

Abstract
The bacterial natural product UK-1 and several structural analogs inhibit replication of the hepatitis C virus in the replicon assay, with IC50 values as low as 0.50 μM. The NS3 helicase has been identified as a possible target of inhibition for several of these compounds, while the remaining inhibitors act via an undetermined mechanism. Gel shift assays suggest that helicase inhibition is a direct result of inhibitor-enzyme binding as opposed to direct RNA binding, and the ATPase activity of NS3 is not affected. The syntheses and biological results are presented herein.
Keywords: Hepatitis C, NS3, Helicase, Inhibitor
The hepatitis C virus (HCV), the causative agent of the hepatitis C infection, has been declared a worldwide health concern by the World Health Organization (WHO).1,2 Estimates predict that a minimum of 3% of the global population, approximately 170 million people, are currently infected.3 Of those diagnosed with the virus, 70–80% of cases are not suppressed by the body’s immune system and chronic liver infection develops.4 This acute infection of the hepatocytes leads to liver cirrhosis, hepatocellular carcinoma, and eventually liver failure.5 Moreover, HCV infection is currently the leading cause of liver transplantation.6
Two direct-acting antiviral (DAA) drugs, Boceprevir and Telaprevir, were approved by the FDA for HCV treatment in 2011. Although these drugs represent the first DAAs approved for HCV therapy, during early phases of development it was found that monotherapy utilizing either of these drugs alone resulted in the rapid development of resistant strains.7,8 As a result, these drugs are only FDA approved when given in combination therapy with two previously approved broad spectrum antivirals, ribavirin and pegylated interferon (PEG-IFN).9,10 The new treatment regimes have increased overall response rates, but the severe side effects and extremely high cost associated with PEG-IFN remain problematic.11 While combination therapy is the most promising approach for future HCV treatment, replacement of broad spectrum antivirals with inhibitors specific for HCV will likely result in much more favorable treatment outcomes.
HCV is a positive sense RNA virus that, following entry into the host hepatocyte, encodes for a single polyprotein of approximately 3000 amino acids.12–14 The polyprotein is processed co- and post-translationally by both host and viral proteases to yield ten viral proteins. The amino terminal third of the peptide contains the four structural proteins core, E1, E2, and p7. The remaining two-thirds of the protein is composed of the nonstructural (NS) proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B.15 Although each NS protein possesses a unique function (or functions), as a whole they are responsible for the synthesis of viral RNA, and thus critical for viral replication.16 For this reason, the NS proteins are commonly pursued as drug targets in HCV research.
NS3 is a protein exhibiting multiple functions and is well characterized both structurally and functionally. NS3 and the adjacent NS4A function as a complex, with NS4A serving as both a cofactor for NS3 and as a membrane anchor.17 The amino terminal domain of NS3 and NS4A form a dimeric serine protease that is responsible for the intramolecular cleavage of NS3 from NS4A, as well as the intermolecular cleavage between all downstream non-structural proteins.18 In addition, the carboxy terminus of NS3 functions as an ATP dependent helicase, and this activity is facilitated by NS4A.19 The exact relevance of the helicase activity as it pertains to HCV viral replication is not known, but it has been shown that a functional helicase is required for in vivo HCV replication.16,20,21 Several functions for the helicase have been proposed, including aiding in the polymerase processivity, assisting with RNA folding, modulating host gene expression, and involvement in genome encapsidation.22,23 There are currently no HCV helicase inhibitors in clinical trials, but numerous strategies for helicase inhibition have been investigated. Since the NS3 helicase activity is dependent upon ATP hydrolysis, various nucleoside analogs have been developed to inhibit the NTPase activity of NS3.24 Other helicase inhibitors include compounds that bind directly to the nucleic acid binding site of the helicase or to unknown allosteric sites.25,26
UK-1 (Figure 1) is a Streptomyces metabolite that exhibits broad spectrum anti-cancer activity and has also been shown to chelate magnesium and zinc.27–29 It was hypothesized that UK-1 and structural analogs could potentially inhibit HIV-1 integrase via magnesium coordination in the enzyme active site. As such, a series of UK-1 analogs (1-6) were synthesized and screened against HIV-1, as well as a number of other viruses. Although no activity against HIV-1 was observed, all of the compounds screened did prove to be effective inhibitors of HCV viral replication in replicons, with IC50 values as low as 0.50μM. In an attempt to determine the mechanism of HCV inhibition, these compounds were also screened against the HCV NS3 helicase, NS3 NTPase, and NS5B polymerase.
