

Abstract
The neurobiology underlying depression in older adults is less extensively evaluated than in younger adults, despite the putative influence of aging on depression neuropathology. Studies using transcranial magnetic stimulation (TMS), a neurophysiological tool capable of probing inhibitory and excitatory cortical neurotransmission, have identified dysfunctional GABAergic inhibitory activity in younger adults with depression. However, GABAergic and glutamatergic cortical neurotransmission have not yet been studied in late-life depression (LLD). Here, we used single- and paired-pulse TMS to measure cortical inhibition and excitation in 92 LLD patients and 41 age-matched healthy controls. To differentiate the influence of age and depression, we also compared these TMS indices to those of 30 younger depressed adults and 30 age- and sex-matched younger healthy adults. LLD patients, older healthy adults, and younger depressed adults demonstrated significantly lower GABAA receptor-mediated cortical inhibition than younger healthy controls. By contrast, no significant differences in cortical inhibition were observed between older adults with and without depression. No significant differences in GABAB receptor-mediated inhibition or cortical excitation were found between the groups. Altogether, these findings suggest that reduced cortical inhibition may be associated with both advancing age and depression, which (i) supports the model of depression as a disease of accelerated aging, and (ii) prompts future investigation into diminished GABAergic neurotransmission in late-life as a biological predisposing factor to the development of depression. Given that cortical neurophysiology was similar in depressed and healthy older adults, future prospective studies need to establish the relative influence of age and depression on cortical inhibition deficits.
Introduction
Depression, whether it occurs early in life and persists for decades or develops for the first time in late life, is one of the most common neuropsychiatric disorders in older adults and is associated with increased rates of suicide [1], premature mortality from comorbid illness [2], and increased health care costs [3]. Although standard treatments are effective for some older adults with depression [4], in general, the prognosis for older adults with early- or late-onset depression is poor [5]. Whereas the majority of research on depression to date has focused on younger adults, there is evidence that the molecular correlates of normal aging resemble those observed in depression [6], and it remains unclear how molecular aging may influence the pathophysiology of late-life depression (LLD), particularly with late-onset [7]. For example, overlapping changes in gene expression [8] and reductions in cortical volume [9, 10] occur in both aging and depression. Going forward, the development of improved treatments for LLD may benefit from a better understanding of the pathophysiological similarities and differences of depression in younger and older adults.
Although all currently approved antidepressants target the monoamine system, studies in younger adults with depression have implicated dysregulation of the two main inhibitory and excitatory neurotransmitter systems: gamma-aminobutyric-acid (GABA) and glutamate [11, 12]. Early studies found abnormal peripheral levels of GABA and glutamate in the cerebrospinal fluid or serum of young adults with depression [13, 14]. Magnetic resonance spectroscopy (MRS) studies have since found mostly downregulated cortical GABA concentrations and abnormal cortical glutamate concentrations in patients under age 60 with depression [15]. Similarly, post-mortem studies of patients with depression have reported reductions in the size and density of cortical GABAergic interneurons [16], downregulation of GABA-related genes [17], and altered glutamate N-methyl-D-aspartate (NMDA) receptor levels [18]. Far fewer studies of depression have investigated GABAergic and glutamatergic systems in older adults. One study of peripheral metabolites in older depressed patients found reduced plasma GABA concentrations in patients compared to age-matched controls [19]. Additionally, a post-mortem study of depressed patients aged 30–86 years found a negative correlation between age and the density and soma size of prefrontal cortex GABAergic neurons [16]. In order to inform treatment development for older adults with depression, a clearer understanding of the role of GABAergic and glutamatergic neurotransmission in LLD is needed.
Transcranial magnetic stimulation (TMS) is a non-invasive brain stimulation method that can be used in conjunction with electromyography (EMG) to reliably probe inhibitory and excitatory neurotransmission in the motor cortex. Whereas MRS is believed to measure tonic inhibitory tone [20], TMS paradigms alone can measure the phasic activation of GABAergic interneurons, thus offering unique insight into the synaptic activity of inhibitory and excitatory systems in humans [21]. For example, short-interval cortical inhibition (SICI) and cortical silent period (CSP) TMS measures can be used to investigate cortical inhibitory neurotransmission [22, 23], with good test–retest reliability [24, 25]. SICI measures suppression of the motor-evoked potential (MEP) amplitude when a subthreshold TMS pulse precedes a suprathreshold TMS pulse by 1–6 ms. The CSP measures the duration of EMG activity suppression following a suprathreshold TMS pulse delivered during muscle contraction.
SICI and CSP indirectly measure GABAA and GABAB receptor-mediated inhibition, respectively. For example, benzodiazepines act as positive allosteric modulators at GABAA receptors and reliably facilitate SICI [26], whereas baclofen, a GABAB receptor agonist, prolongs CSP duration [27]. Likewise, the time course of inhibitory effects observed in SICI are consistent with the fast time course of GABAA-mediated inhibitory postsynaptic potentials (IPSPs) [28], and the CSP duration corresponds to the peak of slow GABAB receptor-mediated IPSPs [29]. SICI and CSP durations have both been found to be low in younger patients with depression and treatment-resistant depression [30–32], reflecting impaired GABAA and GABAB receptor-mediated inhibition. The association between these TMS measures of GABAergic neurotransmission and LLD has yet to be examined.
TMS can also be used to assess cortical excitation through resting motor threshold (RMT) and intracortical facilitation (ICF) paradigms. RMT is thought to index general neuronal membrane excitability [21], whereas pharmacological studies suggest that ICF reflects the activity of excitatory NMDA receptor-dependent glutamatergic circuits. For example, administration of NMDA receptor antagonists has been shown to decrease ICF [26]. Although RMT and ICF have typically been reported to be normal in younger adults with depression, differences in cortical excitability have been found between younger and older healthy adults [33, 34]. As with TMS measures of cortical inhibition, these TMS indices of cortical excitability have yet to be assessed in older adults with depression.
Here, we conducted the first study of TMS measures of cortical inhibition (SICI and CSP) and excitation (RMT and ICF) in a large sample of older adults with early- or late-onset depression. We hypothesized that older adults with depression would display abnormal GABAergic cortical inhibition and glutamatergic cortical excitation compared to age-matched healthy controls. To dissect the influence of age and depression on these TMS measures, we also conducted exploratory comparisons with younger patients with depression, and younger healthy controls.
Materials and methods
Study sample
Older adults with depression who had enrolled in one of two clinical treatment trials at the Centre for Addiction and Mental Health (CAMH) were invited to participate in the study prior to initiating treatment: 92 older adults with depression participated and 41 age-matched healthy controls were recruited from registries and advertisements. For comparison, we also analyzed published data from 30 younger patients with depression [32], and unpublished data from 30 age-matched younger healthy controls.
Older adults with depression were ≥ 60 years of age, met diagnostic criteria for major depressive disorder (MDD), single or recurrent and with early- or late-onset, as diagnosed by the Structured Clinical Interview for the DSM-IV (SCID-IV), and were free of any current comorbid Axis I disorders, with the exception of anxiety disorders. They had a Montgomery-Asberg Depression Rating Scale (MADRS) [35] score ≥ 15 and a Mini-Mental State Examination (MMSE) [36] score > 24. Patients were excluded if they were taking an anticonvulsant. Benzodiazepine use was permitted, however the dose could not exceed lorazepam at 2 mg/d or equivalent. Zopiclone up to 15 mg/d and trazodone up to 50 mg/d were also allowed. Some patients who were being cross-titrated from another antidepressant were on a low dose of this antidepressant at the time of TMS testing. Treatment resistance was assessed in all patients using the Antidepressant Treatment History Form (ATHF) [37].
