

Abstract
Chromosomal instability (CIN) is a hallmark of human cancer and it is associated with poor prognosis, metastasis, and therapeutic resistance. CIN results from errors in chromosome segregation during mitosis leading to structural and numerical chromosomal abnormalities. In addition to generating genomic heterogeneity that acts as a substrate for natural selection, CIN promotes inflammatory signaling by introducing double-stranded DNA into the cytosol, engaging the cGAS-STING anti-viral pathway. These multipronged effects distinguish CIN as a central driver of tumor evolution and as a genomic source for the crosstalk between the tumor and its microenvironment in the course of immune editing and evasion.
The link between chromosomal abnormalities and cancer was first proposed by the German botanist, Theodor Boveri, over a hundred years ago (Boveri, 1914). By following cell division in the sea urchin egg, he occasionally observed aberrant mitoses leading to abnormalities in chromosome numbers in the daughter cells, a state otherwise known as aneuploidy. Since then, mounting evidence has linked numerical and structural chromosomal abnormalities to aggressive tumor behavior.
Aneuploidy versus CIN
Prior to engaging in a discussion of chromosomal instability (CIN) and cancer, an important distinction should be drawn between aneuploidy and CIN. The former denotes a state of abnormal – or non-euploid – chromosome number whereas the latter refers to ongoing chromosome segregation errors throughout consecutive cell divisions (Lengauer et al., 1998). The two often co-occur in human cancer; tumors with high levels of chromosome copy number abnormalities also exhibit evidence of sustained chromosome missegregation. Yet, stable aneuploidies can exist in the absence of CIN as is the case with some hematologic malignancies (Paulsson and Johansson, 2007). This can be due to very low chromosome missegregation rates or strong selective pressure for chromosome copy number (or karyotype) combinations.
While aneuploidy can be readily assessed using widely available experimental techniques such as bulk DNA sequencing, fluorescence in situ hybridization (FISH), or conventional karyotyping, CIN can only be indirectly inferred using these methods. Experimental evaluation of CIN must identify the ongoing rate of chromosome missegregation. This can be achieved by observing the frequency of cells undergoing anaphase, clonal assays, single-cell sequencing with phylogenetic tree reconstruction, or multi-region tumor sequencing taking into account allele-specific copy number information (Bakhoum et al., 2011; Bakker et al., 2016; Jamal-Hanjani et al., 2017; Lengauer et al., 1997).
CIN and Cancer: a complex relationship
It is estimated that 60-80% of human tumors exhibit chromosomal abnormalities suggestive of CIN (Ame et al., 2008; Carter et al., 2012). CIN positively correlates with tumor stage, and is enriched in relapsed as well as metastatic tumor specimens (Bakhoum et al., 2018; Goh et al., 2017; Turajlic et al., 2018). Furthermore, complex aneuploidies and polyploidy resulting from whole genome doubling are features of tumor types with a predilection for metastasis, treatment resistance, and decreased overall survival, such as triple-negative breast cancer, pancreatic and hepatobiliary cancers, lung cancer, anaplastic thyroid cancer, castrate resistant prostate cancer, poorly differentiated sarcomas, gynecologic tumors with serous histologies, glioblastoma, esophagogastric cancers, and microsatellite stable colorectal cancers (Bielski et al., 2018; Carter et al., 2012; Taylor et al., 2018).
The prevalence of CIN reflects the large number of cancer-relevant pathways whose deregulation has been implicated to influence mitotic chromosome segregation (Thompson et al., 2010a). Oncogenic signaling, premitotic replication stress, and defects in centrosome replication, sister chromatid cohesion, spindle assembly checkpoint signaling, or microtubule attachments to chromosomes have all been shown to induce CIN. Furthermore, large panoply of anti-neoplastic therapies used in the upfront treatment or metastatic settings can perturb the fidelity of chromosome segregation during anaphase (Bakhoum et al., 2014; Lee et al., 2016). Additional prevalent sources of chromosome copy number alterations in cancer include homologous recombination defects, telomere dysfunction leading to breakage-fusion-bridge cycle (Maciejowski et al., 2015; Yates et al., 2012). A significant proportion of the defects that induce CIN in cancer converge onto a mitotic phenotype whereby cells undergoing anaphase exhibit chromosomes that lag in the spindle midzone (Cimini et al., 2001). These chromosomes are referred to as lagging chromosomes, and they are the result of their erroneous attachments of microtubules at the kinetochores (Figure 1). Other signs of chromosome missegregation during mitosis include chromatin bridges, ultrafine DNA bridges, and acentric fragments that fail to establish direct attachments to spindle microtubules. In addition to aneuploidy and large scale structural chromosomal alterations, chromosome missegregation can lead to focal yet highly complex rearrangements known as chromothripsis, the formation of double-minute chromosomes which are amenable to massive copy-number amplifications, as well as extrachromosomal DNA (Ly et al., 2017; Stephens et al., 2011; Turner et al., 2017; Zhang et al., 2015) (Figure 1).
Figure 1. Chromosomal instability in cancer.
