.
Conclusions and Implications
VCE‐004.3 is a novel semisynthetic cannabidiol derivative that behaves as a dual PPARγ/CB2 agonist and CB1 receptor modulator that could be considered for the development of novel therapies against different forms of scleroderma.
Abbreviations
- BLM
bleomycin
- CB1
cannabinoid receptor type I
- CB2
cannabinoid receptor type II
- CBD
cannabidiol
- ECS
endocannabinoid system
- LBP
ligand binding pocket
- NHDFs
normal human dermal fibroblasts
- RGZ
rosiglitazone
- SSc
systemic sclerosis
- α‐SMA
α‐smooth muscle actin
Introduction
Systemic sclerosis (SSc) or scleroderma is a chronic multiorgan autoimmune disease of unknown aetiology characterized by vascular, immunological and fibrotic abnormalities. The disease is complex and dynamic, and the interrelationship among the three main hallmarks of SSc results in a wide spectrum of clinical presentations ranging from limited skin involvement [limited cutaneous SSc (lcSSc)] to widespread internal organ fibrosis [diffuse cutaneous SSc (dcSSc)]. The pathogenesis of SSc is not fully understood, and it is believed that inflammation as well as vascular injury in the initial stages drive the autoimmune response and precede fibrosis in the course of the disease (Varga and Abraham, 2007; Pattanaik et al., 2015).
Several lines of evidence have shown that the endocannabinoid system (ECS) may play a role in the physiopathology of SSc. Cannabinoids exert a broad range of biological activity including anti‐inflammatory effects (Katchan et al., 2016), effects on endothelial function (Bouchard et al., 2003) and cell death regulation (Garcia‐Gonzalez et al., 2009). The ECS is composed of cannabinoid receptors, http://www.guidetoimmunopharmacology.org/GRAC/ObjectDisplayForward?objectId=56 and http://www.guidetoimmunopharmacology.org/GRAC/ObjectDisplayForward?objectId=57, their endogenous lipid ligands represented by anandamide (AEA) and 2‐arachidonoylgylcerol (2‐AG) and the enzymes responsible for their degradation (Pacher et al., 2006). CB1 and CB2 receptors are overexpressed in SSc fibroblasts (Garcia‐Gonzalez et al., 2009), and the anti‐inflammatory and anti‐fibrotic actions of cannabinoids have been widely demonstrated both in vitro and in vivo. Thus, inactivation of CB1 receptors decreased the number of infiltrating T‐cells and macrophages in lesioned skin (Marquart et al., 2010). In contrast, CB2 receptor activation limits leukocyte infiltration and tissue fibrosis (Akhmetshina et al., 2009). Thereby, structurally different CB2 agonists such as ajulemic acid, JHW‐133 and VCE‐004.8 have been shown to alleviate skin fibrosis and inflammation in experimental SSc (Akhmetshina et al., 2009; Gonzalez et al., 2012; del Rio et al., 2016).
In addition to binding to classical CB1 and CB2 receptors, some natural and semisynthetic cannabinoids also bind and activate the nuclear hormone receptor http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=595 (Pistis and O'Sullivan, 2017). PPARγ was initially identified through its role in the regulation of glucose and lipid metabolism and cell differentiation, but it is now well established that PPARγ agonists are also exhibit anti‐inflammatory and anti‐fibrotic effects (Clark, 2002; Dantas et al., 2015). In this regard, PPARγ is a regulator of connective tissue homeostasis, and different experimental approaches have shown that PPARγ ligands attenuate hepatic (Galli et al., 2002), renal (Bae et al., 2017) and pulmonary fibrosis (Milam et al., 2008) as well as bleomycin‐induced skin fibrosis (Gonzalez et al., 2012; Wei et al., 2014; del Rio et al., 2016; Ruzehaji et al., 2016).
Different serum autoantibodies against multiple intracellular antigens have been detected and proposed as biomarkers for early and precise diagnosis in SSc (Kayser and Fritzler, 2015). However, how autoantibodies contribute to the prognosis of SSc is not fully understood. Autoantibodies directed against non‐nuclear autoantigens including anti‐PDGF receptor, anti‐endothelial cells, anti‐fibroblast and anti‐angiotensin type 1‐receptor autoantibodies with possible pathogenic properties have been also described (Ho and Reveille, 2003; Baroni et al., 2006; Fineschi et al., 2008; Mihai and Tervaert, 2010; Kayser and Fritzler, 2015).
Hence, targeting the ECS and PPARγ, which are considered potential therapeutic targets in SSc, represents a viable approach to prevent or attenuate inflammation and fibrosis in SSc. In the present study, we demonstrated that the novel http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4150 aminoquinone derivative VCE‐004.3, which targets PPARγ and CB2 receptors, but also antagonizes CB1 receptors, exhibits anti‐inflammatory and anti‐fibrotic effects.
Methods
Cell lines
HEK‐293T, HEK‐293T‐CB1, HEK‐293T‐CB2, NIH‐3T3 and normal human dermal fibroblasts (NHDFs) were cultured in DMEM supplemented with 10% FBS, 2 mM L‐glutamine and 1% penicillin/streptomycin. All cells were maintained at 37°C and 5% CO2 in a humidified atmosphere.
VCE‐004.3 synthesis
VCE‐004.3 was synthesized by oxidizing CBD with the hypervalent iodine reagent SIBX as previously described (del Rio et al., 2016) and then trapping it with n‐pentylamine (four molar equivalents) in ethanol solution (ca. 6 mL·mmol−1 of quinone). The mixture was stirred at room temperature for 18 h, diluted with water (1:5), acidified to pH = 2 with HCl (10% aqueous solution) and then extracted with CH2Cl2 (30 mL). The organic layer was dried (Na2SO4), filtered and concentrated, and the residue was purified by reverse phase chromatography (30 at 100% CH3CN and H2O) to yield (1′R,6′R)‐3‐(pentylamino)‐6‐hydroxy‐3′‐methyl‐4‐pentyl‐6′‐(prop‐1‐en‐2‐yl)‐[1,1′‐bi(cyclohexane)]‐2′,3,6‐triene‐2,5‐dione as a purple‐coloured powder (ca. 70% yield). 1H NMR (CDCl3, 300 MHz) d ppm: 6.43 (bs, 1H), 5.14 (s, 1H), 4.55 (s, 2H), 3.62 (m, 1H), 3.46 (c, J = 6.6 Hz, 2H), 2.72 (m, 1H), 2.48 (t, J = 7.7 Hz, 2H), 2.31‐1.72 (m, 4H), 1.68 (s, 3H), 1.64 (s, 3H), 1.48‐1.24 (m, 12H), 0.90 (m, 6H). MW: 413.61.
Binding assay
PPARγ binding activity was measured by using a PolarScreen™ PPAR Competitor Assay Kit (Life Technologies, Carlsbad, CA, USA) following the manufacturer's instructions. IC50 values were calculated using GraphPad Prism. Binding affinities to cannabinoid receptors were tested by competition studies using commercial membranes containing HEK293 EBNA cells stably expressing human CB1 or CB2 receptors (Perkin‐Elmer Life and Analytical Sciences, Boston, MA, USA). Inhibition of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=734 binding was measured following a previously described procedure (Ragusa et al., 2015). Competition binding data were analysed by using GraphPad Prism® version 5.01 (GraphPad Software Inc., CA, USA).
Docking analysis
Docking analysis was performing using AutoDock4 (Morris et al., 2009) and Vina softwares (Trott and Olson, 2010) with the virtual screening tool PyRx (Wolf, 2009) and PyMOL (Baugh et al., 2011). The receptor model used was the Protein Data Bank (PDB) reference (RCSB PDB accession code), 3B0R (Hughes et al., 2014), and 5U5L (Rajapaksha et al., 2017). The search space for the docking was set according to previous findings about several binding sites for different ligands. After analysis, AutoDock Vina provides the estimated binding affinity value, which is the sum of the intermolecular energy and the torsional free‐energy penalty. A negative value indicates that a bond is thermodynamically stable, whereas a positive value means it is unstable. Search space for the docking was set around the binding sites described previously (Hughes et al., 2014).
Cytotoxicity assay
NIH‐3T3 (1 × 103 cells) were seeded in 96 well plates and after 24 h incubated with VCE‐004.3 for another 6 h in the presence of 0.1 μM YOYO‐1 (Life Technologies). The survival of untreated cells was considered to be 100%. Fluorescence was measured using an Incucyte FLR imaging system (Essen BioScience, Ann Arbor, MI, USA).
Transfections and luciferase assays
HEK‐293T (8 × 104) cells stably expressing human cannabinoid CB1 or CB2 receptors were seeded in 24‐well plates and after 24 h were transiently transfected with the pCRE‐luc plasmid (0.2 μg per well) using Roti©‐Fect (Carl Roth, Karlsruhe, Germany) following the manufacturer's instructions. The cAMP response element (CRE) luciferase reporter contains the firefly luciferase gene under the control of multimerized CRE located upstream of a minimal promoter. Twenty‐four hours after transfection, CB1‐HEK‐293T cells were pretreated for 30 min with VCE‐004.3 and stimulated with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=733 (1 μM) for 6 h. CB2‐HEK‐293T cells were pretreated for 30 min with VCE‐004.3 or WIN55,212 as positive control and stimulated with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5190 (10 μM) for 6 h. NIH‐3T3 (5 × 104) cells were seeded in 24‐well plates and after 24 h were transiently transfected with the indicated constructs (PPARγ‐GAL4, 0.2 μg; PPARα‐GAL4, 0.2 μg; PPARδ‐GAL4, 0.2 μg; GAL4‐luc, 0.8 μg; or CAGA‐luc, 0.5 μg). NIH‐3T3 stably expressing Col1A2‐luc were seeded in 24‐well plates (5 × 104 cells per well) and treated as indicated. After treatments, luciferase activity was measured using a Dual‐Luciferase Assay (Promega, Madison, WI, USA).
Measurement of collagen secretion and accumulation
NHDFs were seeded in 24‐well plates (5 × 104 cells per well), pretreated with VCE‐004.3 for 1 h and stimulated with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5060 for the following 24 h. The deposits of collagen were determined by measuring absorbance at 540 and 605 nm in a Genesis 10 UV spectrophotometer after cells had been stained for 30 min with a mixture of 0.1% Sirius Red and 0.1% Fast Green in saturated picric acid. Collagen secretion was studied by collecting culture medium and measuring soluble collagen using the Sircol Soluble Collagen Assay (Biocolor, County Antrim, UK) following the manufacturer's instructions.