All healthy controls were free of any psychiatric disorders based on the Mini International Neuropsychiatric Interview Version 6.0 (MINI 6.0), and screened negative for any substance use on a urine toxicology screen (MEDTOX® Diagnostics, Inc., Burlington, NC, USA).
The Research Ethics Board at CAMH approved the study procedures in accordance with the Declaration of Helsinki. All participants provided written, informed consent.
TMS-EMG procedures
For EMG recordings, disposable 9 mm surface electrodes were positioned over the right abductor pollicis brevis (APB) muscle belly (active electrode) and the interphalangeal joint of the thumb (reference electrode). The ground electrode was positioned at the proximal end of the right forearm at the inside of the elbow joint. Participants were asked to relax their right hand throughout the experiment, and EMG recordings were monitored in real time to verify the relaxed state of the APB muscle. The EMG signal was amplified, filtered (band pass 2–2.5 kHz) and digitized at 5 kHz.
A figure-of-eight coil with 70 mm diameter coil loops and two Magstim 200 stimulators (Magstim, Whitland, Dyfed, Wales), connected by a Bistim module, were used to administer monophasic TMS pulses. The TMS coil was positioned tangentially on the head, over the left motor cortex, at the optimal location for evoking motor-evoked potentials (MEPs) from the APB muscle at rest. The axis of the coil was held at ~ 45° angle lateral to the midsagittal line, in order to induce a posterior-anterior current flow, optimal for trans-synaptic activation of corticospinal neurons [38]. All TMS methodologies adhered to a recently developed checklist for the collection and reporting of TMS data [39].
RMT was defined as the minimum TMS intensity required to produce a ≥ 50 µV peak-to-peak MEP amplitude in the relaxed APB muscle in at least 5 of 10 consecutive trials. The TMS intensity required to evoke MEPs with a peak-to-peak amplitude of ~1 mV (0.5–1.5 mV) in the relaxed APB muscle was then established for use as the test stimulus (TS) intensity in the paired-pulse paradigms [40].
For SICI and ICF paired-pulse TMS measures, a subthreshold conditioning stimulus (CS) at 80% RMT preceded a suprathreshold TS (intensity required to elicit ~1 mV peak-to-peak MEP amplitude) to the motor cortex with an interstimulus interval (ISI) of 2 ms (SICI) or 10 ms (ICF) [22]. For each participant, 36 trials were conducted, 12 for each condition: (i) TS alone (unconditioned response), (ii) CS preceding TS with 2 ms ISI (SICI conditioned response), and (iii) CS preceding TS with 10 ms ISI (ICF conditioned response). ISIs of 4 ms, 15 ms and 20 ms were also studied in a subset of older adults with depression (n = 40), however ISIs of 2 ms and 10 ms were found to elicit the greatest mean inhibition and facilitation, respectively, and are therefore the focus of this paper. Analyses of the SICI and ICF measures with ISIs of 4 ms, 15 ms and 20 ms are described in the Supplementary Materials and Methods. The conditioned MEP amplitude for SICI and ICF is expressed as a ratio of the conditioned response to the unconditioned response, i.e., SICI or ICF ratio = MEPconditioned/MEPunconditioned [41]. Thus, larger SICI ratios reflect larger conditioned MEPs and lower cortical inhibition.
For the single-pulse CSP paradigm, participants maintained voluntary muscle contraction at 20% of maximum force on a gauge meter using a pinch grip (thumb and index finger). During muscle contraction, 10 single suprathreshold TMS pulses (140% RMT) were delivered to the left motor cortex [23]. An average of the 10 trials was computed off-line, and CSP duration was assessed using the averaged recording [42]. Following a masking procedure, the CSP durations were measured from the onset of the MEP to the return of EMG activity by an experienced rater. Masking was performed using a Matlab script that shuffled and renamed all CSP files in a pseudorandom order. CSP durations were calculated for the shuffled files, which contained no clinical or demographic group indicators. After all CSP durations were extracted, the data were unshuffled. For all TMS paradigms, individual trials were manually checked and excluded if there was excessive noise or artifacts.
Statistical analysis
Demographic characteristics were compared among the groups using a one-way analysis of variance (ANOVA) followed by independent t tests, or using a Chi-Square (χ2) test for categorical variables. The significance level for all analyses was set to an α level of 0.05 (two-tailed). All data are reported as mean ± standard deviation (SD) unless otherwise stated.
Using histograms, boxplots, Q-Q plots, and Shapiro–Wilk test, we determined that RMT and the SICI and ICF ratios were non-normally distributed. RMT and SICI data were log-transformed, and ICF data square-root-transformed, to attain normal distributions. For RMT, CSP, SICI, and ICF measures, the primary analysis consisted of a two-way ANOVA with age (two levels: older and younger) and diagnosis (two levels: depression and no depression) as the between-subjects factors. Secondary analyses consisted of planned pairwise comparisons between subgroups, uncorrected for multiple comparisons. In addition, sensitivity analyses were performed to examine the robustness of the findings in the presence/absence of factors that may influence the TMS measures of GABAergic/glutamatergic functioning: antidepressant or sedative use, treatment resistance, late (≥ 60) or early (< 60) onset of depressive symptoms, and comorbid anxiety disorders (see Supplementary Materials and Methods).
Depending on the distributions of the measures of interest, exploratory Pearson or Spearman correlation analyses were performed. Correlation analyses were performed between each TMS measure and age across and within all groups, and between TMS measures of cortical inhibition and excitation across and within groups. In the LLD subgroup separately, each TMS measure was correlated with MADRS score and age of onset of the first depressive episode. in addition, greater medical burden is closely linked to both old age and depression [43, 44], and may mediate any associations between neurophysiological measures and LLD. Therefore, in the LLD subgroup, each TMS measure was also correlated with the Cumulative Illness Rating Scale for Geriatrics (CIRS-G) score [45].
Results
See Table 1 for a summary of demographic, clinical, and neurophysiological information across groups. Younger healthy controls reported significantly more years of education than older adults with depression (t119 = 2.9, p = 0.005).
Table 1.
