

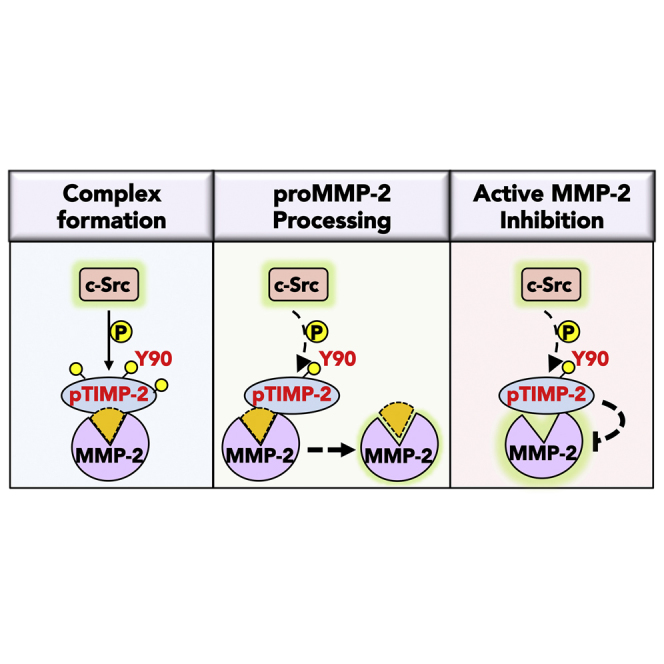
Summary
The tissue inhibitor of metalloproteinases 2 (TIMP-2) is a specific endogenous inhibitor of matrix metalloproteinase 2 (MMP-2), which is a key enzyme that degrades the extracellular matrix and promotes tumor cell invasion. Although the TIMP-2:MMP-2 complex controls proteolysis, the signaling mechanism by which the two proteins associate in the extracellular space remains unidentified. Here we report that TIMP-2 is phosphorylated outside the cell by secreted c-Src tyrosine kinase. As a consequence, phosphorylation at Y90 significantly enhances TIMP-2 potency as an MMP-2 inhibitor and weakens the catalytic action of the active enzyme. TIMP-2 phosphorylation also appears to be essential for its interaction with the latent enzyme proMMP-2 in vivo. Absence of the kinase or non-phosphorylatable Y90 abolishes TIMP-2 binding to the latent enzyme, ultimately hampering proMMP-2 activation. Together, TIMP-2 phosphorylation by secreted c-Src represents a critical extracellular regulatory mechanism that controls the proteolytic function of MMP-2.
Subject Areas: Biochemistry, Enzymology, Molecular Biology
Graphical Abstract
Highlights
-
•
c-Src tyrosine kinase phosphorylates TIMP-2
-
•
Secreted c-Src phosphorylates TIMP-2 extracellularly
-
•
TIMP-2 Y90 phosphorylation promotes extracellular interaction with proMMP-2
-
•
Tyrosine phosphorylation of TIMP-2 regulates proMMP-2 processing and MMP-2 activity
Biochemistry; Enzymology; Molecular Biology
Introduction
The tissue inhibitor of metalloproteinases 2 (TIMP-2) belongs to a family of secreted proteins, the TIMPs, that consists of four members (TIMP-1, -2, -3, and -4). TIMPs inhibit the proteolytic activity of a large family of zinc-dependent endopeptidases, the matrix metalloproteinases (MMPs; Brew and Nagase, 2010, Murphy, 2011). MMPs are implicated in the processing of cell surface protein components and in the degradation of constituents of the extracellular matrix (Bourboulia and Stetler-Stevenson, 2010, Kessenbrock et al., 2010). Consequently, TIMPs are essential in maintaining tissue homeostasis while regulating pericellular and interstitial tissue proteolysis during tissue repair and in diseases such as cancer.
MMP-2 is one of the most studied MMPs, mainly because increased protein expression and hyperactivity correlate with tumor development and progression (Egeblad and Werb, 2002). The MMP-2 proteolytic activity is regulated at several levels, including latent proenzyme activation and active site interaction with the inhibitors TIMPs. Interestingly, TIMP-2 also regulates the activation process of latent proMMP-2 through a mechanism that requires TIMP-2 binding to the C terminus of the proenzyme independent from the active site (Strongin et al., 1995, Visse and Nagase, 2003). Although TIMP-2 binding to proMMP-2 is essential for proMMP-2 activation, the mechanism that promotes this crucial interaction between TIMP-2 and MMP-2 is largely unknown. Furthermore, studies have identified phosphorylation sites on certain TIMPs and MMPs; however, the molecular mechanisms and functional significance of these modifications are unclear (Hornbeck et al., 2015, Sariahmetoglu et al., 2007, Williams and Coppolino, 2011).
In this study, we describe a new mechanism that regulates MMP-2 proteolytic function through phosphorylation of TIMP-2 by secreted c-Src tyrosine kinase. We show that c-Src phosphorylates TIMP-2 and initiates a signaling cascade that triggers the extracellular association of TIMP-2 with latent proMMP-2. Site-specific mutagenesis of TIMP-2 Y90 elucidates the functional role of this phosphorylation on TIMP-2 as a potent MMP-2 inhibitor and establishes an extracellular process that controls proMMP-2 activation.