Figure 1.

UK-1, truncated analogs (1), acid (2), amide (3), and naphthol analogs 4, 5, and 6.
The compounds evaluated are shown in Figure 1. UK-1 and analogs 1-3 were synthesized as previously reported.29,30. The synthesis of 5 is shown in Scheme 1 (for the synthesis of 6, the same methodology was used). This began with carboxylation of 1,5-dihydroxynaphthalene, using magnesium methyl carbonate as previously described.31 The resulting acid was reacted with benzyl chloride, which upon hydrolysis gave 7. The acid was then activated with 1,1′-carbonyldiimidazole (CDI) and coupled to methyl 3-hydroxyanthranilate, giving compound 8. Refluxing 8 in m-xylene with pyridinium p-toluenesulfonate (PPTS) resulted in cyclodehydration as well as monodebenzylation, to give target compound 5. Compound 4 was synthesized similarly, starting with 1-hydroxy-2-naphthoic acid. It should be noted that the position of the benzyl group in 5 (and 6) was not readily determined using 2D NMR techniques. However, fluorescence experiments confirmed the product structure (Supplemental Information: Scheme 2, Figure 3). Briefly, the desired product showed a large Stokes shift consistent with an excited state intramolecular proton transfer (ESIPT) only feasible in isomer 5 (and 6), as opposed to the other possible product of monodebenzylation.32
Scheme 1.

Reagents and conditions: (a) Magnesium methyl carbonate, DMF, 140 °C; (b) BnCl, DMF, K2CO3, reflux; (c) NaOH, MeOH, reflux; (d) i) CDI, THF, R.T. ii) Methyl-3-hydroxyanthranilate, reflux; (e) PPTS, m-xylene, 140 °C.
Potential inhibition of the helicases of Flaviviridae viruses HCV, Japanese encephalitis virus (JEV), and dengue virus (DENV) was investigated using previously described methods.33,34 None of the compounds inhibit JEV or DENV helicases (IC50>700μM), however several of the compounds did inhibit the activity of the HCV helicase (Table 1, Figure 2). UK-1 itself shows weak inhibition using a DNA substrate, but no inhibition with an RNA substrate. Importantly, naphthol derivatives 4-6 show helicase inhibition, with 5 and 6 exhibiting IC50 values in the low micromolar range. None of the compounds inhibit the ATPase activity of the HCV helicase (IC50>1200 μM), eliminating this as a possible mechanism of action. Compounds 5 and 6 do not affect the gel mobility of an EcoRI-digested pT7-7 plasmid, suggesting the inhibition results from direct helicase interaction, rather than simple nucleic acid binding.
Table 1.
Helicase inhibition and viral replication inhibition data for UK-1 and analogs 1-6.
| Compound | Helicasea
|
Repliconc,d | |||
|---|---|---|---|---|---|
| DNA | RNA | ||||
|
| |||||
| IC50(μM)b | IC50(μM)b | EC50 (μM)e | CC50 (μM)f | SI50g | |
| UK-1 | 380 | >1200 | 0.77 | 17 | 22 |
| 1 | >1800 | >1800 | 0.50 | 5.4 | 11 |
| 2 | >1800 | >1800 | 0.53 | 3.5 | 6.6 |
| 3 | >1800 | >1800 | 1.4h | 6.3h | 4.5h |
| 4 | 540 | 590 | 0.53 | 2.5 | 4.7 |
| 5 | 3.8 | 20 | 2.1 | >20 | >10 |
| 6 | 2.6 | 20 | 0.54 | >20 | >37 |
Strand separation of radiolabeled oligonucleotides was monitored using gel electrophoresis (see Supplemental Information).
The concentration of compound required to inhibit 50% of helicase activity.
Experiments were carried out at Southern Research Institute, Frederick, MD.
