

Abstract
Many avenues exist for human pluripotent stem cells (hPSCs) to impact medical care, but they may have their greatest impact on the development of precision medicine. Recent advances in genome editing and stem cell technology have enabled construction of clinically-relevant, genotype-specific “disease-in-a-dish” models. In this review, we outline the use of genome-edited hPSCs in precision disease modeling and drug screening as well as describe methodological advances in scarless genome editing. Scarless genome-editing approaches are attractive for genotype-specific disease modeling as only the intended DNA base-pair edits are incorporated without additional genomic modification. Emerging evidentiary standards for drug development and approval of precision therapies are likely to incorporate more studies utilizing disease models derived from genome-edited hPSCs.
Teaser: Clinically-relevant, genotype-specific “disease-in-a-dish” models represent a path forward for precision medicine
1. Introduction
Improving pre-clinical disease models to more faithfully predict clinical effectiveness and identify toxicity is anticipated to lower the current 90% clinical trial failure rate [1]. This is especially important in the context of precision medicine, because disease models need to be tailored to specific biomarkers or genetic variations. Human pluripotent stem cell (hPSC) based disease models are good candidates to meet this challenge, since they can be tailored through genome editing for the rigorous evaluation of genotype-to-disease phenotype relationships in biologically relevant human cells.
Human induced pluripotent stem cells (hiPSCs) are reprogrammed from routine clinical samples (e.g., blood draws, skin biopsies) and can be reliably expanded in culture [2,3]. Importantly, hiPSCs retain their patient specific genotype throughout the reprogramming process. This feature enables hiPSCs to be differentiated towards disease affected cell types in order to recapitulate a patient’s disease phenotype [4] and to evaluate patient-specific therapeutic response in a controlled cell culture environment [5] (Figure 1A). Stem cell-derived disease models are most applicable for modeling diseases with quantifiable cell autonomous disease phenotypes observed in well-defined cells. With continued advancement of cell differentiation protocols, the “disease-in-a-dish” paradigm has been applied to an array of neurodegenerative, cardiovascular, and other diseases [6].
Figure 1. Paradigms of stem cell disease modeling for therapeutic screening.

(A) Patient-specific disease in a dish models. Unaffected siblings share only ~50% genetic inheritance with affected patients and the field has largely moved towards isogenic controls to validate phenotypic outputs (diseased vs. healthy). These screens are not generalizable (i.e., can only inform treatment for a specific patient) in isolation due to potentially confounding effects of background gene modifiers. (B) hiPSC cohort clinical trial in a dish (“macromedicine”). Cohorts of bio-banked iPSCs, with various disease associated variants/biomarkers and various genetic backgrounds, are differentiated and cell line to phenotypic recovery outcomes are identified for candidate drugs. Computational analysis is performed to identify externally valid (i.e. results can be used to inform treatment for patients not in cohort) drug specific variant/biomarker to phenotypic recovery correlations. Cohort approaches are ideal for diseases with complex inheritance and access to patient hiPSCs. (C) High-throughput, variant-specific therapeutic screening. Disease associated variants can be introduced into a healthy “parental control line” with scarless genome editing to create an array of isogenic cell lines. Direct assessment of variant/biomarker specific therapeutic response relationships can be observed with high-content screening (or other high-throughput) assays. This approach is ideal for rare monogenic diseases where access to patient-specific hiPSCs is limited.
As the process of reprogramming cells has become more efficient, bio-banks of hiPSC cohorts that are disease specific or representative of the general population have been created to conduct so called “clinical trials in a dish” (Figure 1B) (reviewed by [7]). A recent study by Burridge and colleagues [8] revealed that hiPSC derived cardiomyocytes faithfully recapitulated doxyrubicin induced cardiotoxicity phenotypes in breast cancer patients with or without cardiotoxicity after doxyrubicin treatment. Patient derived hiPSC approaches like this are well suited to diseases with multigenic or complex inheritance patterns.
Initial disease models utilized hiPSCs from unaffected siblings to ensure that observed cellular phenotypes were disease specific (Figure 1A, top). However, unaffected siblings are incomplete controls—sibling pairs have roughly 50% shared parental inheritance due to chromosomal segregation and crossover in meiosis. To validate disease-associated variants, the field has utilized genome editing techniques to correct patient mutations to wildtype variants (Figure 1A, bottom). These patient-derived, gene-corrected pairs of cells differ only at the edited locus and are referred to as isogenic cell lines. Isogenic cell lines can be used to model patient specific disease mechanisms [9] and identify personalized therapy [10]. However, the resources required to implement this analysis broadly are prohibitive. To efficiently realize value from precision medicine, methodologies that can predict universal disease variant to treatment outcome correlations should be prioritized.
