

Abstract
Currently, there is a paucity of information regarding the molecular pathogenesis for many high‐consequence pathogens (HCPs) that pose threats to both national and international public health. In spite of this, investigations of the molecular pathogenesis for many HCPs have been limited to gross pathological changes in animal models or global analysis of gene expression. Further, questions remain regarding the ability of animal models of disease to recapitulate human molecular pathogenesis or act as predictors of therapeutic efficacy. Thus, it is likely that medical countermeasure development for HCPs will rely on identifying therapeutic targets that are uniquely modulated during HCP infection. It is also appreciated that many cellular processes can be regulated independently of changes in transcription or translation through phosphorylation events. Cellular kinases, individually or collectively (the kinome), play critical roles in regulating complex biology, underlie various malignancies, and represent high‐priority drug targets. The growing interest in kinases in both basic and translational research has driven efforts to develop technologies that enable characterization of phosphorylation‐mediated signal transduction. To this end, enhanced technical capabilities at the IRF‐Frederick provide the unique capability for characterizing host responses to HCP insult during the course of infection and identify novel targets for therapeutic intervention.
Keywords: kinome, high‐consequence pathogens, filovirus, orthopoxvirus, kinase, signaling pathways
Kinome analysis at the IRF‐Frederick to identify novel information regarding the molecular pathogenesis of high‐consequence pathogens and novel therapeutic targets.
Introduction
High‐consequence pathogens are a global health concern
Currently, there is a paucity of information regarding the molecular pathogenesis for many high‐consequence pathogens (HCPs) that pose direct threats to public health and security both nationally and internationally. For example, variola virus (VARV), the etiologic agent of smallpox, was responsible for ~ 500 million human fatalities in the 20th century prior to its successful eradication in May 1980 (Mahalingam et al., 2004). Following the cessation of routine vaccinia virus (VACV) vaccination, a significant portion of the global population has been left vulnerable to VARV. This has resulted in significant concerns regarding the potential release of VARV, or another closely related orthopoxvirus, such as monkeypox virus (MPXV), to an increasingly vulnerable population. These fears were realized in the continental US, following the unintentional release of MPXV in 2003 from a shipment of Ghanaian rodents destined for exotic pet trade (Damon, 2011). Concerns have also been raised due to the increasing incidence of MPXV infection within Africa (Rimoin et al., 2010). Hemorrhagic fever viruses, including Ebola virus (EBOV), are also considered global health concerns due to the potential for accidental introduction of these highly lethal viruses from endemic regions or intentional manipulation for bioterrorism purposes. These concerns have been exacerbated by recent reports of a case of Marburg virus (MARV) in a tourist returning from Uganda to the Netherlands (Centers for Disease C & Prevention, 2009; Timen et al., 2009). Moreover, concerns regarding virus spread from predominantly rural to urban areas during recent Ebola virus disease outbreaks have further strengthened concerns that these highly lethal viruses could be unintentionally introduced into densely populated areas (Bhaumik, 2012; Wasswa, 2012). Subsequent investigations have demonstrated that EBOV‐infected pigs have the potential to transmit virus under laboratory conditions in an interspecies (nonhuman primates) and intraspecies manner through airborne and contact transmission, respectively (Kobinger et al., 2011; Weingartl et al., 2012). It has been argued that the increasing advances in our understanding of these pathogens, and the disease processes they mediate, may provoke the development of engineered pathogens with enhanced virulence (Petro et al., 2003; Lindler et al., 2005).
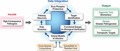
To this end, enhanced technical capabilities for characterizing host and pathogen molecular responses during the course of infection are critically important for understanding microbial pathogenesis and identifying novel targets for therapeutic intervention. The IRF‐Frederick provides the unique capability to combine novel molecular biology approaches with gross pathology analysis, medical imaging, and clinical core functions for the enhanced selection and characterization of animal models of HCPs (Fig. 1).
Figure 1.
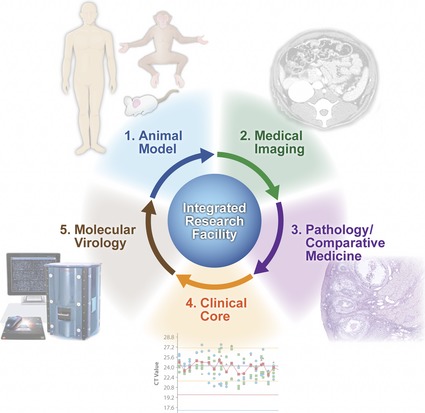
Development and refinement of animal models of human infectious diseases at the IRF‐Frederick. Animal models of infectious disease are characterized and refined through the concerted efforts of the Medical Imaging, Pathology/Comparative Medicine and Clinical core groups at the IRF‐Frederick. Lastly, the molecular virology expertise at the IRF‐Frederick, including systems biology and traditional virology/molecular biology approaches, provides additional capabilities for characterization of high‐consequence pathogens and, ultimately, aid in the refinement of animal infection models.