Patient demographic, clinical, and neurophysiological characteristics
| Older adults with depression (n = 92) | Older healthy adults (n = 41) | Younger adults with depression (n = 30) | Younger healthy adults (n = 30) | Statistic | p value | |
|---|---|---|---|---|---|---|
| Demographic characteristics | ||||||
| Age (years) | 66.8 ± 6.1 | 69.0 ± 8.3 | 44.8 ± 10.5 | 44.9 ± 10.8 | F3,189 = 105.7 | <0.001 |
| Sex | 62 F/30 M | 23 F/18 M | 20 F/10 M | 18 F/12 M | χ2(3) = 1.9 | 0.60 |
| Education (years) | 14.5 ± 2.9 | 15.4 ± 2.3 | NA | 16.2 ± 2.9 | F2,158 = 4.9 | 0.009 |
| Handedness | 66 R/8 La | 40 R/1 L | 30 R/0 L | 28 R/2 L | χ2(6) = 11.3 | 0.08 |
| Clinical characteristics | ||||||
| MADRS score | 25.7 ± 5.4 | NA | NA | NA | − | − |
| HAM-D score | NA | − | 25.6 ± 3.0 | − | − | − |
| MMSE score | 29.0 ± 1.1 | NA | NA | NA | − | − |
| CIRS-G score | 6.5 ± 3.7 | NA | NA | NA | − | − |
| Late-onset (≥ 60 years), no. (%) | 19 (21%) | − | − | − | − | − |
| Treatment-resistant, no. (%)b | 69 (75%) | − | 19 (63%) | − | χ2(1) = 1.5 | 0.22 |
| Comorbid anxiety disorder, no. (%) | 32 (35%) | − | 14 (47%) | − | χ2(1) = 1.4 | 0.24 |
| Medication use, no. (%)c | ||||||
| Antidepressants | 45 (49%) | − | 18 (60%) | − | χ2(1) = 1.1 | 0.29 |
| Benzodiazepines | 27 (29%) | − | 11 (37%) | − | χ2(1) = 0.6 | 0.45 |
| Neurophysiological characteristics | ||||||
| TS intensity (%) | 63.5 ± 15.3 | 63.7 ± 12.7 | NA | 59.5 ± 12.7 | F2,159 = 1.0 | 0.38 |
| TS mean MEP amplitude (mV) | 0.98 ± 0.47 | 0.86 ± 0.34 | 0.95 ± 0.24 | 0.93 ± 0.30 | F3,185 = 0.9 | 0.45 |
Abbreviations: R = right, L = left, MADRS = Montgomery-Asberg Depression Rating Scale, MMSE = Mini-Mental State Examination, HAM-D = Hamilton Rating Scale for Depression, RMT = resting motor threshold (% maximum stimulator output), NA = not available, TS = test stimulus, TS intensity = intensity required to elicit 1 mV peak-to-peak MEP amplitude (% maximum stimulator output), MEP = motor-evoked potential
a Handedness data missing for 18 patients
b Treatment resistance defined as non-response to an adequate antidepressant trial (a score ≥ 3 on the Antidepressant Treatment History Form)
c Medication use at the time of neurophysiological testing
Cortical inhibition
SICI and ICF data were missing for four younger adults with depression, thus 26 younger adults with depression were included in the SICI and ICF analyses. For SICI, significant main effects of age and diagnosis were observed (see Table 2). The interaction between age and diagnosis was not significant (F1,185 = 2.73, p = 0.10). Compared with younger healthy controls, SICI was significantly reduced in older adults with depression (t120 = 3.58, p < 0.001, dCohen = 0.75), older healthy controls (t69 = 2.45, p = 0.017, dCohen = 0.59), and younger adults with depression (t54 = 2.39, p = 0.02, dCohen = 0.64). SICI did not differ significantly between older adults with depression and older healthy controls (t131 = 0.63, p = 0.53) or between older and younger adults with depression (t116 = 0.29, p = 0.78). See Fig. 1 for an illustration of SICI across groups.
Table 2.
TMS measures of cortical inhibition and excitation by group
| Older adults with depression (n = 92) | Older healthy controls (n = 41) | Younger adults with depression (n = 30) | Younger healthy controls (n = 30) | Statistic | p value | |
|---|---|---|---|---|---|---|
| Short-interval cortical inhibition ratio | 0.64 ± 0.49 (L-O: 0.58 ± 0.20) (E-O: 0.66 ± 0.54) |
0.60 ± 0.39 | 0.63 ± 0.41 | 0.37 ± 0.25 | Age: F1,185 = 4.14 DX: F1,185 = 5.61 |
0.043 0.019 |
| Intracortical facilitation ratio | 1.83 ± 0.84 (L-O: 1.90 ± 1.04) (E-O: 1.81 ± 0.79) |
1.77 ± 0.59 | 1.52 ± 0.54 | 1.64 ± 0.72 | Age: F1,185 = 3.63 DX: F1,185 = 0.04 |
0.058 0.84 |
| Cortical silent period duration (ms) | 124.8 ± 33.1 (L-O: 111.4 ± 29.2) (E-O: 128.3 ± 33.3) |
129.4 ± 29.4 | 127.2 ± 33.4 | 135.3 ± 34.1 | Age: F1,187 = 0.62 DX: F1,187 = 1.48 |
0.43 0.23 |
| Resting motor threshold (%) | 49.1 ± 10.5 (L-O: 46.05 ± 7.8) (E-O: 49.9 ± 11.1) |
48.8 ± 7.9 | 47.6 ± 16.6 | 48.0 ± 7.5 | Age: F1,189 = 1.22 DX: F1,189 = 0.004 |
0.27 0.49 |
L-O = Subgroup of older adults with late-onset depression (≥ 60 years, n = 19); E-O = Subgroup of older adults with early-onset depression (< 60 years, n = 73). Resting motor threshold = % maximum stimulator output; DX = Diagnosis of depression. Subgroup data not included in ANOVA statistics in right-most columns
Fig. 1.
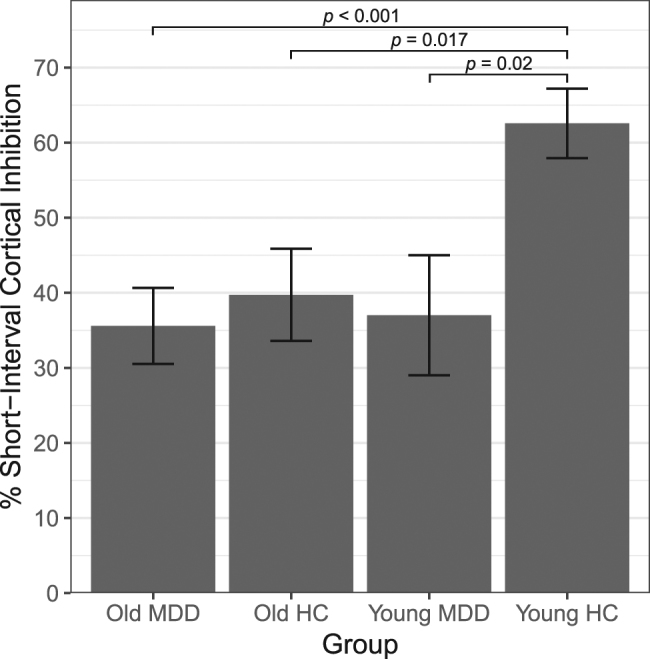
Mean % cortical inhibition in the SICI paradigm by group. % SICI was calculated as (1–SICI ratio) × 100%. Hence, a higher SICI ratio reflects poorer inhibition, whereas a higher % SICI reflects greater inhibition. Error bars represent standard error of the mean. MDD = major depressive disorder patients, HC = healthy controls
Findings of lower SICI in older adults with depression compared with younger healthy controls remained significant in the sensitivity analyses that examined the influence of antidepressant or benzodiazepine/sedative use, treatment resistance, late or early onset of depressive symptoms, and anxiety disorder comorbidity (see Supplementary Materials and Methods).
CSP data were missing for one older adult with depression and one younger adult with depression, thus 91 older patients and 29 younger patients with depression were included in the CSP analyses. For CSP, we did not observe significant main effects of age or diagnosis, or a significant age × diagnosis interaction (F1,187 = 0.11, p = 0.74). See Table 2 for mean SICI ratios and CSP durations across groups.
Cortical excitation
See Table 2 for mean RMT and ICF ratios across groups. No significant age or diagnosis main effects, and no significant age × diagnosis interaction (F1,189 = 0.46, p = 0.50), were observed for RMT. Similarly, no significant main effects or interactions were found for ICF (age × diagnosis interaction: F1,185 = 0.20, p = 0.66).