Results
c-Src Tyrosine Kinase Phosphorylates TIMP-2
To determine whether endogenous TIMP-2 is tyrosine phosphorylated, we isolated naturally secreted TIMP-2 from serum-free cell-conditioned media (CM) of HT1080 human fibrosarcoma cells (Figure 1A). Following SDS-PAGE, we probed with an anti-pan-phosphotyrosine antibody (phos-Tyr, 4G10) and showed that TIMP-2 is tyrosine phosphorylated (Figure 1A and Table S1. [Information of antibodies used]). To identify which tyrosine residue(s) is(are) subject to phosphorylation, we individually mutated all seven of them to the non-phosphorylatable phenylalanine (F) (Figure 1B, Table S2. [Generated constructs] and Table S3. [Sequences of TIMP-2 constructs]; Betts and Russell, 2003). C-terminal His6-tagged phosphodeficient mutants, wild type (WT), and vector control (Vec) were transiently expressed in human embryonic kidney 293H (HEK293H) cells. Following pulldown Ni-NTA from cell extracts, we confirmed that WT TIMP-2 is tyrosine phosphorylated (Figure 1C). Moreover, we detected selective phosphorylation at Y62, Y90, and Y165, as demonstrated from the decreased band intensity of the non-phosphorylatable mutants (Y62F, Y90F, and Y165F) compared with WT TIMP-2 control (Figure 1C). We verified phosphorylation at Y62 as previously reported in mass spectrometry-based proteomic data (Hornbeck et al., 2015). We next simultaneously mutated all three tyrosine residues to F, creating the non-phosphorylatable triple mutant Y62F/Y90F/Y165F (referred to as TF). Following pulldown from HEK293H CM, the single mutants had decreased tyrosine phosphorylation, whereas no detectable phosphorylation was seen in the TF mutant (Figure 1D). These data demonstrate that secreted TIMP-2 is tyrosine phosphorylated and that this post-translational modification selectively occurs at residues Y62, Y90, and Y165.
Figure 1.

c-Src Phosphorylates Human TIMP-2 In Vitro and In Vivo
(A) Endogenous secreted TIMP-2 was immunoprecipitated from 10X concentrated HT1080 cell-conditioned media (CM) using anti-TIMP-2 or IgG (control) and analyzed by immunoblotting for phosphorylation using an anti-pan-phosphotyrosine antibody (phos-Tyr, 4G10).
(B) 3D (PDB: 1BR9) and linear domain structures of human TIMP-2 (hTIMP-2). All seven TIMP-2 tyrosine residues (Y) (black) are shown. Numbering is based on the full-length protein sequence (aa 1–220).
(C and D) (C) TIMP-2 His6-tagged wild type (WT), vector control (Vec), and individual mutant plasmids were transiently expressed in HEK293H cells and pulled down from cell extracts (D) or CM and immunoblotted with indicated antibodies to assess phosphorylation.
(E) Recombinant (rTIMP-2-His6) was used as the substrate in an in vitro kinase assay in the presence of full-length c-Src, v-Src, or c-Abl tyrosine kinases. Following pulldown, immunoblotting was performed to assess tyrosine phosphorylation using phos-Tyr, 4G10 antibody.
(F) TIMP-2 constructs were transiently expressed in SYF and SYF + c-Src cells, pulled down from 10X concentrated CM and immunoblotted to determine TIMP-2 tyrosine phosphorylation.
See also Figure S1.
To determine the tyrosine kinase responsible for this phosphorylation, we hypothesized that phosphorylation occurs at key sequence motifs surrounding the targeted residues. Amino acid isoleucine (I) at the n – 1 position of Y62 and Y90 (n + 1 for Y165) is a common denominator in peptides recognized by c-Src or c-Abl tyrosine kinases (Hubbard and Till, 2000, Pinna and Ruzzene, 1996, Songyang and Cantley, 1995; Figure S1A). To assess TIMP-2 phosphorylation by either tyrosine kinase, we performed an in vitro kinase assay using recombinant unphosphorylated TIMP-2-His6 (rTIMP-2-His6; Figure 1E). Following pulldown Ni-NTA, we found that c-Src and more intensely v-Src phosphorylate rTIMP-2-His6 (Figure 1E). v-Src is known to represent the hyperactive oncogenic version of c-Src because of its truncated inhibitory C-terminal regulatory phosphorylation site (Y527; Roskoski, 2004). We quantitatively measured and confirmed the hyperactivity of v-Src used here (Figure S1B). We also found that c-Abl tyrosine kinase did not phosphorylate TIMP-2 (Figure 1E), and in control experiments using heat shock protein 90 alpha (Hsp90α) as substrate we demonstrate discriminatory phosphorylation by Src kinase but not for c-Abl, confirming our previous work (Beebe et al., 2013, Dunn et al., 2015, Mollapour et al., 2010; Figures 1E and S1C).
