

Abstract
Stroke is one of the leading causes of death and disability worldwide, and the majority of the cases are ischemic stroke. However, it still lacks effective treatment except for thrombolytic therapy in an extremely narrow time window. Increased evidence suggests that histone deacetylase 4 (HDAC4) was dysregulated in ischemic stroke, which plays a key role in the pathogenesis of ischemic stroke and post-stroke recovery by affecting neuronal death, angiogenesis, and neurogenesis. Therefore, we aim to review the dysregulation of HDAC4 in ischemic stroke and the role of dysregulated HDAC4 in the pathogenesis of ischemic stroke. Furthermore, the therapeutic potential of modulating HDAC4 in ischemic stroke is discussed.
Keywords: HDAC4, Ischemic stroke, Cell death, Angiogenesis, Neurogenesis
Background
Stroke is one of the leading causes of death and disability worldwide [1]. In the USA, it is the leading cause of long-term disability, including both physical and cognitive deficits, while it is the leading cause of death in China [2, 3]. The prevalence of stroke continues increasing, and the direct medical costs will reach $184.1 billion in the USA by 2030. In addition, increased risk of neurodegenerative diseases, such as Alzheimer’s disease, was observed in patients who experienced a stroke, which further increases the burden of health care [4]. Ischemic stroke is the major subtype of stroke, accounting for 87% of stroke cases. However, current treatments for ischemic stroke are only limited to thrombolytic therapy within an extremely narrow time window [5]. Thus, developing novel therapeutic approaches for ischemic stroke is urgent.
Histone deacetylases (HDACs) along with histone acetyltransferases (HATs) regulate chromatin remodeling and subsequent gene transcription by controlling the status of histone acetylation. Compared with histone acetylation, histone deacetylation induces a condensed chromatin conformation, contributing to the repression of gene transcription which is involved in diverse physiological processes. Moreover, the function of HDACs is not limited to the histone deacetylation. Recent evidence suggests that HDACs may also contribute to the deacetylation of non-histone proteins [6]. In addition, HDACs also have deacetylase-independent functions, including other modifications of histone, such as methylation [6–8]. Importantly, HDACs are dysregulated in a number of brain disorders, which is implicated in the pathogenesis of these diseases, e.g., ischemic stroke, autism, Alzheimer’s disease, and depressive disorders [9–15]. It suggests that HDACs might be potential targets for the treatment of brain disorders.
Growing evidence indicates that HDAC4 is a specific target for the treatment of ischemic stroke. First, dysregulated HDAC4 was observed in ischemic stroke, which does play a key role in the pathogenesis of ischemic stroke and post-stroke recovery by affecting neuronal death, angiogenesis, and neurogenesis [16–20]. For example, HDAC4 is reduced in ischemic stroke model animals and oxygen-glucose deprivation (OGD)-treated neurons, while increased HDAC4 expression reduces infarct volume in ischemic stroke model animals and increases cell viability of OGD-treated neuronal cells [9, 10, 21, 22]. In addition, HDAC4 has a significant effect on a cognitive function which could be impaired by ischemic stroke [23]. For example, conditional deletion of HDAC4 leads to learning and memory deficits [24–26]. It indicates that HDAC4 might be a target for the treatment of ischemic stroke. Therefore, we aim to review the dysregulation of HDAC4 in ischemic stroke and the role of HDAC4 in the pathogenesis of ischemic stroke and post-stroke recovery. Furthermore, the therapeutic potential of modulating HDAC4 in ischemic stroke is discussed.
Mechanisms of ischemic stroke and post-stroke recovery
Cell death and synaptic impairment
Depending on the severity of reduced blood supply, acute and delayed cell death, i.e., necrosis and apoptosis, occurs in the core region and penumbra region of the ischemic territory, respectively [27]. Necrosis occurs within minutes after stroke, which cannot be rescued. However, apoptosis and impaired synaptic function in the penumbra could be salvageable by proper interventions, suggesting that preventing apoptosis and recovering synaptic function in the penumbra region may be an effective approach to improve post-stroke recovery. Ischemia/reperfusion injury-induced apoptosis and synaptic impairment in the penumbra are mediated by a number of mechanisms, including excitotoxicity, oxidative stress, inflammatory response, and endoplasmic reticulum (ER) stress [28–30]. For example, the dysregulation of synaptic proteins, e.g., subunits of N-methyl-d-aspartic acid (NMDA) receptors, was observed in ischemic stroke, which not only led to synaptic dysfunction but also contributed to excitotoxic cell death [30]. It suggests that suppressing detrimental pathways may have therapeutic potential for ischemic stroke by protecting the penumbra from neuronal death and synaptic impairment.
Angiogenesis
During an acute ischemic stroke, the reduction of blood supply in the ischemic area often activates angiogenesis, a neurovascular remodeling process, which is a compensatory response to the reduction of oxygen. Numerous studies have shown that angiogenesis is positively correlated with the survival rate of patients who experienced an ischemic stroke, indicating that angiogenesis is an endogenous brain repair mechanism [31–33]. Thus, the modulation of vascular growth in the ischemic area could be a therapeutic approach for ischemic stroke. Indeed, the beneficial effects of direct injections or gene transfer of angiogenic factors have been demonstrated by inducing therapeutic angiogenesis in ischemic stroke, myocardial infarction, and limb ischemic injury [34–36]. Enhanced angiogenesis is not only beneficial to the cell survival in the penumbra region but also promotes neurogenesis facilitating post-stroke recovery, which orchestrates post-stroke recovery [37].
Neurogenesis
Neurogenesis, including neural stem cell proliferation, migration, and differentiation, plays a key role in the chronic stage of post-stroke recovery [38]. Increased stem cell proliferation was observed in post-stroke patients and mice model. However, the majority of newly born cells die during the first 2 weeks after their formation. It suggests that improving the survival, migration, and differentiation of newly formed cells is the key of enhancing post-stroke neurogenesis. In addition, repetitive transcranial magnetic stimulation ameliorates cognitive impairment by enhancing neurogenesis in rats with ischemic stroke [39]. Moreover, the consistent efficacy of two approaches, stem cell transplantation and stimulating endogenous neurogenesis, was observed in animal models of ischemic stroke [40, 41]. However, the therapeutic effect of transplantation of stem cell for ischemic stroke needs to be further investigated, and clinical trials are still ongoing [40–42].