Western blot
Cells were seeded in 6‐well plates (1.5 × 105 cells) or in 60 mm plates (3 × 105 cells). After the various treatments, cells were washed with PBS and proteins extracted in 50 μL of lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 10% glycerol and 1% NP‐40) with 10 mM NaF, 1 mM Na3VO4, 10 μg·mL−1 leupeptine, 1 μg·mL−1 pepstatin and aprotinin and 1 μL·mL−1 saturated PMSF; 30 μg of proteins were boiled at 95°C in Laemmli buffer and electrophoresed in 10% SDS/PAGE gels. Separated proteins were transferred (20 V for 30 min) to PVDF membranes and blocked in 0.1% Tween 20 in TBS solution containing 5% skimmed milk for 1 h at room temperature. Membranes were incubated with primary antibodies against pSMAD2 (1:500; AB3849, Merck Millipore, Billerica, MA, USA), SMAD2 (1:1000; Cat # 5339, Cell Signalling, Danvers, MA, USA), p‐http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514 (1:500; sc‐7383, Santa Cruz Biotechnology, Dallas, TX, USA) and ERK 1/2 (1:2000; M5670, Sigma, St Louis, MO, USA) overnight at 4°C. Membranes were washed and incubated with the appropriate HRP‐conjugated secondary antibody for 1 h at room temperature and detected by chemiluminescence system (GE Healthcare Europe GmbH, Freiburg, Germany). Blots were normalized to those obtained with antibodies against α‐tubulin or β‐actin (1:10 000; DM‐1A and AC‐74, Sigma). Densitometric analysis was performed with ImageJ software (NIH, Bethesda, MD, USA).
Cell migration assay
NHDFs (2 × 103 cells) were seeded in complete DMEM in a 96‐well Essen ImageLock plates (Essen BioScience). When cells reached confluence, wounds were made by scratching using the 96‐pin WoundMaker (Essen BioScience). Then, culture medium was replaced by DMEM supplemented with 1% antibiotics and with mitomycin C (10 ng·mL−1) to block cell proliferation. VCE‐004.3 and TGFβ1 10 ng·mL−1 were added in parallel. Wound healing activity was analysed using IncuCyte HD system by taking images every 3 h for 48 h and the percentage of wound confluence calculated with the IncuCyte software.
Fibroblast to myofibroblast differentiation
NIH‐3T3 cells (1.5 × 104) were seeded onto glass coverslips or directly in 6‐well plates. Cells were serum starved (1% FBS) for 24 h, pre‐stimulated with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1056 (rosiglitazone) or VCE‐004.3 for 1 h and incubated with 10 ng·mL−1 TGFβ1 for 24 h. Then, cells were washed with PBS for further Western blot analysis or fixed with 4% formaldehyde for 10 min at 37°C. After fixation, cells were washed twice with PBS and blocked in IHC selective blocking reagent (Merck Millipore) for 30 min. Cells were incubated overnight at 4°C with the primary antibody for α‐smooth muscle actin (α‐SMA) (1:50 in blocking solution, sc‐32 251, Santa Cruz), washed three times with PBS containing 0.1% Tween 20 and incubated with the secondary antibody goat anti‐rat coupled to Alexa 488 (1:1000, Merck Millipore) for 3 h at room temperature. Coverslips were mounted with Vectashield Mounting Medium supplemented with DAPI for nuclear staining. Observations were carried out using a Leica DM2500 microscope and Leica DFC420c camera. Images were acquired as sets of colour images and prepared using ImageJ software.
Animal experiments
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). A total of 90 BALB/C female mice (6–8 weeks old) weighing between 17 and 20 g (Envigo, Valencia, Spain) were used in the studies. Experiments were performed in accordance with European Union guidelines and were approved by the Animal Research Ethic Committee of Córdoba University and the Andalusian Committee for Animal Experimentation (2014PI/016). Mice were housed in groups of nine animals in polycarbonate cages (300 mm × 180 mm × 150 mm) with free access to food and water, in the animal facilities of the University of Córdoba, and maintained in a 12 h light/dark cycle with controlled temperature (20 ± 2°C) and relative humidity (40–50%) (McGrath and Lilley, 2015).
Different administration schedules of s.c. bleomycin (BLM) have been widely used as SSc models. Repetitive injections of s.c. BLM induce skin fibrosis localized to the injection area and lead to systemic symptoms such as lung fibrosis (Beyer et al., 2010). Two different BLM‐induced models were used to assess the effects of VCE‐004.3 in vivo. (i) The inflammatory model of SSc was used to study the preventive effects of VCE‐004.3. Fibrosis was induced by daily s.c. administration of BLM (50 μg per mice; 100 μL) (Mylan, Barcelona, Spain) for 3 weeks. Treatments were administered in parallel and consisted of daily i.p. injections of VCE‐004.3 (20 mg·kg−1), RGZ (5 mg·kg−1) or vehicle (4% DMSO, 6.2% Tween 20, saline; 100 μL). S.c. injections of 0.9% NaCl served as a control. (ii) A pre‐established fibrosis model of SSc was induced to study the curative effect of VCE‐004.3. Fibrosis was induced by daily s.c. injections of BLM (20 μg per mice; 100 μL) for 6 weeks. After 3 weeks of BLM administration, animals were treated in parallel with a daily i.p. injection of VCE‐004.3 (20 mg·kg−1), RGZ (5 mg·kg−1) or vehicle or were given topical application of VCE‐004.3 (250 μM) or vehicle (7:3 polypropylene glycol : ethanol; 100 μL) for the remaining 3 weeks. Mice were acclimatized to manipulators for a week before the experiments in order to reduce stress and allow the detection of subtle changes in behaviour by researchers. Mice weight was evaluated weekly, and no significant changes were observed between experimental groups. No behavioural changes were observed during the protocols. Neither piloerection nor hunched appearances were apparent during the experimental procedures. Mice were killed by cervical dislocation, and the back skin was removed and processed for histological examination or was frozen for further analysis. Macroscopic evaluation of internal organs did not reveal any pathological changes.
Blinding and randomization
Laboratory animals were randomly assigned to experimental groups, and treatments were assessed by a blinded investigator. The order of treatment administration was also randomized. All animal samples were studied, and analysis was carried out in a blinded manner. For immunohistochemical studies, three random fields of each skin biopsy were photographed and evaluated by two independent observers.
Histochemical analysis
Skin sections (5 μm‐thick) were stained with Masson's trichrome, picrosirius red or toluidine blue. Myofibroblasts were detected in skin sections by immunofluorescent labelling with α‐SMA (1:50, sc‐32 251, Santa Cruz) and Alexa 488 (A‐11008; Life Technologies). For quantification, images were taken with a LSM 5 Exciter confocal microscope (Zeiss, Jena, Germany) and quantified in 10 to 15 randomly chosen fields at 250 magnification using ImageJ software (http://rsb.info.nih.gov/.ij). For immunohistochemical detection of macrophages, T‐cells and ERK1/2 activation, we used F4/80 (1:50; MCA497, Bio‐Rad), CD3 (1:100, sc20047, Santa Cruz) and p‐ERK 1/2 (1:100; sc‐7383, Santa Cruz) antibodies respectively. Slides were developed with diaminobenzidine chromogen (Merck Millipore) and counterstained with Harris haematoxylin solution. Slides were photographed, digitized using a Leica DFC420c camera and analysed using ImageJ software.
Real‐time PCR
Total RNA was isolated from mice frozen skin tissue using QIAzol Lysis Reagent (Qiagen, Hilden, Germany) and purified with RNeasy Mini Kit (Qiagen). Total RNA (1 μg) was retrotranscribed using the iScript™ cDNA Synthesis Kit (Bio‐Rad), and the cDNA generated was analysed by real‐time PCR using the iQTM SYBR Green Supermix (Bio‐Rad) using a CFX96 Real‐Time PCR Detection System (Bio‐Rad). The oligonucleotide primers used are listed in Supporting Information Table S1. Gene expression was normalized to GADPH mRNA levels in each sample and expressed using the 2−ΔΔCt method.
Patients and IgG antibodies purification
In total, 12 patients, six with lSSc and six with dSSc, were included in the study after ethics committee approval. All subjects provided written informed consent. Samples were obtained from peripheral venous blood. IgGs were purified using a pool of six patients with lcSSc and six patients with dSSc using high affinity chromatography of G‐sepharose protein (MabTrap, GE Healthcare Bio‐Sciences AB, Uppsala, Sweden) following the manufacturer's recommendations.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). In vitro results are shown as mean ± SD and in vivo data as mean ± SEM. Statistical analysis was carried out using the SPSS v.19 software for Windows (IBM Corporation, New York, USA). Results were tested for normal distribution using Kolmogorov–Smirnov normality test. Then, data were analysed using one‐way ANOVA followed by Tukey's post hoc test when F achieved was P < 0.05, and there was no significant variance in homogeneity. Some results were normalized to control to avoid unwanted sources of variation. Differences between not normally distributed results were studied by the Kruskall–Wallis followed by Dunn's post hoc test using the GraphPad Prism v.5 for Windows. Minimal statistical significance was set at P < 0.05.
Materials
Rosiglitazone (RGZ) was from Cayman Chem (Ann Arbor, MI, USA), TGFβ1 was obtained from Immunotools GmbH (Friesoythe, Germany) and non‐immunized human IgG was from Jackson Immunoresearch (West Grove, PA, USA). All other reagents were purchased from Sigma Co (St Louis, MO, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018) and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c).
Results
VCE‐004.3 is a CBD quinone derivative with selective PPARγ and CB 2 agonism and CB 1 antagonism
We have previously shown that VCE‐004.8, a dual PPARγ/CB2 agonist, alleviates dermal fibrosis through PPARγ and CB2 receptor‐dependent pathways in a mice model of SSc (del Rio et al., 2016). As part of our programme of chemical synthesis searching for CBD quinone derivatives, we selected the novel compound VCE‐004.3 (Figure 1A) and investigated its mechanism of action and efficacy in dermal inflammation and fibrotic events. Firstly, we evaluated the binding affinity to PPARγ and found that VCE‐004.3 was able to bind PPARγ with an IC50 of 3.5 μM, which is a significantly lower affinity than the binding of RGZ (Figure 1B). VCE‐004.3 was not cytotoxic and induced PPARγ transcriptional activity in a concentration‐dependent manner in NIH‐3T3 cells transiently co‐transfected with GAL4‐luc and PPARγ‐GAL4 plasmids (Figure 1C). However, VCE‐004.3 did not activate PPARα (Figure 1D) or PPARδ (Figure 1E) transcription, indicating that VCE‐004.3 is a selective agonist of the PPARγ isoform.
Figure 1.