To test the in vivo phosphorylation of TIMP-2, we transiently expressed WT TIMP-2-His6 and mutants in a triple kinase knockout (c-Src, Yes, and Fyn) mouse embryonic fibroblast cell line SYF and in cells with wild-type c-Src reintroduced (SYF + c-Src; Figure 1F). Pulldown experiments confirmed tyrosine phosphorylation of WT TIMP-2 in the SYF + c-Src but not in the parental SYF cell CM (Figure 1F). Since phosphorylation at Y62F, Y90F, and Y165F is reduced, and TF lacks phosphorylation in the SYF + c-Src CM, the overall findings suggest that c-Src targets all three tyrosine residues.
Secreted c-Src Phosphorylates TIMP-2 in the Extracellular Space
TIMP-2 contains an amino-terminal signal sequence that directs the newly synthesized protein to the ER, followed by secretion via the ER/Golgi pathway (Benham, 2012; Figure S1A). c-Src is a cytosolic kinase known to phosphorylate substrates at sites localized within the cell. To elucidate the cellular compartment where c-Src phosphorylates TIMP-2, we first assessed c-Src secretion from cells. CM was collected from mammalian cell lines and following normalization to total cellular protein levels, samples were analyzed by immunoblotting (Figures S2A and S2B). c-Src protein was present at varying levels in the extracts and CM from all cell lines tested (Figures S2A and S2B). Absence of cytosolic GAPDH in the CM also verifies lack of cytoplasmic fractions as a result of cell injury. Next, we asked if TIMP-2 could have been phosphorylated before secretion. HEK293H cells were transiently transfected with WT TIMP-2-His6, followed by 12-hr serum starvation and treatment with brefeldin A, an inhibitor that blocks conventional secretion by disrupting protein transport from ER to Golgi (Figure 2A). As expected, brefeldin A hindered TIMP-2 secretion but not the secretion of c-Src (Figure 2A). Tyrosine phosphorylation was also abolished in TIMP-2 isolated from cell extracts (Figure 2A). These data indicate that tyrosine phosphorylation occurs following TIMP-2 transport to Golgi or extracellularly. As c-Src secretion remains unaffected, we hypothesized that the phosphorylation occurs following secretion. To test this, we serum starved HT1080 cells for 18 hr and then supplemented the CM with rTIMP-2-His6 (Figure 2B). We detected both TIMP-2 and c-Src in the CM, 2 and 8 hr post treatment (Figure 2B). Notably, the exogenously added rTIMP-2-His6 is also detected in cell extracts, with protein levels increasing over time, indicating that a certain amount of free rTIMP-2-His6 becomes cell associated (Figure 2B). It is, therefore, not surprising to find that both cell-associated and free TIMP-2 are tyrosine phosphorylated (Figure 2B). To strengthen our findings on extracellular phosphorylation, we assessed the effects of anti-c-Src antibodies as possible inhibitors of TIMP-2 tyrosine phosphorylation (Table S1. [Information of antibodies used]). We pre-treated HT1080 cultures with an anti-c-Src antibody (mAb1) or rabbit IgG control before the addition of rTIMP-2-His6. As predicted, TIMP-2 tyrosine phosphorylation was impaired in both cell extracts and the CM (Figure 2C). Taken together, our data suggest that TIMP-2 and c-Src are secreted through different secretory pathways and that TIMP-2 is phosphorylated by c-Src outside the cell.
Figure 2.

c-Src Phosphorylates TIMP-2 Extracellularly
(A) HEK293H cells were transiently transfected with vector (Vec) control or wild-type (WT) TIMP-2-His6. Cells were serum starved in the presence (+) and absence (−) of brefeldin A for 12 hr, and cell extracts and CM were collected for immunoblotting and pulldown Ni-NTA to determine c-Src secretion and tyrosine phosphorylation of TIMP-2.
(B) HT1080 cells were serum starved for 24 hr and then treated with rTIMP-2-His6 for 2 and 8 hr. Cell extracts and CM were collected for immunoblotting and pulldown Ni-NTA analyses to determine phosphorylation of TIMP-2.
(C) Anti-c-Src monoclonal antibody (mAb1) or Rabbit IgG control were added in the CM for 1 hr before addition of recombinant TIMP-2-His6 for 8 hr. Cell extracts and CM were collected for analyses. GAPDH was analyzed for equal loading in all blots.
See also Figure S2.