Mapping the functional host response: kinomics
The molecular pathogenesis for many high consequence of emerging pathogens remains largely uncharacterized, despite the associated health risks and potential economic or critical infrastructure burdens associated with HCP disease outbreaks (whether intentional or unintentional). Indeed, a large proportion of investigations of HCP pathogenesis have been limited to global surveys of host gene expression or virus life cycle from experimental animal infections or clinical samples from infected patients (Gupta et al., 2001; Geisbert et al., 2003; Rubins et al., 2004; Towner et al., 2004; Esteves et al., 2007; Hartman et al., 2008). For example, EBOV particle attachment and entry into human macrophages resulted in the induction of pro‐inflammatory mediators [including interleukin (IL)‐6, IL‐8, and tumor necrosis factor (TNF)‐α] as demonstrated by microarray analysis (Wahl‐Jensen et al., 2011). Kash et al. (2006) have also demonstrated that EBOV infection of human hepatocytes resulted in the suppression of antiviral responses, and Yen et al., (2011)identified discernable differences in the immune responses of nonhuman primates (NHPs) treated with coagulation inhibitors as compared to untreated animals in a lethal EBOV infection model. Connor and colleagues have also demonstrated through transcriptional profiling that Lassa virus infection resulted in the early induction of interferon‐responsive and Toll‐like receptor‐mediated signaling networks in aerosol‐exposed NHPs (Malhotra et al., 2013). Further, analyses of host gene expression during MPXV infection by Alkhalil et al. (2010) and Rubins et al. (2011) demonstrated that infection resulted in the global suppression of host gene expression. A more recent comparison of orthopoxvirus modulation of host gene expression programs during the course of in vitro infection was reported by Bourquain and colleagues and demonstrated that ~ 96% of the cellular transcripts assayed were unresponsive to infection with cowpox virus (CPXV), VACV, or MPXV (Bourquain et al., 2013). In addition, the host responses to CPXV, VACV, and MPXV were significantly different, suggesting that viruses from the same family are likely adapted to a particular mechanism of pathogenesis. This limits the ability to infer host responses for virus families from individual viruses. Further, Rubins et al. (2008) also investigated the gene expression programs of both VACV and MPXV providing the first map of pathogen‐specific responses during the course of infection for orthopoxviruses.
While such studies of global gene expression have been informative, many questions remain regarding the molecular pathogenesis of HCPs. In particular, there is considerable debate as to whether it is most appropriate to assess biologic responses at the transcriptional, translational, or post‐translational levels. Although the proliferation of various high‐throughput methodologies has provided platforms for assessing biologic responses at each of these levels, the associated costs and expertise required for such investigations have in many instances limited their application. There has also been an increasing appreciation for that many cellular processes are regulated independently of changes in transcription or translation through post‐translational modifications with kinase‐mediated protein phosphorylation being the best characterized. Indeed, virtually all signal transduction processes are regulated by kinase‐mediated protein phosphorylation independent of biologic complexity (from prokaryotes to eukaryotes) (Hunter, 2000). In support of this, dysregulated kinase activities have been implicated in a growing number of human malignancies with > 250 kinase genes mapping to disease loci (Knuutila et al., 1998). As a testament to the biologic importance of kinases, there have been over 500 kinases identified in the human genome (≈ 2% of the human genome), and it is estimated that ≈ 30% of the human proteome is modified by kinase‐mediated phosphorylation (Hunter, 1995). Individual kinase activities may also be more reliable predictors of functional cellular changes than changes in gene or protein expression considering their central role in such broad cellular processes as growth and development, metabolism, and innate immunity (Arsenault et al., 2011; Kindrachuk & Napper, 2013). Thus, there is increasing interest in characterizing and quantifying the global activation state of host kinases or the kinome. Additionally, the integration of systems biology approaches, including pathway over‐representation analysis (ORA), provides enhanced capabilities to identify events (i.e. signaling networks; individual kinases) that are critical to disease progression or resolution.
From the perspective of therapeutic design and development, kinases are logical drug targets considering their regulatory role in cell processes and conserved catalytic cleft (Arsenault et al., 2011). As a testament to this, there are currently 25 kinase inhibitors with US Food and Drug Administration (FDA) licensure (encompassing indications that include cancer, Rheumatoid arthritis, myeloid fibrosis and transplant rejection) and a continually increasing number that are entering preclinical trials. In addition, kinases are the most frequently targeted gene class in cancer therapy, second only to the G protein‐coupled receptors as therapeutic targets (Cohen, 2002; Hopkins & Groom, 2002). Kinase inhibitors have been employed as treatments for malignancies as diverse as leukemia and gastrointestinal stromal tumors (imatinib) (Druker et al., 1996, 2001), diabetic retinopathy (ruboxistaurin) (Danis & Sheetz, 2009), atopic dermatitis (safingol) (Eglen & Reisine, 2009), and cerebral ischemia (fasudil) (Yamashita et al., 2007). There is also an increasing impetus to consider the repurposing of FDA‐approved kinase inhibitors for use as anti‐infective therapies. As the associated costs of moving a new drug from bench to bedside are estimated at > $1 billion, the use of approved kinase inhibitors in novel applications is enticing. Further, the National Institutes of Health Center for Advancing Translational Sciences (NCATS) has recently adopted a new strategy aimed at repurposing drugs with pre‐existing FDA approval for treatment of additional malignancies (Allison, 2012). In support of this, Reeves et al. (2005) have demonstrated that imatinib, an Abl‐family kinase inhibitor, promoted survival in VACV‐infected mice and reduced viral dissemination by five orders of magnitude. Although imatinib had a negligible effect on VACV yield, the authors demonstrated that the release of enveloped extracellular virus (EEV) was inhibited by drug treatment in vitro and in vivo. Subsequent studies by the authors demonstrated that imatinib provided protection from lethal VACV infection in mice when delivered prophylactically or therapeutically. Further, Reeves et al. (2011) demonstrated a similar inhibitory effect for imatinib against MPXV and VARV EEV release and viral spread providing evidence for a broad antiorthopoxvirus effect of the drug. Napier et al. (2011) have also demonstrated that these anti‐infective activities are not limited to orthopoxvirus family members as imatinib reduced bacterial loads and granulomatous lesion numbers when added prophylactically or therapeutically in a mouse model of mycobacterial infection. Interestingly, the authors also demonstrated that coadministration of imatinib with either rifampicin or rifabutin acted synergistically to reduce mycobacterial load in vivo.