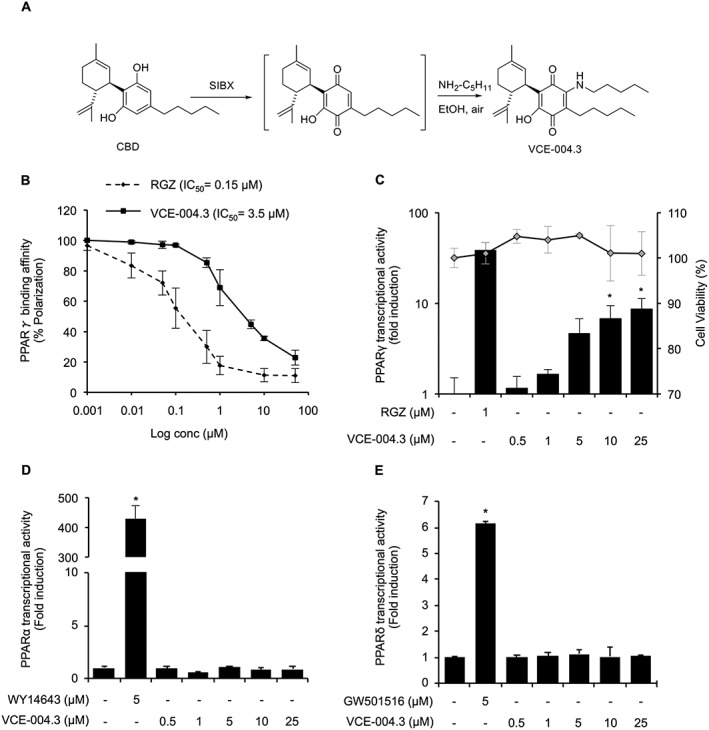
VCE‐004.3 is a selective PPARγ agonist. (A) Schematic for synthesis of VCE‐004.3. (B) The binding affinity of VCE‐004.3 and RGZ to PPARγ. Results are presented as a logarithmic scale, and IC50 values were calculated as the 50% inhibition of percentage of fluorescence polarization (n = 5). (C) PPARγ transcriptional activity induced by VCE‐004.3 its cytotoxicity in NIH‐3T3 fibroblasts. Cells were co‐transfected with PPARγ‐GAL4 and GAL4‐luc constructs, stimulated with VCE‐004.3 or RGZ as a positive control for 6 h and lysed for luciferase activity. Results are presented in a bar chart and shown as fold induction compared to control (n = 10). To test cytotoxicity, cells were treated with the compound for 6 h in the presence of YOYO‐1, and fluorescence intensity was measured and presented in a dot chart (n = 5). (D and E) HEK‐293T cells were co‐transfected with PPARα‐GAL4 (D) or PPARδ‐GAL4 (E) and GAL4‐luc, treated with VCE‐004.3 for 6 h and lysed for luciferase activity (n = 5). Results are shown as mean ± SD. *P < 0.05 versus control.
To further characterize the effects of VCE‐004.3 on PPARγ transcriptional activities, we performed experiments to study the behaviour of VCE‐004.3 in the presence of RGZ. To this end, GAL4‐PPARγ/GAL4‐luc transfected NIH‐3T3 cells were preincubated with increasing concentrations of VCE‐004.3 and then treated with 1 μM RGZ. VCE‐004.3 decreased the RGZ‐induced PPARγ transactivation (Figure 2A), suggesting that VCE‐004.3 could prevent RGZ binding by competing with the same binding site on PPARγ. Moreover, the stimulating effect on PPARγ was eliminated after removal of VCE‐004.3 by washing cells with PBS, suggesting that VCE‐004.3 binds to PPARγ in a reversible and non‐covalent manner (Figure 2B).
Figure 2.
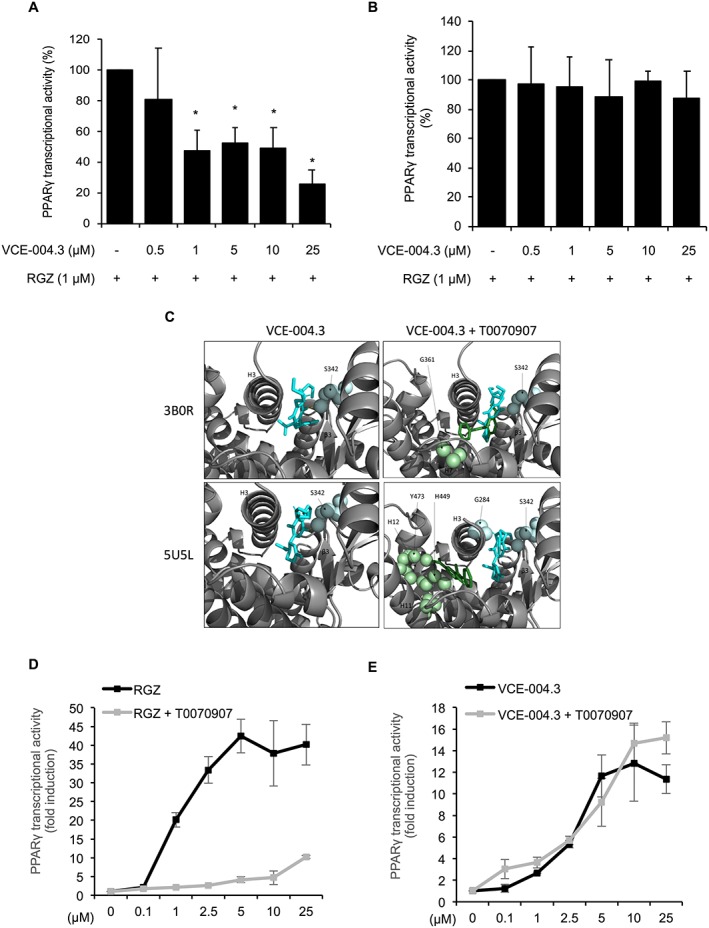
VCE‐004.3 competed with RGZ for the canonical pocket and binds to an alternate site in PPARγ. NIH‐3T3 cells were transfected with PPARγ‐GAL4 plus GAL4‐luc. (A) Cells were pretreated with VCE‐004.3 for 1 h and then incubated for 6 h in the presence of RGZ (n = 5). (B) Cells were pretreated with VCE‐004.3 for 1 h and then washed with PBS and stimulated with RGZ for 6 h (n = 5). (C) PPARγ LBD structures 3B0R and 5U5L bound to VCE‐004.3 (blue) with and without GW9662 (orange). (D and E) NIH‐3T3 cells were transfected with PPARγ‐GAL4 plus GAL4‐luc, pretreated with T0070907 (5 μM) for 30 min and stimulated with RGZ (D) or VCE‐004.3 (E) for 6 h (n = 5). Cells were lysed and tested for luciferase activity. Results are shown as mean ± SD. *P < 0.05 versus untreated cells.
It was previously reported that synthetic PPARγ ligands bind to the canonical ligand binding pocket (LBP) and also to an alternate site on this nuclear receptor, an effect that cannot be blocked by covalently bound synthetic antagonists, demonstrating non‐overlapping binding with the canonical pocket (Hughes et al., 2014). Therefore, we performed docking experiments using crystal structures 3B0R and 5U5L deposited in the PDB. As depicted in Figure 2C, molecular docking with 3B0R indicated that VCE‐004.3 binds Leu340 in Ω‐loop β3 and Tyr373 in Helix 1/2 (alternative site) with a predicted K i of 108.38 nM. In the presence of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3442 in the canonical site, VCE‐004.3 binds to Ser342 in the Ω‐loop β3–β4 and Leu340 in β3 with a predicted K i of 569.80 nM. In the case of 5U5L, VCE‐004.3 was found to bind Ser342 in the Ω‐loop β3–β4 and Gly346 in β4 with a predicted K i of 1.12 μM, and in the presence of GW9662, we found that VCE‐004.3 binds to Leu340 in β3 and Ser342 in Ω‐loop β3–β4 with a predicted K i of 17.72 nM.
To confirm that VCE‐004.3 is functional through the alternative binding site, we studied VCE‐004.3‐mediated PPARγ transcriptional activity in the presence of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=3444, an irreversible PPARγ antagonist that covalently binds to Cys285 in the PPARγ LBP canonical binding site. As expected, T0070907 was very effective at blocking RGZ‐induced PPARγ transactivation (Figure 2D), which is consistent with the fact that RGZ activates PPARγ by acting mainly through the canonical binding site (Hughes et al., 2014). By contrast, T0070907 did not block the transcriptional activity of VCE‐004.3 (Figure 2E). Altogether, our results strongly suggest that, in contrast to RGZ, VCE‐004.3 is a PPARγ modulator (m‐PPARγ) acting preferentially through the PPARγ LBP alternative binding site. In this sense, human mesenchymal stem cells treated with VCE‐004.3 showed fewer and smaller lipid droplets and lower expression levels of adipogenic differentiation markers such as PPARγ, ADIPOQ and CEBPA compared to cells treated with the PPARγ full agonist (PPARγ‐fa) RGZ (Supporting Information Figure S1).
Next, we studied the effect of VCE‐004.3 on CB1 and CB2 receptors. VCE‐004.3 was able to bind CB1 receptors with a pKi value of 5.61 ± 0.15 (Figure 3A) and antagonized the activity of WIN55,212 in HEK293T‐CB1 cells transfected with the pCRE‐luc reporter plasmid (Figure 3B). VCE‐004.3 also bound to CB2 receptor‐containing membranes with a pKi value of 6.69 ± 0.10 (Figure 3C) behaving as CB2 agonist since it inhibited forskolin‐induced luciferase activity in HEK293T‐CB2 cells transfected with the pCRE‐luc reporter plasmid (Figure 3D). The effects of WIN55,212 and VCE‐004.3 on CB2 receptor activation were prevented by the CB2 antagonists http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=750 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=751 (data not shown). In summary, VCE‐004.3 is a dual PPARγ/CB2 agonist with CB1 antagonistic activity.
Figure 3.
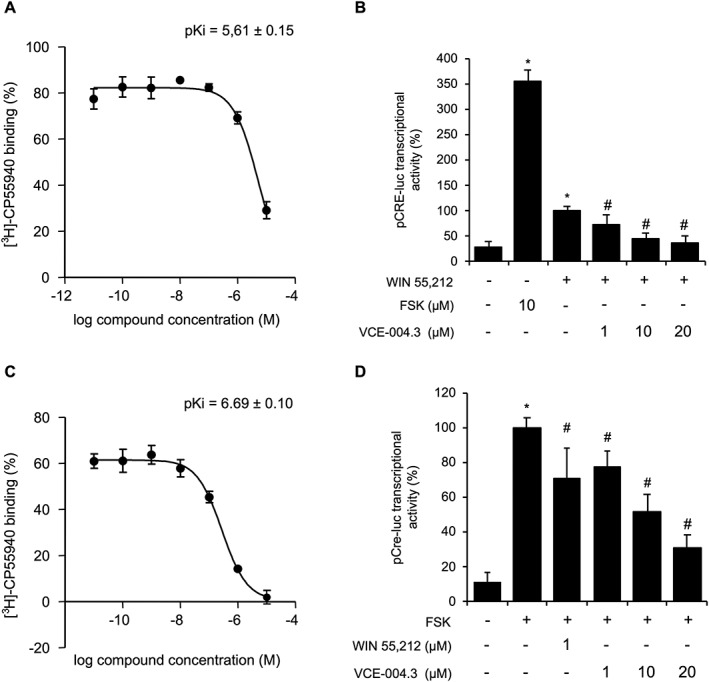
VCE‐004.3 is a functional CB1 antagonist and CB2 agonist. (A) VCE‐004.3 binding affinity to CB1 receptor membranes. Results are expressed as mean ± SEM (n = 5). (B) CB1 receptor antagonism. HEK293‐T‐CB1 cells transfected with pCRE‐luc plasmid were pretreated for 30 min with VCE‐004.3, stimulated with WIN55,212 (1 μM) for 6 h and lysed for luciferase activity. Results are expressed as mean ± SD (n = 5). (C) Binding affinity of VCE‐004.3 to CB2 receptor membranes. Results are presented as mean ± SD (n = 5). (D) VCE‐004.3 is a functional CB2 receptor agonist. HEK293‐T cells stably expressing CB2 receptors were transfected with pCRE‐luc plasmid and pretreated for 30 min with VCE‐004.3 or WIN55,212 as positive control. Then, cells were stimulated with forskolin (FSK; 10 μM) for 6 h and lysed for luciferase activity. Results are shown as mean ± SD (n = 5). *P < 0.05 versus untreated cells. # P < 0.05 versus positive control.