c-Src-Mediated Phosphorylation of TIMP-2 Y90 Is Essential for Binding to proMMP-2
TIMP-2 interaction with proMMP-2 is crucial for cell surface activation of the latent enzyme (Brown et al., 1993, Morgunova et al., 2002, Strongin et al., 1995, Visse and Nagase, 2003). We next examined if TIMP-2 tyrosine phosphorylation participates in the formation of the proenzyme-inhibitor complex. Using an anti-TIMP-2 antibody we immunoprecipitated endogenous TIMP-2 from SYF CM and found no interaction between TIMP-2 and proMMP-2 (Figure 3A and Table S1. [Information of antibodies used]). In contrast, the TIMP-2:proMMP-2 complex was observed in SYF + c-Src CM by co-immunoprecipitation using anti-TIMP-2 antibody. We next generated and transiently transfected SYF cells with a phosphomimetic triple mutant Y62E/Y90E/Y165E (referred to as TE; Table S2. [Generated constructs], Table S3. [Sequences of TIMP-2 constructs], Table S4. [PCR mutagenesis primer sequences]). In contrast to WT and TF, pulldown of TIMP-2 TE led to co-pulldown of proMMP-2 in the SYF CM (Figure 3B top). Notably, TIMP-2 TE appears to associate more with proMMP-2 than with WT TIMP-2, suggesting that the degree of in vivo tyrosine phosphorylation regulates TIMP-2 interaction with MMP-2 (Figures 3B bottom and S3A). As anticipated, WT TIMP-2 from the SYF + c-Src CM also bound to proMMP-2 (Figure 3B bottom). We obtained further evidence from pulldown experiments performed in transiently transfected HEK293H cells and in SYF and SYF + c-Src cells exogenously treated with rTIMP-2-His6 (Figures S3A and S3B). Taken together, we show that tyrosine phosphorylation of TIMP-2 is required for its interaction with proMMP-2 in vivo.
Figure 3.

Phosphorylation of Y90 Is Essential for TIMP-2:proMMP-2 Interaction
(A) SYF and SYF + c-Src cells were serum starved for 18 hr. Immunoprecipitation (IP) of endogenous TIMP-2 from the cell CM was followed by co-immunoprecipitation (co-IP) of 72 kDa proMMP-2 to determine interaction.
(B) SYF (top) and SYF + c-Src (bottom) were transiently transfected with Vec control, WT TIMP-2-His6, and mutants (TF and TE). Pulldown was performed from the CM followed by immunoblotting and co-pulldown to assess protein interaction.
(C) HEK293H cells were transiently transfected with the indicated plasmids. Vec control, WT, and non-phosphorylatable (F) and phosphomimetic (E) TIMP-2 mutants were pulled down from the CM. Interaction of TIMP-2 proteins with secreted 72 kDa proMMP-2 was determined by co-pulldown and immunoblotting.
See also Figure S3.
We next asked if phosphorylation of a single tyrosine residue was essential to promote TIMP-2 complex with proMMP-2. Following transient transfection of HEK293H cells with single mutants, our data revealed that TIMP-2 Y90F does not complex with endogenous proMMP-2, whereas the phosphomimetic Y90E displayed enhanced interaction with the proenzyme (Figure 3C). Our data suggest that phosphorylation of TIMP-2 Y90 is critical to the regulation of the extracellular interaction of TIMP-2 with proMMP-2.
TIMP-2 Y90 Phosphorylation Regulates MMP-2 Function
The classic model of proMMP-2 activation involves TIMP-2 serving as a scaffold that tethers latent 72 kDa proMMP-2 to the plasma membrane (Brown et al., 1993, Strongin et al., 1995, Visse and Nagase, 2003). A membrane-bound active protease MT1-MMP mediates processing of proMMP-2 that generates the intermediate 64 kDa MMP-2 followed by autocatalysis that results in fully active free 62 kDa MMP-2 (Kinoshita et al., 1998, Strongin et al., 1995). As the interaction between TIMP-2 and proMMP-2 precedes the proteolytic processing of the proenzyme in vivo (Brown et al., 1993, Strongin et al., 1995), we predicted that Y90 phosphorylation would be critical in this process. We therefore tested WT TIMP-2-His6, Y90F, and Y90E proteins for their ability to activate proMMP-2 in vivo. Proteins purified from HEK293H CM were verified by Coomassie blue staining for purity and by reverse zymography for their ability to inhibit MMP-2-mediated gelatin degradation (Figures S4A–S4C and Table S5. [Buffer composition]).
We next treated HT1080 cells with the purified proteins to stimulate proMMP-2 activation, following the flow diagram (Figure 4A). Gelatin zymography confirmed that WT TIMP-2 facilitated the activation of proMMP-2, shown by the conversion of the 72 kDa proMMP-2 to the 64 and 62 kDa species (Figure 4B). We also confirmed previous studies showing that at high amounts TIMP-2 inhibits proMMP-2 activation (Caterina et al., 2000, Wang et al., 2000). Similar to WT TIMP-2, Y62F/Y165F (Y90 is readily phosphorylated) and Y90E (phosphomimetic) also assist in zymogen activation. However, TIMP-2 Y90F is unable to promote the activation of proMMP-2 (Figure 4B). Similar to our earlier findings in HEK293H CM, TIMP-2 Y90F and proMMP-2 did not interact in the HT1080 cell CM (Figure 4C and Table S1. [Information of antibodies used]). These data suggest that phosphorylation of TIMP-2 Y90 is critical for TIMP-2:proMMP-2 complex formation and cellular activation of proMMP-2.
Figure 4.

TIMP-2 Tyr90 Regulates MMP-2 Function
(A) Flowchart followed in proMMP-2 activation studies.
(B) HEK293H-purified WT TIMP-2 and indicated mutant proteins were added at increasing concentrations in the CM of HT1080 cells. Media were collected and analyzed by gelatin zymography. MMP-2 forms shown include the 72 kDa proMMP-2, 64 kDa intermediate form, and 62 kDa fully active form. Reduction of the 72 kDa form and increase of the 64 kDa and 62 kDa forms indicate proMMP-2 activation.