Characteristics of HDAC4
HDACs are a large family of enzymes, regulating chromatin remodeling and subsequent gene transcription mainly by controlling the status of histone acetylation. According to the sequence homology, HDACs are grouped into class I (HDAC1, 2, 3, and 8), class II (IIa: HDAC4, 5, 7, and 9; IIb: HDAC6 and 10), class III (SIRT1–7), and class IV (HDAC11). The HDAC4 protein consists of a long N-terminal domain and a highly conserved C-terminal catalytic domain [15]. Compared with most of HDACs, HDAC4 is usually trapped in the cytoplasm. Its shuttling between the cytoplasm and nucleus is tightly controlled by both the phosphorylation status of HDAC4 and its interacting partners, such as calcium/calmodulin-dependent kinase II (CaMKII), protein phosphotase 2A (PP2A), protein kinase C (PKC), and tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein (14-3-3) [43–46]. For example, HDAC4 is the substrate of CaMKII, which can export HDAC4 to the cytoplasm [47].
Compared with other HDACs, HDAC4 per se features weak histone deacetylase activity. It may also contribute to the histone deacetylation via interacting with HDAC3 and HDAC5, respectively [48–50]. Moreover, HDAC4 does have histone deacetylase-independent functions. For example, HDAC4 is involved in histone methylation contributing to the regulation of gene transcription [8]. In addition, HDAC4 could regulate gene transcription by interacting with multiple transcriptional factors, including runt-related transcription factor 2 (Runx2), myocyte enhancer factor 2 (MEF2), serum response factor (SRF), heterochromatin protein 1(HP1), nuclear factor kappa B (NF-κB), and activating transcription factor 4 (ATF4). [51–53]. Furthermore, HDAC4 is implicated in regulating protein SUMOylation by interacting with the SUMO-conjugating enzyme Ubc9 (Ubc9) [54]. Thus, HDAC4 may contribute to a number of physiological and pathological processes via histone deacetylase-dependent and deacetylase-independent pathways [51, 52].
Dysregulation of HDAC4 in ischemic stroke
HDAC4 is highly expressed in the brain, mainly in neurons [9]. Recent studies indicate that HDAC4 is dysregulated in ischemic stroke, which may play a pivotal role in the pathogenesis of ischemic stroke and post-stroke recovery. Compared with sham treatment, middle cerebral artery occlusion (MCAO)/reperfusion significantly reduces the expression of HDAC4 in the cortex of rats, which is mediated by NADPH oxidase [9, 10]. Consistently, the HDAC4 expression is significantly reduced in the cardiomyocytes following ischemia/reperfusion injury [55]. However, the expression of HDAC4 is increased in oligodendrocyte progenitor cells in the brains of ischemic stroke model rats [56].
A number of microRNAs targeting HDAC4 were altered in ischemic stroke, which may also contribute to the dysregulation of HDAC4 in ischemic stroke. For example, miR-9 and miR-124 are markedly increased in both serum and CSF of patients with ischemic stroke [57, 58]. However, Liu et al. showed that serum miR-124 and miR-9 were reduced in patients with ischemic stroke, although the sample size was small [59]. In addition, the reduction of miR-9 was detected in the brain of ischemic stroke model mice [60]. Moreover, miR-206 and miR-29b, two microRNAs targeting HDAC4, are significantly increased in ischemic rat brains and in OGD-treated primary neurons [61–63]. It suggests that the combination effect of dysregulated microRNAs may contribute to the reduction of HDAC4 in ischemic stroke.
In addition to HDAC4 expression, nuclear shuttling of HDAC4 is altered in ischemic stroke, which plays an important role in the pathogenesis of stroke and post-stroke recovery. Increased HDAC4 nuclear shuttling was observed in the neurons of ischemic stroke model mice/ats and in oxygen-glucose deprivation (OGD)-treated neurons, while the overexpression of calcium/calmodulin-dependent protein kinase IV (CaMKIV) reduced the levels of nuclear HDAC4 in ischemic stroke [11, 64]. However, increased cytoplasmic HDAC4 expression was detected in oligodendrocyte progenitor cells in the brains of ischemic stroke model rats [56].
The role of HDAC4 in ischemic stroke and underlying mechanisms
HDAC4 in neuronal death and synaptic impairment
Accumulated evidence indicates that HDAC4 plays an important role in the post-stroke recovery by modulating neuronal death and synaptic plasticity (Fig. 1). First, HDAC4 deficiency causes a progressive loss of neurons in the cerebellum of mice, while the forcing expression of HDAC4 protects neurons from cell death [16]. Moreover, the HDAC4-C-terminal fragment is crucial to rescue HDAC4 knockdown-induced cell death and a reduction of synaptic strength in mouse brains [51]. Zhang et al. showed that reduced HDAC4 expression is associated with blood-brain barrier (BBB) breakdown contributing to ischemia/reperfusion injury-induced infarct in ischemic stroke model rats, while increased HDAC4 expression ameliorates BBB injury, contributing to the reduced infarct volume [10]. Consistently, class IIa histone deacetylase-specific inhibitor increases mortality and infarct volume in the brains of ischemic stroke model rats and exacerbates neuronal remodeling impairment, such as reduced dendritic and axonal and myelination densities [65]. However, pan-HDAC inhibitors have a protective effect on stroke [66, 67]. Moreover, HDAC4 increases cell viability of OGD-treated cells via reducing high-mobility group protein 1(HMGB1) expression [9]. In addition, a proteomics analysis indicated that HDAC4 is a regulator of proteins involved in neuronal excitability and synaptic plasticity [68]. Silencing HDAC4 expression results in the impairment of synaptic plasticity and learning and memory deficits in both mice and Drosophila, although one report showed that HDAC4 knockdown with siRNA improved the survival of OGD-treated neurons [11, 26, 69]. Currently, mechanisms of reduced HDAC4 in ischemia/reperfusion injury-induced neuronal death and synaptic impairment remain elusive. However, a number of studies indicate that the effect of HDAC4 on neuronal death and synaptic impairment might be mediated by its partners, e.g., Runx2, MEF2, SRF, HP1, NF-κB, and ATF4, contributing to the processes of ER stress, inflammation, and oxidative stress response [16, 51–53, 70, 71]. For example, HDAC4 overexpression causes ATF4 retention in the cytoplasm, inhibiting ER stress-induced apoptosis, while HDAC4 reduction exacerbates ER stress-induced apoptosis [53].
Fig. 1.