VCE‐004.3 prevented collagen transcription and synthesis induced by TGFβ
It has been shown that PPARγ ligands inhibit TGFβ‐induced collagen gene transcription by targeting the p300 transcriptional coactivator (Ghosh et al., 2009). Thus, we studied the effect of VCE‐004.3 on collagen transcription in NIH‐3T3 cells stably expressing the Col1A2‐luc plasmid, which encodes the gene promoter of the pro‐alpha2 chain component of type I collagen fused to the luciferase gene. Pretreatment with either RGZ or VCE‐004.3 resulted in a significant inhibition of collagen transcription induced by TGFβ1 stimulation (Figure 4A). The binding of TGFβ to its receptor entails the activation of SMAD2/SMAD3 by phosphorylation, induction of its transcriptional activity and stimulation of the Col1A2 promoter (Shi and Massague, 2003). Pre‐incubation of NIH‐3T3 cells transfected with CAGA‐luc plasmid with VCE.004.3 significantly reduced SMAD's transcriptional activity (Figure 4B). To further investigate the effect of VCE‐004.3 on TGFβ1 signalling, we studied both the canonical and the ERK1/2 non‐canonical pathways. As depicted in Figure 4C, VCE‐004.3 did not interfere with TGFβ1‐induced SMAD2 phosphorylation, but it inhibited the phosphorylation of ERK1/2 proteins in NIH‐3T3 cells. Similar results were found with RGZ. Next, to correlate the effect of VCE‐004.3 on Col1A2 transcriptional activity with collagen secretion and deposition, NHDFs were preincubated with increasing concentrations of VCE‐004.3 and stimulated with TGFβ1 for 24 h. VCE‐004.3 significantly inhibited TGFβ1‐induced collagen release and accumulation in NHDFs (Figure 4D, E).
Figure 4.
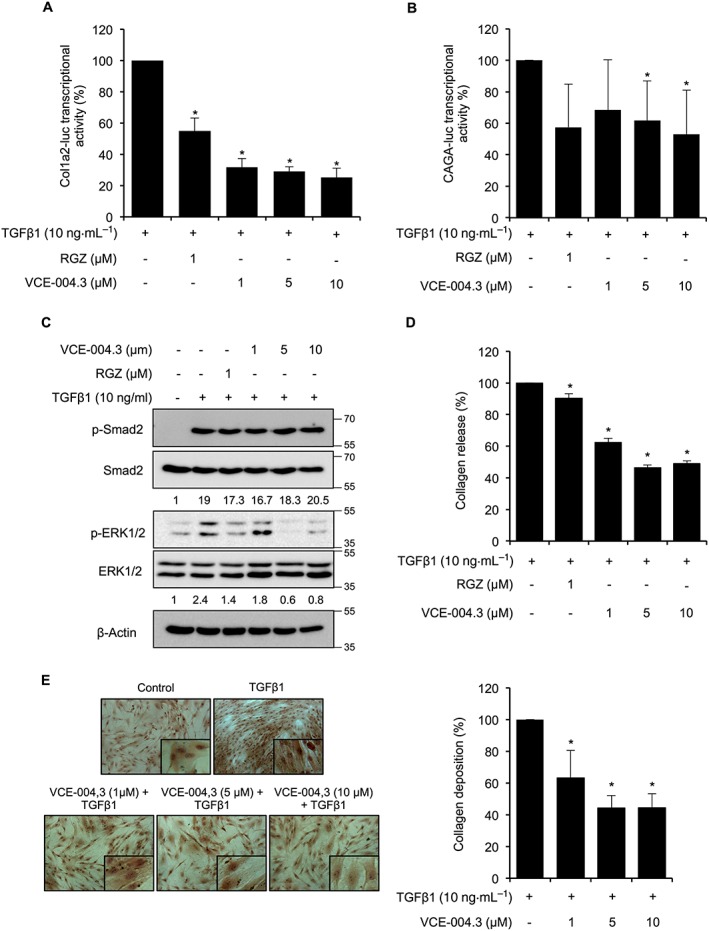
Effect of VCE‐004.3 on collagen gene transcription and synthesis in vitro. (A) NIH‐3T3‐Col1A2‐luc cells were pretreated with VCE‐004.3 for 1 h and stimulated with TGFβ1 for the following 24 h (n = 5). (B) NIH‐3T3 cells were transfected with CAGA‐luc plasmid, pretreated with VCE‐004.3 for 1 h and stimulated with TGFβ1 for 6 h (n = 5). Cells were tested for luciferase activity. (C) Serum‐starved NIH‐3T3 cells were preincubated with the compound for 1 h and stimulated with TGFβ1 for 2 h. Protein expression was studied by Western blot, and values under images represent mean fold induction of signal intensities after β‐actin normalization (n = 5). (D and E) NHDFs were serum starved (1% FBS) for 24 h. Then, cells were pretreated with VCE‐004.3 for 1 h and stimulated with TGFβ1 for 24 h. Soluble collagen in the culture medium was measured using the Sircol Assay (D), and collagen deposits were studied by the Sirius Red method (E). Results are presented as mean percentage of inhibition ± SD taking TGFβ1 alone as 100%. *P < 0.05 versus TGFβ1.
VCE‐004.3 prevented BLM‐induced dermal inflammation and fibrosis in vivo
The BLM mice model is widely used to evaluate potential anti‐inflammatory and anti‐fibrotic therapies for SSc (Wu et al., 2009; Gonzalez et al., 2012; del Rio et al., 2016; Ruzehaji et al., 2016). BLM causes inflammation in a short period of time, characterized by an elevation of pro‐inflammatory cytokines and growth factors that peak around day 14 of BLM exposure (Avouac, 2014). In this model of inflammation, BLM administration for 3 weeks resulted in increased dermal thickness versus the control group, which was attenuated by RGZ and VCE‐004.3 treatments. However, neither RGZ nor VCE‐004.3 was able to restore the loss of adipose tissue layer induced by BLM administration (Figure 5A). In addition, RGZ as well as VCE‐004.3 were able to reduce collagen accumulation in the skin after BLM administration (Figure 5B). Next, we studied the expression of cytokines associated with inflammation and fibrosis. As expected, BLM‐treated mice had significantly higher expression levels of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4996, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4998 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4406 in the skin compared with control mice. This up‐regulation was significantly reduced in mice treated with either RGZ or VCE‐004.3 (Figure 5C). Despite no significant changes in the expression levels of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974, Tgfβ and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4980 observed after BLM challenge, the expression of these cytokines was less in mice treated with RGZ or VCE‐004.3 (Figure 5C).
Figure 5.
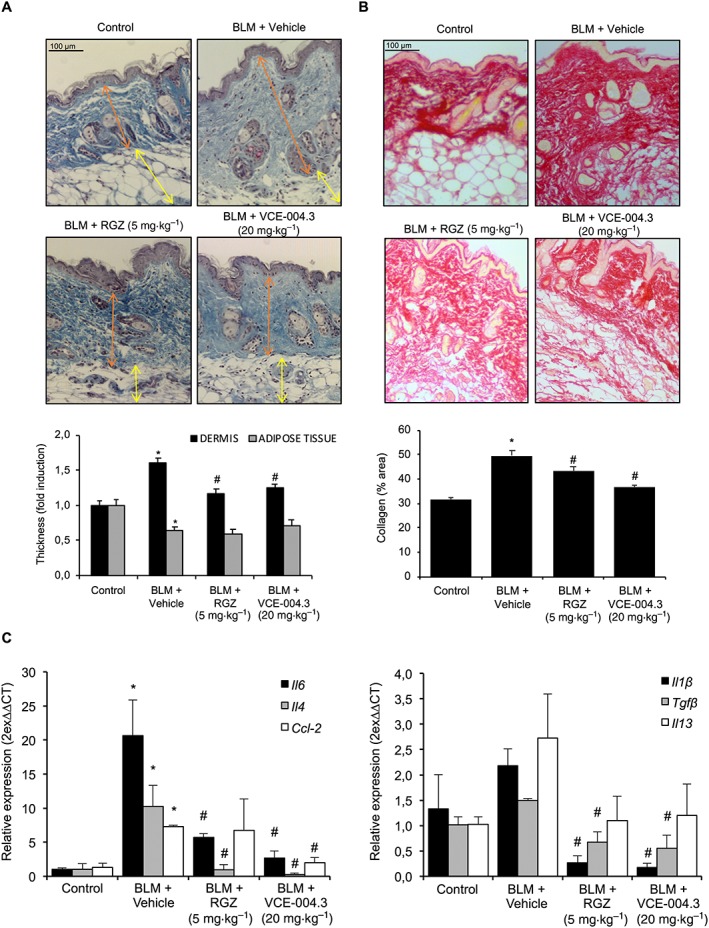
VCE‐004.3 alleviated skin inflammation and collagen accumulation induced by BLM. Mice were injected with BLM for 3 weeks and treated in parallel with i.p. injections of RGZ, VCE‐004.3 or vehicle. (A) Representative images of Masson's trichrome staining of skin sections and their respective measurement of the thickness of the layer of dermal and subcutaneous adipose tissue. (B) Representative images of collagen staining by picrosirius red dye and their quantification. (C) Gene expression of profibrotic and pro‐inflammatory genes including Il‐6, Tgfβ, Il‐4, Ccl2, Il‐1β and Il‐13 were measured by q‐RT‐PCR. Results are presented as mean ± SEM referred to control group (n = 9 animals per group). *P < 0.05 versus control; # P < 0.05 versus BLM‐treated mice.