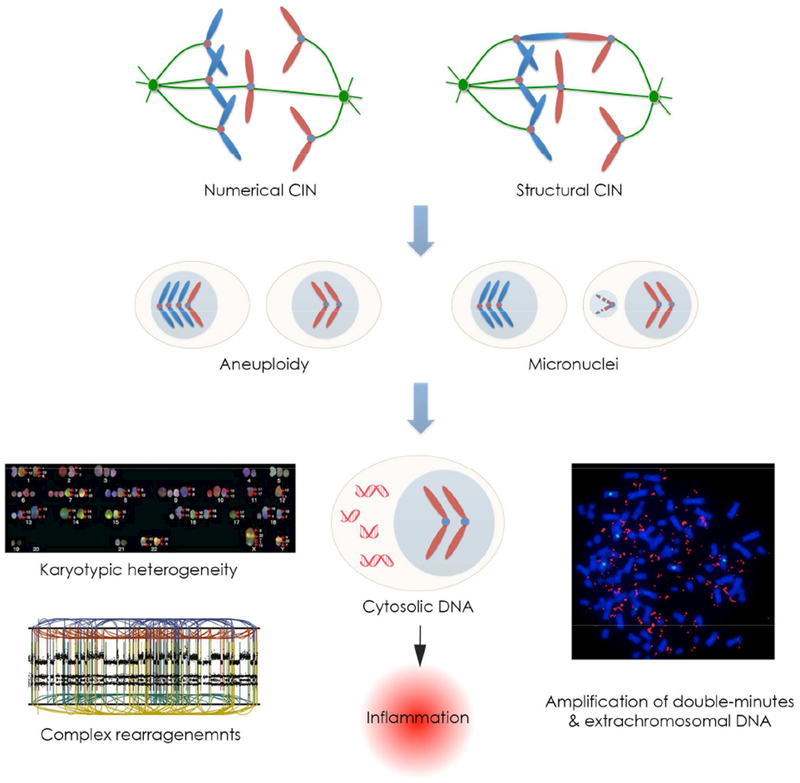
Multiple defects found in cancer can lead to both numerical and structural chromosomal abnormalities, which manifest as errors in chromosome segregation during mitosis. These errors lead to aneuploidy, karyotype heterogeneity, as well as the formation of micronuclei. Chromosomes enclosed in micronuclei are subjected to increased DNA damage and can become exposed to the cytoplasm after micronuclear envelope rupture. This mechanism has been proposed to promote massive structural chromosomal rearrangements, known as chromothripsis, the formation of extrachromosomal DNA as well as double minutes, which can be subjected to strong selective pressures and be present in hundreds of copies per cell. Furthermore, the presence of cytoplasmic double-stranded DNA (dsDNA) promotes inflammatory signaling through the activation of the cytosolic dsDNA sensing, cGAS-STING pathway.
Despite the pervasiveness of CIN in human cancer, its role in tumor evolution is complex and seemingly paradoxical (Birkbak et al., 2011). On one hand, CIN and complex aneuploidies correlate with resistance to antineoplastic agents, such as taxol, both in tumor-derived cell lines as well as in clinical settings (Bakhoum et al., 2011; Carter et al., 2006; Swanton et al., 2009). Metastatic lesions and circulating tumor cells exhibit evidence for CIN and increased chromosome copy number heterogeneity (Bakhoum et al., 2018; Gutenberg et al., 2010; Pailler et al., 2015; Warth et al., 2009). The presence of chromosome missegregation in primary tumors is associated with a higher likelihood of distant spread, relapse, and oncogene-independence in breast and lung cancers as well as Diffuse Large B-cell Lymphoma (DLBCL) (Bakhoum et al., 2011; 2018; Sotillo et al., 2010).
Conversely, excessive levels of CIN forebode enhanced sensitivity to cytotoxic therapies such as cisplatin and 5-Fluorouracil (5-FU) in ovarian, rectal, and breast cancers (Jamal-Hanjani et al., 2015; Roylance et al., 2011; Swanton et al., 2009; Zaki et al., 2014). Inducing whole-chromosome missegregation sensitizes transplanted human glioblastoma tumors to radiation treatment (Bakhoum et al., 2015).
This seemingly paradoxical relationship stems from the complexity of the phenotypes imparted by CIN not only on cancer cells but also on the tumor microenvironment. CIN generates chromosome copy number heterogeneity that serves as a substrate for natural selection enhancing tumor fitness and facilitating immune evasion, drug resistance, and metastasis (Chen et al., 2012; Davoli et al., 2017; Laughney et al., 2015; Mueller et al., 2018; Notta et al., 2016; Pavelka et al., 2010; Potapova et al., 2013). In parallel, chromosome segregation errors impart a number of cellular burdens, including loss of genetic material, activation of DNA damage signaling as well as proteotoxic stress all of which can impact viability, especially in normal or diploid cells (Santaguida et al., 2017; Sheltzer et al., 2017; Torres et al., 2007) (Figure 2).
Figure 2. The multifaceted role of chromosomal instability (CIN) in cancer.
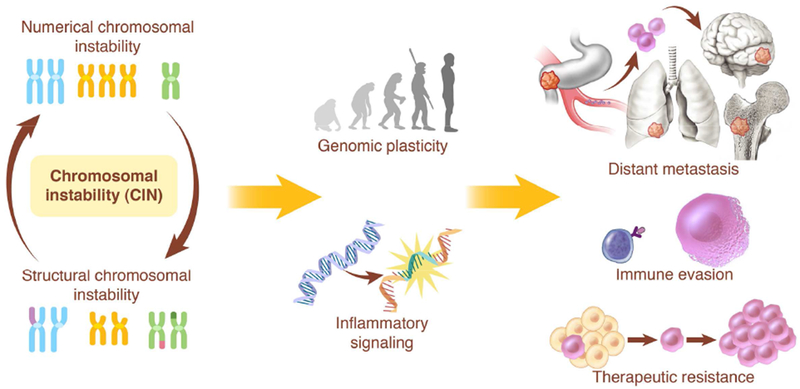
Chromosome missegregation can influence tumor evolution through the generation of genomic copy number heterogeneity that serves as the substrate for natural selection. In parallel, ongoing segregation errors can impart a variety of cellular stresses including transcriptional changes stemming from the leakage of genomic double-stranded DNA into the cytoplasm leading to inflammatory signaling. The multi-pronged effects of chromosome segregation enables genomic plasticity and supports tumor evolution, facilitating processes such as metastasis, immune evasion, and therapeutic resistance. In order to fully understand the consequences of CIN, one must first appreciate the diverse effects of chromosome missegregation on the tumor and its microenvironment.