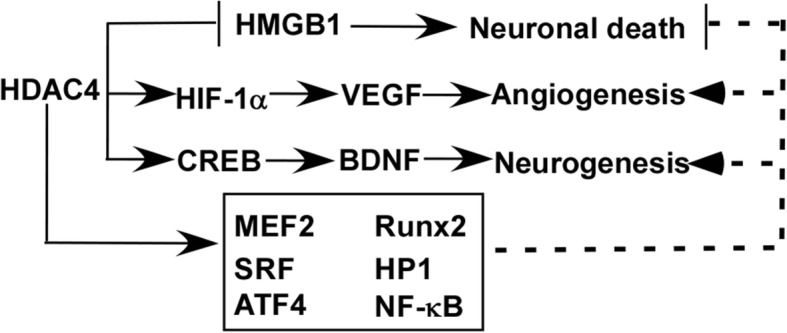
The role of HDAC4 in ischemic stroke and underlying mechanisms. HDAC4 could inhibit neuronal death via reducing HDMGB1expression and release, while it promotes angiogenesis and neurogenesis via HIF-1α-VEGF signaling and CREB-BDBF signaling, respectively. The interacting partners of HDAC4, MEF2, Runx2, SRF, HP1, ATF4, F-κB, etc. might also mediate its role in the neuronal death, angiogenesis, and neurogenesis in ischemic stroke. The solid line represents known mechanisms, while the dash line represents possible mechanisms
In addition to HDAC4 levels, nuclear shuttling of HDAC4 also contributes to neuronal death and synaptic impairment in ischemic stroke. Nuclear HDAC4 represses the expression of constituents of synapses leading to the impairment of synaptic architecture and strength in mice [51]. In addition, the neuroprotective effect of CaMKIV on OGD neurons is mediated by reducing nuclear HDAC4 [11]. Moreover, mice carrying nuclear HDAC4 mutant exhibit deficits in neurotransmission, learning, and memory [51]. Yuan et al. reported that ischemic stroke-induced nuclear shuttling of HDAC4 strongly facilitated OGD-induced neuronal death and exacerbated infarct volume and functional deficits in ischemic model mice [11]. In addition, accumulation of nuclear HDAC4 exerts neurotoxicity in models of Parkinson’s disease [72].
HDAC4 in angiogenesis
Post-stroke angiogenesis has a beneficial effect on cell survival and stroke recovery. Qian et al. reported that siRNA-induced HDAC4 reduction suppressed hypoxia-inducible factor-1α (HIF-1α) expression, which inhibited HIF-1α-associated vascular endothelial growth factor (VEGF) expression in ischemia/reperfusion injury [73, 74]. It suggests that HDAC4 alteration may regulate the angiogenesis in ischemic stroke via HIF-1α-VEGF signaling (Fig. 1). Moreover, HDAC4 phosphorylation is also the key regulator of angiogenesis. Phosphorylation of HDAC4 is remarkably upregulated in the endothelial cells under hypoxic conditions while blocking the phosphorylation of HDAC4 inhibits endothelial cell migration and tube formation, which is associated with the suppression of HIF-1α-VEGF signaling [75]. Consistently, Liu et al. showed that phosphorylation of HDAC4 was associated with the induction of HIF-1α-VEGF signaling, promoting angiogenesis in ischemic stroke model mice and cells [75]. GO6976, an inhibitor of HDAC4, blocks the phosphorylation of HDAC4 and inhibits the tube formation and migration of endothelial cells [75]. It suggests that HDAC4 phosphorylation facilitates angiogenesis in ischemic stroke. Moreover, HDAC4 may be involved in angiogenesis via its interacting partners, such as NF-κB [76]. Furthermore, Madelaine et al. identified miR-9 inhibition as a positive regulator of neurogenesis and angiogenesis [77]. As HDAC4 is a target of miR-9, it may contribute to the effect of miRNA-9 inhibition on angiogenesis and neurogenesis, suggesting that HDAC4 might be a potential target for the treatment of ischemic stroke.
HDAC4 in neurogenesis
Growing evidence indicates that HDAC4 may contribute to neurogenesis via regulating the expression and function of multiple molecules. First, HDAC4 regulates the activity and expression of cAMP response element-binding protein (CREB) and brain-derived neurotrophic factor (BDNF), respectively, which play a key role in neurogenesis after ischemic stroke [18–20, 78] (Fig. 1). For example, increased CREB activity and BDNF expression promote post-ischemic stroke neurogenesis and neuroregeneration in rats. However, nuclear shuttling of HDAC4 suppresses the transcriptional activity of CREB by reducing the interaction among acetyltransferase, CBP, and CREB, leading to the reduction of BDNF. [18–20]. In addition, HDAC4 might be another key mediator of the effect of miRNA-9 on neurogenesis in ischemic stroke as HDAC4 is the target of miRNA-9 [77, 79]. Moreover, HDAC4 may be implicated in neurogenesis by regulating the activity of its partners, such as Runx2, MEF2, SRF, HP1, NF-κB, and ATF4. For example, MEF2 promotes neurogenesis while nuclear HDAC4 suppresses the activity of MEF2 [79–81]. The above evidence suggests that the alteration of HDAC4 expression and nuclear shuttling in ischemic stroke may play a pivotal role in post-stroke recovery by affecting neurogenesis.
Clinical perspectives
HDAC4, a unique target for ischemic stroke treatment
HDAC4 is a unique target for the treatment of ischemic stroke compared with other HDACs, such as HDAC2 [16–20]. For example, HDAC4 features different characteristics and plays an opposite role in ischemic stroke compared with HDAC2 (Table 1). HDAC4 and HDAC2 genes are located at chromosome 2q37 and chromosome 6q21, respectively, encoding1084 and 488 amino acids, respectively. HDAC4 contains both intrinsic nuclear localization signal and nuclear export signal, while HDAC2 only contains a nuclear localization signal [82–84]. Thus, HDAC2 is mainly localized in the nucleus, while HDAC4 enriches in the cytoplasm and shuttles between the cytoplasm and nucleus [82–84]. Compared with HDAC2, HDAC4 per se features weak histone deacetylase activity as the critical tyrosine residue within the catalytic domain is substituted by histidine [85]. Compared with HDAC2, HDAC4 interacts with multiple partners, e.g., Runx, MEF2, SRF, HP1, NF-κB, 14-3-3, and Ubc9 [16, 51–53, 70, 71]. HDAC4’s partners may mediate HDAC4’ function in ischemic stroke as the partners are involved in the key processes of ischemic stroke, i.e., neuronal death, angiogenesis, and neurogenesis [53, 76, 79–81] (Fig. 1). Conditional deletion of HDAC4 leads to learning and memory deficits, while global HDACs inhibitors or HDAC2 reduction significantly improves learning and memory function in mice [24–26]. Importantly, reduced HDAC4 expression and increased nuclear shuttling are detected in ischemic stroke model cells and animals, while multiple HDACs, including HDAC2, are increased in ischemic stroke models [9–11, 17, 21, 64]. Moreover, increased HDAC4 expression reduces infarct volume in ischemic stroke model animals and increases cell viability of OGD-treated neurons, while reduced HDAC2 expression promotes neuronal survival and functional recovery in ischemic stroke model animals [9, 10, 21, 22]. Consistently, pan-HDACs inhibitors and the specific inhibitor of class І HDACs, including HDAC2, alleviate stroke-induced neurological deficits facilitating post-stroke recovery in mice. However, the specific class IIa inhibitor increases mortality and infarct volume in the brains of ischemic stroke model rats, exacerbates neuronal remodeling impairment, and has no rescue effect on neurological deficits [21, 22, 65].