Mast cell degranulation as well as macrophage and lymphocyte accumulation are the hallmarks of BLM‐induced fibrosis and play important roles during the inflammatory phase of SSc (Yamamoto et al., 1999). Toluidine blue staining showed an increased number of degranulated mast cells in the lesioned skin of the BLM group. Although we did not observe any effect in the RGZ‐treated group, treatment with VCE‐004.3 significantly decreased mast cell degranulation to levels comparable to the control group. Moreover, immunohistochemical examination demonstrated that VCE‐004.3 significantly reduced the infiltration of F4/80+ macrophages and CD3+ lymphocytes compared with BLM untreated mice (Figure 6).
Figure 6.
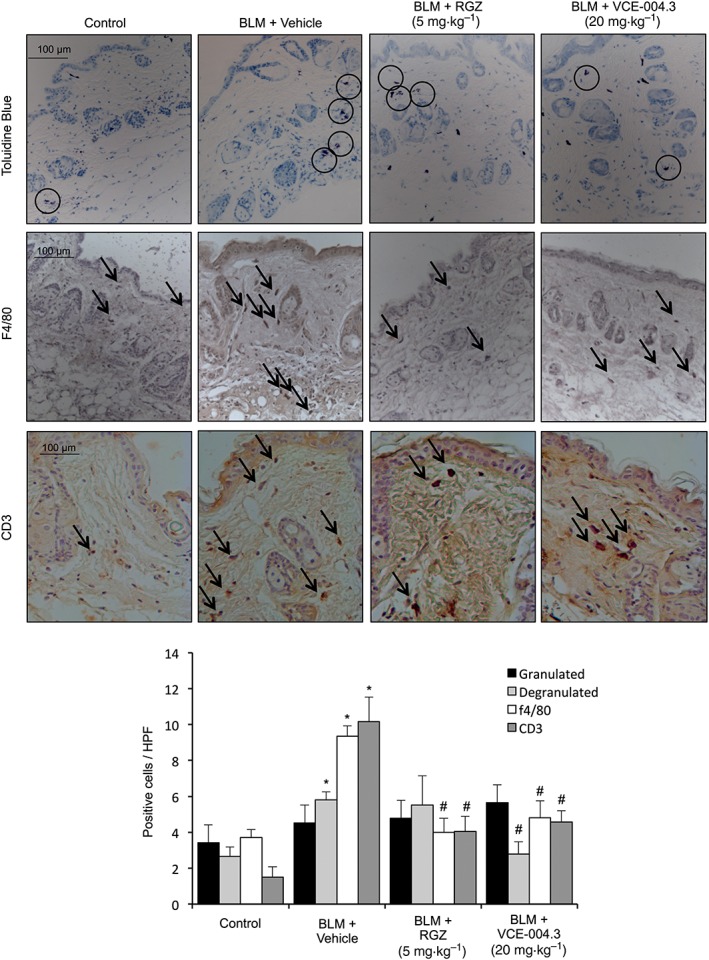
VCE‐004.3 reduced inflammatory cell infiltration in the skin. Mice were injected with BLM for 3 weeks and treated in parallel with i.p. injections of RGZ, VCE‐004.3 or vehicle. Representative images of the mast cell degranulation process are shown, as detected by toluidine blue staining and F4/80+ macrophages and CD3+ T‐lymphocyte infiltration in the skin detected by immunostaining and their corresponding quantification (n = 9 animals per group). Results represent the mean ± SEM. *P < 0.05 versus control; # P < 0.05 versus BLM‐treated mice.
It is well known that TGFβ drives the conversion of fibroblasts into myofibroblasts, and the accumulation of myofibroblasts in fibrotic skin is a hallmark of SSc (Gilbane et al., 2013; Marangoni et al., 2015). Myofibroblasts are characterized by the de novo synthesis of α‐SMA fibres, and we found a substantial reduction of spindle‐shaped α‐SMA+ cells populating the fibrotic dermis from the RGZ and VCE‐004.3 treated groups (Figure 7A). Moreover, VCE‐004.3, as well as RGZ, prevented the changes in cell morphology including cellular hypertrophy and cell shape variations from stellate to bipolar cells associated with TGFβ1‐induced myofibroblast differentiation (Figure 7B). This was accompanied by a reduction in the steady levels of α‐SMA protein; this effect was statistically significant at the dose of 10 μM (Figure 7C) and indicated that PPARγ ligands such as VCE‐004.3 attenuate TGFβ‐induced myofibroblast differentiation.
Figure 7.
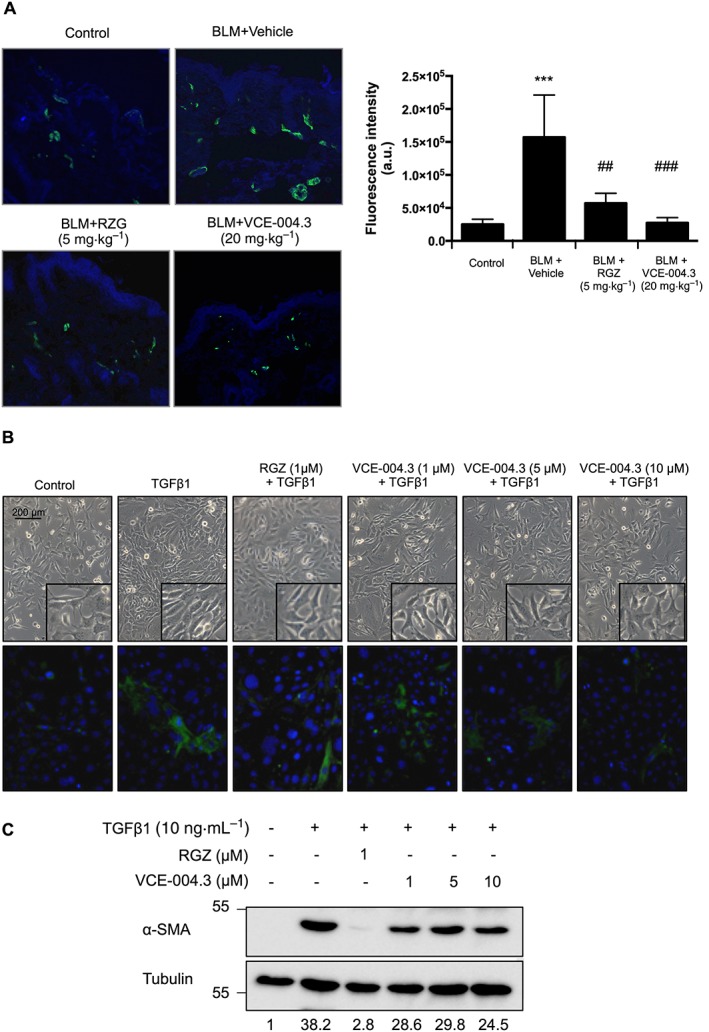
VCE‐004.3 prevented BLM‐induced myofibroblast accumulation and fibroblast to myofibroblast differentiation. (A) Mice were injected with BLM for 3 weeks and treated in parallel with i.p. injections of RGZ, VCE‐004.3 or vehicle. Representative images of α‐SMA+ cells in skin (green) detected by immunostaining and their corresponding quantification are shown (n = 6 animals per group). (B and C) NIH‐3T3 differentiation into myofibroblasts. Cells were incubated in low serum conditions (1% FBS) for 24 h. Then, cells were pretreated with VCE‐004.3 for 1 h and stimulated with TGFβ1 for 24 h (B, upper panel). Cells were immunostained for α‐SMA (green) and their nuclei stained with DAPI (blue) (B, bottom panel). (C) α‐SMA protein expression was determined by Western blot. Values under the gel indicate α‐SMA protein signal intensities after normalization to tubulin signal intensities (n = 5).
It has been previously shown that disruption of the MAPK/ERK pathway reduces inflammation and fibrosis in vivo (Galuppo et al., 2011). Therefore, we evaluated the status of phosphorylated ERK1/2 in the skin after BLM administration. Mice subjected to BLM administration showed a significant increase in ERK1/2 phosphorylation while treatment with VCE‐004.3 as well as RGZ resulted in a significant decrease in p‐ERK1/2 immunostaining (Figure 8).
Figure 8.
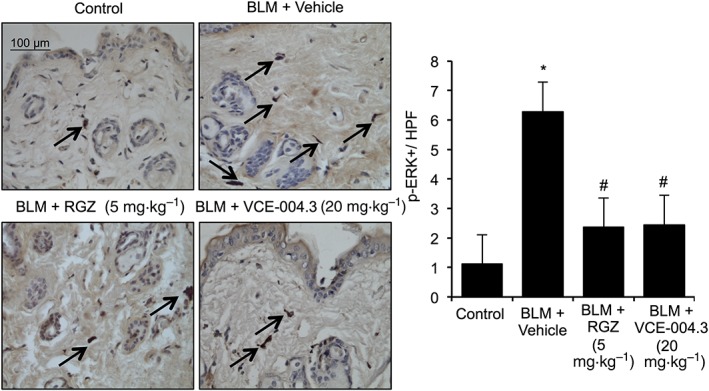
VCE‐004.3 inhibited BLM‐induced ERK1/2 phosphorylation in vivo. Mice were injected with BLM for 3 weeks and treated in parallel with i.p. injections of RGZ, VCE‐004.3 or vehicle. Representative images of p‐ERK+ cells detected by immunostaining and their respective quantification. Values are presented as mean ± SEM (n = 9 animals per group). *P < 0.05 versus control; # P < 0.05 versus BLM‐treated mice.
VCE‐004.3 alleviated established fibrosis in mice
To evaluate the efficacy of VCE‐004.3 in a model of established skin fibrosis, mice were challenged with BLM for 6 weeks, and the pharmacological treatments were carried out during the last 3 weeks of BLM injections. Animals were treated with BLM alone, BLM plus RGZ (5 mg·kg−1 i.p), BLM plus VCE‐004.3 (20 mg·kg−1 i.p.) or BLM plus VCE‐004.3 (250 nM topically). Consistent with previous studies, dermal thickness increased more than twofold after 6 weeks of BLM challenge in parallelwith a strong reduction in the adipose tissue. Treatment for 3 weeks with VCE‐004.3 in the absence of BLM did not influence the structures of the skin. In contrast, treatment with either RGZ or VCE‐004.3 (i.p. or topical) of BLM‐challenged mice significantly reversed dermal thickness, although they could restore the adipose layer thickness to the control level (Figure 9A). According to this reduction of dermal thickness, RGZ and VCE‐004.3 prevented collagen accumulation in the skin (Figure 9B). In a similar experiment, co‐treatment with the CB2 antagonist AM630 attenuated the effect of VCE‐004.3 to a certain extent. However, the PPARγ antagonist T0070907 could not block the effect of the compound, since no significant changes in either dermal thickness or collagen accumulation were observed when compared with VCE‐004.3‐treated animals (Supporting Information Figure S2). This result confirmed that the biological activity of VCE‐004.3 in vivo is mediated by targeting PPARγ at the alternative binding site as well as its effect on CB2 receptors.
Figure 9.