Concerns remain regarding the application of kinase inhibitors to infectious disease therapies due to the potential immunosuppressive effects following prolonged therapeutic administration. However, this must also be tempered with the appreciation that: (1) kinase inhibitor treatments for HCP infections would likely be short term in nature; and (2) the pathology of various HCP family members has been associated with overactivation of the innate immune system. Thus, kinase inhibitors that both suppress viral replication (directly or indirectly) and reduce the pathological effects associated with overactivation of innate immunity may offer a dual purpose in short term and/or immediate treatment strategies for HCP infections.
High‐throughput peptide arrays for characterizing kinome responses
The increasing interest in characterizing the global roles for kinases and kinase‐mediated signal transduction in human disease has potentiated the development of novel research platforms for these purposes (Jalal et al., 2007). It should also be appreciated that the evolutionary conservation of kinases and their respective substrates provides the opportunity to apply similar methodologies for kinome analysis across animals of multiple animal in vitro and in vivo. Traditional phosphoproteomic technologies have been limited by the confining nature of the technological requirements for such analyses as well as the relative scarcity of phosphorylated proteins within a given protein sample (≈ 1–2% of the total population of an individual protein). In contrast, investigation of the host kinome based on well‐defined and conserved enzymatic phosphorylation events seems a logical alternative (Arsenault et al., 2011). Incorporation of high‐throughput analyses based on kinase‐mediated phosphotransfer events, such as peptide kinome arrays, provides a functional mechanism for characterizing the modulation of host cell signaling networks during disease pathogenesis (Cohen, 2002; Hopkins & Groom, 2002). Kinome arrays utilize the principles of kinase substrate specificity as this is dictated by the residues adjacent (± 4) to the phosphoacceptor site (Kreegipuu et al., 1998). Indeed, the application of synthetic peptides for kinase analysis is supported by reports that kinases recognize and phosphorylate linear peptide targets with comparable V max and K m values as those for the native proteins (Zhu et al., 2000). Thus, the synthesis of short linear peptide sequences followed by their covalent linkage to a solid platform (glass slides) provides an economically viable high‐throughput platform for investigating host kinome responses (Kindrachuk & Napper, 2013). Detailed reviews regarding commonly used strategies for peptide synthesis and covalent linkage can be found elsewhere (Houseman et al., 2002; Jalal et al., 2007). As such, peptide arrays comprising of hundreds to thousands of immobilized, specific peptide targets for kinases have been reported. From the perspective of kinase target selection, publically available databases such as PhosphoSite (http://www.phosphosite.org) or Phospho.ELM (http://phospho.elm.eu.org) provide manually curated, literature‐based phosphorylation sites for a broad selection of proteins.
For typical kinome array analyses, cellular lysates from comparative samples (infected/stimulated vs. uninfected/unstimulated) are applied to the kinome array followed by phosphotransfer from active kinases in the cell supernatants to their corresponding peptide targets (Fig. 2). Although the use of gamma phosphorous‐32 (γ‐32P) for such analyses limited the high‐throughput aspect of kinome analysis and increased concerns regarding radioactive waste removal (in particular within high‐containment laboratory environments), the incorporation of phospho‐specific fluorescent stains, such as the Pro‐Q Diamond Phosphoprotein Stain, has removed these constraints. In addition, postimaging extraction of kinome array data was hindered by the lack of software analysis packages suitable for the extraction of kinome array data. To remedy this, Li et al. (2012) developed the Platform for Integrated Intelligent Kinome Analysis (PIIKA) pipeline (http://saphire.usask.ca/saphire/piika/index.html) for kinome array data analysis. Through PIIKA, a relative degree of activity or phosphorylation (fold change) under different experimental conditions is assigned based on a user‐selected comparative (disease vs. normal; treated vs. untreated; infected vs. uninfected, etc.). Following data extraction from PIIKA, subsequent analysis of these data sets through pathway ORA databases provides a computational method for dissecting biologic information from the kinome array data by identifying over‐represented functional signaling networks or pathways within the kinome data sets. Publically available pathway analysis databases such as InnateDB (http://www.innatedb.com) (Lynn et al., 2008) or the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa & Goto, 2000) provide a mechanism for performing such analyses. Commercially available software, such as the Ingenuity Pathway Analysis software suite, provides alternative mechanisms for deriving biologic data from kinome analyses. We recommend reviews by Khatri et al. (2012) and Ramanan et al. (2012) to the reader for more in‐depth analyses on the subject.