Table 1.
Difference between HDAC4 and HDAC2
| HDAC4 | HDAC2 | |
|---|---|---|
| Features | ||
| Gene locus (chromosome) | 2q37 | 6q21 |
| Number of amino acids | 1084 | 488 |
| Nuclear localization signal | + | + |
| Nuclear export signal | + | – |
| Subcellular distribution | Cytoplasm/nucleus | Nucleus |
| Histone deacetylase activity | Weak | Strong |
| Effect on cognitive function | Beneficial | Impaired |
| Ischemic stroke | ||
| Altered expression | Reduced | Increased |
| Altered distribution | Increased nuclear shuttling | – |
| Rescue effect of class-specific inhibitor on neurological deficits | – | + |
| Effect on infarct size | Reduced | Increased |
The alteration and function of HDAC4 are opposite to those of HDAC2 in ischemic stroke models, indicating that increasing HDAC4 expression is a unique target for the treatment of ischemic stroke compared with inhibiting HDAC2 and other HDACs to treat ischemic stroke. Although it is inconclusive that increasing HDAC4 expression could offer a better ischemic stroke therapy compared with HDAC2 inhibition, co-regulating HDAC4 and HDAC2 or other HDACs might have better therapeutic potential. The combination effect of increasing the HDAC4 level and inhibiting the activity of HDAC2 or other HDACs needs to be further investigated.
Current status of HDACs-based treatment
Currently, thrombolysis with tissue plasminogen activator remains the only globally approved treatment for ischemic stroke [5]. No HDAC-based approach or agent has been approved for ischemic stroke treatment, although four pan-HDAC inhibitors, vorinostat, romidepsin, belinostat, and panobinostat, are approved by the US FDA for the treatment of cutaneous T cell lymphoma, peripheral T cell lymphoma, and multiple myeloma, respectively [86, 87]. More than 350 clinical trials involving HDAC inhibitors (https://www.clinicaltrials.gov/) have been carried out or are ongoing against various diseases, including cancers, Alzheimer’s disease, schizophrenia, asthma, and chronic obstructive pulmonary disease (COPD). However, no HDAC-based clinical trial has been carried out for ischemic stroke. Moreover, it still lacks HDAC4-based preclinical studies on larger animals, although the therapeutic effect of HDAC4 has been observed in neurons, rats, and mice. Therefore, further investigation is needed before HDAC4-based clinical trials.
Potential of HDAC4-based therapy for ischemic stroke
Accumulated evidence suggested that increasing HDAC4 expression may have therapeutic potential for ischemic stroke treatment. Several approaches of regulating HDAC4 level could be translated into the clinic (Fig. 2). Adenovirus- and adeno-associated virus-mediated HDAC4 overexpression has been applied in vitro and in vivo, indicating that virus-based HDAC4 overexpression could be a potential gene therapy for ischemic stroke treatment [16, 88–91]. However, further preclinical investigation is needed to determine the therapeutic effect on larger animals other than rodents. In addition, the efficacy and safety need to be evaluated.
Fig. 2.
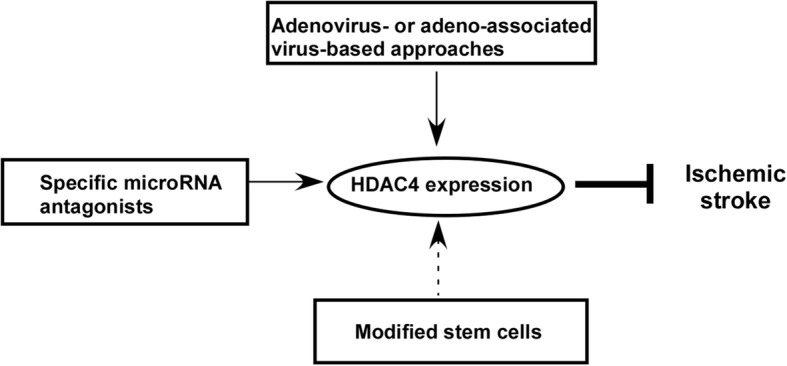
Potential of HDAC4-based therapy for ischemic stroke. Adenovirus- or adeno-associated virus-mediated HDAC4 overexpression and microRNA-based upregulation of HDAC4 have the potential to be translated into the clinic for ischemic stroke treatment. The therapeutic potential of HDAC4-modified stem cells remains elusive
MicroRNA-based therapies hold great promise in various diseases. Significant alteration of microRNAs targeting HDAC4 has been detected in ischemic stroke patients and model animals, indicating that modulating microRNAs targeting HDAC4 could be a therapeutic approach. A number of microRNAs targeting HDAC4 are increased, e.g., miR-9, miR-124, miR-29b, and miR-206, suggesting that restoring or downregulating their levels may subsequently increase HDAC4 expression. Specific microRNA antagonists, including anti-miRs, locked nucleic acids, and antagomirs, could restore HDAC4 expression or increase HDAC4 expressions. Among them, antagomirs can be delivered without any vector or vehicle assistance. A recent study showed that intranasal administration of an antagomir specifically targeting miR-206 significantly improved memory function in the model mice of Alzheimer’s disease [92]. It suggested that the non-invasive intranasal administration of specific antagomirs could be an effective approach to increase HDAC4 expression for ischemic stroke treatment. Further preclinical investigation needs to be done to determine the specificity, efficacy, and safety of this approach. The combination effect of targeting various microRNAs needs to be investigated.
Both preclinical studies and clinical trials indicated that stem cell-based therapies would be an effective approach for the treatment of many kinds of diseases, including ischemic stroke [93, 94]. In addition to numerous preclinical studies, a variety of stem cell-based clinical trials for the treatment of ischemic stroke have been carried out or are ongoing, including neural stem cells, mesenchymal stem cells, embryonic stem cells, and induced pluripotent stem cells (https://www.clinicaltrials.gov/). For example, the consistent efficacy of neural stem cell transplantation for ischemic stroke treatment was observed in both preclinical studies and clinical trials, e.g., the trial of Pilot Investigation of Human Neural Stem Cells in Chronic Ischemic Stroke Patients (PISCES) [40, 41]. The results of the PISCES trial might be more conclusive with the enrolment of additional patients and the introduction of a placebo control group in the phase 2 trial (NCT02117635) [40]. Whether HDAC4-modified stem cells could have a better therapeutic effect in patients with ischemic stroke needs to be further investigated. First, the alteration of HDAC4 in different types of stem cells is unclear as only one report showed that both total HDAC4 and cytoplasmic HDAC4 was increased in oligodendrocyte progenitor cells of ischemic stroke model rats [56]. In addition, the role of HDAC4 in different types of stem cells and underlying mechanisms remain elusive.