Correlations with TMS measures
Across all groups, a weak positive correlation was observed between age and the SICI ratio (Spearman’s rho (rs) = 0.17, p = 0.019), reflecting an association between diminished cortical inhibition and advancing age. The correlation between age and SICI remained significant in the subgroup of healthy controls (rs = 0.29, p = 0.016), but not in those with depression (rs = 0.07, p = 0.47). The CIRS-G score, which reflects chronic medical illness burden, was also positively correlated with the SICI ratio in the subgroup of older adults with depression (rs = 0.26, p = 0.014). No other significant correlations with the TMS measures were observed.
Exploration of the relationships between TMS measures also found no significant correlations between SICI and ICF (rs = 0.097), SICI and CSP (rs = −0.092), or ICF and CSP (rs = 0.007) across groups. In only patients with depression, a small but significant positive correlation was observed between SICI and ICF ratios (rs = 0.21, p = 0.021), indicating a possible association between impaired cortical inhibition and elevated cortical excitation in depression. By contrast, the correlation between SICI and ICF was not observed in healthy adults (rs = −0.08, p = 0.51).
Discussion
To our knowledge, this is the first TMS study investigating cortical inhibition and excitation in older adults with depression. Compared with younger healthy adults (56.1% mean cortical inhibition), we demonstrated that cortical inhibition is reduced in (i) older adults with early- or late-onset depression, (ii) older healthy adults and (iii) younger adults with depression (35.6, 39.7, and 41.7% mean inhibition, respectively). The diminished cortical inhibition observed in these three groups was specific to SICI, which reflects GABAA receptor-mediated inhibition, and not to CSP, which reflects GABAB receptor-mediated inhibition. The observed SICI reductions are congruent with earlier reports of lower SICI in adults with depression by Bajbouj et al. (left hemisphere: 35.2% inhibition, compared with 69.3% inhibition in healthy controls) [30] and Lefaucheur et al. (left hemisphere: 36% inhibition, compared with 77.4% in healthy controls) [31]. Contrary to our hypotheses, we did not discern any differences in cortical inhibition (SICI and CSP) between older adults with depression and age-matched healthy adults. However, our findings of (i) reduced GABAA receptor-mediated neurotransmission in older vs. younger adults and (ii) an association between decreasing inhibition and advancing age, are consistent with previous studies that have shown lower SICI in older, compared to younger, healthy adults [46, 47], and a similar linear relationship between SICI and age [47]. Incongruent reports of elevated SICI in older adults might be accounted for by small sample sizes in earlier reports and methodological differences between studies, including different sample demographics, CS intensities, ISIs, and locations of EMG measurement [33, 48]. Taken together, our findings suggest that GABAA receptor-mediated inhibitory neurotransmission is diminished during both aging and depression, and that the influence of aging and depressive states on TMS measures of cortical inhibition cannot be differentiated in LLD.
Various biological factors may have contributed to the observed reductions in cortical inhibition with both advancing age and depression. First, we observed a weak negative correlation between cortical inhibition (SICI) and cortical excitation (ICF) in depressed patients; it is possible that the observed inhibition deficits involve an abnormal inhibition/excitation balance in adults with depression across the lifespan. Accordingly, an abnormal GABA/glutamate signaling balance has been shown in a rodent model of depression, accompanied by a shift in the GABA/glutamate balance with antidepressant treatment [49]. Although speculative, it is possible that an abnormal balance of cortical inhibitory and excitatory neurotransmission could contribute to the observed imbalance between resting-state functional networks in depressed patients, which in turn could drive the imbalance between internal and external mental contents that is observed in depression [50]. We also found that the burden of comorbid physical illness was weakly associated with reduced GABAA receptor-mediated inhibitory neurotransmission in older adults with depression. It is important to consider the effects that systemic health in LLD may have on cortical inhibition and brain functioning in general. Depression is associated with higher rates of age-related illness [43, 44], and there is some evidence that age-related pathologies, such as hypoxia, can impair GABAergic inhibitory neurotransmission [51]. Depression has similarly been associated with accelerated biological aging [52]; aging can affect other biological processes, including muscle mass, motor units, and nerve conduction velocity [53–55], which may influence TMS measures of GABAergic neurotransmission in LLD. Likewise, stressful and traumatic experiences, which typically accumulate with advancing age and are risk factors for the development of depression [56], have been shown to influence GABAergic neurotransmission [57]. As such, the functional consequences of biological shifts associated with aging and depression are likely driven by the interaction of many different mechanisms, rather than GABAergic inhibition alone. Furthermore, deficits in TMS measures of cortical inhibition have been observed in a wide range of psychiatric disorders [58]. Thus, rather than being specific to depressive states, the deficits in cortical inhibition observed here in depression may be more generally associated with psychopathology across the lifespan.
In contrast to SICI, the present study did not find significant effects of age or depression on CSP duration. Changes in CSP have been reported following administration of benzodiazepines, which are positive modulators of GABAA receptors [26]. CSP may therefore reflect the tonic conductance of a subtype of GABAA receptors, in addition to GABAB receptor-mediated neurotransmission, whereas SICI more likely reflects the phasic, i.e., transient, activation of GABAA receptors at the synapse [59]. Nevertheless, abnormally short CSPs have been reported previously in older healthy adults [60] and younger patients with depression [30–32]. However, the mean CSP duration reported here in 30 younger healthy adults (134.2 ± 36.9 ms) appears to be considerably shorter than the mean CSP duration observed by our group previously in a sample of 25 healthy controls of similar age (158.5 ± 31.7 ms [32]). The lack of significant CSP differences between groups in the present study may be related to the shorter mean CSP in the current sample of healthy controls as compared to what was previously demonstrated. The lower healthy control CSP durations reported in the current study as compared with earlier reports could reflect the high between-subject variability of the CSP measure [61]; as such, caution should be taken when interpreting between-subject differences in CSP.
We similarly observed no significant differences in cortical excitation measures (RMT and ICF) between any groups. Our results are congruent with the majority of TMS studies assessing indices of phasic glutamatergic functioning, which have found similar cortical excitation in MDD patients and controls [30, 32, 58]. Notably, RMT is primarily dependent on ion channel conductivity, voltage-gated Na+/K+ channels [21, 62], and is unaffected by GABA [21], glutamate [63, 64], and dopamine [65]. By contrast, ICF is closely related to NMDA receptor-mediated glutamatergic function [21, 26]. NMDA receptors are a target in the treatment of depression [66], however not all patients benefit from treatment with NMDA antagonists [67]. Moreover, although some MRS studies have found abnormally low cortical concentrations of glutamate in depression [68], MRS measures of glutamate-related metabolites reflect a mix of physiological and non-physiological components of the glutamatergic system, rather than NMDA receptor-mediated cortical neurotransmission per se; as such, ICF does not correlate with MRS measures of glutamate concentrations [69]. Going forward, an investigation of the ability for cortical excitability to change, by indexing neuroplasticity using the TMS paradigm paired associative stimulation, may elicit a clearer understanding of pathophysiological mechanisms related to cortical excitation in LLD.
This work also complements the TMS literature investigating the influence of aging on various aspects of neurophysiology. We observed significant reductions in GABAA receptor-mediated neurotransmission in the motor cortex with advancing age, yet interestingly no significant differences between younger and older adults in other TMS measures of cortical inhibition and excitation (CSP, ICF, and RMT). Previous studies have similarly found no significant differences in cortical excitation with age [46–48], and although earlier TMS studies of cortical inhibition in older healthy adults have yielded mixed results [34], the present work involved one of the largest samples of older adults tested in a single study to date. There is evidence that other aspects of motor cortex neurophysiology are diminished in older compared with younger adults, including neuroplasticity [70, 71], interhemispheric inhibition [72, 73], and inhibitory and excitatory connectivity between ipsilateral motor regions [74]. The functional consequences of specific changes in the physiology of the motor cortex have yet to be fully characterized, yet may shed some light into age-related decline in motor control [75]. For example, contrary to the current findings, a variant of the ICF paradigm that may specifically reflect excitatory processes prior to and during particular types of grasp has been shown to be abnormal in old age and to correlate with hand dexterity [76]. Moreover, factors that can normalize impaired cortical physiology, such as an active lifestyle [73], may be useful in preventing age-related decline in functioning.