More recently, an intriguing aspect of CIN has been identified whereby chromosome segregation errors as well as replication stress can activate innate immune signaling through the introduction of genomic double-stranded DNA (dsDNA) into the cytosol and engagement of the cGAS-STING cytosolic dsDNA sensing antiviral pathway (Bakhoum et al., 2018; Coquel et al., 2018; Harding et al., 2017; Mackenzie et al., 2017). This new dimension adds to the complexity of understanding the role of CIN in tumor evolution and reveals that the consequences of CIN are not only tumor-cell autonomous but also involve their crosstalk with the immune microenvironment. Therefore, to better understand the role of CIN in tumor evolution, it is critical to deconstruct the respective effects of genomic copy number heterogeneity from those of the transcriptional responses to cytosolic DNA; while they often act in concert to promote therapeutic resistance, immune evasion and metastasis, in some contexts they can act in opposite directions (Figure 2).
CIN as a genomic source for innate immune activation
The link between karyotypic abnormalities and immune activation was first suggested Senovilla et al. upon observing T-lymphocyte-mediated delay in tumor growth in colorectal CT26 tumors that were pharmacologically manipulated to induce aneuploidy (Senovilla et al., 2012). In this model enhanced immunogenicity was attributed to increased extracellular calreticulin exposure by cancer cells likely resulting from endoplasmic reticulum stress.
Subsequently, a number of groups have identified a direct mechanism by which chromosome segregation errors can lead to the activation of immune signaling pathways; when chromosomes lag during anaphase, they often form structures termed micronuclei (Crasta et al., 2012; Kato and Sandberg, 1968). Envelopes surrounding these micronuclei are rupture-prone leading to exposure of their genomic content to the cytoplasm (Hatch et al., 2013) (Figure 3). When double-stranded DNA (dsDNA) from micronuclei comes into contact with the cytoplasm during interphase, this leads to the activation of the cGAS-STING (cyclic GMP-AMP synthase-Stimulator of Interferon Genes) pathway (Bakhoum et al., 2018; Harding et al., 2017; Mackenzie et al., 2017; Yang et al., 2017). Cytosolic dsDNA is first sensed by cGAS leading to the catalytic production of the cyclic dinucleotide, cGAMP, which in turn stabilizes STING, promoting its perinuclear localization at the ER-membrane (Cai et al., 2014; Ishikawa and Barber, 2008; Ishikawa et al., 2009; Sun et al., 2013) (Figure 3A). STING mediates the transcriptional activation of inflammatory pathways including type I interferon, the senescence associated-secretory phenotype (SASP), among others. Using fluorescence-activated cell sorting (FACS) followed by single-cell RNA sequencing of RNAseH2V-mouse embryonic fibroblasts (MEFs), Mackenzie et al. have elegantly demonstrated that pro-inflammatory Interferon-stimulated genes (ISGs), such as CCL5 and CXCL10 are induced exclusively in cells containing micronuclei (Mackenzie et al., 2017). Subsequently, chromosome-tracking experiments have demonstrated that the same chromosomes that undergo missegregation are the ones that ultimately end up as fragmented cytoplasmic chromatin (Bakhoum et al., 2018), providing a direct link between chromosome missegregation and innate immune signaling (Figure 3A).
Figure 3. CIN activates cGAS-STING signaling.
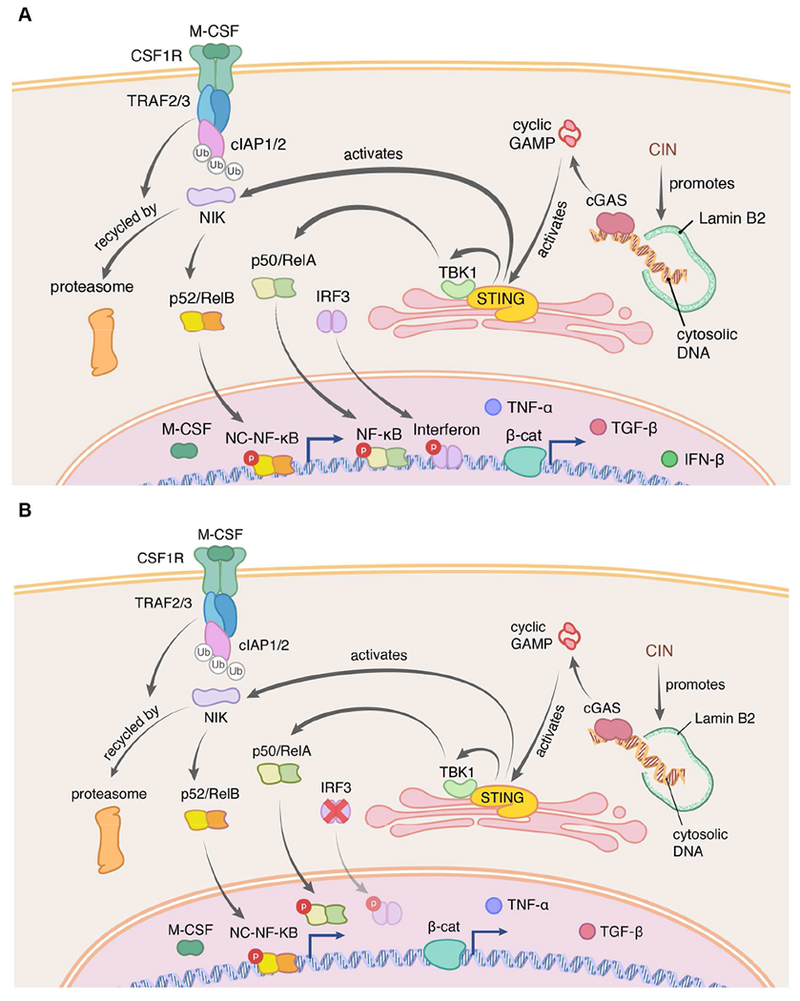
Chromosome segregation errors lead to the formation of micronuclei, which often rupture in S-phase exposing double-stranded DNA (dsDNA) to the cytosol. The presence of cytosolic dsDNA activates the anti-viral cGAS-STING pathway which in normal cells promotes type I interferon production (A). Chromosomally unstable cancer cells however largely suppress type I interferon signaling through multiple mechanisms, yet they maintain the ability to exhibit alternative inflammatory STING-dependent signaling such as NF-κB (B). Chronic NF-κB activation has been shown to mediate the senescence-associated secretory phenotype (SASP) as well as cellular migration and metastasis.