Conclusions
HDAC4 expression was reduced in ischemic stroke, which may contribute to the pathogenesis of ischemic stroke by promoting neuronal death and inhibiting angiogenesis and neurogenesis. The increased HDAC4 expression could inhibit neuronal death via reducing HMGB1 expression and release and promote angiogenesis and neurogenesis via HIF-1α-VEGF signaling and CREB-BDBF signaling, respectively. The interacting partners of HDAC4, MEF2, Runx2, SRF, HP1, ATF4, and NF-κB might also mediate its role in inhibiting neuronal death and promoting angiogenesis and neurogenesis in ischemic stroke. Importantly, it remains to find similar pattern and mechanisms in patients with ischemic stroke as most studies are performed in cultured neurons and animal models. Currently, a number of approaches to regulate HDAC4 level have the potential to be translated into the clinic, such as adenovirus-/adeno-associated virus-mediated HDAC4 overexpression and microRNA-based upregulation of HDAC4. Although a variety of stem cell-based clinical trials for the treatment of ischemic stroke has been carried out or are ongoing, the therapeutic potential of HDAC4-modified stem cells remains elusive. Therefore, modulating HDAC4 expression could be translated into the clinic as an effective treatment for ischemic stroke. However, the therapeutic potential of HDAC4-modified stem cells needs to be further investigated in preclinical studies.
Funding
The present work was funded by the National Natural Science Foundation of China (81771147), Natural Science Foundation of Shandong Province (ZR2016HM30), Young Teachers Research Support Foundation of Jining Medical University (JY2016KJ028Y), and Teachers Research Support Foundation of Jining Medical University (JY2017JS001).
Abbreviations
- NF-κB
Nuclear factor kappa B
- 14-3-3
Andtyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein
- BBB
Blood-brain barrier
- BDNF
Brain-derived neurotrophic factor
- CaMK IV
Calcium/calmodulin-dependent protein kinase IV
- CaMKII
Calcium/calmodulin-dependent kinase II
- CREB
cAMP response element-binding protein
- CSF
Cerebrospinal fluid
- ER
Endoplasmic reticulum
- HDAC
Histone deacetylase
- HP1
Heterochromatin protein 1
- MCAO
Middle cerebral artery occlusion
- MEF2
Myocyte enhancer factor 2
- NADPH
Nicotinamide adenine dinucleotide phosphate-oxidase
- NMDA
N-methyl-d-aspartic acid
- OGD
Oxygen-glucose deprivation
- PKC
Protein kinase C
- PP2A
Protein phosphotase 2A
- Runx2
Runt-related transcription factor 2
- SIRT1
Sirtuin 1
- SRF
Serum response factor
Authors’ contributions
QK, YH, XL, and XW wrote the manuscript. BJ wrote and revised the manuscript. YW formulated and revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Bingyuan Ji, Email: jby2006@126.com.
Yili Wu, Email: yili_wu2004@yahoo.ca, Email: wuyili@mail.jnmc.edu.cn.
References
- 1.Wang R, Ying Z, Zhao J, Zhang Y, Lu H, Deng Y, et al. Lys(203) and Lys(382) are essential for the proteasomal degradation of BACE1. Curr Alzheimer Res. 2012;9(5):606–615. doi: 10.2174/156720512800618026. [DOI] [PubMed] [Google Scholar]
- 2.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart Disease and Stroke Statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133(4):e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 3.Wang H, Naghavi M, Allen C, Barber RM, Bhutta ZA, Carter A, et al. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1459–1544. doi: 10.1016/S0140-6736(16)31012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu Y, Xu Q, Song W. Oxidative stress and Alzheimer’s disease. In: Laher I, editor. Systems biology of free radicals and antioxidants. Berlin: Springer Berlin Heidelberg; 2014. p. 2147-74.
- 5.Chapman SN, Mehndiratta P, Johansen MC, McMurry TL, Johnston KC, Southerland AM. Current perspectives on the use of intravenous recombinant tissue plasminogen activator (tPA) for treatment of acute ischemic stroke. Vasc Health Risk Manag. 2014;10:75–87. doi: 10.2147/VHRM.S39213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lardenoije R, Iatrou A, Kenis G, Kompotis K, Steinbusch HW, Mastroeni D, et al. The epigenetics of aging and neurodegeneration. Prog Neurobiol. 2015;131:21–64. doi: 10.1016/j.pneurobio.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han X, Niu J, Zhao Y, Kong Q, Tong T, Han L. HDAC4 stabilizes SIRT1 via sumoylation SIRT1 to delay cellular senescence. Clin Exp Pharmacol Physiol. 2016;43(1):41–46. doi: 10.1111/1440-1681.12496. [DOI] [PubMed] [Google Scholar]
- 8.Hohl M, Wagner M, Reil JC, Muller SA, Tauchnitz M, Zimmer AM, et al. HDAC4 controls histone methylation in response to elevated cardiac load. J Clin Invest. 2013;123(3):1359–1370. doi: 10.1172/JCI61084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He M, Zhang B, Wei X, Wang Z, Fan B, Du P, et al. HDAC4/5-HMGB1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J Cell Mol Med. 2013;17(4):531–542. doi: 10.1111/jcmm.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang QY, Wang ZJ, Sun DM, Wang Y, Xu P, Wu WJ, et al. Novel therapeutic effects of leonurine on ischemic stroke: new mechanisms of BBB integrity. Oxidative Med Cell Longev. 2017;2017:7150376. doi: 10.1155/2017/7150376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan H, Denton K, Liu L, Li XJ, Benashski S, McCullough L, et al. Nuclear translocation of histone deacetylase 4 induces neuronal death in stroke. Neurobiol Dis. 2016;91:182–193. doi: 10.1016/j.nbd.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Morris B, Etoubleau C, Bourthoumieu S, Reynaud-Perrine S, Laroche C, Lebbar A, et al. Dose dependent expression of HDAC4 causes variable expressivity in a novel inherited case of brachydactyly mental retardation syndrome. Am J Med Genet A. 2012;158A(8):2015–2020. doi: 10.1002/ajmg.a.35463. [DOI] [PubMed] [Google Scholar]
- 13.Otsuki K, Uchida S, Hobara T, Yamagata H, Watanabe Y. Epigenetic regulation in depression. Nihon Shinkei Seishin Yakurigaku Zasshi. 2012;32(4):181–186. [PubMed] [Google Scholar]