(C) TIMP-2-His6 proteins were transiently transfected in HT1080 cells followed by pulldown and co-pulldown for proMMP-2 to assess protein interaction.
(D–F) (D) HEK293H-purified WT TIMP-2, (E) Y90F, (F) and Y90E were tested for their ability to inhibit active MMP-2. Enzyme and inhibitors were pre-incubated at 25°C for 15 min before substrate addition. Experiments were performed at least twice. Error bars represent mean ± SD of three technical replicates.
(G) WT TIMP-2, Y90F, and Y90E Ki were determined to assess TIMP-2 inhibitory potency and represent the mean average of at least two independent experiments. A Student's t-test was performed between the WT and each of the two mutants. p Values were estimated to assess statistical significance (**p < 0.01).
See also Figure S4.
Active MMP-2 proteolytic activity is also regulated through direct contact with TIMP-2. We next evaluated the impact of Y90 phosphorylation on TIMP-2 ability to inhibit the 62 kDa active MMP-2. We performed steady-state kinetics on the active protease in the presence of purified WT TIMP-2, Y90F, and 90E. Double-reciprocal Lineweaver-Burk plots verified competitive inhibition (Figures S4D–S4F). The initial velocity (vo) of substrate cleavage was fit to the Michaelis-Menten kinetic model, and the inhibition constant Ki for each TIMP-2 species was determined using the classic model for tight-binding non-competitive inhibition (Figures 4D–4F). We found that TIMP-2 Y90E inhibits active 62 kDa MMP-2 with a Ki of 0.154 × 10−9 M (154 pM), an almost 5-fold increase in active site inhibition (Figures 4F and 4G). The Ki value shown for WT TIMP-2:62 kDa MMP-2 is also in agreement with previous studies (Willenbrock et al., 1993, Wingfield et al., 1999). These data suggest that phosphorylation of Y90 modulates the binding affinity of the inhibitor to active MMP-2.
Discussion
Phosphorylation is considered as one of the fundamental molecular mechanisms that regulate intracellular events from protein-protein interactions and activity to signal transduction and disease development (Nishi et al., 2011). Here we show that secreted TIMP-2 is tyrosine phosphorylated at three tyrosine residues, two of which (Y62 and Y165) are unique to TIMP-2 protein, suggesting that their phosphorylation could modulate extracellular functions specific for TIMP-2 (Figure 1). We concentrated on tyrosine kinases that recognize consensus phosphorylation sites on their targeted substrates and discovered that c-Src tyrosine kinase phosphorylates TIMP-2 (Figure 1). Anti-c-Src antibodies block phosphorylation, supporting the idea that this process occurs extracellularly (Figure 2). Lack of phosphorylation at Y90 compromises proenzyme processing and activation, likely due to disruption of TIMP-2 interaction with proMMP-2 (Figures 3 and 4). Phosphorylation at Y90 enhances the inhibitory function of TIMP-2 against active MMP-2 that is seemingly mediated by an enhanced affinity reflected in the 5-fold increase in Ki observed for phosphomimetic Y90E TIMP-2 (Figure 4). Combined, these findings support a potential role for c-Src in extracellular signaling and protein function (Figure S4G).
How does phosphorylation of Y90 regulate the interaction between TIMP-2 and proMMP-2? The structures of TIMP-2 and proMMP-2 are already solved, and, indeed, Y90 is located far away from the known interface between the two proteins (Morgunova et al., 2002). However, we believe our current knowledge on the structure of free human TIMP-2 or the formation of TIMP-2:proMMP-2 complexes has been largely built upon structural data and biophysical studies, in which bacterial or low eukaryotic expression systems are utilized to produce recombinant proteins devoid of any post-translational modifications (Brew and Nagase, 2010, Morgunova et al., 1999, Morgunova et al., 2002, Tuuttila et al., 1998). Our data argue that in the extracellular space, protein complexes are dependent upon specific signals, including post-translational modifications, in contrast to the high-affinity protein-protein interactions that readily occur in in vitro systems. Furthermore, it is quite possible that in the cellular context, inter-domain or additional local protein interactions that facilitate changes of TIMP-2 structural properties and assist in the binding to proMMP-2 following phosphorylation may take place. Further studies are warranted to address these questions.
Our data also suggest that c-Src kinase mediates the extracellular phosphorylation of TIMP-2. We identified c-Src released in the cell CM, corroborating emerging studies showing that c-Src is enriched in the extracellular vesicles (DeRita et al., 2016, Di Noto et al., 2013). Brefeldin A inhibited TIMP-2 but not c-Src secretion, and ER-retained TIMP-2 appears not to be tyrosine phosphorylated. Although it is possible that the TIMP-2 post-translational modification occurs after protein transport to Golgi (Figure 2A), there is no evidence to support c-Src localization within the Golgi network (Reinecke and Caplan, 2014, Sato et al., 2009). Alternatively, it is possible that both proteins somehow co-localize at the plasma membrane where phosphorylation could take place following their release. Nevertheless, we demonstrated that anti-c-Src antibodies block phosphorylation of exogenous TIMP-2 (Figure 2C).