CIN as a trigger of tumor immune editing
Under normal conditions, the cGAS-STING pathway functions as an innate cellular defense mechanism against viral infections. Once engaged, STING activates TANK-binding kinase 1 (TBK1) promoting the phosphorylation and nuclear translocation of transcription factors such as interferon regulatory factor 3 (IRF3) and nuclear-factor kappa-light-chain-enhancer of activated B-cells (NF-κB) (Abe and Barber, 2014) (Figure 3A). IRF3 and NF-κB mediate the transcription of ISGs and a host of other inflammatory genes leading to cell death, senescence, and immune-mediated clearance of infected cells (Dou et al., 2017; Galluzzi et al., 2018; Glück et al., 2017; Takahashi et al., 2018). In fact, accumulation of cytosolic dsDNA, either through endogenous DNA damage or as a result of downregulation of cytoplasmic nucleases such as Trex1, underlies auto-immune conditions such as Aicardi-Goutières syndrome and systemic lupus erythematosus (Crow et al., 2006; Lee-Kirsch et al., 2007; Stetson et al., 2008).
The connection between CIN and the cGAS-STING pathway raises the exciting possibility that chromosome segregation errors could serve as a trigger of immune editing during the early steps of tumorigenesis. Indeed, the generation of complex karyotypes in otherwise diploid retinal pigment epithelial (RPE1) cells activates inflammatory pathways and enhances natural-killer (NK) cell mediated killing (Santaguida et al., 2017). Furthermore, inflammatory signaling downstream of cGAS-STING in response to cytoplasmic DNA is a key mechanism in the immune-mediated clearance of cells harboring oncogenic RAS (Dou et al., 2017). Such a proposition is congruent with the relatively low abundance of chromosome copy number alterations in early stage tumors, as well as the negative effect of experimentally inducing aneuploidy or chromosome missegregation during early tumorigenesis (Rowald et al., 2016; Sheltzer et al., 2017).
A potential role for CIN in tumor immune editing is not necessarily at odds with the view that chromosome copy number heterogeneity enhances the fitness of expanding clones during tumor development once tolerance has been established. Indeed, recurrent patterns of chromosome copy number alterations (CNAs) responds, in part, to metabolic selection pressures and increase the baseline fitness of tumor cells (Graham et al., 2017). Even, in the pre-malignant state, structural and numerical chromosomal alterations help drive early clonal expansions in the peripheral blood (Loh et al., 2018). Furthermore, a pan-cancer genomic analysis based on the potency and distribution of oncogenes and tumor suppressor genes suggested that cumulative haploinsufficiencies and triplosensitivities drive aneuploidy patterns and shape the cancer genome in primary tumors (Davoli et al., 2013). Interestingly, these aneuploidies are dependent on the tissue that has given rise to the tumor and they bolster the underlying genetic network that controls proliferation in a tissue-specific manner (Sack et al., 2018). These findings might explain tumor type-specificity for the observed patterns of chromosome gains and losses such as the amplification of chromosomes 1 and 8 in breast cancer or chromosome 17 loss in ovarian cancer (Goh et al., 2017; Tavassoli et al., 1993).
In addition to karyotypic changes that enhance fitness through incremental chromosome gains and losses, chromosome missegregation can promote massive chromosomal rearrangements in the span of a single cell division (Ly et al., 2017; Zhang et al., 2015). The resulting abnormalities, which include chromothripsis, amplification of double minutes, and extrachromosomal DNA, can be subjected to selective pressures leading to dramatic changes in copy numbers of individual genomic loci (Stephens et al., 2011; Turner et al., 2017) (Figure 1). These punctuated genomic alterations have been proposed to underlie disease progression in pancreatic cancer (Notta et al., 2016). Collectively, these findings pinpoint a state of equipoise between the deleterious and beneficial consequences of CIN in the early steps of tumorigenesis.