- 14.Shen X, Chen J, Li J, Kofler J, Herrup K. Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. eNeuro. 2016;3:1. doi: 10.1523/ENEURO.0124-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y, Hou F, Wang X, Kong Q, Han X, Bai B. Aberrant expression of histone deacetylases 4 in cognitive disorders: molecular mechanisms and a potential target. Front Mol Neurosci. 2016;9:114. doi: 10.3389/fnmol.2016.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D’Mello SR. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008;68(8):1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen YT, Zang XF, Pan J, Zhu XL, Chen F, Chen ZB, et al. Expression patterns of histone deacetylases in experimental stroke and potential targets for neuroprotection. Clin Exp Pharmacol Physiol. 2012;39(9):751–758. doi: 10.1111/j.1440-1681.2012.05729.x. [DOI] [PubMed] [Google Scholar]
- 18.Sen T, Sen N. Isoflurane-induced inactivation of CREB through histone deacetylase 4 is responsible for cognitive impairment in developing brain. Neurobiol Dis. 2016;96:12–21. doi: 10.1016/j.nbd.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang G, Zhang T, Li N, Wu L, Gu J, Li C, et al. Tetramethylpyrazine nitrone activates the BDNF/Akt/CREB pathway to promote post-ischaemic neuroregeneration and recovery of neurological functions in rats. Br J Pharmacol. 2018;175(3):517–531. doi: 10.1111/bph.14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeong CH, Kim SM, Lim JY, Ryu CH, Jun JA, Jeun SS. Mesenchymal stem cells expressing brain-derived neurotrophic factor enhance endogenous neurogenesis in an ischemic stroke model. Biomed Res Int. 2014;2014:129145. doi: 10.1155/2014/129145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang Y, Lin YH, Ni HY, Dong J, Yuan HJ, Zhang Y, et al. Inhibiting histone deacetylase 2 (HDAC2) promotes functional recovery from stroke. J Am Heart Assoc. 2017;6:10. doi: 10.1161/JAHA.117.007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin YH, Dong J, Tang Y, Ni HY, Zhang Y, Su P, et al. Opening a new time window for treatment of stroke by targeting HDAC2. J Neurosci. 2017;37(28):6712–6728. doi: 10.1523/JNEUROSCI.0341-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han Z, Dong X, Zhang C, Wu Y, Yuan Z, Wang X. Polymorphism of HDAC9 gene is associated with increased risk of acute coronary syndrome in Chinese Han population. Biomed Res Int. 2016;2016:3746276. doi: 10.1155/2016/3746276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB: CBP-dependent transcriptional activation. J Neurosci. 2007;27(23):6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459(7243):55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim MS, Akhtar MW, Adachi M, Mahgoub M, Bassel-Duby R, Kavalali ET, et al. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J Neurosci. 2012;32(32):10879–10886. doi: 10.1523/JNEUROSCI.2089-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Y, Wang X, Zhou X, Cheng B, Li G, Bai B. Temporal expression of Apelin/Apelin receptor in ischemic stroke and its therapeutic potential. Front Mol Neurosci. 2017;10:1. doi: 10.3389/fnmol.2017.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67(2):181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S, Zis O, Ly PT, Wu Y, Zhang M, Cai F, et al. Down-regulation of MIF by NFkappaB under hypoxia accelerated neuronal loss during stroke. FASEB J. 2014; 10.1096/fj.14-253625. [DOI] [PMC free article] [PubMed]
- 30.Gutierrez-Vargas JA, Munoz-Manco JI, Garcia-Segura LM, Cardona-Gomez GP. GluN2B N-methyl-D-aspartic acid receptor subunit mediates atorvastatin-induced neuroprotection after focal cerebral ischemia. J Neurosci Res. 2014;92(11):1529–1548. doi: 10.1002/jnr.23426. [DOI] [PubMed] [Google Scholar]
- 31.Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke. 1994;25(9):1794–1798. doi: 10.1161/01.STR.25.9.1794. [DOI] [PubMed] [Google Scholar]
- 32.Arenillas JF, Sobrino T, Castillo J, Davalos A. The role of angiogenesis in damage and recovery from ischemic stroke. Curr Treat Options Cardiovasc Med. 2007;9(3):205–212. doi: 10.1007/s11936-007-0014-5. [DOI] [PubMed] [Google Scholar]
- 33.Zhang ZG, Chopp M. Promoting brain remodeling to aid in stroke recovery. Trends Mol Med. 2015;21(9):543–548. doi: 10.1016/j.molmed.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu J, Liu Q, Jiang Y, Wu L, Xu G, Liu X. Enhanced angiogenesis promoted by human umbilical mesenchymal stem cell transplantation in stroked mouse is Notch1 signaling associated. Neuroscience. 2015;290:288–299. doi: 10.1016/j.neuroscience.2015.01.038. [DOI] [PubMed] [Google Scholar]
- 35.Seto SW, Chang D, Jenkins A, Bensoussan A, Kiat H. Angiogenesis in ischemic stroke and angiogenic effects of Chinese herbal medicine. J Clin Med. 2016;5:6. doi: 10.3390/jcm5060056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y, Zhang X, Cui L, Chen R, Zhang Y, Zhang C, et al. Salvianolic acids enhance cerebral angiogenesis and neurological recovery by activating JAK2/STAT3 signaling pathway after ischemic stroke in mice. J Neurochem. 2017; 10.1111/jnc.14140. [DOI] [PubMed]
- 37.Ruan L, Wang B, ZhuGe Q, Jin K. Coupling of neurogenesis and angiogenesis after ischemic stroke. Brain Res. 2015;1623:166–173. doi: 10.1016/j.brainres.2015.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindvall O, Kokaia Z. Neurogenesis following stroke affecting the adult brain. Cold Spring Harb Perspect Biol. 2015;7:11. doi: 10.1101/cshperspect.a019034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo F, Lou J, Han X, Deng Y, Huang X. Repetitive transcranial magnetic stimulation ameliorates cognitive impairment by enhancing neurogenesis and suppressing apoptosis in the hippocampus in rats with ischemic stroke. Front Physiol. 2017;8:559. doi: 10.3389/fphys.2017.00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borlongan CV. Age of PISCES: stem-cell clinical trials in stroke. Lancet. 2016;388(10046):736–738. doi: 10.1016/S0140-6736(16)31259-4. [DOI] [PubMed] [Google Scholar]