Of note, the majority of older adults with depression studied here were treatment-resistant (75%). Some studies have implicated greater GABAergic deficits in patients with treatment-resistant depression, as compared with non-treatment-resistant patients [32, 77]. However, our findings of reduced SICI in older adults with depression, as compared with younger healthy controls, were observed for subgroups of treatment-resistant and non-treatment-resistant older patients alike (see Supplementary Materials and Methods).
Some limitations of our study should be considered. First, the presented TMS findings from the motor cortex may not directly reflect brain regions involved in the pathology of depression. Future studies of LLD neuropathology should assess cortical inhibition directly in the DLPFC, using TMS combined with EEG [78]. Second, although TMS currently offers the only in vivo measures of phasic GABAergic activity in humans, TMS measures of cortical inhibition are indirect indices of GABAergic inhibitory neurotransmission. In addition, future work should include a more comprehensive clinical characterization of the control groups, such as inclusion of CIRS, MMSE, and depression scores, and further neuropsychological characterization of the LLD patients. At last, owing to our inclusion of older adults with depression that presented with cardiac or cerebrovascular risk factors, it is possible that some of the patients studied here suffered from vascular depression. Although several clinical studies show a strong correlation between LLD and cardiac risk factors, there is evidence that patients with vascular depression have different neurophysiological profiles than patients with nonvascular MDD [79]. However, the majority of the patients in the current study had recurrent MDD with onset before the age of 60, whereas vascular depression is more closely associated with late-onset MDD (onset ≥ 60 years) [80, 81].
Overall, our findings, taken together with previous reports of impaired cortical inhibition in younger adults with depression, are consistent with the age-by-disease model of LLD, which posits that age-related biological changes that contribute to the impairment of specific neuronal and glial processes promote vulnerability to the development of depressive symptoms in late-life [6, 7]. Previous literature reveals a robust and heterogeneous effect of age on numerous biological pathways that overlap with those implicated in depression pathology, including, but not limited to, glial-mediated inflammation, oxidative stress, and calcium regulation [6]. There is evidence that physiological changes in the brains of older adults and the brains of younger depressed adults are similar. Contributions of genetic, endocrine, epigenetic, and environmental factors may determine whether and when pathophysiological thresholds are reached and depressive symptoms are expressed [7].
Alternatively, the current finding that reductions in cortical inhibition are similar in depression and old age are in line with accumulating evidence that depression is a disease of accelerated biological aging [52]. Major depressive disorder is associated with higher rates of medical comorbidities, including heart disease [43], metabolic syndrome [82], and Alzheimer’s disease [83], which commonly occur in old age. Moreover, cellular biomarkers of aging and age-related disease have been observed in depression. For example, shorter telomere length, which promotes apoptosis and is typically observed in old age, has been associated with depression [84]. More specifically, an increase in the number of toxic processes, such as oxidative stress, coupled with a decline in protective factors, such as antioxidants, (i) can cause cellular damage, and (ii) have been implicated in both age-related diseases and depression [52]. Deficits in cortical inhibitory neurotransmission in depression, shown here to mimic reductions that occur in old age, therefore lend further support for the involvement of premature biological aging in depression.
In conclusion, our study is the first to examine TMS measures of cortical inhibition and excitation in LLD. We investigated a relatively large sample of older adults with early- or late-onset depression, and one of the largest samples of healthy older adults to date. Our findings indicate that cortical inhibition measures cannot differentiate between the changes associated with aging or with depression in late life. However, our findings of SICI reduction (i.e., cortical inhibition deficits), taken together with comparable previous findings of SICI deficits in younger patients with depression [30, 31], suggest that both advancing age and MDD involve reduced GABAergic inhibitory neurotransmission. These overlapping cortical inhibition reductions mirror other biological changes that occur with both age and depression, such as changes in the expression of genes coding for GABA interneuron-related peptides [85]. These observations are consistent with the conceptualization of depression as a disease of accelerated aging, and with the age-by-disease model of LLD [6, 7] and may provide targets for both the prevention and treatment of LLD. Future research should aim to clarify whether reductions in GABAergic cortical inhibition in old age, as observed here, constitute a biological risk factor for the development of depression in late-life.
Electronic supplementary material
Acknowledgements
This study was funded in part by a Brain and Behaviour Research Foundation New Investigator Award (DMB), the Canadian Institutes of Health Research and the National Institutes of Health (R01MH083643, R34MH101365). We would also like to acknowledge the Temerty Centre and the Canada Foundation for Innovation for providing TMS equipment.
Competing interests
JIL, AB, and RZ report no biomedical interests or conflicts. BHM currently receives research funding from Brain Canada, the Centre for Addiction and Mental Health (CAMH) Foundation, the Canadian Institutes of Health Research (CIHR), Patient-Centered Outcomes Research Institute (PCORI), and the US National Institute of Health (NIH). During the last five years, he also received research support from Bristol-Myers Squibb (medications for a NIH-funded clinical trial), Eli-Lilly (medications for a NIH-funded clinical trial), and Pfizer (medications for a NIH-funded clinical trial). He directly own stocks of General Electric (less than $5000). EJL reports research funding (current/past) from Janssen, Alkermes, Acadia, Takeda, Lundbeck, Barnes Jewish Foundation, PCORI, and Taylor Family Institute for Innovative Psychiatric Research. CFR has received research support from the NIH, PCORI, the Center for Medicare and Medicaid Services, the American Foundation for Suicide Prevention, the Brain and Behavior Research Foundation, and the Commonwealth of Pennsylvania. Bristol Meyers Squib and Pfizer have provided pharmaceutical supplies for his NIH sponsored research. JFK received medication supplies from Indivior to support this investigator-initiated trial. He has also received medication supplies from Pfizer for investigator-initiated work. JFK receives research funding from NIH and PCORI. TKR receives research support from Brain Canada, Brain and Behavior Research Foundation, Canada Foundation for Innovation, the CIHR, Ontario Ministry of Health and Long-Term Care, Ontario Ministry of Research and Innovation, the NIH, and the W. Garfield Weston Foundation. YN has received equipment-in-kind support for an investigator-initiated study from Magventure Inc. He has also received research support from Otsuka Pharmaceutical Co., Ltd, Shionogi & Co., Ltd., and Meiji Seika Pharma Co., Ltd. YN has received research support from Meiji Yasuda Mental Health Foundation. ES has received support from NIH, CIHR, the Brain & Behavior Research Foundation (NARSAD) and the Campbell Family Mental Health Research Institute of CAMH. ES is co-inventor on a US provisional patent application that covers compounds modulating the function of GABA neurons. ZJD has received within the last 3 years both research and equipment in-kind support for an investigator-initiated study through Brainsway Inc and Magventure Inc. ZJD has also received monies for participation on an advisory board from Sunovion Inc. Finally, ZJD owns > $10,000 (CAD) in stock of Biogen Inc. DMB has received research support from CIHR, NIH, Brain Canada and the Temerty Family through the CAMH Foundation and the Campbell Research Institute. He receives research support and in-kind equipment support for an investigator-initiated study from Brainsway Ltd. and he is the site principal investigator for three sponsor-initiated studies for Brainsway Ltd. He also receives in-kind equipment support from Magventure for an investigator-initiated study. He receives medication supplies for an investigator-initiated trial from Indivior.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Joint first authors: Jennifer I. Lissemore, Apoorva Bhandari
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41386-018-0093-x).