Results are the averages of 2–4 independent dose-response experiments using a combination of luciferase reporter-based and RNA assays unless noted.
The concentration of compound that inhibits 50% of viral RNA replication.
The concentration of compound that kills 50% of the cells.
The ratio of EC50 to CC50.
Result is from a single luciferase reporter-based assay.
Figure 2. Inhibition of the unwinding activity of HCV helicase using DNA substrate.
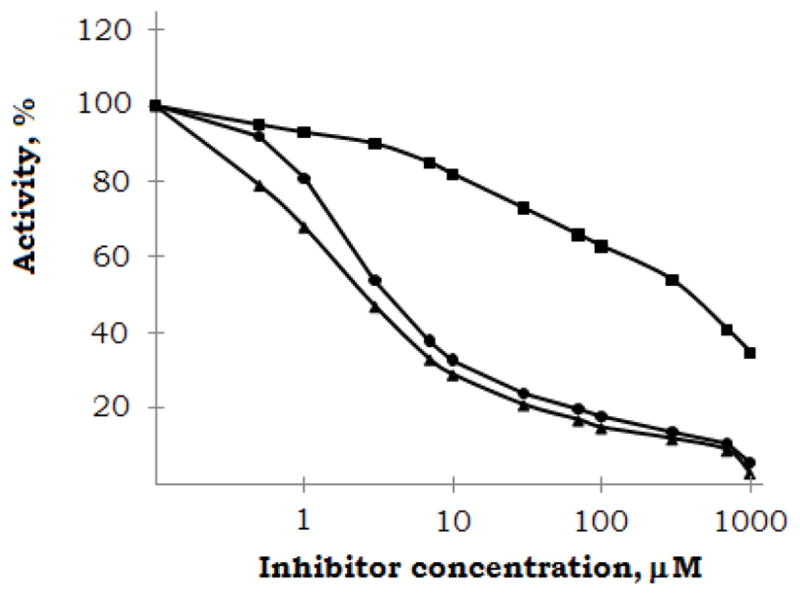
Strand separation of radiolabeled oligonucleotides was monitored using gel electrophoresis. UK-1 (■), 5 (●), and 6 (▲). Results presented are representatives of three independent experiments.
To determine if the compounds are active against HCV replication in cells, they were screened in the replicon assay (Table 1). Quite interestingly, all seven compounds were active, with EC50 values in the low- to sub-micromolar range. While the mechanism of viral inhibition for compounds 5 and 6 may result from helicase inhibition, this is not the case for 1-3 and seems unlikely for weak helicase inhibitors UK-1 and 4. This suggests that within this group of compounds, there is a second, as yet undetermined mechanism of inhibition. The compounds were then screened against the HCV RNA-dependent RNA polymerase NS5B, and very little inhibition was observed (inhibition ≤ 30% at 100 μM). There was no significant difference in activities between analogs 1-3, despite expected differences in cell permeability and susceptibility to cellular esterases. Compounds 5 and 6 exhibit cell toxicity values greater than 20 μM, giving selectivity indices greater than 10 and 37, respectively. All other compounds showed measureable toxicity under the assay conditions, although the selectivity index for UK-1 is still greater than 20.
All noted compounds are significantly better inhibitors in replicons than in the helicase assay. This discrepancy could result from the fact that the helicase experiments were conducted with the helicase and NTPase domains of NS3 (NS3h). The helicase activity of full length NS3 is greater than that of NS3h alone.35–37 It has also been shown that adjacent NS4A acts as a cofactor for NS3 and increases helicase activity.38 It is therefore possible that the inhibitors are more active in the presence of full length NS3/NS4A than NS3h alone. This could explain the increased inhibitory activity of 5 and 6 in replicons versus in the helicase assay. Alternatively, based on the similar activities of all inhibitors in replicons, these compounds could potentially share the same target, which is not the NS3 helicase. To further explore this possibility, evidence for a direct interaction between 5 (and 6) with purified NS3 is being sought, as well as potential interactions with other HCV proteins.i
In summary, several novel inhibitors of HCV viral replication have been identified which exhibit minimal cytotoxicity. Two of these compounds (5 and 6) inhibit the NS3 helicase with micromolar IC50 values and are highly selective; no inhibition is observed with the closely related helicases of DENV and JEV. It has been speculated that viral inhibition may be a result of direct interaction with the helicase, as neither ATPase inhibition nor direct RNA binding is observed. A second group of compounds effectively inhibits HCV RNA replication in the replicon assay via an as yet to be determined mechanism. Experiments are currently underway to further investigate and verify the proteins target(s) for all inhibitors.