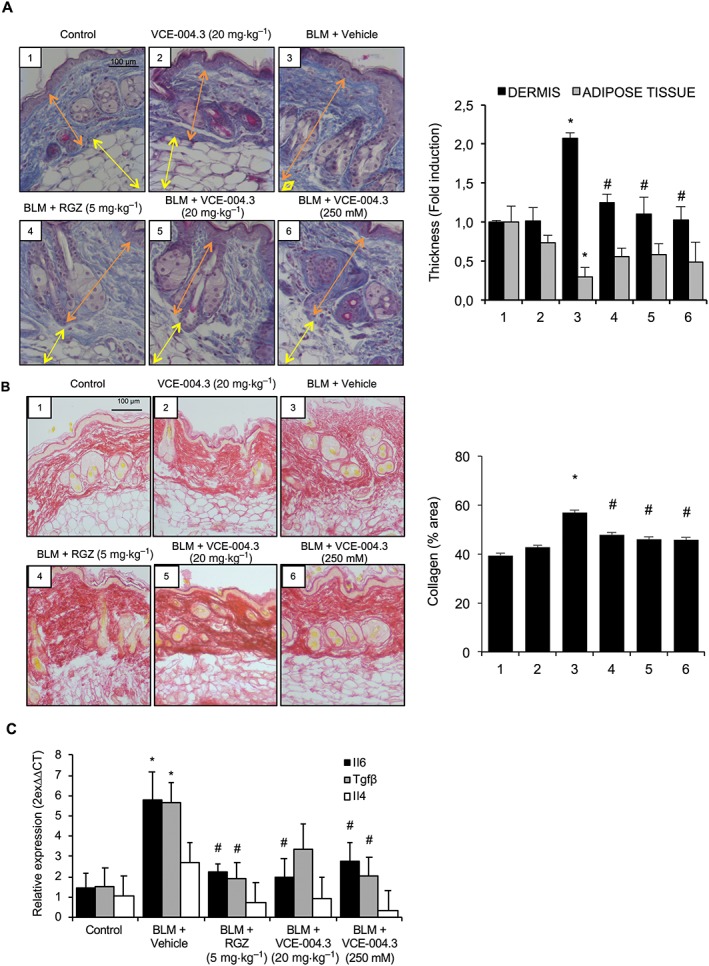
Systemic and topical treatment with VCE‐004.3 reversed fibrosis in a BLM‐induced model of established skin fibrosis. Mice were injected with BLM for 6 weeks and treated with the compound during the last 3 weeks of BLM challenge. (A) Representative images of Masson's trichrome staining of skin sections and their respective measurements of the thickness of dermal and subcutaneous adipose layers. (B) Representative images of collagen staining by picrosirius red dye and their quantification. (C) Expression of fibrosis‐related genes in the skin. Values are presented as mean ± SEM (n = 9 animals per group). *P < 0.05 versus control; # P < 0.05 versus BLM‐treated mice.
The mRNA levels of Il‐4, Il‐6 and Tgfβ were also investigated in this model. Expression levels of Il‐6 were significantly reduced with all the pharmacological treatments. Il‐6 mRNA was increased in the fibrotic model, but to a much lesser extent than in the inflammatory model, indicating that the cytokine signature is different in the two approaches. Accordingly, Il‐4 mRNA in the skin was not significantly increased after 6 weeks of BLM challenge, but in any case, the expression levels were reduced in both RGZ and VCE‐004.3 treated mice. The expression of Tgfβ was strongly up‐regulated in the BLM group, and this effect was partially inhibited in VCE‐004.3 (i.p.) treated mice and significantly reduced after RGZ and topical VCE‐004.3 treatments (Figure 9C).
Effect of VCE‐004.3 on ERK1/2 activation and fibroblast migration
It is well accepted that autoantibodies could play a key role in the pathogenesis of SSc. Stimulatory autoantibodies targeting the PDGF receptor have been detected in 100% of SSc patients and demonstrated to trigger an intracellular cascade that involves ROS, Ha‐Ras and ERK1/2 activation, eventually leading to fibroblast activation and increased collagen gene expression (Baroni et al., 2006; Luchetti et al., 2016). In addition, different studies have demonstrated that MAPKs are activated in response to fibrogenic stimuli and contribute to myofibroblast differentiation and function (Leask, 2012). To study the effect of VCE‐004.3 on ERK1/2 phosphorylation, NHDFs were preincubated with the compound under investigation, and then stimulated with human recombinant PDGF‐BB, or with IgG purified from patients diagnosed with lcSSc or dcSSc, with the steady state levels for total and phosphorylated ERK1/2 protein being analysed by Western blots. VCE‐004.3 clearly prevented ERK1/2 phosphorylation in all the condition tested (Figure 10A–C).
Figure 10.
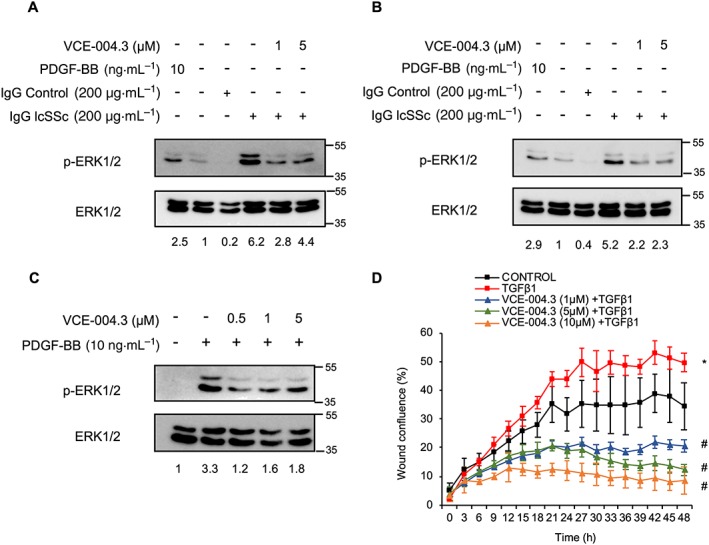
VCE‐004.3 prevented stimulation of ERK signalling by SSc autoantibodies and fibroblast migration in vitro. NHDFs were serum‐starved (1% FBS) for 24 h and then preincubated with VCE‐004.3 for 1 h and stimulated with lSSc (A) or dSSc (B) IgG for 15 min. (C) NIH‐3T3 cells were serum‐starved (1% FBS) for 24 h and then preincubated with VCE‐004.3 for 15 min and stimulated with PDGF‐BB for 5 min. Protein expression was determined by Western blot, and values under the gel indicate mean fold induction of signal intensities. (D) NHDF monolayers were scratched and treated with VCE‐004.3 at the indicated doses in the presence of TGFβ1. Results are presented as percentage of wound closure (confluence) ± SD (n = 5). *P < 0.05 versus control; # P < 0.05 versus TGFβ1‐treated cells.
Migration of skin fibroblast plays a crucial role in SSs (Gilbane et al., 2013), and we tested if the anti‐fibrotic effect of VCE‐004.3 could be associated with reduced fibroblast migration. Thus, NHDF cell monolayers were scratched, and the effect of VCE‐004.3 on TGFβ1‐induced wound healing was analysed over the time. Our results show that VCE‐004.3 strongly inhibited the migration of fibroblasts induced by TGFβ1 (Figure 10D).
Discussion
We and others have previously investigated the anti‐fibrotic effects of synthetic cannabinoids that activate both PPARγ and CB2 receptors in animal models of skin fibrosis (Gonzalez et al., 2012; del Rio et al., 2016). In this study, we report for the first time that a novel CBD derivative, VCE‐004.3, which is a dual PPARγ/CB2 agonist that also behaves as a CB1 antagonist, exhibits both anti‐inflammatory and anti‐fibrotic effects.
Glitazones like RGZ are full agonists of PPARγ (PPARγ‐fa) that are widely used in clinical practice for glycaemic control. Nevertheless, this kind of PPARγ‐fa may cause several adverse effects, including fluid retention, congestive heart failure and bladder cancer (Ciudin et al., 2012). Accordingly, the search for PPARγ modulators (PPARγ‐m) as safer alternatives to PPARγ‐fa has grown, with the expectation of separating mechanism‐associated side effects from insulin‐sensitizing activity. It has been demonstrated that the level of activation of PPARγ is directly related to adipogenesis but not to insulin sensitivity (Doshi et al., 2010). PPARγ‐m can bind the receptor in a different manner than PPARγ‐fa, leading conformational changes that alter coactivator recruitment (Doshi et al., 2010). In this context, VCE‐004.3 can be considered as a PPARγ‐m, since it is less potent as an activator PPARγ than RGZ, and is less adipogenic. Here, we showed that VCE‐004.3 activates PPARγ transcriptional activity and competes for the RGZ binding pocket, which is located within the PPARγ LBP. However, the effect of VCE‐004.3 on PPARγ transactivation could not be blocked by T0070907 showing that both compounds bind simultaneously to distinct PPARγ binding sites, as confirmed by docking studies. These results are consistent with previous studies that reported that covalent antagonists do not block ligands that interact with the alternate binding site (Hughes et al., 2014). Moreover, synthetic PPARγ agonists that bind to the alternate site induce PPARγ‐driven anti‐diabetic activities (Hughes et al., 2014). Thus, it was not unexpected to find that T0070907 did not prevent the anti‐fibrotic response of VCE‐004.3 in our in vivo studies. Since PPARγ ligands that bind to canonical binding site are also effective in BLM‐induced fibrosis (Wu et al., 2009; Wei et al., 2014), we can assume that, due to the versatile nature of the LBP of PPARγ, PPARγ‐ms (partial agonist) are also effective at preventing inflammation and fibrosis.
It has been previously shown that the canonical pathway activated by TGFβ drives the phosphorylation of SMADs that is upstream of the fibrogenic process (Walton et al., 2017). Our results show that VCE‐004.3 reduces TGFβ‐induced SMAD2/3 transcriptional activation and collagen synthesis but does not affect SMAD2/3 phosphorylation. Since ligand‐mediated PPARγ activation does not interfere with the phosphorylation of SMADs, it is likely that VCE‐004.3 competes with SMAD proteins for the interaction with p300 co‐activator, thus preventing collagen synthesis induced by TGFβ (Ghosh et al., 2009). However, other TGFβ‐dependent signalling pathways such as the Ras/MEK/ERK cascade have also been suggested to contribute to SSc pathogenesis (Leask, 2012). VCE‐004.3 also down‐regulates ERK1/2 signalling, a major pathway controlling cell growth, proliferation, migration, apoptosis and myofibroblast differentiation (McCubrey et al., 2007). Indeed, hyperactivation of ERK1/2 was observed in fibroblasts from SSc patients, and MEK/ERK signalling is required for α‐SMA stress fibre assembly (Chen et al., 2011). Interestingly, a recent report has shown that skin fibroblasts express CB2 receptors and that these are further up‐regulated under inflammatory conditions (Bort et al., 2017). It is therefore possible that VCE‐004.3 inhibits ERK1/2 activation both in vitro and in vivo by acting directly through CB2 receptors. Accordingly, the anti‐fibrotic activity of VCE.004.3 in vivo was partially prevented by the CB2 antagonist AM630.