Overcoming CIN-induced immune activation
The prevalence of CIN in human cancer indicates that at some point during tumor evolution, tumor cells grow tolerant to chromosome segregation errors and acquire the ability to evade immune recognition. Experimental evidence suggest that tumors can achieve this tolerance through a number of mechanisms, including adaptively rewiring their response to cytosolic DNA signaling as well as through the acquisition of genomic copy number heterogeneity that eschew the deleterious components of innate immune activation.
Are cGAS and STING lost in chromosomally unstable cancer cells?
The immune-stimulatory effect of cGAS-STING activation during viral infection has led to the widespread notion that cGAS and STING are themselves frequently lost in cancer (Konno et al., 2018; Xia et al., 2016a; 2016b). This was initially prompted by the observation that a large proportion of cancer cell lines fail to suppress replication-defective oncolytic viruses, a function that is dependent on cGAS-STING activity (Xia et al., 2016a) as well as the fact that cancer cell lines often do not induce ISGs in response to transfection with cGAMP or dsDNA (Lau et al., 2015; Stetson et al., 2008). However, such defects can result either from the loss of cGAS or STING themselves or a blockade in interferon signaling downstream of STING (Figure 3B).
Pan-cancer genomic data argue against widespread inactivation of cGAS and STING through mutational processes or copy number alterations. A survey of over 10,000 tumors included in the PanCancer TCGA database reveals that genes encoding cGAS and STING are rarely mutated in cancer – with 0.6% and 0.5% of tumors exhibiting mutations in either CGAS or TMEM173, the STING encoding gene, respectively. This is in agreement with Konno et al. who found that less than 1% of tumors exhibit mutations in these genes and, when experimentally tested, approximately half of these mutations did not impact protein function (Konno et al., 2018). Furthermore, CGAS and TMEM173 are infrequently altered at the copy number level, with a larger proportion of tumors exhibiting amplifications rather than deletions (Figure 4).
Figure 4. CGAS and TMEM173 (STING) are not frequently lost in cancer.
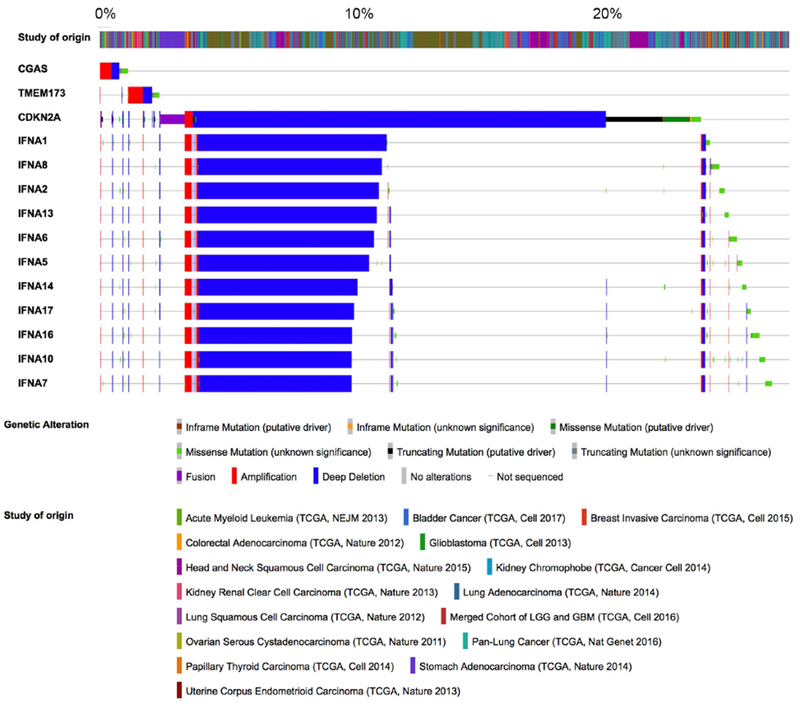
Oncoprints showing mutations and copy number alterations in CGAS, TMEM173, CDKN2A, and the interferon gene cluster in TCGA tumors. Most tumors maintain genomically intact copies of CGAS and TMEM173. A sizeable minority exhibits deep deletions in the interferon gene cluster on chromosome 9p suggesting a genetic mechanism for silencing type I interferon signaling in the presence of chronic cGAS-STING activation.
Protein and mRNA expression level data paint a mixed picture. Immunohistochemical analysis in colorectal cancer and melanoma reveals lower cGAS and STING protein levels in advanced stages (Xia et al., 2016a; 2016b). These losses are also seen in some cancer-derived cell lines and are accompanied by failure to induce type I interferon signaling upon transfection with dsDNA. Similarly, in gastric cancer, reduction in STING mRNA and protein levels correlates with increased tumor stage (Song et al., 2017), which has led some to propose that loss of these proteins is means of escaping tumor immune recognition (Xia et al., 2016b).
On the other hand, elevated levels of STING protein in tumor cells have been reported in genetically engineered mouse models and patient samples of pancreatic ductal adenocarcinoma (Baird et al., 2016). Human head-and-neck squamous cell carcinoma tumor cells express higher levels of STING compared to their normal tissue counterpart (Liang et al., 2015). This is congruent with detectable levels of cGAS and STING proteins in a number of aggressive cancer-derived cell lines such as B16 melanoma, Yac1 lymphoma, multiple myeloma, TRAMP-C2, DU145, PC-3 prostate, head and neck squamous cell carcinoma, and MCF-7, 4T1 and MDA-MB-231 breast cancer cells (Bakhoum et al., 2018; Chandra et al., 2014; Gaston et al., 2016; Ho et al., 2016; Lam et al., 2014; Liang et al., 2015; Takashima et al., 2016; Tang et al., 2016). Furthermore, many tumors types, including, but not limited to, breast, lung, pancreatic, thyroid and head and neck cancers, exhibit, on average, decreased methylation of the cGAS and STING promoters accompanied by increased mRNA levels compared with their normal tissue counterparts (Konno et al., 2018). Understandably, the promoter methylation status in tumor tissues displays increased variability compared to normal tissues and a small proportion of tumor samples displays hypermethylated promoters. This proportion differed among tumor types with some, such as colorectal cancer, exhibiting a larger percentage of tumors with hypermethylated promoters raising the prospects that epigenetic silencing of cGAS-STING signaling might be dependent on the tissue of origin; the colon is an immune rich environment that imparts unique selective pressures on a nascent tumor. Nonetheless, in sum these data suggest that the majority of tumors retain functional copies of cGAS and STING (Figure 5A).