Figure 2.
Overview of kinome peptide array experimental procedure. Host cells/tissues are infected in vitro or in vivo with pathogen of interest (I). Following infection for the desired time course, cells/tissues are isolated (pelleting by centrifugation or homogenized, respectively) followed by cell lysis (II). Cell debris is removed by high‐speed centrifugation, and supernatants are spotted on the kinome arrays (III). Arrays are incubated to allow for phosphotransfer from activated kinases in the supernatants to the specific peptide targets on the arrays followed by washing, staining, and imaging. (IV). Data are extracted from the spot intensities, and fold‐change differences in phosphorylation are derived using PIIKA followed by functional network analysis (ex. InnateDB or Ingenuity Pathway Analysis) (V).
Profiling host kinome responses to monkeypox virus infection
With the cessation of routine VACV immunization following the declaration of global smallpox eradication in 1980, a significant portion of the global population has been left vulnerable to VARV. Thus, concerns have been raised regarding the potential impact of an outbreak of VARV virus in an increasingly vulnerable population. Additionally, the increasing incidence of MPXV (Rimoin et al., 2010) lends further credence to these concerns and highlights the importance for the design and development of novel antiviral therapeutic strategies for orthopoxvirus infections. MPXV is comprised of two distinct clades that are genetically, clinically, and geographically distinct. Central African MPXV has associated case fatality rates of approximately 10% in nonvaccinated individuals, whereas West African MPXV, the virus responsible for the 2003 outbreak in the US, has not caused fatalities (Jezek et al., 1988). Animal infection models in multiple animal species have demonstrated similar differences in virulence between viruses of the different MPXV clades (Sbrana et al., 2007; Hutson et al., 2009, 2010; Osorio et al., 2009; Saijo et al., 2009). However, there has been little information regarding the molecular processes (host or viral) responsible for these virulence differences.
To address this shortcoming, we employed kinome analysis to characterize the differential host responses to viruses of the MPXV clades. Bowick et al. (2007) had previously employed systems kinome analysis in the first investigation comparing differential host responses to virulent and attenuated viruses in their investigation of Pichindé virus pathogenesis. Through a combination of kinome analysis, pathway ORA, and molecular biologic techniques, we demonstrated in human monocytes that West African MPXV up‐regulated growth factor‐ and apoptosis‐mediated host cell responses as compared to Congo Basin MPXV. These results were confirmed by fluorescence‐activated cell sorting analysis as West African MPXV infection resulted in a significant increase in apoptosis in human monocytes as compared to Congo Basin MPXV. We also identified a subset of kinases that were differentially modulated by the two MPXV clades, including Akt and p53. Chemical inhibition of Akt phosphorylation significantly reduced Congo Basin MPXV virus titers, whereas West African MPXV virus yield was not affected. Although West African and Congo Basin MPXV proteins share 99.4 % amino acid sequence similarity, the differences in sequence are localized to proteins associated with modification of host responses (Likos et al., 2005) and are therefore likely responsible for the differences seen in our kinome results.
Future perspectives: characterizing host responses to infection with species‐specific kinome analysis
For many infectious diseases, and in particular those mediated by HCPs, a large portion of our knowledge of the disease process has relied on gross pathology and molecular pathogenesis data extracted from animal models of disease. In this regard, animal models have been employed extensively for investigations of HCP pathogenesis and as well as for the identification of novel therapeutics and evaluation of their associated efficacies/toxicities. Murine models have been employed extensively for examining HCP pathogenesis due to their relative cost, limited genetic background (and thus increased experimental reproducibility), and the availability of analytical reagents. Recently, Seok et al. (2013) reported that the genomic responses of laboratory mice in acute inflammatory disease models correlated poorly with those of human patients. Although the authors recognized that these prior studies were likely impeded by inadequate study designs and data sets, it is likely that many investigations have also been hindered by an assumed conservation of functional host responses between humans and nonhuman animal species. Moreover, there has been a relative paucity of therapeutic candidates that have translated from animal models into approved use for humans. Hackam & Redelmeier (2006) recently noted that only one‐third of these studies translated into randomized clinical trials, and of these, only one‐tenth of the therapeutic candidates acquired FDA approval. Further, Van der Worp et al. (2010) have postulated that the actual success rates were likely much lower as these statistics were derived based on publications from high‐impact journals.