The earliest events of SSc include obliterative vasculopathy, which induces inflammation and autoimmunity. Indeed, in early dcSSc, the dermis exhibits inflammatory infiltrates mainly composed of monocytes and activated lymphocytes (Varga and Abraham, 2007). Accordingly, we used a model characterized by early inflammation induced by 3 weeks of BLM administration in which treatment with the compound is carried out in parallel with the BLM injections (Gonzalez et al., 2012). Our data indicate that VCE‐004.3 prevents dermal thickening as well as inflammatory cell infiltration and the expression of pro‐inflammatory and profibrotic cytokines. Previous studies have shown that PPARγ activation by RGZ mediates immunomodulatory effects that prevent BLM‐induced skin fibrosis (Wu et al., 2009). Moreover, macrophage activation as well as T‐cell proliferation are also regulated by PPARγ activation (Clark et al., 2000; Harris and Phipps, 2001). Indeed, activators of PPARγ and CB2 receptors inhibited M1 polarization and influenced macrophage M2 differentiation (Bouhlel et al., 2007; Guillot et al., 2014). Therefore, it is conceivable that PPARγ activation and modulation of the cannabinoid system by VCE‐004.3 could limit leukocyte infiltration and macrophage activation in the skin after BLM administration (Akhmetshina et al., 2009; Marquart et al., 2010).
The BLM‐induced model of SSc is also useful to evaluate the progression from an inflammatory process to a fibrotic state. In this model, skin fibrosis is measured by determining dermal thickness, myofibroblast counts and collagen content, assessed by the hydroxyproline assay. Alternatively, the content of collagen in skin biopsies can be also evaluated by picrosirius staining, a semi‐quantitative method, since a highly significant correlation between picrosirius staining and the results of hydroxyproline assays used to measure skin collagen have recently been found (Caetano et al., 2016). Progressive fibrosis of the skin and other internal organs caused by excessive accumulation of collagen and other ECM is one of the three main hallmarks of SSc (Pattanaik et al., 2015). Many compounds that have been tested using the BLM model were investigated in a preventive schedule to see if an anti‐inflammatory agent can hinder the subsequent fibrosis. In the event, treatment with VCE‐004.3 (topical and i.p.) could reduce dermal thickness and collagen content as well as the expression of cytokines involved in collagen production after the development of fibrosis, thus alleviating the previously established damage. CB1 and CB2 receptors have been shown to play opposite roles in experimental models of skin fibrosis. While CB1 receptor activation has a detrimental effect on the disease, the genetic or pharmacological inactivation of this receptor alleviates skin fibrosis. In addition, CB2 agonists have been shown to prevent BLM‐induced fibrosis (Akhmetshina et al., 2009; Marquart et al., 2010; Gonzalez et al., 2012; del Rio et al., 2016). Thus, VCE‐004.3 may have a beneficial effect on skin fibrosis by acting as a dual CB1 antagonist and CB2 agonist. While the CB2 agonism effect on BLM‐induced skin fibrosis has been confirmed using AM630, the potential CB1 antagonism requires further research. We found that VCE‐004.3 binds to CB1 receptors with relatively low affinity but inhibits CB1‐dependent cAMP signalling with a potency similar to other CB1 antagonists. This Janus‐behaviour (Dhopeshwarkar et al., 2017) could, in principle, be due to either to binding to a different site to [3H]‐CP55940 or to it behaving as a potential negative allosteric modulator. Experiments are on course to elucidate the mechanism of action of VCE‐004.3 on the functionality of CB1 receptors.
As in many autoimmune diseases, beta cell activation and generation of autoantibodies against different targets are also present in SSc patients, and this plays a critical role in the pathogenesis of the disease. Among others, agonistic PDGF receptor autoantibodies have been shown to change fibroblast phenotype into SSc features through Ha‐Ras‐ERK1/2 and ROS cascades (Baroni et al., 2006). In addition, the inhibiting the activation of MAPKs has been shown to suppress the progression of fibrosis (Galuppo et al., 2011). Treatment with VCE‐004.3 prevented ERK1/2 activation in normal fibroblasts after they were exposed to stimulatory autoantibodies isolated from lcSSc and dcSSc patients, indicating that the anti‐fibrotic effects of VCE‐004.3 may be also mediated by ERK1/2. Interestingly, the presence of autoantibodies has been also demonstrated in the BLM model (Yamamoto, 2006), and it is, therefore, possible that VCE‐004.3 inhibits ERK1/2 activation in vivo by targeting both TGFβ‐ and autoantibody‐induced ERK1/2 phosphorylation.
Finally, the transdermal administration of cannabinoids is now well established (Lodzki et al., 2003; Valiveti et al., 2004), and in this study, we have shown that topical application of VCE‐004.3 also inhibited BLM‐induced skin fibrosis; therefore, this compound could be also formulated for the treatment of localized forms of cutaneous fibrosis.
Cannabinoids like CBD are multitarget compounds and many semisynthetic derivatives have been designed to improve the bioactivities and druggability of the natural products. In this context, VCE‐004.3 is a novel cannabinoid derivative that acts as a dual PPARγ/CB2 agonist and as a negative modulator of CB1 receptors. Because of this, it qualifies as a candidate for the development of novel therapies against different forms of scleroderma.
Author contributions
C.D.R., I.C. and E.M. contributed to the conception and design of the study. C.D.R., I.C., B.P., C.N., A.G.M. and M.L.B. performed in vitro and in vivo experiments. C.D.R. and B.P. conducted the statistical analysis. M.G.C. and J.F.R. performed the binding studies. R.O.C., C.P.S. and C.L.P. selected patients and isolated IgG from plasma. C.P. and M.A.C. performed the in silico analysis. G.A. supervised the chemical synthesis of VCE‐004.3. C.D.R. and E.M. wrote the manuscript. All the authors contributed to the analysis and interpretation of data, critically reviewed and approved the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effect of VCE‐004.3 on MSC differentiation into adipocytes. (A) MSC underwent adipocytic differentiation in the presence of VCE‐004.3 or RGZ as positive control. Representative images showing OilRed O staining (upper panel) and cell number (bottom left) and Oilred positive cells (bottom right) were counted. (B) Gene expression of PPAR‐γ2, ADIPOQ and CEBPA in MSCs differentiated for 14 days. Data represent the percentage of increase over AM considered as the 100% of adipogenic induction (n = 5) *P < 0.05 versus AM.
Figure S2 Pretreatment with the CB2 antagonist AM630 but not the PPARγ antagonist T007907 partially abrogated the effect of VCE‐004.3 on BLM‐induced chronic model of SSc. Mice were injected with BLM for six weeks and treated with the compound in the presence of either AM630 or T0070907 during the last three weeks of BLM challenge. A. Representative images of Masson's trichrome staining of skin sections and their respective measurement of dermal and subcutaneous adipose layer thickness. B. Representative images of picrosirius red dye in mice skin and their quantification. Values are represented as mean ± SEM (n = 9 animals per group). *P < 0.05 versus control; #P < 0.05 versus BLM‐treated mice; †P < 0.05 versus BLM + VCE‐004.3.
Acknowledgements
This work was partially supported by the MINECO grant RTC‐2015‐3364 cofounded by the European Development Regional Fund in the Framework of the Operative Program ‘Reinforcement of research, technological development and innovation’. E.M. was also supported by the MINECO grant SAF2014‐53763‐P. B.P. is a predoctoral fellow supported by the i‐PFIS program, Instituto de Salud Carlos III (IFI15/00022; European Social Fund “investing in your future”).
del Rio, C. , Cantarero, I. , Palomares, B. , Gómez‐Cañas, M. , Fernández‐Ruiz, J. , Pavicic, C. , García‐Martín, A. , Luz Bellido, M. , Ortega‐Castro, R. , Pérez‐Sánchez, C. , López‐Pedrera, C. , Appendino, G. , Calzado, M. A. , and Muñoz, E. (2018) VCE‐004.3, a cannabidiol aminoquinone derivative, prevents bleomycin‐induced skin fibrosis and inflammation through PPARγ‐ and CB2 receptor‐dependent pathways. British Journal of Pharmacology, 175: 3813–3831. 10.1111/bph.14450.