A more scalable genome-edited disease modeling approach involves introduction of disease specific mutations into well-characterized healthy hPSC lines with known genetic background (Figure 1C). Using this approach, an array of unique disease-associated variants can be rationally engineered into the hPSCs. This eliminates the need to acquire hiPSCs from patients with rare variants where the ability to perform a new screen for each individual is not feasible. Instead gene-variant targeted therapies can be screened in advance. Wang et al. demonstrated the value of this approach by introducing disease associated variants of Barth syndrome in normal or unaffected iPSCs [11]. For diseases with primarily monogenic inheritance, this technique enables validation of the causality of disease associated variants and high-throughput screening of genotype-specific therapies.
In this review, we detail methods for scarless, or knock-in/footprint free, precision genome editing of hPSCs. Scarless methods are named to distinguish them from “genetic scarring” methods that permanently integrate additional sequences into the genome that may impact precise disease phenotypes in undefined ways [12,13]. We end with a forecast of how genome-edited hPSC derived models could integrate into precision drug development.
2. Precision Genome Editing with CRISPR
CRISPR has emerged as the workhorse of the genome editing field. The most common variant employs a CRISPR associated endonuclease, Cas9, and a short, single-guide RNA (sgRNA) to enable targeted double strand breaks (DSBs) in genomic DNA [14,15] (Figure 2). DSBs are primarily repaired via non-homologous end joining (NHEJ) and homology directed repair (HDR). NHEJ requires no template for repair and results in a wide spectrum of insertion and deletion (indel) mutations. These mutations often result in loss of gene function, but effects of undefined gene products may confound disease modeling in unanticipated ways [16]. Conversely, HDR requires a repair template (usually the homologous chromosome) which is used for error-free repair in eukaryotic cells [17].
Figure 2. Schematic of Cas9-induced DSB repair and focus areas for improving precision editing.
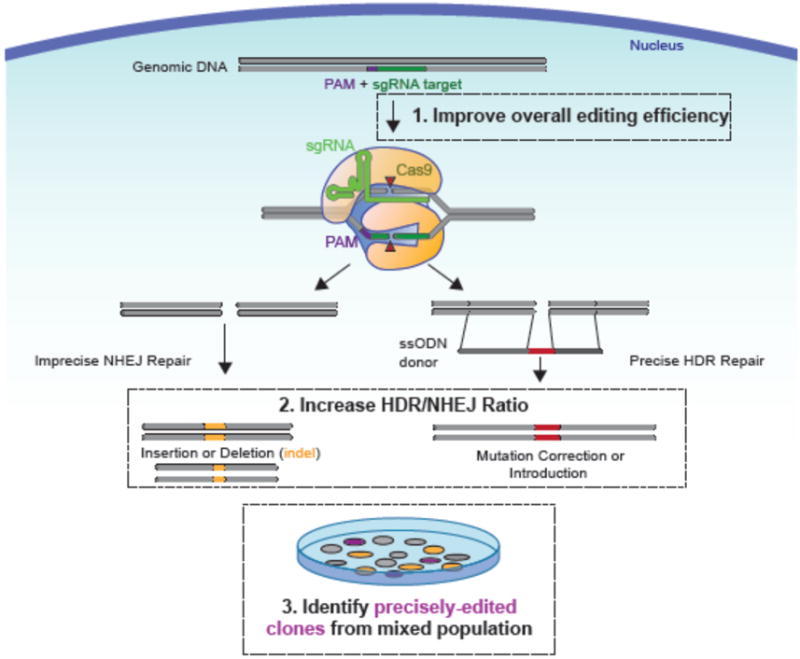
Cas9 endonuclease is targeted to a specific locus in the human genome via a single guide RNA (sgRNA). Cas9 makes double strand breaks (DSBs) that are repaired via error prone non-homologous end joining (NHEJ) or precise homology directed repair (HDR). Scarless gene editing is facilitated by encoding desired edits on a plasmid or a double or single stranded linear donor DNA template (single stranded oligonucleotide DNA, “ssODN” shown here). NHEJ occurs more frequently than HDR in hPSCs. To facilitate increased precise and scarless HDR mediated genome editing, the field has focused primarily on: 1) increasing overall (NHEJ and HDR) genome editing, 2) increasing ratio of HDR to NHEJ, and 3) improving selection of genome-edited clones.
Delivering synthetic “donor” DNA, along with the CRISPR/Cas9 system, can co-opt the cell’s intrinsic HDR machinery for introduction or correction of mutations, or introduction of synthetic genes [18,19]. Unfortunately, the overall efficiency of HDR is low (<10%) in hPSCs, so integrated drug selection cassettes were initially used to improve the efficiency of identifying edited clones [20]. However, a portion of the cassette sequence often remains integrated which may impact mRNA processing and/or protein translation in undefined ways [12,13]. While there are clear advantages of scarless genome-editing for precision hPSC disease modeling, a major limitation has been the low overall efficiency of these methods. To address this challenge, the field had focused in several major areas (Figure 2, see also Table 1): increasing overall (NHEJ and HDR) genome editing (section 2.1), increasing ratio of HDR to NHEJ (section 2.2), and improving selection of genome-edited clones (section 2.3).