Thus, a likely practical solution to this is the design of species‐specific molecular platforms for: (1) annotating physiological processes in a species‐specific fashion; and (2) guiding the selection of appropriate animal hosts for disease models on the basis of the conservation of molecular responses to those of humans. Further, in reference to the FDA Animal Rule (Services, U.D.o.H.a.H., F.a.D. Administration, 2002), the incorporation of conserved molecular responses to that of humans for guiding animal model selection would presumably increase confidence in the study of therapeutics for diseases in which human clinical studies are not ethical. Thus, the design of species‐specific kinome arrays seems a logical approach in particular for species for which reagent availability is scarce. Here, Napper and colleagues have demonstrated the ability to create species‐specific kinome arrays for diverse organisms including humans, cows, and horses (Arsenault et al., 2011; Kindrachuk & Napper, 2013). Although much of the early design of species‐specific kinome arrays was guided through manual sequence homology searches using BLAST, improved bioinformatics applications have provided significant improvements in the prediction of phosphorylation sites for nontypical species. In particular, this manual approach to peptide target design was time‐consuming, subjective, and limited to homologues of human phosphorylation sites (Trost et al., 2013). Trost et al. (2013) have recently reported the creation of dapple (http://saphire.usask.ca), a software pipeline for the homology‐based prediction of phosphorylation sites. dapple provides an automated interface that circumvents the limitations of such manual approaches to peptide target prediction while also providing a mechanism for increasing the accuracy of the predictions and as well the diversity of the species which can be examined (Fig. 3). Through collaboration, our laboratory has designed kinome peptide arrays for multiple animal species of interest in the investigation of high‐containment pathogens. The ability to incorporate routine kinome analysis, whether it is from the perspective of the host or pathogen, provides a mechanism for identification of novel biomarkers of disease, characterize molecular pathogenesis, and identify novel targets for therapeutic intervention (Fig. 4).
Figure 3.
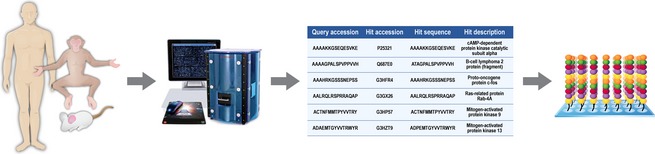
Designing species‐specific kinome peptide arrays for characterizing host responses to infection. Kinase phosphorylation targets within host proteins are identified based upon homology with previously characterized phosphorylation sites (human, mouse, etc.) through bioinformatic analyses. Peptides representing phosphorylation sites from the species of interest are synthesized and covalently linked to microscope slides to produce kinome peptide arrays that encompass hundreds to thousands of kinase targets.
Figure 4.
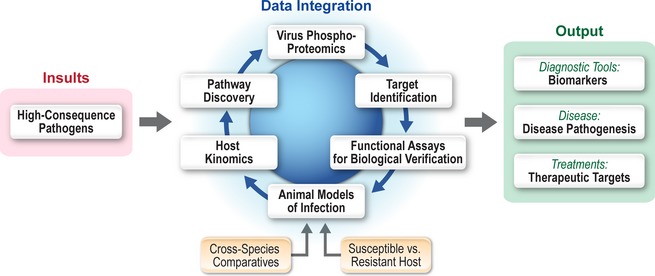
Model of outcome‐based molecular characterization of high‐consequence pathogens. For many high‐consequence pathogens, there is a paucity of information regarding molecular disease mechanisms. Thus, molecular approaches for characterization of these diseases at the IRF‐Frederick focus on molecular processes within the host and pathogen. This model provides output data in regard to the molecular characterization of infectious disease pathogenesis, identification of biomarkers associated with disease, and novel insight into potential therapeutic intervention strategies.
Kinomics at the IRF‐Frederick
The IRF‐Frederick is the first maximum containment facility to integrate common medical imaging modalities with comparative medicine/pathology, clinical core services, and molecular diagnostics within a maximum containment (BSL‐4) environment. Within the IRF‐Frederick, the strategic mission is to manage, coordinate, and facilitate the research of emerging infectious diseases for the development of vaccines, countermeasures, and the improved medical outcomes of patients. In this regard, kinomics research at the IRF‐Frederick will provide researchers with the ability to temporally characterize the functional cellular response of host species to high‐consequence pathogens. Further, based on the unique blend of medical imaging capabilities and maximum containment laboratories, the IRF‐Frederick provides an unprecedented capability to verify the biologic relevance from such molecular analyses both in vitro and in vivo. The integration of the various medical imaging modalities to evaluate the progression of disease in a single animal with kinome analysis provides an opportunity to verify the biologic relevance of the molecular responses through the course of disease or therapeutic intervention and to facilitate the development of medical countermeasures.
In addition, as investigations of HCP molecular pathogenesis have been limited by reagent availability for nonhuman animal species, the design and development of species‐specific kinome analysis at the IRF‐Frederick will provide a unique opportunity for characterizing these molecular responses and identifying novel targets for therapeutic intervention. Taken together, it is envisioned that kinomics will provide the opportunity for informed selection of appropriate animal models that best mimic human molecular disease and as well the identities of conserved biomarkers that are associated with increased susceptibility or resistance to infection.
Conclusions
Given the emerging importance of cellular kinases as causes, indicators, and therapeutic targets of disease, it seems certain that there will be ongoing effort to develop tools and strategies which enable characterization of the kinome in a high‐throughput, cost‐effective fashion. In our opinion, peptide arrays represent the most practical and robust approach to date for achieving these goals.