- 41.Lu J, Manaenko A, Hu Q. Targeting adult neurogenesis for poststroke therapy. Stem Cells Int. 2017;2017:5868632. doi: 10.1155/2017/5868632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagpal A, Choy FC, Howell S, Hillier S, Chan F, Hamilton-Bruce MA, et al. Safety and effectiveness of stem cell therapies in early-phase clinical trials in stroke: a systematic review and meta-analysis. Stem Cell Res Ther. 2017;8(1):191. doi: 10.1186/s13287-017-0643-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mielcarek M, Landles C, Weiss A, Bradaia A, Seredenina T, Inuabasi L, et al. HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 2013;11(11):e1001717. doi: 10.1371/journal.pbio.1001717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishino TG, Miyazaki M, Hoshino H, Miwa Y, Horinouchi S, Yoshida M. 14-3-3 regulates the nuclear import of class IIa histone deacetylases. Biochem Biophys Res Commun. 2008;377(3):852–856. doi: 10.1016/j.bbrc.2008.10.079. [DOI] [PubMed] [Google Scholar]
- 45.Wu Q, Yang X, Zhang L, Zhang Y, Feng L. Nuclear accumulation of histone deacetylase 4 (HDAC4) exerts neurotoxicity in models of Parkinson’s disease. Mol Neurobiol. 2016; 10.1007/s12035-016-0199-2. [DOI] [PubMed]
- 46.Litke C, Bading H, Mauceri D. Histone deacetylase 4 shapes neuronal morphology via a mechanism involving regulation of expression of vascular endothelial growth factor D. J Biol Chem. 2018;293(21):8196–8207. doi: 10.1074/jbc.RA117.001613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Qin G, Zhao TC. HDAC4: mechanism of regulation and biological functions. Epigenomics. 2014;6(1):139–150. doi: 10.2217/epi.13.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci U S A. 1999;96(9):4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee HA, Song MJ, Seok YM, Kang SH, Kim SY, Kim I. Histone deacetylase 3 and 4 complex stimulates the transcriptional activity of the mineralocorticoid receptor. PLoS One. 2015;10(8):e0136801. doi: 10.1371/journal.pone.0136801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ginnan R, Sun LY, Schwarz JJ, Singer HA. MEF2 is regulated by CaMKIIdelta2 and a HDAC4-HDAC5 heterodimer in vascular smooth muscle cells. Biochem J. 2012;444(1):105–114. doi: 10.1042/BJ20120152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sando R, 3rd, Gounko N, Pieraut S, Liao L, Yates J, 3rd, Maximov A. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell. 2012;151(4):821–834. doi: 10.1016/j.cell.2012.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet. 2013;14(5):347–359. doi: 10.1038/nrg3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang P, Sun Q, Zhao C, Ling S, Li Q, Chang YZ, et al. HDAC4 protects cells from ER stress induced apoptosis through interaction with ATF4. Cell Signal. 2014;26(3):556–563. doi: 10.1016/j.cellsig.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 54.Schwartz S, Truglio M, Scott MJ, Fitzsimons HL. Long-term memory in Drosophila is influenced by the histone deacetylase HDAC4 interacting with the SUMO-conjugating enzyme Ubc9. Genetics. 2016; 10.1534/genetics.115.183194. [DOI] [PMC free article] [PubMed]
- 55.Kang B, Li W, Xi W, Yi Y, Ciren Y, Shen H, et al. Hydrogen sulfide protects cardiomyocytes against apoptosis in ischemia/reperfusion through MiR-1-regulated histone deacetylase 4 pathway. Cell Physiol Biochem. 2017;41(1):10–21. doi: 10.1159/000455816. [DOI] [PubMed] [Google Scholar]
- 56.Kassis H, Chopp M, Liu XS, Shehadah A, Roberts C, Zhang ZG. Histone deacetylase expression in white matter oligodendrocytes after stroke. Neurochem Int. 2014;77:17–23. doi: 10.1016/j.neuint.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sorensen SS, Nygaard AB, Carlsen AL, Heegaard NHH, Bak M, Christensen T. Elevation of brain-enriched miRNAs in cerebrospinal fluid of patients with acute ischemic stroke. Biomark Res. 2017;5:24. doi: 10.1186/s40364-017-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ji Q, Ji Y, Peng J, Zhou X, Chen X, Zhao H, et al. Increased brain-specific MiR-9 and MiR-124 in the serum exosomes of acute ischemic stroke patients. PLoS One. 2016;11(9):e0163645. doi: 10.1371/journal.pone.0163645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, Zhang J, Han R, Liu H, Sun D, Liu X. Downregulation of serum brain specific microRNA is associated with inflammation and infarct volume in acute ischemic stroke. J Clin Neurosci. 2015;22(2):291–295. doi: 10.1016/j.jocn.2014.05.042. [DOI] [PubMed] [Google Scholar]
- 60.Wei N, Xiao L, Xue R, Zhang D, Zhou J, Ren H, et al. MicroRNA-9 mediates the cell apoptosis by targeting Bcl2l11 in ischemic stroke. Mol Neurobiol. 2016;53(10):6809–6817. doi: 10.1007/s12035-015-9605-4. [DOI] [PubMed] [Google Scholar]
- 61.Shi G, Liu Y, Liu T, Yan W, Liu X, Wang Y, et al. Upregulated miR-29b promotes neuronal cell death by inhibiting Bcl2L2 after ischemic brain injury. Exp Brain Res. 2012;216(2):225–230. doi: 10.1007/s00221-011-2925-3. [DOI] [PubMed] [Google Scholar]
- 62.Winbanks CE, Wang B, Beyer C, Koh P, White L, Kantharidis P, et al. TGF-beta regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J Biol Chem. 2011;286(16):13805–13814. doi: 10.1074/jbc.M110.192625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu FJ, Lim KY, Kaur P, Sepramaniam S, Armugam A, Wong PT, et al. microRNAs involved in regulating spontaneous recovery in embolic stroke model. PLoS One. 2013;8(6):e66393. doi: 10.1371/journal.pone.0066393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kassis H, Shehadah A, Chopp M, Roberts C, Zhang ZG. Stroke induces nuclear shuttling of histone deacetylase 4. Stroke. 2015;46(7):1909–1915. doi: 10.1161/STROKEAHA.115.009046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kassis H, Shehadah A, Li C, Zhang Y, Cui Y, Roberts C, et al. Class IIa histone deacetylases affect neuronal remodeling and functional outcome after stroke. Neurochem Int. 2016;96:24–31. doi: 10.1016/j.neuint.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu XS, Chopp M, Kassis H, Jia LF, Hozeska-Solgot A, Zhang RL, et al. Valproic acid increases white matter repair and neurogenesis after stroke. Neuroscience. 2012;220:313–321. doi: 10.1016/j.neuroscience.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baltan S, Murphy SP, Danilov CA, Bachleda A, Morrison RS. Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J Neurosci. 2011;31(11):3990–3999. doi: 10.1523/JNEUROSCI.5379-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neuner SM, Wilmott LA, Hoffmann BR, Mozhui K, Kaczorowski CC. Hippocampal proteomics defines pathways associated with memory decline and resilience in ‘normal’ aging and Alzheimer’s disease mouse models. Behav Brain Res. 2016; 10.1016/j.bbr.2016.06.002. [DOI] [PMC free article] [PubMed]