References
- 1.Van Orden K, Conwell Y. Suicides in late life. Curr Psychiatry Rep. 2011;13:234–41. doi: 10.1007/s11920-011-0193-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ganguli M, Dodge HH, Mulsant BH. Rates and predictors of mortality in an aging, rural, community-based cohort: the role of depression. Arch Gen Psychiatry. 2002;59:1046–52. doi: 10.1001/archpsyc.59.11.1046. [DOI] [PubMed] [Google Scholar]
- 3.Katon WJ, Lin E, Russo J, Unützer J. Increased medical costs of a population-based sample of depressed elderly patients. Arch Gen Psychiatry. 2003;60:897–903. doi: 10.1001/archpsyc.60.9.897. [DOI] [PubMed] [Google Scholar]
- 4.Alexopoulos GS, Meyers BS, Young RC, Kakuma T, Feder M, Einhorn A, et al. Recovery in geriatric depression. Arch Gen Psychiatry. 1996;53:305–12. doi: 10.1001/archpsyc.1996.01830040039008. [DOI] [PubMed] [Google Scholar]
- 5.Beekman AT, Geerlings SW, Deeg DJ, Smit JH, Schoevers RS, de Beurs E, et al. The natural history of late-life depression: a 6-year prospective study in the community. Arch Gen Psychiatry. 2002;59:605–11. doi: 10.1001/archpsyc.59.7.605. [DOI] [PubMed] [Google Scholar]
- 6.Sibille E. Molecular aging of the brain, neuroplasticity, and vulnerability to depression and other brain-related disorders. Dialog Clin Neurosci. 2013;15:53–65. doi: 10.31887/DCNS.2013.15.1/esibille. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKinney BC, Sibille E. The age-by-disease interaction hypothesis of late-life depression. Am J Geriatr Psychiatry. 2013;21:418–32. doi: 10.1016/j.jagp.2013.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Douillard-Guilloux G, Guilloux JP, Lewis DA, Sibille E. Anticipated brain molecular aging in major depression. Am J Geriatr Psychiatry. 2013;21:450–60. doi: 10.1016/j.jagp.2013.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–98. doi: 10.1016/S0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 10.Resnick SM, Pham DL, Kraut MA, Zonderman AB, Davatzikos C. Longitudinal magnetic resonance imaging studies of older adults: a shrinking brain. J Neurosci. 2003;23:3295–301. doi: 10.1523/JNEUROSCI.23-08-03295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry. 2011;16:383–406. doi: 10.1038/mp.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanacora G, Treccani G, Popoli M. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. 2012;62:63–77. doi: 10.1016/j.neuropharm.2011.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerner RH, Hare TA. CSF GABA in normal subjects and patients with depression, schizophrenia, mania, and anorexia nervosa. Am J Psychiatry. 1981;138:1098–101. doi: 10.1176/ajp.138.9.1259. [DOI] [PubMed] [Google Scholar]
- 14.Kim J, Schmid-Burgk W, Claus D, Kornhuber H. Increased serum glutamate in depressed patients. Eur Arch Psychiatry Clin Neurosci. 1982;232:299–304. doi: 10.1007/BF00345492. [DOI] [PubMed] [Google Scholar]
- 15.Sanacora G, Gueorguieva R, Epperson C, et al. Subtype-specific alterations of γ-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–13. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- 16.Rajkowska G, O’Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32:471–82. doi: 10.1038/sj.npp.1301234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guilloux JP, Douillard-Guilloux G, Kota R, Wang X, Gardier A, Martinowich K, et al. Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry. 2012;17:1130–42. doi: 10.1038/mp.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajkowska G. Pathology in astroglia, glutamate, and GABA in major depressive disorder: evidence from studies of human postmortem tissue. In: Popoli M, Diamond D, Sanacora G, editors. Synaptic Stress and Pathogenesis of Neuropsychiatric Disorders. New York: Springer; 2014. pp. 245–64. [Google Scholar]
- 19.Paige LA, Mitchell MW, Krishnan KRR, Kaddurah‐Daouk R, Steffens DC. A preliminary metabolomic analysis of older adults with and without depression. Int J Geriatr Psychiatry. 2007;22:418–23. doi: 10.1002/gps.1690. [DOI] [PubMed] [Google Scholar]
- 20.Dyke K, Pépés SE, Chen C, Kim S, Sigurdsson HP, Draper A, et al. Comparing GABA-dependent physiological measures of inhibition with proton magnetic resonance spectroscopy measurement of GABA using ultra-high-field MRI. Neuroimage. 2017;152:360–70. doi: 10.1016/j.neuroimage.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ziemann U, Lonnecker S, Steinhoff BJ, Paulus W. Effects of antiepileptic drugs on motor cortex excitability in humans: a transcranial magnetic stimulation study. Ann Neurol. 1996;40:367–78. doi: 10.1002/ana.410400306. [DOI] [PubMed] [Google Scholar]
- 22.Kujirai T, Caramia MD, Rothwell JC, Day BL, Thompson PD, Ferbert A, et al. Corticocortical inhibition in human motor cortex. J Physiol. 1993;471:501–19. doi: 10.1113/jphysiol.1993.sp019912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cantello R, Gianelli M, Civardi C, Mutani R. Magnetic brain stimulation: the silent period after the motor evoked potential. Neurology. 1992;42:1951–59. doi: 10.1212/WNL.42.10.1951. [DOI] [PubMed] [Google Scholar]
- 24.Fritz C, Braune HJ, Pylatiuk C, Pohl M. Silent period following transcranial magnetic stimulation: a study of intra- and inter-examiner reliability. Electromyogr Mot Control. 1997;105:235–40. doi: 10.1016/S0924-980X(97)96675-3. [DOI] [PubMed] [Google Scholar]
- 25.Maeda F, Gangitano M, Thall M, Pascual-Leone A. Inter- and intra-individual variability of paired-pulse curves with transcranial magnetic stimulation (TMS) Clin Neurophysiol. 2002;113:376–82. doi: 10.1016/S1388-2457(02)00008-1. [DOI] [PubMed] [Google Scholar]
- 26.Paulus W, Classen J, Cohen LG, Large CH, Di Lazzaro V, Nitsche M, et al. State of the art: pharmacologic effects on cortical excitability measures tested by transcranial magnetic stimulation. Brain Stimul. 2008;1:151–63. doi: 10.1016/j.brs.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Siebner HR, Dressnandt J, Auer C, Conrad B. Continuous intrathecal baclofen infusions induced a marked increase of the transcranially evoked silent period in a patient with generalized dystonia. Muscle Nerve. 1998;21:1209–12. doi: 10.1002/(SICI)1097-4598(199809)21:9<1209::AID-MUS15>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 28.Davies C, Davies S, Collingridge G. Paired-pulse depression of monosynaptic GABA‐mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol. 1990;424:513–31. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connors B, Malenka R, Silva L. Two inhibitory postsynaptic potentials, and GABAA and GABAB receptor-mediated responses in neocortex of rat and cat. J Physiol. 1988;406:443–68. doi: 10.1113/jphysiol.1988.sp017390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bajbouj M, Lisanby SH, Lang UE, Danker-Hopfe H, Heuser I, Neu P. Evidence for impaired cortical inhibition in patients with unipolar major depression. Biol Psychiatry. 2006;59:395–400. doi: 10.1016/j.biopsych.2005.07.036. [DOI] [PubMed] [Google Scholar]