We determined that latent enzyme processing depends on the phosphorylation of TIMP-2 by c-Src. Our findings that TIMP-2:proMMP-2 interaction is coupled to TIMP-2 tyrosine phosphorylation provides an explanation. TIMP-2 Y90 phosphorylation could support conformational changes exposing interaction surfaces that facilitate favorable and stronger complex formation with proMMP-2 in vivo (Batra et al., 2013, Sharabi et al., 2014, Zou et al., 2016). Expression of the phosphomimetic TIMP-2 TE in SYF cells was sufficient to restore TIMP-2 interaction with proMMP-2 (Figure 3B). Lack of TIMP-2 phosphorylation would also hinder activation (Bernardo and Fridman, 2003, Butler et al., 1998, English et al., 2006, Lafleur et al., 2003, Morrison et al., 2001). Indeed, the inability of TIMP-2 Y90F to interact with proMMP-2 was critical for generating the intermediate 64 kDa MMP-2 (Figures 3C, 4B, and 4C). It is also possible that phosphorylation of TIMP-2 may be implicated in MT1-MMP:TIMP-2 complex formation that would change the dynamics of the MT1-MMP:TIMP-2:proMMP-2 complex (Brew and Nagase, 2010, Rapti et al., 2006, Williamson et al., 2001). Thus, the overall mechanism of proMMP-2 activation warrants further investigation.
TIMP-2 also interacts with the active site of MMP-2, resulting in enzyme inhibition (Brew and Nagase, 2010). Structural changes on TIMP-2 Y90E may have increased its affinity for the active site of the enzyme, explaining its lower Ki (Figure 4F). We could also speculate that as TIMP-2 displays biphasic binding for 62 kDa MMP-2, Y90E may bind strongly to the C-terminal domain of active MMP-2 before binding and inhibiting the active site of the enzyme (Olson et al., 1997). Future studies utilizing TIMP-2 phosphomimetics and MMP-2 domain mutants will provide further insight into the role of TIMP-2 phosphorylation in the binding and inhibition of MMP-2.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We gratefully acknowledge Dr. Peg AtKisson for providing critical comments on the manuscript. We wish to thank Dr. Paul Soloway for providing us with the MEF TIMP-2 knock-out (TIMP-2 −/− + ras/myc) cell line. This work was supported by funds from SUNY Upstate Medical University, The Upstate Foundation, SUNY Research Foundation, and Upstate Breast Cancer Research Funds to D.B. and M.M., The Carol M. Baldwin Research Grant to D.B. and M.M., and Associated Medical Schools of New York to D.B.
Author Contributions
Conceptualization, D.B. and M.M.; Methodology, D.B. and M.M.; Investigation, J.S.-P., A.J.B.-W., M.R.W., R.B., B.W., M.M., and D.B.; Writing – Original draft, D.B.; Writing – Review and Editing, J.S.-P., A.J.B.-W., M.M., W.G.S.-S., G.B., and D.B.; Funding Acquisition, M.M. and D.B.; Resources, M.M., W.G.S.-S., G.B., and D.B.; Supervision, D.B.
Declaration of Interests
D.B., M.M., and G.B. have filed for a patent based partially on results presented in this manuscript. The other authors declare no competing interests.
Published: March 23, 2018
Footnotes
Supplemental Information includes Transparent Methods, four figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.02.004.
Supplemental Information
References
- Batra J., Soares A.S., Mehner C., Radisky E.S. Matrix metalloproteinase-10/TIMP-2 structure and analyses define conserved core interactions and diverse exosite interactions in MMP/TIMP complexes. PLoS One. 2013;8:e75836. doi: 10.1371/journal.pone.0075836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe K., Mollapour M., Scroggins B., Prodromou C., Xu W., Tokita M., Taldone T., Pullen L., Zierer B.K., Lee M.J. Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget. 2013;4:1065–1074. doi: 10.18632/oncotarget.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham A.M. Protein secretion and the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2012;4:a012872. doi: 10.1101/cshperspect.a012872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo M.M., Fridman R. TIMP-2 (tissue inhibitor of metalloproteinase-2) regulates MMP-2 (matrix metalloproteinase-2) activity in the extracellular environment after pro-MMP-2 activation by MT1 (membrane type 1)-MMP. Biochem. J. 2003;374:739–745. doi: 10.1042/BJ20030557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts M.J., Russell R.B. Amino acid properties and consequences of substitutions. In: Barnes M.R., Gray I.C., editors. Bioinformatics for Geneticists. John Wiley & Sons, Ltd; 2003. pp. 291–316. [Google Scholar]