Table 1. Summary of recent strategies for precision genome editing.
Examples were selected based on relevance to scarless genome editing in hPSCs and are not exhaustive.
| Method | Advantages | Disadvantages | Examples |
|---|---|---|---|
| 1. Approaches for improving overall genome editing efficiency | |||
| A. Increase Cas9 expression | |||
| Stable inducible Cas9 expression | - Cas9 can be integrated into AAVS or other safe harbor loci under control of a Tet On promoter. - High Efficiency NHEJ and HDR. |
- Not scarless. - Concern for higher probability of off-target DSBs. - Line must be generated before other editing. |
[24,25] |
| Non-permanent constitutive or inducible Cas9 expression | - Cas9 expressed between piggyBac or on episomal plasmid. - High efficiency and scarless after Cas9 removed. |
- Concern for higher probability of off-target DSBs. - Line must be generated before other editing. |
[22,26] |
| Enrich for Transient Cas9 Expression | - Plasmid vectors enable enrichment for cells with high Cas9 expression. - Do not need to create Cas9 cell line before each experiment. |
- Some concern for higher probability of off-target DSBs. - Lower efficiency vs inducible stable Cas9. |
[31,46] |
| 2. Approaches for increasing HDR/NHEJ ratio | |||
| A. Cell cycle control | |||
| Controlled timing of Cas9 delivery |
- Cell cycle checkpoint blockers are used to sync cells. - Cas9 protein can be delivered to cells when synced in a pro-HDR state. |
- Titration of checkpoint blockers is needed for each cell line. - HDR Efficiency gains are only 1-2%. |
[37] |
| Cell cycle selective degradation of Cas9 | - Geminin peptide conjugated to Cas9 causes Cas9 degradation in G1 (high NHEJ) phase of cell cycle. - Can be transiently expressed from plasmid. |
- Requires cloning of specialized Cas9 vector (not commercially available). | [38] |
| B. Enrichment for HDR biased cells | |||
| Co-insertion of integrated selectable marker | - Integration of puromycin resistance gene at safe harbor locus in addition to the target edit. - Selection for one HDR event is correlated with additional HDR events. |
- Selectable marker must be removed for scarless editing. - Additional Cas9 and sgRNA must be targeted to safe harbor site. - Potential off-target DSBs. |
[39,40] |
| C. Repair pathway modulation | |||
| Factors for inhibition of NHEJ or promotion of HDR | - Co-delivery of some small molecules or other factors (SCR7, L755507, etc.) shown to increase HDR/NHEJ ratio. | - Effectiveness varies from cell line to cell line. - Toxicity of some factors reported in specific hPSC lines. |
[16,41–43] |
| D. Improved design of donor DNA | |||
| Use of single stranded DNA donors (ssODN) vs. double stranded DNA | - Insertion of sequences up to ~30nt. - ssODNs can be synthesized rapidly without cloning. - ssDNA has less probability of off-target integration vs dsDNA donors. |
- ssODNs incorporated via HDR, not efficient in post-mitotic cells. - Not suitable for large constructs, synthesis of long ssODNs for synthetic gene insertion is only recently reported. |
[62] |
| Use of asymmetric homology arms for ssODN | - ssODNs with one short arm (~30 nt) and one longer arm (~70 nt) have higher integration efficiency due to 5′-3′ exonuclease activity or strand release by Cas9. | - short ssDNA less stable than dsDNA (improve stability with phosphorothiorate bonds but increase cytotoxicity). | [27] [44] |
| 3. Approaches for identifying precisely-edited clones from a mixed population | |||
| A. Deep sequencing/high-throughput sequencing (HTS) based | |||
| HTS Library Preparation | - Commercially available DNA barcoding library kits enable genotyping of 96 or more clonal populations on single HTS lane. | - Most HTS reads only 100-200 nt in length; possibility of incorrectly genotyping clones due to undetected large deletions. | [46] |
| B. Non-deep sequencing based | |||
| Digital droplet PCR | - Identification of rare sequences in mixed population. - Enables targeted subcloning to isolate precisely-edited clones. |
- Requires specialized equipment. - Primer used for detection of precise edit must be optimized for each locus. |
[47,48] |
| Capillary electrophoresis of labeled amplicons | - Uses Sanger sequencing capillaries to identify indels and/or base changes. - Can be less costly vs ddPCR or HTS. |
- Difficult to detect rare edited alleles in a mixed population compared to HTS or ddPCR. | [49,50] |
| C. Scarless integration of selectable markers | |||
| piggyBac Transposon | - piggyBac flanked sequences can be used to completely remove pos/neg (fluorophore + puroΔtk) selection cassette. - FACS can enable isolation of polyclonal precise edited populations |
- Methods require a second treatment step to remove selection cassette. - piggyBac repeat sequences may increase off-target integration; careful donor design is needed. |
[52,53] |
| D. Prevention of additional on target edits | |||
| CORRECT Method | - Integrate blocking mutation along with intended edit to prevent Cas9 re-cutting. - Homozygous vs heterozygous insertion can be predicted by distance of intended edit from DSB site. |