Figure 5. Immune suppressive nature of CIN-induced inflammation.
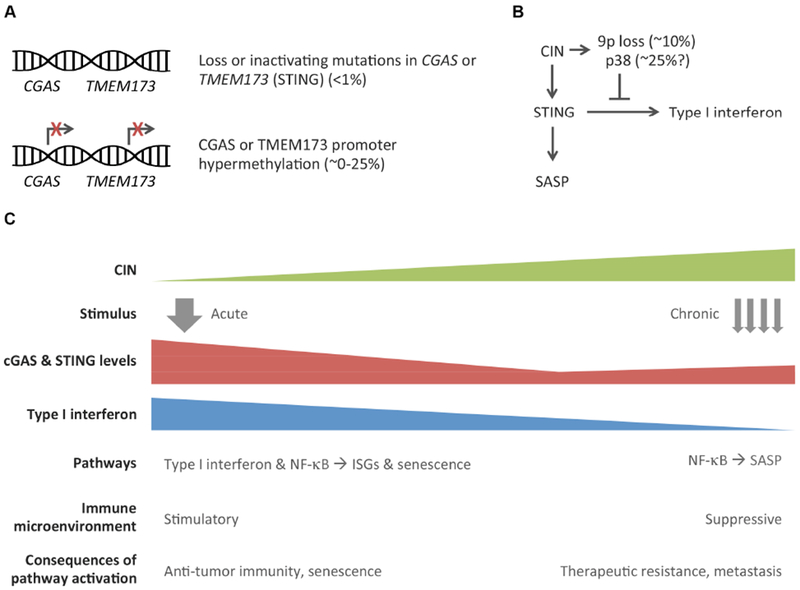
Whereas most tumors retain functional copies of the cGAS and STING coding genes (A) tumor cells have been shown capable of suppressing type I interferon signaling through a variety of mechanisms (B). Chromosome segregation errors can promote chromosome 9p loss, which harbors the interferon gene cluster. Furthermore, they activate the p38 MAP-kinase pathway, which has been found to selectively inhibit ISG induction without affecting other STING-dependent inflammatory signaling, such as the SASP. Therefore, the consequences of cGAS-STING activation in cancer are context-dependent (C); acute STING activation in normal cells or near-diploid tumors is likely to exhibit an anti-tumor effect through the activation of type I interferon signaling, cellular senescence, and ensuing T-cell mediated immunity. As tumors become progressively chromosomally unstable, they adapt to tolerate chronic cGAS-STING signaling in response to cytosolic DNA by downregulating downstream interferon signaling and instead maintaining alternative pathways that promote tumorigenesis, senescence bypass, therapeutic resistance, and metastasis.
Tumor cell-intrinsic suppression of type I interferon signaling
The presence of cGAS and STING in cancer however is not synonymous with induction of type I interferon, which appears to be suppressed to varying degrees in the majority of tumor cell lines despite the presence of cytosolic DNA. An analysis of over 1000 cell lines found in the cancer cell line encyclopedia (CCLE) by Dou et al. has revealed no significant correlation between STING mRNA levels and ISGs (Dou et al., 2017). Similar results are seen in MDA-MB-231 cells where cGAS-STING activation fails to promote type I interferon signaling or ISG induction (Bakhoum et al., 2018) (Figure 3B).
The mechanisms by which this suppression occurs remain poorly understood. One possibility is through the activation of the p38 MAP-kinase stress pathway, a known inhibitor of interferon signaling downstream of STING (Figure 5B). During the late stages of viral infection, p38-mediated phosphorylation of USP21, a deubiquitinating enzyme, negatively regulates STING (Chen et al., 2017). The p38 pathway is thought to be active in chromosomally unstable tumor cells in response to the ongoing stress resulting from chromosome missegregation and endogenous DNA damage (Thompson et al., 2010b). Interestingly, pharmacologic perturbation of p38 selectively influences type I interferon signaling downstream of STING while other STING-related pathways remain unaffected (Dou et al., 2017).
A second plausible mechanism for silencing interferon signaling is through modulation of STING signaling strength. The downstream consequences of STING activation are highly dependent on its protein expression levels (Gulen et al., 2017). For instance, lymphocytes express elevated levels of STING and as a result, they are exquisitely sensitive to exogenous addition of cGAMP (Cerboni et al., 2017; Gulen et al., 2017). This cell-type-specific sensitivity is attributed to STING expression level; bone marrow-derived macrophages and dendritic cells, which are normally activated by cytosolic DNA, exhibit resistance to STING activation. And, experimentally increasing STING expression in myeloid cells to levels comparable to those of T-cells renders the former sensitive to cGAMP. Thus, moderate reductions in STING protein levels or activity might enable some tumor cells to avert lethal cell-intrinsic consequences while maintaining a pro-survival program through reduced, yet chronic, STING signaling.