- Bourboulia D., Stetler-Stevenson W.G. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): positive and negative regulators in tumor cell adhesion. Semin. Cancer Biol. 2010;20:161–168. doi: 10.1016/j.semcancer.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew K., Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim. Biophys. Acta. 2010;1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P.D., Kleiner D.E., Unsworth E.J., Stetler-Stevenson W.G. Cellular activation of the 72 kDa type IV procollagenase/TIMP-2 complex. Kidney Int. 1993;43:163–170. doi: 10.1038/ki.1993.27. [DOI] [PubMed] [Google Scholar]
- Butler G.S., Butler M.J., Atkinson S.J., Will H., Tamura T., Schade van Westrum S., Crabbe T., Clements J., d'Ortho M.P., Murphy G. The TIMP2 membrane type 1 metalloproteinase “receptor” regulates the concentration and efficient activation of progelatinase A. A kinetic study. J. Biol. Chem. 1998;273:871–880. doi: 10.1074/jbc.273.2.871. [DOI] [PubMed] [Google Scholar]
- Caterina J.J., Yamada S., Caterina N.C., Longenecker G., Holmback K., Shi J., Yermovsky A.E., Engler J.A., Birkedal-Hansen H. Inactivating mutation of the mouse tissue inhibitor of metalloproteinases-2(Timp-2) gene alters proMMP-2 activation. J. Biol. Chem. 2000;275:26416–26422. doi: 10.1074/jbc.M001271200. [DOI] [PubMed] [Google Scholar]
- DeRita R.M., Zerlanko B., Singh A., Lu H., Iozzo R.V., Benovic J.L., Languino L.R. c-Src, insulin-like growth factor I receptor, G-protein-coupled receptor kinases and focal adhesion kinase are enriched into prostate cancer cell exosomes. J. Cell Biochem. 2016;118:66–73. doi: 10.1002/jcb.25611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noto G., Paolini L., Zendrini A., Radeghieri A., Caimi L., Ricotta D. C-src enriched serum microvesicles are generated in malignant plasma cell dyscrasia. PLoS One. 2013;8:e70811. doi: 10.1371/journal.pone.0070811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn D.M., Woodford M.R., Truman A.W., Jensen S.M., Schulman J., Caza T., Remillard T.C., Loiselle D., Wolfgeher D., Blagg B.S.J. c-Abl mediated tyrosine phosphorylation of Aha1 activates its co-chaperone function in cancer cells. Cell Rep. 2015;12:1006–1018. doi: 10.1016/j.celrep.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M., Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- English J.L., Kassiri Z., Koskivirta I., Atkinson S.J., Di Grappa M., Soloway P.D., Nagase H., Vuorio E., Murphy G., Khokha R. Individual Timp deficiencies differentially impact pro-MMP-2 activation. J. Biol. Chem. 2006;281:10337–10346. doi: 10.1074/jbc.M512009200. [DOI] [PubMed] [Google Scholar]
- Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard S.R., Till J.H. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K., Plaks V., Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita T., Sato H., Okada A., Ohuchi E., Imai K., Okada Y., Seiki M. TIMP-2 promotes activation of progelatinase A by membrane-type 1 matrix metalloproteinase immobilized on agarose beads. J. Biol. Chem. 1998;273:16098–16103. doi: 10.1074/jbc.273.26.16098. [DOI] [PubMed] [Google Scholar]
- Lafleur M.A., Tester A.M., Thompson E.W. Selective involvement of TIMP-2 in the second activational cleavage of pro-MMP-2: refinement of the pro-MMP-2 activation mechanism. FEBS Lett. 2003;553:457–463. doi: 10.1016/s0014-5793(03)01094-9. [DOI] [PubMed] [Google Scholar]
- Mollapour M., Tsutsumi S., Donnelly A.C., Beebe K., Tokita M.J., Lee M.J., Lee S., Morra G., Bourboulia D., Scroggins B.T. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol. Cell. 2010;37:333–343. doi: 10.1016/j.molcel.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgunova E., Tuuttila A., Bergmann U., Isupov M., Lindqvist Y., Schneider G., Tryggvason K. Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science. 1999;284:1667–1670. doi: 10.1126/science.284.5420.1667. [DOI] [PubMed] [Google Scholar]
- Morgunova E., Tuuttila A., Bergmann U., Tryggvason K. Structural insight into the complex formation of latent matrix metalloproteinase 2 with tissue inhibitor of metalloproteinase 2. Proc. Natl. Acad. Sci. USA. 2002;99:7414–7419. doi: 10.1073/pnas.102185399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison C.J., Butler G.S., Bigg H.F., Roberts C.R., Soloway P.D., Overall C.M. Cellular activation of MMP-2 (gelatinase A) by MT2-MMP occurs via a TIMP-2-independent pathway. J. Biol. Chem. 2001;276:47402–47410. doi: 10.1074/jbc.M108643200. [DOI] [PubMed] [Google Scholar]
- Murphy G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011;12:233. doi: 10.1186/gb-2011-12-11-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi H., Hashimoto K., Panchenko A.R. Phosphorylation in protein-protein binding: effect on stability and function. Structure. 2011;19:1807–1815. doi: 10.1016/j.str.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M.W., Gervasi D.C., Mobashery S., Fridman R. Kinetic analysis of the binding of human matrix metalloproteinase-2 and -9 to tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2. J. Biol. Chem. 1997;272:29975–29983. doi: 10.1074/jbc.272.47.29975. [DOI] [PubMed] [Google Scholar]