- Requires second editing step with new ssODN and sgRNA or new ssODN with modified PAM Cas9 variant for scarless editing. | [54] |
2.1 Increasing Overall Genome Editing Efficiency
The level of intracellular Cas9 expression appears to represent a universal limiting step in genome editing [21–23]. Improvements to Cas9 delivery, or enrichment of cells with high Cas9 expression, have yielded improved editing outcomes. Cas9 can be stably integrated into a parental cell line or can be transiently delivered to cells during each new editing workflow. Generating a parental cell line with conditional or inducible Cas9 expression [22,24–27] has been shown to yield NHEJ efficiencies of up to 60% and precise HDR efficiencies up to 40%. The limitation of these methods is that the Cas9 construct is either permanently integrated [24,25] or must be later removed with a subsequent reagent delivery and/or clonal selection step [22,26] to achieve scarless editing. These methods are valuable when a single parental cell line will be used to generate many unique genotype specific cell lines.
Conversely, transient delivery of Cas9 is more applicable when a variety of cell lines must be edited (e.g., creating isogenic controls for bio-banked hiPSCs [7]), where prior generation of many stable lines would be too cumbersome. Cas9 can be delivered as a recombinant protein pre-complexed with sgRNA or encoded as mRNA or within a plasmid. Plasmid reagents have been used most extensively for hiPSC disease modeling applications, because co-expression of selectable markers from the plasmid can be used to enrich for Cas9 expressing cells. Plasmids that encode a viral 2A “ribosomal skip” peptide can be used to express GFP or a puromycin resistance gene in stoichiometric proportion to Cas9 [28–30]. These plasmids enable enrichment of Cas9 expressing cells based on fluorescence assisted cell sorting (FACS) or puromycin selection. Single cell FACS can be challenging in hPSCs because of contamination risks and the variable sensitivity of hPSC lines to singularization [31]. In cell lines recalcitrant to FACS, transient puromycin selection serves as viable alternative [30].
Importantly, extended exposure to high levels of Cas9—as expected in stable expression lines or stringent enrichment methods—has been shown to increase the probability of off-target DSB formation. Off-target DSBs are an important consideration, as undefined indels throughout the genome can confound phenotypic outputs of disease models. Fortunately, sgRNA design algorithms [32,33], high fidelity [34,35], and nickase [36] Cas9 variants have been shown to decrease off-target editing, in some cases, at the expense of some on-target efficacy.
2.2 Improving Ratio of HDR to NHEJ
In hPSCs, imprecise NHEJ is heavily favored over HDR during repair of Cas9 induced DSBs. Improving the HDR to NHEJ ratio improves the efficiency of precise editing workflows and has been accomplished by harnessing or manipulating intrinsic DSB repair processes. Delivering Cas9 protein only during the S and G2 phase of the cell cycle, when HDR pathways are most active, was shown to increase HDR efficiency; however, the parameters for cell cycle control varied considerably from cell line to cell line [37]. A related approach conjugates a peptide to Cas9 which targets Cas9 for selective degradation during the G1 phase of the cell cycle, when NHEJ predominates [38]. More recently, enrichment for cells that are presumably in an HDR biased state was performed by co-inserting a drug selectable marker at a secondary safe harbor locus [39,40]. Co-insertion methods appear to be more effective than direct manipulation of the cell cycle, but they require generation of DSBs at additional genomic loci and can result in permanent integration of the selection cassette. Identification of small molecule or other targeted factors that inhibit NHEJ or promote HDR has also demonstrated increased HDR/NHEJ editing ratios in several reports [16,41–43]. However, it is not clear if these factors are tolerated or are universally effective across hPSC lines [27,41].
Other efforts have focused on optimizing donor DNA construction and improving its co-localization with the nuclease to stimulate HDR. Early work demonstrated that optimal single-stranded oligonucleotide (ssODN) donor length is approximately 70–120 nt, and that changes encoded in the donor DNA are more likely to be integrated when the changes are near the DSB site [31]. Work by Richardson and colleagues demonstrated that sidedness of the DNA strand used as the ssODN template and asymmetric lengths of homology arms can be used to increase HDR efficiency [44]. Later reports reinforce the value of asymmetric homology arms, but did not see improvements by selecting the template strand [27]. Prevention of ssODN degradation with the use of phosphorothioate-modified oligonucleotides has also been shown to enhance HDR efficiency in cultured cells, presumably by stabilizing the ssODN within cells and during delivery [45].