Supplementary Material
Acknowledgments
This research was financed in part by NIGMS Initiative for Maximizing Student Development Grant (r25-GM55036) (D.N.W), U.S. Department of Education, Ronald E. McNair Post Baccalaureate Achievement Program Grant #P217A030141 (S.M), and EMBARC Grant #DBI-0453294 (P.S and M.T). Thanks to Professor Neerja Kaushik-Basu, Department of Biochemistry and Molecular Biology, UMDNJ-New Jersey Medical School for running NS5B assays and a special thanks to Dr. Ramachandra S. Hosmane for his advice and guidance.
Footnotes
Examples of helicase and gel shift assays are included in the Supplemental Information (Figures 4–6)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.WHO. J Viral Hepatitis. 1999;6:35–47. [PubMed] [Google Scholar]
- 2.Lavanchy D. Liver Int. 2009;29:74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 3.Alter J. World J Gastroenterol. 2007;13:2436–2431. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alter J, Mast E. Gastroenterol Clin North Am. 1994;23:437–455. [PubMed] [Google Scholar]
- 5.Lauer M, Walker D. N Engl J Med. 2001;345:41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- 6.Brown S, Gaglio J. Liver Transpl. 2003;9:S10–S13. doi: 10.1053/jlts.2003.50244. [DOI] [PubMed] [Google Scholar]
- 7.Sarazin C, Kieffer TL, Bartels D. Gastroenterology. 2007;132:1767–1777. doi: 10.1053/j.gastro.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 8.Susser S, Welsch C, Wang Y. Hepatology. 2009;50:1709–1718. doi: 10.1002/hep.23192. [DOI] [PubMed] [Google Scholar]
- 9.Bacon R, Gordon C, Lawitz E. N Engl J Med. 2011;364:1207–1217. doi: 10.1056/NEJMoa1009482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobson M, McHutchinson G, Dusheiko G. N Engl J Med. 2011;364:2405–2416. doi: 10.1056/NEJMoa1012912. [DOI] [PubMed] [Google Scholar]
- 11.Soriano V, Labarga P, Fernandez-Montero J, Benito J, Poveda E, Rallon N, Sanchez C, Vispo E, Barreiro P. Antiviral Res. 2012 doi: 10.1016/j.antiviral.2012.10.011. . (Article in Press) [DOI] [PubMed]
- 12.Frick DN. Curr Issues Mol Biol. 2007;9:1–20. [PMC free article] [PubMed] [Google Scholar]
- 13.Sakamoto N, Watanabe M. J Gastroenterol. 2009;44:643–649. doi: 10.1007/s00535-009-0084-0. [DOI] [PubMed] [Google Scholar]
- 14.Wardell AD, Errington W, Ciaramella G, Merson J, McGarvey MJ. J General Virology. 1999;80:701–709. doi: 10.1099/0022-1317-80-3-701. [DOI] [PubMed] [Google Scholar]
- 15.Choo L, Richman H, Han H, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby A, Barr J, Weiner J, Bradley W, Kuo G, Houghton M. Proc Natl Acad Sci. 1991;88:2451–2455. doi: 10.1073/pnas.88.6.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan S, He Y. Hepatitis C: Antiviral Drug Discovery and Development. Caister Academic Press; UK: 2011. [Google Scholar]