A third mechanism of averting cell-intrinsic type I interferon production is through loss of the interferon gene cluster on chromosome 9p (Figure 5B). This has been observed in melanoma and acute lymphocytic leukemia, among others (Heyman et al., 1993; Linsley et al., 2014; Litvin et al., 2015). Tumors with 9p loss exhibit lower basal activity of the interferon pathway and reduced T-cell infiltration, providing a mechanism by which locus-specific aneuploidy can enable chromosomally unstable cells to exist in the presence of cytosolic DNA while averting full-fledged immune attack. A survey of over 6,000 primary tumors in the TCGA database revealed that deep deletions of genes encoded by interferon gene cluster occurs in a significant proportion of tumors that have genomically intact copies of the cGAS and STING-coding genes (Figure 4).
Beyond Type I interferon: alternative CIN-induced inflammatory signaling
While tumor cells might exhibit suppression type I interferon signaling in the presence of cytosolic DNA, STING activation promotes other inflammatory pathways in a tumor cell-autonomous manner. In contrast to the absence of significant correlation between STING and ISGs, there is a positive correlation between cGAS and STING mRNA levels and those of genes involved in the SASP (Dou et al., 2017), in line with previously reported role for the cGAS-STING pathway in cellular senescence (Glück et al., 2017; Yang et al., 2017). STING activates a number of pro-inflammatory transcription factors beyond IRF3, including the canonical and non-canonical NF-κB pathways (Abe and Barber, 2014). While these factors have overlapping functions, their differential regulation downstream of STING can lead to distinct outcomes. For instance, the canonical NF-κB pathway induces SASP inflammatory factors upon cytosolic DNA signaling in primary IMR90 human lung fibroblasts (Dou et al., 2017; Glück et al., 2017). On the other hand, the noncanonical NF-κB pathway is upregulated upon chronic stimulation of cGAS-STING in MDA-MB-231 cells and plays a critical role in their ability to migrate and spread to distant organs (Bakhoum et al., 2018). These pathway-specific functions appear to be independent of the interferon-regulatory factors.
The mechanisms regulating inflammation downstream of cGAS-STING in cancer are still poorly understood. Yet, an important theme that emerges is that of the distinction between acute and chronic inflammation (Figure 5C). Unlike acute inflammatory responses, persistent inflammation is known to promote an immune-suppressive pro-metastatic tumor microenvironment (Coussens and Werb, 2002). Similarly, chronic engagement of the cGAS-STING pathway has pro-tumorigenic effects; TMEM173 knockout mice exhibit reduced carcinogen-induced tumor formation (Ahn et al., 2014). And while acute STING-mediated SASP represents a barrier against tumorigenesis, chronic SASP-related inflammation is associated with senescence evasion in transformed immortalized IMR90 cells (Dou et al., 2017). Likewise, chronic cGAS-STING mediate metastatic behavior in MDA-MB-231 breast cancer cells as well as a mesenchymal, treatment-resistant, phenotype in H69M lung cancer tumor models (Bakhoum et al., 2018; Cañadas et al., 2018).
The respective contributions of cell-intrinsic and cell-extrinsic cytosolic DNA signaling during tumor evolution remain to be fully elucidated. It might only require a small subset of tumor cells to experience chromosome missegregation events leading to SASP-related cytokine production in order to recruit immune cells to the tumor microenvironment. An inflamed microenvironment can in turn further propagate CIN in tumor cells, either through direct genotoxic stress or induction of EMT such that this cycle is maintained in a feed-forward fashion (Comaills et al., 2016; Suarez-Carmona et al., 2017).
Evidence for CIN-induced immune suppressive inflammatory phenotype can also be found in genomic data of human tumors (Davoli et al., 2017; Taylor et al., 2018; Thorsson et al., 2018). By carefully annotating the composition of the immune microenvironment, Thorsson et al. found that aneuploidy positively correlates with overall tumor leukocyte fraction in line with activation of inflammatory pathways. However, the composition of the aneuploid tumor microenvironment is dominated by macrophages and is characterized by an immune suppressive phenotypes that comprised the activation of tumor growth-factor-β (TGF-β) (Thorsson et al., 2018). In addition, there are a number of interesting correlations between genomic copy number alterations and tumor immune composition. For instance, 1p amplification is associated with increased leukocyte infiltration, whereas loss of 19q, which harbors TGFB1, reduces the tumor leukocyte fraction. Furthermore, amplifications of chromosome 2, 20q, 22q, as well as deletions of 5q, 9p, chr19 are associated with changes in macrophage polarity (Thorsson et al., 2018). These interesting associations highlight the need to develop mouse models that enable the dissection of the bidirectional cross talk between tumor karyotypes and the immune microenvironment.
The versatility of CIN-induced inflammation and cGAS-STING signaling in cancer cells demonstrates that tumor cells can alter their circuitry downstream of STING, in a context-dependent manner, to minimize the lethal consequences of cell-autonomous interferon signaling while permitting alternative inflammatory pathways that sustain tumor growth, therapeutic resistance, and metastasis. Furthermore, the distinction between acute and chronic activation states of cytosolic dsDNA sensing are critical to interpreting experimental results as well as data derived from clinical tumor samples.