- 31.Lefaucheur JP, Lucas B, Andraud F, Hogrel JY, Bellivier F, Del Cul A, et al. Inter-hemispheric asymmetry of motor corticospinal excitability in major depression studied by transcranial magnetic stimulation. J Psychiatr Res. 2008;42:389–98. doi: 10.1016/j.jpsychires.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 32.Levinson AJ, Fitzgerald PB, Favalli G, Blumberger DM, Daigle M, Daskalakis ZJ. Evidence of cortical inhibitory deficits in major depressive disorder. Biol Psychiatry. 2010;67:458–64. doi: 10.1016/j.biopsych.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 33.McGinley M, Hoffman RL, Russ DW, Thomas JS, Clark BC. Older adults exhibit more intracortical inhibition and less intracortical facilitation than young adults. Exp Gerontol. 2010;45:671–8. doi: 10.1016/j.exger.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhandari A, Radhu N, Farzan F, Mulsant BH, Rajji TK, Daskalakis ZJ, et al. A meta-analysis of the effects of aging on motor cortex neurophysiology assessed by transcranial magnetic stimulation. Clin Neurophysiol. 2016;127:2834–45. doi: 10.1016/j.clinph.2016.05.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–9. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- 36.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 37.Sackeim HA. The definition and meaning of treatment-resistant depression. J Clin Psychiatry. 2001;62:10–17. [PubMed] [Google Scholar]
- 38.Kaneko K, Kawai S, Fuchigami Y, Morita H, Ofuji A. The effect of current direction induced by transcranial magnetic stimulation on the corticospinal excitability in human brain. Electroencephalogr Clin Neurophysiol. 1996;101:478–82. doi: 10.1016/s0013-4694(96)96021-x. [DOI] [PubMed] [Google Scholar]
- 39.Chipchase L, Schabrun S, Cohen L, Hodges P, Ridding M, Rothwell J, et al. A checklist for assessing the methodological quality of studies using transcranial magnetic stimulation to study the motor system: an international consensus study. Clin Neurophysiol. 2012;123:1698–704. doi: 10.1016/j.clinph.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rossini PM, Barker AT, Berardelli A, Caramia MD, Caruso G, Cracco RQ, et al. Non-invasive electrical and magnetic stimulation of the brain, spinal cord and roots: Basic principles and procedures for routine clinical application. Report of an IFCN committee. Electroencephalogr Clin Neurophysiol. 1994;91:79–92. doi: 10.1016/0013-4694(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 41.Daskalakis ZJ, Christensen BK, Fitzgerald PB, Chen R. Transcranial magnetic stimulation: a new investigational and treatment tool in psychiatry. J Neuropsychiatry Clin Neurosci. 2002;14:406–15. doi: 10.1176/jnp.14.4.406. [DOI] [PubMed] [Google Scholar]
- 42.Daskalakis ZJ, Molnar GF, Christensen BK, Sailer A, Fitzgerald PB, Chen R. An automated method to determine the transcranial magnetic stimulation-induced contralateral silent period. Clin Neurophysiol. 2003;114:938–44. doi: 10.1016/S1388-2457(03)00038-5. [DOI] [PubMed] [Google Scholar]
- 43.Nicholson A, Kuper H, Hemingway H. Depression as an aetiologic and prognostic factor in coronary heart disease: a meta-analysis of 6362 events among 146 538 participants in 54 observational studies. Eur Heart J. 2006;27:2763–74. doi: 10.1093/eurheartj/ehl338. [DOI] [PubMed] [Google Scholar]
- 44.Brown ES, Varghese FP, McEwen BS. Association of depression with medical illness: does cortisol play a role? Biol Psychiatry. 2004;55:1–9. doi: 10.1016/S0006-3223(03)00473-6. [DOI] [PubMed] [Google Scholar]
- 45.Miller MD, Paradis CF, Houck PR, Mazumdar S, Stack JA, Rifai AH, et al. Rating chronic medical illness burden in geropsychiatric practice and research: application of the Cumulative Illness Rating Scale. Psychiatry Res. 1992;41:237–48. doi: 10.1016/0165-1781(92)90005-N. [DOI] [PubMed] [Google Scholar]
- 46.Cueva AS, Galhardoni R, Cury RG, Parravano DC, Correa G, Araujo H, et al. Normative data of cortical excitability measurements obtained by transcranial magnetic stimulation in healthy subjects. Clin Neurophysiol. 2016;46:43–51. doi: 10.1016/j.neucli.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 47.Peinemann A, Lehner C, Conrad B, Siebner HR. Age-related decrease in paired-pulse intracortical inhibition in the human primary motor cortex. Neurosci Lett. 2001;313:33–6. doi: 10.1016/S0304-3940(01)02239-X. [DOI] [PubMed] [Google Scholar]
- 48.Smith AE, Ridding MC, Higgins RD, Wittert GA, Pitcher JB. Age-related changes in short-latency motor cortex inhibition. Exp Brain Res. 2009;198:489–500. doi: 10.1007/s00221-009-1945-8. [DOI] [PubMed] [Google Scholar]
- 49.Shabel SJ, Proulx CD, Piriz J, Malinow R. GABA/glutamate co-release controls habenula output and is modified by antidepressant treatment. Science. 2014;345:1494–8. doi: 10.1126/science.1250469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Northoff G, Sibille E. Why are cortical GABA neurons relevant to internal focus in depression? A cross-level model linking cellular, biochemical, and neural network findings. Mol Psychiatry. 2014;19:966–77. doi: 10.1038/mp.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luhmann HJ, Kral T, Heinemann U. Influence of hypoxia on excitation and GABAergic inhibition in mature and developing rat neocortex. Exp Brain Res. 1993;97:209–24. doi: 10.1007/BF00228690. [DOI] [PubMed] [Google Scholar]
- 52.Wolkowitz OM, Epel ES, Reus VI, Mellon SH. Depression gets old fast: do stress and depression accelerate cell aging? Depress Anxiety. 2010;27:327–38. doi: 10.1002/da.20686. [DOI] [PubMed] [Google Scholar]
- 53.Doherty TJ, Vandervoort AA, Brown WF. Effects of ageing on the motor unit: a brief review. Can J Appl Physiol. 1993;18:331–58. doi: 10.1139/h93-029. [DOI] [PubMed] [Google Scholar]
- 54.Dorfman LJ, Bosley TM. Age-related changes in peripheral and central nerve conduction in man. Neurology. 1979;29:38–44. doi: 10.1212/WNL.29.1.38. [DOI] [PubMed] [Google Scholar]
- 55.Mesrati F, Vecchierini MF. F-waves: Neurophysiology and clinical value. Clin Neurophysiol. 2004;34:217–43. doi: 10.1016/j.neucli.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 56.Kendler Kenneth S, Karkowski Laura M, Carol, Prescott A. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry. 1999;156:837–41. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- 57.Biggio G, Concas A, Corda MG, Giorgi O, Sanna E, Serra M. Gabaergic and dopaminergic transmission in the rat cerebral cortex: Effect of stress, anxiolytic and anxiogenic drugs. Pharmacol Ther. 1990;48:121–42. doi: 10.1016/0163-7258(90)90077-F. [DOI] [PubMed] [Google Scholar]