- 17.Failla C, Tomei L, De Francesco R. J Virol. 1994;68:3753–3760. doi: 10.1128/jvi.68.6.3753-3760.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobson H. J Virol. 1993;67:3835–3844. doi: 10.1128/jvi.67.7.3835-3844.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng W, Dumont S, Tinoco I, Bustamante C. Proc Natl Acad Sci USA. 2007;104:13954–13959. doi: 10.1073/pnas.0702315104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lam M, Frick N. J Virol. 2006;80:404–411. doi: 10.1128/JVI.80.1.404-411.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolykhalov A, Mihalik K, Feinstone M, Rice M. J Virol. 2000;74:2046–2051. doi: 10.1128/jvi.74.4.2046-2051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajkowitsch L, Chen D, Stampfl S, Semrad K, Waldsich C, Mayer O, Jantsch F, Konrat R, Schroeder R. RNA Biol. 2007;4:118–130. doi: 10.4161/rna.4.3.5445. [DOI] [PubMed] [Google Scholar]
- 23.Jarvis C, Newport W, Von Hippel H. J Biol Chem. 1991;266:1830–1840. [PubMed] [Google Scholar]
- 24.Belon A, Frick N. J Mol Biol. 2009;388:851–864. doi: 10.1016/j.jmb.2009.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim L, Morgenstern A, Griffith P, Dwyer D, Thomson A, Murcko A, Lin C, Caron R. Structure. 1998;6:89–100. doi: 10.1016/s0969-2126(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 26.Stankiewicz-Drogon A, Palchykovska G, Kostina G, Alexeeva V, Shved D, Boguszewska-Chachulska Bioorg Med Chem. 2008;16:8846–8852. doi: 10.1016/j.bmc.2008.08.074. [DOI] [PubMed] [Google Scholar]
- 27.Ueki M, Ueno K, Miyadoh S, Abe Shibata K, Tanguchi M, Oi SJ. Antibiotics. 1993;46:1089–1094. doi: 10.7164/antibiotics.46.1089. [DOI] [PubMed] [Google Scholar]
- 28.Reynolds M, DeLuca M, Kerwin SM. Bioorg Chem. 1999;27:326–337. [Google Scholar]
- 29.Kumar D, Jacob MR, Reynolds MB, Kerwin SM. Bioorg Med Chem Lett. 2002;10:3997–4004. doi: 10.1016/s0968-0896(02)00327-9. [DOI] [PubMed] [Google Scholar]
- 30.McKee M, Kerwin S. Bioorg Med Chem. 2008;16:1775–1783. doi: 10.1016/j.bmc.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cate LA. Synthesis. 1983:385–386. [Google Scholar]
- 32.Henary MM, Fahrni CJ. J Phys Chem A. 2002;106:5210–5220. [Google Scholar]
- 33.Borowski P, Kuehl R, Mueller O, Hwang L-H, Schulze zur Wiesch J. Eur J Biochem. 1999;266:715–723. doi: 10.1046/j.1432-1327.1999.00854.x. [DOI] [PubMed] [Google Scholar]
- 34.Gallinari P, Brennan D, Nardi C, Brunetti M, Tomei L, Steinkühler C, De Francesco R. J Virology. 1998;72:6758–6769. doi: 10.1128/jvi.72.8.6758-6769.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beran R, Serebrov V, Pyle A. J Biol Chem. 2007;282:34913–34920. doi: 10.1074/jbc.M707165200. [DOI] [PubMed] [Google Scholar]
- 36.Frick D, Rypma R, Lam A, Gu B. J Biol Chem. 2004:1269–1280. doi: 10.1074/jbc.M310630200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajagopal V, Gurjar M, Levin M, Patel S. J Bio Chem. 2010:17821–17832. doi: 10.1074/jbc.M110.114785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beran R, Lindenbach B, Pyle A. J Virol. 2009:3268–3275. doi: 10.1128/JVI.01849-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Still WC, Kahn M, Mitra A. J Org Chem. 1978;43(14):2923–2925. [Google Scholar]
- 40.Borowski P, Niebuhr A, Mueller O, Bretner M, Felczak K, Kulikowski T, Schmitz H. J Virol. 2001;75:3220–3229. doi: 10.1128/JVI.75.7.3220-3229.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korba B, Montero A, Farrar K, Gaye K, Mukerjee S, Ayers M, Rossignol JF. Antiviral Res. 2008;77:56–63. doi: 10.1016/j.antiviral.2007.08.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.