- 69.Fitzsimons HL, Schwartz S, Given FM, Scott MJ. The histone deacetylase HDAC4 regulates long-term memory in Drosophila. PLoS One. 2013;8(12):e83903. doi: 10.1371/journal.pone.0083903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luan B, Goodarzi MO, Phillips NG, Guo X, Chen YD, Yao J, et al. Leptin-mediated increases in catecholamine signaling reduce adipose tissue inflammation via activation of macrophage HDAC4. Cell Metab. 2014;19(6):1058–1065. doi: 10.1016/j.cmet.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang Y, Qin X, Liu S, Li J, Zhu X, Gao T, et al. Peroxisome proliferator-activated receptor gamma is inhibited by histone deacetylase 4 in cortical neurons under oxidative stress. J Neurochem. 2011;118(3):429–439. doi: 10.1111/j.1471-4159.2011.07316.x. [DOI] [PubMed] [Google Scholar]
- 72.Wu Q, Yang X, Zhang L, Zhang Y, Feng L. Nuclear accumulation of histone deacetylase 4 (HDAC4) exerts neurotoxicity in models of parkinson’s disease. Mol Neurobiol. 2017;54(9):6970–6983. doi: 10.1007/s12035-016-0199-2. [DOI] [PubMed] [Google Scholar]
- 73.Qian DZ, Kachhap SK, Collis SJ, Verheul HM, Carducci MA, Atadja P, et al. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006;66(17):8814–8821. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 74.Granger A, Abdullah I, Huebner F, Stout A, Wang T, Huebner T, et al. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 2008;22(10):3549–3560. doi: 10.1096/fj.08-108548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu J, Zhou X, Li Q, Zhou SM, Hu B, Hu GW, et al. Role of phosphorylated HDAC4 in stroke-induced angiogenesis. Biomed Res Int. 2017;2017:2957538. doi: 10.1155/2017/2957538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu Q, Liu LZ, Yin Y, He J, Li Q, Qian X, et al. Regulatory circuit of PKM2/NF-kappaB/miR-148a/152-modulated tumor angiogenesis and cancer progression. Oncogene. 2015;34(43):5482–5493. doi: 10.1038/onc.2015.6. [DOI] [PubMed] [Google Scholar]
- 77.Madelaine R, Sloan SA, Huber N, Notwell JH, Leung LC, Skariah G, et al. MicroRNA-9 couples brain neurogenesis and angiogenesis. Cell Rep. 2017;20(7):1533–1542. doi: 10.1016/j.celrep.2017.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang C, Zuo F, Wang Y, Lu H, Yang Q, Wang J. Progesterone changes VEGF and BDNF expression and promotes neurogenesis after ischemic stroke. Mol Neurobiol. 2016. Epub ahead of print. 10.1007/s12035-015-9651-y. [DOI] [PMC free article] [PubMed]
- 79.Davila JL, Goff LA, Ricupero CL, Camarillo C, Oni EN, Swerdel MR, et al. A positive feedback mechanism that regulates expression of miR-9 during neurogenesis. PLoS One. 2014;9(4):e94348. doi: 10.1371/journal.pone.0094348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Latchney SE, Jiang Y, Petrik DP, Eisch AJ, Hsieh J. Inducible knockout of Mef2a, -c, and -d from nestin-expressing stem/progenitor cells and their progeny unexpectedly uncouples neurogenesis and dendritogenesis in vivo. FASEB J. 2015;29(12):5059–5071. doi: 10.1096/fj.15-275651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Z, McKercher SR, Cui J, Nie Z, Soussou W, Roberts AJ, et al. Myocyte enhancer factor 2C as a neurogenic and antiapoptotic transcription factor in murine embryonic stem cells. J Neurosci. 2008;28(26):6557–6568. doi: 10.1523/JNEUROSCI.0134-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang AH, Yang XJ. Histone deacetylase 4 possesses intrinsic nuclear import and export signals. Mol Cell Biol. 2001;21(17):5992–6005. doi: 10.1128/MCB.21.17.5992-6005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408(6808):106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McKinsey TA, Zhang CL, Olson EN. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol. 2001;21(18):6312–6321. doi: 10.1128/MCB.21.18.6312-6321.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007;104(44):17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 87.Faria Freitas M, Cuendet M, Bertrand P. HDAC inhibitors: a 2013–2017 patent survey. Expert Opin Ther Pat. 2018:1–17. 10.1080/13543776.2018.1459568. [DOI] [PubMed]
- 88.Gaur V, Connor T, Sanigorski A, Martin SD, Bruce CR, Henstridge DC, et al. Disruption of the class IIa HDAC corepressor complex increases energy expenditure and lipid oxidation. Cell Rep. 2016;16(11):2802–2810. doi: 10.1016/j.celrep.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 89.Zhang LX, DeNicola M, Qin X, Du J, Ma J, Tina Zhao Y, et al. Specific inhibition of HDAC4 in cardiac progenitor cells enhances myocardial repairs. Am J Physiol Cell Physiol. 2014;307(4):C358–C372. doi: 10.1152/ajpcell.00187.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Khalil W, Xia H, Bodempudi V, Kahm J, Hergert P, Smith K, et al. Pathologic regulation of collagen I by an aberrant protein phosphatase 2A/histone deacetylase C4/MicroRNA-29 signal axis in idiopathic pulmonary fibrosis fibroblasts. Am J Respir Cell Mol Biol. 2015;53(3):391–399. doi: 10.1165/rcmb.2014-0150OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen C, Wei X, Wang S, Jiao Q, Zhang Y, Du G, et al. Compression regulates gene expression of chondrocytes through HDAC4 nuclear relocation via PP2A-dependent HDAC4 dephosphorylation. Biochim Biophys Acta. 2016;1863(7 Pt A):1633–1642. doi: 10.1016/j.bbamcr.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee ST, Chu K, Jung KH, Kim JH, Huh JY, Yoon H, et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann Neurol. 2012;72(2):269–277. doi: 10.1002/ana.23588. [DOI] [PubMed] [Google Scholar]
- 93.Marei HE, Hasan A, Rizzi R, Althani A, Afifi N, Cenciarelli C, et al. Potential of stem cell-based therapy for ischemic stroke. Front Neurol. 2018;9:34. doi: 10.3389/fneur.2018.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Trounson A, McDonald C. Stem cell therapies in clinical trials: progress and challenges. Cell Stem Cell. 2015;17(1):11–22. doi: 10.1016/j.stem.2015.06.007. [DOI] [PubMed] [Google Scholar]