2.3 Improving Selection of Genome-Edited Clones
A major obstacle for isolating precisely genome-edited clones is identifying them from a mixed population. Many workflows utilize high-throughput or deep sequencing library preparation methods to sequence many unique clones in parallel on a single lane [46]. Non-deep sequencing techniques include digital droplet PCR (ddPCR) [47,48] and fluorescent gel capillary based methods [49,50]. These methods require working with a high number of clonal cell lines—typically over a hundred— but they can be combined with methods that increase editing efficiency or HDR/NHEJ ratios to improve workflows [51]. Recently, PiggyBac transposon [52,53] methods have been used to integrate selectable markers that can be used to enrich for precisely-edited hPSCs before being completely removed to yield scarless editing. These methods are efficient for selecting precisely edited clones, but the requirement for temporary introduction of a selection cassette transiently disrupts endogenous gene expression, which prevents their use in essential genes or those involved in maintenance of pluripotency.
A major hurdle for precise editing of clones has been isolating clones without additional on-target indels. This is especially challenging when heterozygous mutant lines (on allele mutated, the other allele unedited/WT) are desired. One solution, presented by Paquet and colleagues [54], involves a two-step process where both the intended mutation and a blocking mutation are made with the first ssODN repair. The blocking mutation prevents re-cutting of the repaired template and can be removed by a subsequent editing step using a newly designed sgRNA, or the same sgRNA with a Cas variant with different PAM specificity. This method requires repeated editing steps, but may be necessary for generation of precise heterozygous mutants.
3. Outlook
The development of more representative disease models and genome editing methods with higher throughput and efficiency could advance these models further. First, organoid and engineered co-culture technologies [55] could recapitulate tissue/organ function more faithfully than 2D cultures. For example, Dekkers and colleagues found that drug treatment response of cystic fibrosis (CF) patient-derived gut organoids mimicked the response of different patients with those same mutations in an in-human CF clinical trial [56]. Second, high-throughput genome editing methods, including genome-wide loss of function screens, primarily rely on imprecise NHEJ-mediated DNA repair (reviewed in [57]). These techniques provide valuable high level data on gene function, but are primarily limited to survival or cell death phenotypes. Further, most employ non-hPSC cell lines to ensure technical viability (as these tend to have higher gene-editing efficiency) at the expense of biological relevance. Findlay and colleagues presented a strategy for high-throughput precision HDR mediated genome editing to determine the effect of precise gene variants on transcription level for the BRCA1 gene in immortalized HEK 293T cells [58]. Precision HDR within cancer cell lines also enabled target validation for several small molecules [59]. Future improvements in scarless editing efficiency within hPSCs may enable such high-throughput editing to be performed within stem cell derived models.
Investment in hPSC disease modeling strategies is contingent not only on technical advances, but also regulatory acceptance of these models. In the US, the FDA has signaled that cell based models may play an increasing role in future drug approvals, especially for rare diseases, by supplementing or even substituting for clinical trial data [60]. A dramatic example is the on label approval of the CF drug ivacaftor for twenty three additional CF mutations (ten originally) based on cell based assays alone [61]. Reliance on cell-based assays in lieu of clinical trial data will likely be limited to rare cases; tradeoffs in drug access and patient safety must also be carefully considered. More likely, genome-edited stem cell derived disease models will play a role in generating preclinical data for precision medicine trials. There is an immediate role in drug repurposing, where the therapeutic potential of approved compounds or those previously shown to be safe in Phase I trials could be screened in genotype-specific models. Consideration of smaller or modified clinical trials based on precisely edited hPSC-derived disease model data could incentivize therapeutic development for rare or biomarker specific subsets of common diseases. Given the recent advances in generating genome-edited disease models, it is highly likely that evidence from such models will be increasingly used in the drug discovery pipeline for precision therapies.
Box 1. Key Terms.
Precision medicine- disease prevention and treatment that considers differences in patient’s genes, environments, and lifestyles.
Biomarker-a genomic variant or phenotypic trait that is informative for predicting specific disease progression or treatment outcome.
Pluripotent-capable of giving rise to all cell types of the body.
Reprogram-the process of reverting mature somatic cells to a pluripotent or stem-like state. This can now be accomplished using non-integrating vectors.
Isogenic cell lines-cell lines whose genetic makeup only differs at a specific locus.
Scarless-a targeted genome modification where only the intended DNA base-pair edits are incorporated without permanent integration of additional DNA sequences.