Although outbreaks of HCPs are often sporadic, they remain a serious global health concern. These concerns are exacerbated by both a knowledge gap regarding HCP molecular pathogenesis and a scarcity of therapeutic options beyond supportive care. This is particularly troubling considering the recent outbreaks of Sudan virus and Bundibugyo virus in Uganda and the DRC (Albarino et al., 2013), and Sin Nombre virus in California (Centers for Disease C & Prevention, 2012), and the recent emergence of Middle East respiratory syndrome coronavirus (De Groot et al., 2013). Thus, it is inherently important to characterize molecular pathogenesis and identify potential therapeutic targets or strategies for these emerging infectious diseases. Kinome analysis with peptide arrays provides a high‐throughput mechanism for investigating HCP pathogenesis (Arsenault et al., 2011). In addition, the introduction of systems approaches to specifically mine large data sets provides the opportunity to gain further biologic perspective from kinome data (Li et al., 2012).
Due to the complexity of host immune responses, particularly during infection and disease progression, it is prudent to investigate temporal responses at biologic levels that are closest to functional phenotypes. Kinome analysis satisfies this goal by monitoring the activation state of functional signaling networks. Further, it may also provide opportunity to identify unique molecular signatures or biomarkers in the way of signaling networks or individual kinases that are broadly conserved across various pathogens. This is of particular importance from the perspective of therapeutic treatment strategies, as the identification of conserved host targets modulated by multiple HCPs would provide focused targets for the design and development of broad range antiviral therapeutics.
Taken together, kinome analysis at the IRF‐Frederick provides a unique opportunity for comparing the global, functional host response to various HCPs. In particular, the kinomics research program at the IRF‐Frederick provides a unique platform for investigating the delicate molecular interplay between the host and pathogen during infection within a BSL‐4 environment.
Disclaimer
The content of this publication does not necessarily reflect the views or policies of the US Department of Health and Human Services or of the institutions and companies affiliated with the authors. JHK performed this work as an employee of Tunnell Governments Services, Inc., a subcontractor to Battelle Memorial Institute; and JK and JW performed this work as employees of Battelle Memorial Institute, all under its prime contract with NIAID, under Contract No. HHSN272200700016I.
This MiniReview describes the novel BSL‐4/ABSL4 imaging facility available at Ft. Detrick, MD.
References
- Albarino CG, Shoemaker T & Khristova ML et al (2013) Genomic analysis of filoviruses associated with four viral hemorrhagic fever outbreaks in Uganda and the Democratic Republic of the Congo in 2012. Virology 442: 97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhalil A, Hammamieh R, Hardick J, Ichou MA, Jett M & Ibrahim S (2010) Gene expression profiling of monkeypox virus‐infected cells reveals novel interfaces for host‐virus interactions. Virol J 7: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison M (2012) NCATS launches drug repurposing program. Nat Biotechnol 30: 571–572. [DOI] [PubMed] [Google Scholar]
- Arsenault R, Griebel P & Napper S (2011) Peptide arrays for kinome analysis: new opportunities and remaining challenges. Proteomics 11: 4595–4609. [DOI] [PubMed] [Google Scholar]
- Bhaumik S (2012) Twin Ebola outbreaks in Africa: Uganda and Democratic Republic of Congo affected. Natl Med J India 25: 317. [PubMed] [Google Scholar]
- Bourquain D, Dabrowski PW & Nitsche A (2013) Comparison of host cell gene expression in cowpox, monkeypox or vaccinia virus‐infected cells reveals virus‐specific regulation of immune response genes. Virol J 10: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowick GC, Fennewald SM & Scott EP et al (2007) Identification of differentially activated cell‐signaling networks associated with pichinde virus pathogenesis by using systems kinomics. J Virol 81: 1923–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease C & Prevention (2009) Imported case of Marburg hemorrhagic fever ‐ Colorado, 2008. MMWR Morb Mortal Wkly Rep 58: 1377–1381. [PubMed] [Google Scholar]
- Centers for Disease C & Prevention (2012) Hantavirus pulmonary syndrome in visitors to a national park–Yosemite Valley, California, 2012. MMWR Morb Mortal Wkly Rep 61: 952. [PubMed] [Google Scholar]
- Cohen P (2002) Protein kinases–the major drug targets of the twenty‐first century? Nat Rev Drug Discov 1: 309–315. [DOI] [PubMed] [Google Scholar]
- Damon IK (2011) Status of human monkeypox: clinical disease, epidemiology and research. Vaccine 29(suppl 4): D54–D59. [DOI] [PubMed] [Google Scholar]
- Danis RP & Sheetz MJ (2009) Ruboxistaurin: PKC‐beta inhibition for complications of diabetes. Expert Opin Pharmacother 10: 2913–2925. [DOI] [PubMed] [Google Scholar]