- 58.Radhu N, de Jesus DR, Ravindran LN, Zanjani A, Fitzgerald PB, Daskalakis ZJ. A meta-analysis of cortical inhibition and excitability using transcranial magnetic stimulation in psychiatric disorders. Clin Neurophysiol. 2013;124:1309–20. doi: 10.1016/j.clinph.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 59.Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci. 2005;6:215–29. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- 60.Oliviero A, Profice P, Tonali P, Pilato F, Saturno E, Dileone M, et al. Effects of aging on motor cortex excitability. Neurosci Res. 2006;55:74–7. doi: 10.1016/j.neures.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 61.Orth M, Rothwell JC. The cortical silent period: Intrinsic variability and relation to the waveform of the transcranial magnetic stimulation pulse. Clin Neurophysiol. 2004;115:1076–82. doi: 10.1016/j.clinph.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 62.Chen R, Samii A, Canos M, Wassermann EM, Hallett M. Effects of phenytoin on cortical excitability in humans. Neurology. 1997;49:881–3. doi: 10.1212/WNL.49.3.881. [DOI] [PubMed] [Google Scholar]
- 63.Ziemann U, Corwell B, Cohen LG. Modulation of plasticity in human motor cortex after forearm ischemic nerve block. J Neurosci. 1998;18:1115–23. doi: 10.1523/JNEUROSCI.18-03-01115.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liepert J, Schwenkreis P, Tegenthoff M, Malin JP. The glutamate antagonist riluzole suppresses intracortical facilitation. J Neural Transm (Vienna) 1997;104:1207–14. doi: 10.1007/BF01294721. [DOI] [PubMed] [Google Scholar]
- 65.Ziemann U, Tergau F, Bruns D, Baudewig J, Paulus W. Changes in human motor cortex excitability induced by dopaminergic and anti-dopaminergic drugs. Electroencephalogr Clin Neurophysiol. 1997;105:430–7. doi: 10.1016/S0924-980X(97)00050-7. [DOI] [PubMed] [Google Scholar]
- 66.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4. doi: 10.1016/S0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 67.Zarate CA, Jr, Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry. 2006;163:153–5. doi: 10.1176/appi.ajp.163.1.153. [DOI] [PubMed] [Google Scholar]
- 68.Luykx J, Laban K, Van Den Heuvel M, Boks M, Mandl R, Kahn R, et al. Region and state specific glutamate downregulation in major depressive disorder: a meta-analysis of 1 H-MRS findings. Neurosci Biobehav Rev. 2012;36:198–05. doi: 10.1016/j.neubiorev.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 69.Stagg C, Bestmann S, Constantinescu A, Moreno Moreno L, Allman C, Mekle R, et al. Relationship between physiological measures of excitability and levels of glutamate and GABA in the human motor cortex. J Physiol. 2011;589:5845–55. doi: 10.1113/jphysiol.2011.216978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rogasch NC, Dartnall TJ, Cirillo J, Nordstrom MA, Semmler JG. Corticomotor plasticity and learning of a ballistic thumb training task are diminished in older adults. J Appl Physiol. 2009;107:1874–83. doi: 10.1152/japplphysiol.00443.2009. [DOI] [PubMed] [Google Scholar]
- 71.Tecchio F, Zappasodi F, Pasqualetti P, Gennaro LD, Pellicciari MC, Ercolani M, et al. Age dependence of primary motor cortex plasticity induced by paired associative stimulation. Clin Neurophysiol. 2008;119:675–82. doi: 10.1016/j.clinph.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 72.Davidson T, Tremblay F. Age and hemispheric differences in transcallosal inhibition between motor cortices: an ispsilateral silent period study. BMC Neurosci. 2013;14:62. doi: 10.1186/1471-2202-14-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McGregor KM, Zlatar Z, Kleim E, Sudhyadhom A, Bauer A, Phan S, et al. Physical activity and neural correlates of aging: a combined TMS/fMRI study. Behav Brain Res. 2011;222:158–68. doi: 10.1016/j.bbr.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ni Z, Isayama R, Castillo G, Gunraj C, Saha U, Chen R. Reduced dorsal premotor cortex and primary motor cortex connectivity in older adults. Neurobiol Aging. 2015;36:301–3. doi: 10.1016/j.neurobiolaging.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 75.Shiffman LM. Effects of aging on adult hand function. Am J Occup Ther. 1992;46:785–92. doi: 10.5014/ajot.46.9.785. [DOI] [PubMed] [Google Scholar]
- 76.Clark J, Loftus A, Hammond G. Age-related changes in short-interval intracortical facilitation and dexterity. Neuroreport. 2011;22:499–503. doi: 10.1097/WNR.0b013e3283487480. [DOI] [PubMed] [Google Scholar]
- 77.Price RB, Shungu DC, Mao X, Nestadt P, Kelly C, Collins KA, et al. Amino acid neurotransmitters assessed by proton magnetic resonance spectroscopy: Relationship to treatment resistance in major depressive disorder. Biol Psychiatry. 2009;65:792–800. doi: 10.1016/j.biopsych.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Daskalakis ZJ, Farzan F, Barr MS, Maller JJ, Chen R, Fitzgerald PB. Long-interval cortical inhibition from the dorsolateral prefrontal cortex: a TMS-EEG study. Neuropsychopharmacology. 2008;33:2860–9. doi: 10.1038/npp.2008.22. [DOI] [PubMed] [Google Scholar]
- 79.Concerto C, Lanza G, Cantone M, Pennisi M, Giordano D, Spampinato C, et al. Different patterns of cortical excitability in major depression and vascular depression: a transcranial magnetic stimulation study. BMC Psychiatry. 2013;13:300. doi: 10.1186/1471-244X-13-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.de Groot J, de Leeuw F, Oudkerk M, Hofman A, Jolles J, Breteler MB. Cerebral white matter lesions and depressive symptoms in elderly adults. Arch Gen Psychiatry. 2000;57:1071–6. doi: 10.1001/archpsyc.57.11.1071. [DOI] [PubMed] [Google Scholar]
- 81.O’Brien J, Desmond P, Ames D, Schweitzer I, Harrigan S, Tress B. A magnetic resonance imaging study of white matter lesions in depression and Alzheimer’s disease. Br J Psychiatry. 1996;168:477–85. doi: 10.1192/bjp.168.4.477. [DOI] [PubMed] [Google Scholar]
- 82.McIntyre RS, Soczynska JK, Konarski JZ, Woldeyohannes HO, Law CWY, Miranda A, et al. Should depressive syndromes be reclassified as “Metabolic Syndrome Type II. Ann Clin Psychiatry. 2007;19:257–64. doi: 10.1080/10401230701653377. [DOI] [PubMed] [Google Scholar]
- 83.Speck CE, Kukull WA, Brenner DE, Bowen JD, McCormick WC, Ten L, et al. History of depression as a risk factor for Alzheimer’s disease. Epidemiology. 1995;6:366–9. doi: 10.1097/00001648-199507000-00006. [DOI] [PubMed] [Google Scholar]
- 84.Verhoeven JE, Revesz D, Epel ES, Lin J, Wolkowitz OM, Penninx BW. Major depressive disorder and accelerated cellular aging: Results from a large psychiatric cohort study. Mol Psychiatry. 2014;19:895–901. doi: 10.1038/mp.2013.151. [DOI] [PubMed] [Google Scholar]
- 85.Tripp A, Kota RS, Lewis DA, Sibille E. Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiol Dis. 2011;42:116–24. doi: 10.1016/j.nbd.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.