References
- Akhmetshina A, Dees C, Busch N, Beer J, Sarter K, Zwerina J et al (2009). The cannabinoid receptor CB2 exerts antifibrotic effects in experimental dermal fibrosis. Arthritis Rheum 60: 1129–1136. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avouac J (2014). Mouse model of experimental dermal fibrosis: the bleomycin‐induced dermal fibrosis. Methods Mol Biol 1142: 91–98. [DOI] [PubMed] [Google Scholar]
- Bae KH, Seo JB, Jung YA, Seo HY, Kang SH, Jeon HJ et al (2017). Lobeglitazone, a novel peroxisome proliferator‐activated receptor γ agonist, attenuates renal fibrosis caused by unilateral ureteral obstruction in mice. Endocrinol Metab (Seoul) 32: 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroni SS, Santillo M, Bevilacqua F, Luchetti M, Spadoni T, Mancini M et al (2006). Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N Engl J Med 354: 2667–2676. [DOI] [PubMed] [Google Scholar]
- Baugh EH, Lyskov S, Weitzner BD, Gray JJ (2011). Real‐time PyMOL visualization for Rosetta and Pyrosetta. PLoS One 6: e21931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer C, Schett G, Distler O, Distler JH (2010). Animal models of systemic sclerosis: prospects and limitations. Arthritis Rheum 62: 2831–2844. [DOI] [PubMed] [Google Scholar]
- Bort A, Alvarado‐Vazquez PA, Moracho‐Vilrriales C, Virga KG, Gumina G, Romero‐Sandoval A et al (2017). Effects of JWH015 in cytokine secretion in primary human keratinocytes and fibroblasts and its suitability for topical/transdermal delivery. Mol Pain 13 1744806916688220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard JF, Lepicier P, Lamontagne D (2003). Contribution of endocannabinoids in the endothelial protection afforded by ischemic preconditioning in the isolated rat heart. Life Sci 72: 1859–1870. [DOI] [PubMed] [Google Scholar]
- Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S et al (2007). PPARγ activation primes human monocytes into alternative M2 macrophages with anti‐inflammatory properties. Cell Metab 6: 137–143. [DOI] [PubMed] [Google Scholar]
- Caetano GF, Fronza M, Leite MN, Gomes A, Frade MA (2016). Comparison of collagen content in skin wounds evaluated by biochemical assay and by computer‐aided histomorphometric analysis. Pharm Biol 54: 2555–2559. [DOI] [PubMed] [Google Scholar]
- Chen Y, Leask A, Abraham DJ, Kennedy L, Shi‐Wen X, Denton CP et al (2011). Thrombospondin 1 is a key mediator of transforming growth factor β‐mediated cell contractility in systemic sclerosis via a mitogen‐activated protein kinase kinase (MEK)/extracellular signal‐regulated kinase (ERK)‐dependent mechanism. Fibrogenesis Tissue Repair 4: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciudin A, Hernandez C, Simo R (2012). Update on cardiovascular safety of PPARγ agonists and relevance to medicinal chemistry and clinical pharmacology. Curr Top Med Chem 12: 585–604. [DOI] [PubMed] [Google Scholar]
- Clark RB (2002). The role of PPARs in inflammation and immunity. J Leukoc Biol 71: 388–400. [PubMed] [Google Scholar]
- Clark RB, Bishop‐Bailey D, Estrada‐Hernandez T, Hla T, Puddington L, Padula SJ (2000). The nuclear receptor PPARγ and immunoregulation: PPARγ mediates inhibition of helper T cell responses. J Immunol 164: 1364–1371. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantas AT, Pereira MC, De Melo Rego MJ, Darocha LF Jr, Pitta Ida R, Marques CD et al (2015). The role of PPARγ in systemic sclerosis. PPAR Res 2015: 124624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rio C, Navarrete C, Collado JA, Bellido ML, Gomez‐Canas M, Pazos MR et al (2016). The cannabinoid quinol VCE‐004.8 alleviates bleomycin‐induced scleroderma and exerts potent antifibrotic effects through peroxisome proliferator‐activated receptor‐γ and CB2 pathways. Sci Rep 6 21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhopeshwarkar A, Murataeva N, Makriyannis A, Straiker A, Mackie K (2017). Two Janus cannabinoids that are both CB2 agonists and CB1 antagonists. J Pharmacol Exp Ther 360: 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi LS, Brahma MK, Bahirat UA, Dixit AV, Nemmani KV (2010). Discovery and development of selective PPARγ modulators as safe and effective antidiabetic agents. Expert Opin Investig Drugs 19: 489–512. [DOI] [PubMed] [Google Scholar]
- Fineschi S, Goffin L, Rezzonico R, Cozzi F, Dayer JM, Meroni PL et al (2008). Antifibroblast antibodies in systemic sclerosis induce fibroblasts to produce profibrotic chemokines, with partial exploitation of toll‐like receptor 4. Arthritis Rheum 58: 3913–3923. [DOI] [PubMed] [Google Scholar]
- Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati‐Baroni G et al (2002). Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro . Gastroenterology 122: 1924–1940. [DOI] [PubMed] [Google Scholar]
- Galuppo M, Esposito E, Mazzon E, Di Paola R, Paterniti I, Impellizzeri D et al (2011). MEK inhibition suppresses the development of lung fibrosis in the bleomycin model. Naunyn Schmiedebergs Arch Pharmacol 384: 21–37. [DOI] [PubMed] [Google Scholar]
- Garcia‐Gonzalez E, Selvi E, Balistreri E, Lorenzini S, Maggio R, Natale MR et al (2009). Cannabinoids inhibit fibrogenesis in diffuse systemic sclerosis fibroblasts. Rheumatology (Oxford) 48: 1050–1056. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Bhattacharyya S, Wei J, Kim S, Barak Y, Mori Y et al (2009). Peroxisome proliferator‐activated receptor‐γ abrogates Smad‐dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J 23: 2968–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbane AJ, Denton CP, Holmes AM (2013). Scleroderma pathogenesis: a pivotal role for fibroblasts as effector cells. Arthritis Res Ther 15: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez EG, Selvi E, Balistreri E, Akhmetshina A, Palumbo K, Lorenzini S et al (2012). Synthetic cannabinoid ajulemic acid exerts potent antifibrotic effects in experimental models of systemic sclerosis. Ann Rheum Dis 71: 1545–1551. [DOI] [PubMed] [Google Scholar]
- Guillot A, Hamdaoui N, Bizy A, Zoltani K, Souktani R, Zafrani ES et al (2014). Cannabinoid receptor 2 counteracts interleukin‐17‐induced immune and fibrogenic responses in mouse liver. Hepatology 59: 296–306. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS guide to pharmacology in 2018: updates and expansion to encompass the new guide to immunopharmacology. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SG, Phipps RP (2001). The nuclear receptor PPARγ is expressed by mouse T lymphocytes and PPARγ agonists induce apoptosis. Eur J Immunol 31: 1098–1105. [DOI] [PubMed] [Google Scholar]
- Ho KT, Reveille JD (2003). The clinical relevance of autoantibodies in scleroderma. Arthritis Res Ther 5: 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TS, Giri PK, De Vera IM, Marciano DP, Kuruvilla DS, Shin Y et al (2014). An alternate binding site for PPARγ ligands. Nat Commun 5: 3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katchan V, David P, Shoenfeld Y (2016). Cannabinoids and autoimmune diseases: a systematic review. Autoimmun Rev 15: 513–528. [DOI] [PubMed] [Google Scholar]
- Kayser C, Fritzler MJ (2015). Autoantibodies in systemic sclerosis: unanswered questions. Front Immunol 6: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A (2012). MEK/ERK inhibitors: proof‐of‐concept studies in lung fibrosis. J Cell Commun Signal 6: 59–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodzki M, Godin B, Rakou L, Mechoulam R, Gallily R, Touitou E (2003). Cannabidiol‐transdermal delivery and anti‐inflammatory effect in a murine model. J Control Release 93: 377–387. [DOI] [PubMed] [Google Scholar]
- Luchetti MM, Moroncini G, Jose Escamez M, Svegliati Baroni S, Spadoni T, Grieco A et al (2016). Induction of scleroderma fibrosis in skin‐humanized mice by administration of anti‐platelet‐derived growth factor receptor agonistic autoantibodies. Arthritis Rheumatol 68: 2263–2273. [DOI] [PubMed] [Google Scholar]
- Marangoni RG, Korman BD, Wei J, Wood TA, Graham LV, Whitfield ML et al (2015). Myofibroblasts in murine cutaneous fibrosis originate from adiponectin‐positive intradermal progenitors. Arthritis Rheumatol 67: 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquart S, Zerr P, Akhmetshina A, Palumbo K, Reich N, Tomcik M et al (2010). Inactivation of the cannabinoid receptor CB1 prevents leukocyte infiltration and experimental fibrosis. Arthritis Rheum 62: 3467–3476. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F et al (2007). Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773: 1263–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihai C, Tervaert JW (2010). Anti‐endothelial cell antibodies in systemic sclerosis. Ann Rheum Dis 69: 319–324. [DOI] [PubMed] [Google Scholar]
- Milam JE, Keshamouni VG, Phan SH, Hu B, Gangireddy SR, Hogaboam CM et al (2008). PPAR‐γ agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin‐induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 294: L891–L901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS et al (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30: 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Batkai S, Kunos G (2006). The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev 58: 389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanaik D, Brown M, Postlethwaite BC, Postlethwaite AE (2015). Pathogenesis of systemic sclerosis. Front Immunol 6: 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistis M, O'Sullivan SE (2017). The role of nuclear hormone receptors in cannabinoid function. Adv Pharmacol 80: 291–328. [DOI] [PubMed] [Google Scholar]
- Ragusa G, Gomez‐Canas M, Morales P, Hurst DP, Deligia F, Pazos R et al (2015). Synthesis, pharmacological evaluation and docking studies of pyrrole structure‐based CB2 receptor antagonists. Eur J Med Chem 101: 651–667. [DOI] [PubMed] [Google Scholar]
- Rajapaksha H, Bhatia H, Wegener K, Petrovsky N, Bruning JB (2017). X‐ray crystal structure of rivoglitazone bound to PPARγ and PPAR subtype selectivity of TZDs. Biochim Biophys Acta 1861: 1981–1991. [DOI] [PubMed] [Google Scholar]
- Ruzehaji N, Frantz C, Ponsoye M, Avouac J, Pezet S, Guilbert T et al (2016). Pan PPAR agonist IVA337 is effective in prevention and treatment of experimental skin fibrosis. Ann Rheum Dis 75: 2175–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J (2003). Mechanisms of TGF‐β signaling from cell membrane to the nucleus. Cell 113: 685–700. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiveti S, Hammell DC, Earles DC, Stinchcomb AL (2004). Transdermal delivery of the synthetic cannabinoid WIN 55,212‐2: in vitro/in vivo correlation. Pharm Res 21: 1137–1145. [DOI] [PubMed] [Google Scholar]
- Varga J, Abraham D (2007). Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 117: 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton KL, Johnson KE, Harrison CA (2017). Targeting TGF‐β mediated SMAD signaling for the prevention of fibrosis. Front Pharmacol 8: 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Zhu H, Komura K, Lord G, Tomcik M, Wang W et al (2014). A synthetic PPAR‐γ agonist triterpenoid ameliorates experimental fibrosis: PPAR‐γ‐independent suppression of fibrotic responses. Ann Rheum Dis 73: 446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf LK (2009). New software and websites for the chemical enterprise. Chem Eng News 87: 48. [Google Scholar]
- Wu M, Melichian DS, Chang E, Warner‐Blankenship M, Ghosh AK, Varga J (2009). Rosiglitazone abrogates bleomycin‐induced scleroderma and blocks profibrotic responses through peroxisome proliferator‐activated receptor‐γ. Am J Pathol 174: 519–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T (2006). The bleomycin‐induced scleroderma model: what have we learned for scleroderma pathogenesis? Arch Dermatol Res 297: 333–344. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Takagawa S, Katayama I, Yamazaki K, Hamazaki Y, Shinkai H et al (1999). Animal model of sclerotic skin. I: local injections of bleomycin induce sclerotic skin mimicking scleroderma. J Invest Dermatol 112: 456–462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of VCE‐004.3 on MSC differentiation into adipocytes. (A) MSC underwent adipocytic differentiation in the presence of VCE‐004.3 or RGZ as positive control. Representative images showing OilRed O staining (upper panel) and cell number (bottom left) and Oilred positive cells (bottom right) were counted. (B) Gene expression of PPAR‐γ2, ADIPOQ and CEBPA in MSCs differentiated for 14 days. Data represent the percentage of increase over AM considered as the 100% of adipogenic induction (n = 5) *P < 0.05 versus AM.
Figure S2 Pretreatment with the CB2 antagonist AM630 but not the PPARγ antagonist T007907 partially abrogated the effect of VCE‐004.3 on BLM‐induced chronic model of SSc. Mice were injected with BLM for six weeks and treated with the compound in the presence of either AM630 or T0070907 during the last three weeks of BLM challenge. A. Representative images of Masson's trichrome staining of skin sections and their respective measurement of dermal and subcutaneous adipose layer thickness. B. Representative images of picrosirius red dye in mice skin and their quantification. Values are represented as mean ± SEM (n = 9 animals per group). *P < 0.05 versus control; #P < 0.05 versus BLM‐treated mice; †P < 0.05 versus BLM + VCE‐004.3.