- De Groot RJ, Baker SC & Baric RS et al (2013) Middle East respiratory syndrome coronavirus (MERS‐CoV): announcement of the Coronavirus Study Group. J Virol 87: 7790–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Tamura S & Buchdunger E et al (1996) Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr‐Abl positive cells. Nat Med 2: 561–566. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M & Resta DJ et al (2001) Efficacy and safety of a specific inhibitor of the BCR‐ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 344: 1031–1037. [DOI] [PubMed] [Google Scholar]
- Eglen RM & Reisine T (2009) The current status of drug discovery against the human kinome. Assay Drug Dev Technol 7: 22–43. [DOI] [PubMed] [Google Scholar]
- Esteves GH, Simoes AC, Souza E, Dias RA, Ospina R & Venancio TM (2007) New insights about host response to smallpox using microarray data. BMC Syst Biol 1: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert TW, Young HA, Jahrling PB, Davis KJ, Larsen T, Kagan E & Hensley LE (2003) Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus‐induced cytolysis of endothelial cells. Am J Pathol 163: 2371–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Mahanty S, Ahmed R & Rollin PE (2001) Monocyte‐derived human macrophages and peripheral blood mononuclear cells infected with ebola virus secrete MIP‐1alpha and TNF‐alpha and inhibit poly‐IC‐induced IFN‐alpha in vitro . Virology 284: 20–25. [DOI] [PubMed] [Google Scholar]
- Hackam DG & Redelmeier DA (2006) Translation of research evidence from animals to humans. JAMA 296: 1731–1732. [DOI] [PubMed] [Google Scholar]
- Hartman AL, Ling L, Nichol ST & Hibberd ML (2008) Whole‐genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J Virol 82: 5348–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AL & Groom CR (2002) The druggable genome. Nat Rev Drug Discov 1: 727–730. [DOI] [PubMed] [Google Scholar]
- Houseman BT, Huh JH, Kron SJ & Mrksich M (2002) Peptide chips for the quantitative evaluation of protein kinase activity. Nat Biotechnol 20: 270–274. [DOI] [PubMed] [Google Scholar]
- Hunter T (1995) Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80: 225–236. [DOI] [PubMed] [Google Scholar]
- Hunter T (2000) Signaling–2000 and beyond. Cell 100: 113–127. [DOI] [PubMed] [Google Scholar]
- Hutson CL, Olson VA & Carroll DS et al (2009) A prairie dog animal model of systemic orthopoxvirus disease using West African and Congo Basin strains of monkeypox virus. J Gen Virol 90: 323–333. [DOI] [PubMed] [Google Scholar]
- Hutson CL, Carroll DS & Self J et al (2010) Dosage comparison of Congo Basin and West African strains of monkeypox virus using a prairie dog animal model of systemic orthopoxvirus disease. Virology 402: 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalal S, Kindrachuk J & Napper S (2007) Phosphoproteome and kinome analysis: unique perspectives on the same problem. Curr Anal Chem 3: 1–15. [Google Scholar]
- Jezek Z, Grab B, Paluku KM & Szczeniowski MV (1988) Human monkeypox: disease pattern, incidence and attack rates in a rural area of northern Zaire. Trop Geogr Med 40: 73–83. [PubMed] [Google Scholar]
- Kanehisa M & Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash JC, Muhlberger E & Carter V et al (2006) Global suppression of the host antiviral response by Ebola‐ and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J Virol 80: 3009–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, Sirota M & Butte AJ (2012) Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol 8: e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindrachuk J & Napper S (2013) Sample preparation and profiling: probing the kinome for biomarkers and therapeutic targets: peptide arrays for global phosphorylation‐mediated signal transduction Comprehensive Biomarker Discovery and Validation for Clinical Application (Horvatovich P. & Bischoff R, eds), pp. 162–195. Royal Society of Chemistry, Cambridge, UK. [Google Scholar]
- Knuutila S, Bjorkqvist AM & Autio K et al (1998) DNA copy number amplifications in human neoplasms: review of comparative genomic hybridization studies. Am J Pathol 152: 1107–1123. [PMC free article] [PubMed] [Google Scholar]
- Kobinger GP, Leung A & Neufeld J et al (2011) Replication, pathogenicity, shedding, and transmission of Zaire ebolavirus in pigs. J Infect Dis 204: 200–208. [DOI] [PubMed] [Google Scholar]
- Kreegipuu A, Blom N, Brunak S & Jarv J (1998) Statistical analysis of protein kinase specificity determinants. FEBS Lett 430: 45–50. [DOI] [PubMed] [Google Scholar]
- Li Y, Arsenault RJ, Trost B, Slind J, Griebel PJ, Napper S & Kusalik A (2012) A systematic approach for analysis of peptide array kinome data. Sci Signal 5: pl2. [DOI] [PubMed] [Google Scholar]
- Likos AM, Sammons SA & Olson VA et al (2005) A tale of two clades: monkeypox viruses. J Gen Virol 86: 2661–2672. [DOI] [PubMed] [Google Scholar]
- Lindler L, Choffnes E & Korch G (2005) Definition and overview of emerging threats Biological Weapons Defense: Infectious Disease and Counterbioterrorism (Lindler L, Lebeda F. & Korch G, eds), pp. 351–360. Humana Press, New Jersey. [Google Scholar]
- Lynn DJ, Winsor GL & Chan C et al (2008) InnateDB: facilitating systems‐level analyses of the mammalian innate immune response. Mol Syst Biol 4: 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam S, Damon IK & Lidbury BA (2004) 25 years since the eradication of smallpox: why poxvirus research is still relevant. Trends Immunol 25: 636–639. [DOI] [PubMed] [Google Scholar]