CRISPR-short for clustered regularly interspaced short palindromic repeats.
Non-homologous end joining (NHEJ)-an error prone DNA repair process where double strand breaks are directly ligated, commonly resulting in insertion or deletion mutations.
Homology Directed Repair (HDR)-a precise DNA repair process in which cells repair double strand breaks in DNA by using a homologous DNA template (usually the homologous chromosome). This process can be co-opted to engineer-in specific sequences by delivering synthetic DNA donors with homology arms.
Acknowledgments
This work was supported by the NSF [CBET-1350178 to K.S., CBET-1645123 to K.S.]; the NIH [1R35GM119644-01 to K.S., 4T32HG002760-14 to B.S., 1F30EY027699-01 to B.S.]; the VitreoRetinal Surgery Foundation to B.S, the John Merck Fund [Translational Research Program to K.S.]; Brain & Behavior Research Foundation [K.S.]; Burroughs Wellcome Fund [K.S.]. Additionally, we thank all members of the Saha lab for helpful discussion and comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clinical Development Success Rates 2006-2015 - BIO, Biomedtracker, Amplion, 2016. (n.d.). https://www.bio.org/sites/default/files/Clinical%20Development%20Success%20Rates%202006-2015%20-%20BIO,%20Biomedtracker,%20Amplion%202016.pdf accessed November 13, 2017.
- 2.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 4.Singh R, Shen W, Kuai D, Martin JM, Guo X, Smith MA, Perez ET, Phillips MJ, Simonett JM, Wallace KA, Verhoeven AD, Capowski EE, Zhang X, Yin Y, Halbach PJ, Fishman GA, Wright LS, Pattnaik BR, Gamm DM. iPS cell modeling of Best disease: Insights into the pathophysiology of an inherited macular degeneration. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds469. dds469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh R, Kuai D, Guziewicz KE, Meyer J, Wilson M, Lu J, Smith M, Clark E, Verhoeven A, Aguirre GD, Gamm DM. Pharmacological Modulation of Photoreceptor Outer Segment Degradation in a Human iPS Cell Model of Inherited Macular Degeneration. Mol Ther. 2015;23:1700–1711. doi: 10.1038/mt.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol. 2016;17:170–182. doi: 10.1038/nrm.2015.27. [DOI] [PubMed] [Google Scholar]
- 7.Warren CR, Cowan CA. Humanity in a Dish: Population Genetics with iPSCs. Trends Cell Biol. 2017 doi: 10.1016/j.tcb.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, Holmström A, Chang AC, Coronado MJ, Ebert AD, Knowles JW, Telli ML, Witteles RM, Blau HM, Bernstein D, Altman RB, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med. 2016;22:547–556. doi: 10.1038/nm.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, Soldner F, Sunico CR, Nagar S, Talantova M, Lee B, Lopez K, Nutter A, Shan B, Molokanova E, Zhang Y, Han X, Nakamura T, Masliah E, Yates JR, Nakanishi N, Andreyev AY, Okamoto S, Jaenisch R, Ambasudhan R, Lipton SA. Isogenic Human iPSC Parkinson’s Model Shows Nitrosative Stress-Induced Dysfunction in MEF2-PGC1α Transcription. Cell. 2013;155:1351–1364. doi: 10.1016/j.cell.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sala L, Yu Z, Ward-van Oostwaard D, van Veldhoven JP, Moretti A, Laugwitz K-L, Mummery CL, IJzerman AP, Bellin M. A new hERG allosteric modulator rescues genetic and drug-induced long-QT syndrome phenotypes in cardiomyocytes from isogenic pairs of patient induced pluripotent stem cells. EMBO Mol Med. 2016;8:1065–1081. doi: 10.15252/emmm.201606260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, Yuan H, Jiang D, Zhang D, Zangi L, Geva J, Roberts AE, Ma Q, Ding J, Chen J, Wang DZ, Li K, Wang J, Wanders RJA, Kulik W, Vaz FM, Laflamme MA, Murry CE, Chien KR, Kelley RI, Church GM, Parker KK, Pu WT. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20:616. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Z, Verma N, González F, Shi ZD, Huangfu D. A CRISPR/Cas-Mediated Selection-free Knockin Strategy in Human Embryonic Stem Cells. Stem Cell Rep. 2015;4:1103–1111. doi: 10.1016/j.stemcr.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao W, Siegel D, Biton A, Tonqueze OL, Zaitlen N, Ahituv N, Erle DJ. CRISPR–Cas9-mediated functional dissection of 3′-UTRs. Nucleic Acids Res. 2017;45:10800–10810. doi: 10.1093/nar/gkx675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Driscoll M, Jeggo PA. The role of double-strand break repair — insights from human genetics. Nat Rev Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- 18.Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood. 2011;118:4599–4608. doi: 10.1182/blood-2011-02-335554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, Meng X, Miller JC, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD, Holmes MC, Naldini L. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 21.Dow LE, Fisher J, O’Rourke KP, Muley A, Kastenhuber ER, Livshits G, Tschaharganeh DF, Socci ND, Lowe SW. Inducible in vivo genome editing with CRISPR-Cas9. Nat Biotechnol. 2015;33:390–394. doi: 10.1038/nbt.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang G, Yang L, Grishin D, Rios X, Ye LY, Hu Y, Li K, Zhang D, Church GM, Pu WT. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat Protoc. 2017;12:88–103. doi: 10.1038/nprot.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steyer B, Carlson-Stevermer J, Angenent-Mari N, Khalil A, Harkness T, Saha K. High content analysis platform for optimization of lipid mediated CRISPR-Cas9 delivery strategies in human cells. Acta Biomater. 2016;34:143–158. doi: 10.1016/j.actbio.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.González F, Zhu Z, Shi ZD, Lelli K, Verma N, Li QV, Huangfu D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao J, Wu L, Zhang SM, Lu M, Cheung WKC, Cai W, Gale M, Xu Q, Yan Q. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 2016;44:e149–e149. doi: 10.1093/nar/gkw660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie Y, Wang D, Lan F, Wei G, Ni T, Chai R, Liu D, Hu S, Li M, Li D, Wang H, Wang Y. An episomal vector-based CRISPR/Cas9 system for highly efficient gene knockout in human pluripotent stem cells. Sci Rep. 2017;7:2320. doi: 10.1038/s41598-017-02456-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang X, Potter J, Kumar S, Ravinder N, Chesnut JD. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J Biotechnol. 2017;241:136–146. doi: 10.1016/j.jbiotec.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 28.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 2013;12:393–394. doi: 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steyer B, Bu Q, Cory E, Jiang K, Duong S, Sinha D, Steltzer S, Gamm D, Chang Q, Saha K. Scarless Genome Editing of Human Pluripotent Stem Cells via Transient Puromycin Selection. Stem Cell Rep. 2018 doi: 10.1016/j.stemcr.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang L, Guell M, Byrne S, Yang JL, Angeles ADL, Mali P, Aach J, Kim-Kiselak C, Briggs AW, Rios X, Huang PY, Daley G, Church G. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013;41:9049–9061. doi: 10.1093/nar/gkt555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim HK, Min S, Song M, Jung S, Choi JW, Kim Y, Lee S, Yoon S, Henry Kim H. Deep learning improves prediction of CRISPR–Cpf1 guide RNA activity. Nat Biotechnol. 2018 doi: 10.1038/nbt.4061. [DOI] [PubMed] [Google Scholar]
- 34.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529 doi: 10.1038/nature16526. nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casini A, Olivieri M, Petris G, Montagna C, Reginato G, Maule G, Lorenzin F, Prandi D, Romanel A, Demichelis F, Inga A, Cereseto A. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat Biotechnol. 2018 doi: 10.1038/nbt.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 2014;3:e04766. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Howden SE, McColl B, Glaser A, Vadolas J, Petrou S, Little MH, Elefanty AG, Stanley EG. A Cas9 Variant for Efficient Generation of Indel-Free Knockin or Gene-Corrected Human Pluripotent Stem Cells. Stem Cell Rep (nd) doi: 10.1016/j.stemcr.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitzelfelt KA, McDermott-Roe C, Grzybowski MN, Marquez M, Kuo CT, Riedel M, Lai S, Choi MJ, Kolander KD, Helbling D, Dimmock DP, Battle MA, Jou CJ, Tristani-Firouzi M, Verbsky JW, Benjamin IJ, Geurts AM. Efficient Precision Genome Editing in iPSCs via Genetic Co-targeting with Selection. Stem Cell Rep. 2017;8:491–499. doi: 10.1016/j.stemcr.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shy BR, MacDougall MS, Clarke R, Merrill BJ. Co-incident insertion enables high efficiency genome engineering in mouse embryonic stem cells. Nucleic Acids Res. 2016;44:7997–8010. doi: 10.1093/nar/gkw685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song J, Yang D, Xu J, Zhu T, Chen YE, Zhang J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat Commun. 2016;7:10548. doi: 10.1038/ncomms10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- 43.Yu C, Liu Y, Ma T, Liu K, Xu S, Zhang Y, Liu H, La Russa M, Xie M, Ding S, Qi LS. Small Molecules Enhance CRISPR Genome Editing in Pluripotent Stem Cells. Cell Stem Cell. 2015;16:142–147. doi: 10.1016/j.stem.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34:339–344. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- 45.Renaud JB, Boix C, Charpentier M, De Cian A, Cochennec J, Duvernois-Berthet E, Perrouault L, Tesson L, Edouard J, Thinard R, Cherifi Y, Menoret S, Fontanière S, de Crozé N, Fraichard A, Sohm F, Anegon I, Concordet JP, Giovannangeli C. Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell Rep. 2016;14:2263–2272. doi: 10.1016/j.celrep.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 46.Byrne SM, Church GM. Crispr-mediated Gene Targeting of Human Induced Pluripotent Stem Cells. Curr Protoc Stem Cell Biol. 2015;35:5A.8.1–22. doi: 10.1002/9780470151808.sc05a08s35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyaoka Y, Chan AH, Judge LM, Yoo J, Huang M, Nguyen TD, Lizarraga PP, So PL, Conklin BR. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods. 2014;11:291–293. doi: 10.1038/nmeth.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Findlay SD, Vincent KM, Berman JR, Postovit LM. A Digital PCR-Based Method for Efficient and Highly Specific Screening of Genome Edited Cells. PLOS ONE. 2016;11:e0153901. doi: 10.1371/journal.pone.0153901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Z, Steentoft C, Hauge C, Hansen L, Thomsen AL, Niola F, Vester-Christensen MB, Frödin M, Clausen H, Wandall HH, Bennett EP. Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 2015;43:e59. doi: 10.1093/nar/gkv126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramlee MK, Yan T, Cheung AMS, Chuah CTH, Li S. High-throughput genotyping of CRISPR/Cas9-mediated mutants using fluorescent PCR-capillary gel electrophoresis. Sci Rep. 2015;5 doi: 10.1038/srep15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lonowski LA, Narimatsu Y, Riaz A, Delay CE, Yang Z, Niola F, Duda K, Ober EA, Clausen H, Wandall HH, Hansen SH, Bennett EP, Frödin M. Genome editing using FACS enrichment of nuclease-expressing cells and indel detection by amplicon analysis. Nat Protoc. 2017;12:581–603. doi: 10.1038/nprot.2016.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arias-Fuenzalida J, Jarazo J, Qing X, Walter J, Gomez-Giro G, Nickels SL, Zaehres H, Schöler HR, Schwamborn JC. FACS-Assisted CRISPR-Cas9 Genome Editing Facilitates Parkinson’s Disease Modeling. Stem Cell Rep. 2017 doi: 10.1016/j.stemcr.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eggenschwiler R, Moslem M, Fráguas MS, Galla M, Papp O, Naujock M, Fonfara I, Gensch I, Wähner A, Beh-Pajooh A, Mussolino C, Tauscher M, Steinemann D, Wegner F, Petri S, Schambach A, Charpentier E, Cathomen T, Cantz T. Improved bi-allelic modification of a transcriptionally silent locus in patient-derived iPSC by Cas9 nickase. Sci Rep. 2016;6 doi: 10.1038/srep38198. srep38198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- 55.Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. 2014;32 doi: 10.1038/nbt.2989. nbt.2989. [DOI] [PubMed] [Google Scholar]
- 56.Dekkers JF, Berkers G, Kruisselbrink E, Vonk A, de Jonge HR, Janssens HM, Bronsveld I, Avande Graaf E, Nieuwenhuis EES, Houwen RHJ, Vleggaar FP, Escher JC, de Rijke YB, Majoor CJ, Heijerman HGM, de Winter-de Groot KM, Clevers H, van der Ent CK, Beekman JM. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci Transl Med. 2016;8:344ra84. doi: 10.1126/scitranslmed.aad8278. [DOI] [PubMed] [Google Scholar]
- 57.Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR– Cas9. Nat Rev Genet. 2015;16 doi: 10.1038/nrg3899. nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Findlay GM, Boyle EA, Hause RJ, Klein JC, Shendure J. Saturation editing of genomic regions by multiplex homology-directed repair. Nature. 2014;513:120–123. doi: 10.1038/nature13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smurnyy Y, Cai M, Wu H, McWhinnie E, Tallarico JA, Yang Y, Feng Y. DNA sequencing and CRISPR-Cas9 gene editing for target validation in mammalian cells. Nat Chem Biol. 2014;10:623–625. doi: 10.1038/nchembio.1550. [DOI] [PubMed] [Google Scholar]
- 60.O. of the Commissioner, Press Announcements - FDA expands approved use of Kalydeco to treat additional mutations of cystic fibrosis. (n.d.). https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm559212.htm (accessed November 4, 2017)
- 61.KALYDECO (ivacaftor) Label. (n.d.). https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203188s019lbl.pdf (accessed November 4, 2017)
- 62.Li H, Beckman KA, Pessino V, Huang B, Weissman JS, Leonetti MD. Design and specificity of long ssDNA donors for CRISPR-based knock-in, bioRxiv. 2017:178905. doi: 10.1101/178905. [DOI] [Google Scholar]