- Pinna L.A., Ruzzene M. How do protein kinases recognize their substrates? Biochim. Biophys. Acta. 1996;1314:191–225. doi: 10.1016/s0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]
- Rapti M., Knauper V., Murphy G., Williamson R.A. Characterization of the AB loop region of TIMP-2. Involvement in pro-MMP-2 activation. J. Biol. Chem. 2006;281:23386–23394. doi: 10.1074/jbc.M604423200. [DOI] [PubMed] [Google Scholar]
- Reinecke J., Caplan S. Endocytosis and the Src family of non-receptor tyrosine kinases. Biomol. Concepts. 2014;5:143–155. doi: 10.1515/bmc-2014-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R., Jr. Src protein-tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004;324:1155–1164. doi: 10.1016/j.bbrc.2004.09.171. [DOI] [PubMed] [Google Scholar]
- Sariahmetoglu M., Crawford B.D., Leon H., Sawicka J., Li L., Ballermann B.J., Holmes C., Berthiaume L.G., Holt A., Sawicki G. Regulation of matrix metalloproteinase-2 (MMP-2) activity by phosphorylation. FASEB J. 2007;21:2486–2495. doi: 10.1096/fj.06-7938com. [DOI] [PubMed] [Google Scholar]
- Sato I., Obata Y., Kasahara K., Nakayama Y., Fukumoto Y., Yamasaki T., Yokoyama K.K., Saito T., Yamaguchi N. Differential trafficking of Src, Lyn, Yes and Fyn is specified by the state of palmitoylation in the SH4 domain. J. Cell Sci. 2009;122:965–975. doi: 10.1242/jcs.034843. [DOI] [PubMed] [Google Scholar]
- Sharabi O., Shirian J., Grossman M., Lebendiker M., Sagi I., Shifman J. Affinity- and specificity-enhancing mutations are frequent in multispecific interactions between TIMP2 and MMPs. PLoS One. 2014;9:e93712. doi: 10.1371/journal.pone.0093712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z., Cantley L.C. Recognition and specificity in protein tyrosine kinase-mediated signalling. Trends Biochem. Sci. 1995;20:470–475. doi: 10.1016/s0968-0004(00)89103-3. [DOI] [PubMed] [Google Scholar]
- Strongin A.Y., Collier I., Bannikov G., Marmer B.L., Grant G.A., Goldberg G.I. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- Tuuttila A., Morgunova E., Bergmann U., Lindqvist Y., Maskos K., Fernandez-Catalan C., Bode W., Tryggvason K., Schneider G. Three-dimensional structure of human tissue inhibitor of metalloproteinases-2 at 2.1 A resolution. J. Mol. Biol. 1998;284:1133–1140. doi: 10.1006/jmbi.1998.2223. [DOI] [PubMed] [Google Scholar]
- Visse R., Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- Wang Z., Juttermann R., Soloway P.D. TIMP-2 is required for efficient activation of proMMP-2 in vivo. J. Biol. Chem. 2000;275:26411–26415. doi: 10.1074/jbc.M001270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willenbrock F., Crabbe T., Slocombe P.M., Sutton C.W., Docherty A.J., Cockett M.I., O'Shea M., Brocklehurst K., Phillips I.R., Murphy G. The activity of the tissue inhibitors of metalloproteinases is regulated by C-terminal domain interactions: a kinetic analysis of the inhibition of gelatinase A. Biochemistry. 1993;32:4330–4337. doi: 10.1021/bi00067a023. [DOI] [PubMed] [Google Scholar]
- Williams K.C., Coppolino M.G. Phosphorylation of membrane type 1-matrix metalloproteinase (MT1-MMP) and its vesicle-associated membrane protein 7 (VAMP7)-dependent trafficking facilitate cell invasion and migration. J. Biol. Chem. 2011;286:43405–43416. doi: 10.1074/jbc.M111.297069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson R.A., Hutton M., Vogt G., Rapti M., Knauper V., Carr M.D., Murphy G. Tyrosine 36 plays a critical role in the interaction of the AB loop of tissue inhibitor of metalloproteinases-2 with matrix metalloproteinase-14. J. Biol. Chem. 2001;276:32966–32970. doi: 10.1074/jbc.M101843200. [DOI] [PubMed] [Google Scholar]
- Wingfield P.T., Sax J.K., Stahl S.J., Kaufman J., Palmer I., Chung V., Corcoran M.L., Kleiner D.E., Stetler-Stevenson W.G. Biophysical and functional characterization of full-length, recombinant human tissue inhibitor of metalloproteinases-2 (TIMP-2) produced in Escherichia coli. Comparison of wild type and amino-terminal alanine appended variant with implications for the mechanism of TIMP functions. J. Biol. Chem. 1999;274:21362–21368. doi: 10.1074/jbc.274.30.21362. [DOI] [PubMed] [Google Scholar]
- Zou H., Wu Y., Brew K. Thermodynamic basis of selectivity in the interactions of tissue inhibitors of metalloproteinases N-domains with matrix Metalloproteinases-1, -3, and -14. J. Biol. Chem. 2016;291:11348–11358. doi: 10.1074/jbc.M116.720250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.