- Malhotra S, Yen JY & Honko AN et al (2013) Transcriptional profiling of the circulating immune response to lassa virus in an aerosol model of exposure. PLoS Negl Trop Dis 7: e2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napier RJ, Rafi W & Cheruvu M et al (2011) Imatinib‐sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe 10: 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio JE, Iams KP, Meteyer CU & Rocke TE (2009) Comparison of monkeypox viruses pathogenesis in mice by in vivo imaging. PLoS ONE 4: e6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petro JB, Plasse TR & McNulty JA (2003) Biotechnology: impact on biological warfare and biodefense. Biosecur Bioterror 1: 161–168. [DOI] [PubMed] [Google Scholar]
- Ramanan VK, Shen L, Moore JH & Saykin AJ (2012) Pathway analysis of genomic data: concepts, methods, and prospects for future development. Trends Genet 28: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves PM, Bommarius B & Lebeis S et al (2005) Disabling poxvirus pathogenesis by inhibition of Abl‐family tyrosine kinases. Nat Med 11: 731–739. [DOI] [PubMed] [Google Scholar]
- Reeves PM, Smith SK, Olson VA, Thorne SH, Bornmann W, Damon IK & Kalman D (2011) Variola and monkeypox viruses utilize conserved mechanisms of virion motility and release that depend on abl and SRC family tyrosine kinases. J Virol 85: 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimoin AW, Mulembakani PM & Johnston SC et al (2010) Major increase in human monkeypox incidence 30 years after smallpox vaccination campaigns cease in the Democratic Republic of Congo. P Natl Acad Sci USA 107: 16262–16267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubins KH, Hensley LE & Jahrling PB et al (2004) The host response to smallpox: analysis of the gene expression program in peripheral blood cells in a nonhuman primate model. P Natl Acad Sci USA 101: 15190–15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubins KH, Hensley LE, Bell GW, Wang C, Lefkowitz EJ, Brown PO & Relman DA (2008) Comparative analysis of viral gene expression programs during poxvirus infection: a transcriptional map of the vaccinia and monkeypox genomes. PLoS ONE 3: e2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubins KH, Hensley LE, Relman DA & Brown PO (2011) Stunned silence: gene expression programs in human cells infected with monkeypox or vaccinia virus. PLoS ONE 6: e15615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo M, Ami Y & Suzaki Y et al (2009) Virulence and pathophysiology of the Congo Basin and West African strains of monkeypox virus in non‐human primates. J Gen Virol 90: 2266–2271. [DOI] [PubMed] [Google Scholar]
- Sbrana E, Xiao SY, Newman PC & Tesh RB (2007) Comparative pathology of North American and central African strains of monkeypox virus in a ground squirrel model of the disease. Am J Trop Med Hyg 76: 155–164. [PubMed] [Google Scholar]
- Seok J, Warren HS & Cuenca AG et al (2013) Genomic responses in mouse models poorly mimic human inflammatory diseases. P Natl Acad Sci USA 110: 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Services, U.D.o.H.a.H., F.a.D. Administration (ed) (2002) Federal Register. 37988–37998.
- Timen A, Koopmans MP & Vossen AC et al (2009) Response to imported case of Marburg hemorrhagic fever, the Netherland. Emerg Infect Dis 15: 1171–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towner JS, Rollin PE & Bausch DG et al (2004) Rapid diagnosis of Ebola hemorrhagic fever by reverse transcription‐PCR in an outbreak setting and assessment of patient viral load as a predictor of outcome. J Virol 78: 4330–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B, Arsenault R, Griebel P, Napper S & Kusalik A (2013) dapple: a pipeline for the homology‐based prediction of phosphorylation sites. Bioinformatics 29: 1693–1695. [DOI] [PubMed] [Google Scholar]
- Van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O'Collins V & Macleod MR (2010) Can animal models of disease reliably inform human studies? PLoS Med 7: e1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl‐Jensen V, Kurz S & Feldmann F et al (2011) Ebola virion attachment and entry into human macrophages profoundly effects early cellular gene expression. PLoS Negl Trop Dis 5: e1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasswa H (2012) Uganda gears up to contain Ebola epidemic as fears of spread cause panic. BMJ 345: e5210. [DOI] [PubMed] [Google Scholar]
- Weingartl HM, Embury‐Hyatt C, Nfon C, Leung A, Smith G & Kobinger G (2012) Transmission of Ebola virus from pigs to non‐human primates. Sci Rep 2: 811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita K, Kotani Y & Nakajima Y et al (2007) Fasudil, a Rho kinase (ROCK) inhibitor, protects against ischemic neuronal damage in vitro and in vivo by acting directly on neurons. Brain Res 1154: 215–224. [DOI] [PubMed] [Google Scholar]
- Yen JY, Garamszegi S & Geisbert JB et al (2011) Therapeutics of Ebola hemorrhagic fever: whole‐genome transcriptional analysis of successful disease mitigation. J Infect Dis 204(suppl 3): S1043–S1052. [DOI] [PubMed] [Google Scholar]
- Zhu H, Klemic JF & Chang S et al (2000) Analysis of yeast protein kinases using protein chips. Nat Genet 26: 283–289. [DOI] [PubMed] [Google Scholar]