Aneuploidy as a vehicle for immune evasion
Other means by which karyotypic abnormalities can directly promote immune evasion is through direct interference with antigen presentation. Copy number loss of heterozygosity in the human-leukocyte antigen (LOHHLA) occurs in nearly 40% of non-small cell lung cancers (NSCLC) (McGranahan et al., 2017). Interestingly, LOHHLA is associated with a high subclonal neoantigen burden as well as APOBEC-mediated mutagenesis. These tumors also exhibit increased T-cell infiltration suggesting strong selection for aneuploid clones that fine-tune their MHC-class I dosage. Similar observations have been noted by Chowell et al. in melanoma, whereby somatic LOH at the HLA locus is associated with poor prognosis and a worse response to immune checkpoint blockade (Chowell et al., 2017). Along the same veins, multiregion sequencing in high-grade serous ovarian cancer reveals similar LOH in tumor regions that are most replete with CD8 T-cells presenting evidence for dynamic spatial selection (Zhang et al., 2018).
CIN as a driver of metastasis
The role of CIN in metastasis has long been suspected given the emergence of karyotypic complexity during the later stages of tumor progression. The ability to genetically engineer cancer cells with varying rates of chromosome missegregation has enabled the decoupling of the inflammatory consequences of CIN from that of aneuploidy: by comparing chromosomally unstable tumor cells to their chromosomally stable – yet still aneuploid – counterparts (Bakhoum et al., 2018). Interestingly, in cells with CIN, chronic cGAS-STING activation has been shown to promote migration, invasion and metastasis. These phenotypes are mediated by the noncanonical NF-κB pathway downstream of STING (Figure 3B). Suppression of CIN, depletion of STING, or the noncanonical NF-κB transcription factors reduces metastasis in chromosomally unstable tumor cells.
The ability of tumor cells to co-opt the noncanonical NF-κB pathway provides insight into the mechanism of CIN-driven cancer metastasis. This pathway is a developmental program involved in monocyte and lymphocyte maturation and its activation follows relatively slow kinetics (Sun, 2011; Vogel et al., 2013), making it an ideal candidate to respond to the ongoing presence of cytosolic DNA (Figure 5). Interestingly, parallels to this scenario are observed during normal inflammation and raise the intriguing possibility that tumor cells engage in immune mimicry as they respond to chronic activation of cytosolic DNA signaling. First, cells from the myeloid lineage are activated in response to cytosolic DNA – originating from tumor cells or from viral infections – leading to increased migratory behavior (Gulen et al., 2017; Kis-Toth et al., 2011). Second, polyploidy is coincident with macrophage activation in granulomas suggesting a potential role for cytosolic DNA in this process. Third, transient nuclear rupture during leukocytes migration and extravasation leads to cGAS-STING activation (Denais et al., 2016; Herrtwich et al., 2016). It is therefore, tempting to postulate that cytosolic DNA signaling is an evolutionary conserved cue for migratory behavior during inflammation that is co-opted by tumor cells to spread to distant organs.
In addition to inflammatory signaling, aneuploidy can independently facilitate metastatic progression through gene dosage alteration (Campbell et al., 2010; Notta et al., 2016). This has been tested by Mueller et al. using KRAS-driven human cancer cells as well as genetically engineered murine pancreatic tumors (Mueller et al., 2018). Increased mutant-KRAS gene dosage led to pancreatic cancer metastasis, whereby cells with the highest number of copies exhibited aggressive undifferentiated phenotypes. In other settings, prospective multiregion sequencing of human renal cell carcinoma (RCC) revealed a significant increase in copy number heterogeneity in metastatic samples with enrichment for loss of chromosomes 9p and 14q (Turajlic et al., 2018). This raises the prospects that 9p loss is a pre-requisite to tolerate cGAS-STING signaling arising from increased copy number alterations, promoting immune escape and metastasis in RCC. This work highlights the need to generate genetically engineered mouse models that can better probe the relationship between CIN and metastasis in immune competent environments.
CIN and therapeutic response and resistance
It has long been appreciated that CIN facilitates treatment resistance by generating heterogeneity at the level of gene dosage. Yet, to maintain equipoise between genomic chaos and the acquisition of heterogeneity, cancer cells must restrict chromosome missegregation rates within a limited range that maximizes their viability (Burkard and Weaver, 2017; Laughney et al., 2015). This notion underlies, in part, the anti-neoplastic effects of therapies that promote CIN. For instance, widely used agents such as taxol, PARP inhibitors, and ionizing radiation are amongst the strongest inducers of chromosome segregation errors (Bakhoum et al., 2015; Lee et al., 2016). In addition to their well-characterized mechanisms of action, their effect on chromosome segregation invokes a contribution of the cGAS-STING pathway and anti-tumor immunity. To this end, Zierhut et al. have demonstrated that STING is an important determinant of mitotic cell death when breast cancer cell lines are treated with taxol in vitro (Zierhut and Funabiki, 2017). Similarly, recent reports link tumor response to PARP inhibitors to their ability to engage cGAS-STING signaling as well as anti-tumor immunity in murine transplantable ovarian and colorectal cancer models (Shen, 2018). In addition to their action on replication forks, PARP inhibitors perturb mitotic chromosome segregation providing a direct link for the generation of cytosolic dsDNA (Schoonen et al., 2017). Finally, cytosolic DNA signaling in tumor cells can promote systemic anti-tumor immunity after radiation treatment where regression of transplantable breast and melanoma tumors occurs in a T-cell dependent manner (Harding et al., 2017; Vanpouille-Box et al.,2017)
By the same token, sustained cGAS-STING activation can engender therapeutic resistance. Host STING-induced chronic type I interferon signaling during prolonged exposure to fractionated radiation therapy promotes tumor radio-resistance in transplanted MC38 colon tumors. This treatment resistance is mediated by the mobilization of myeloid-derived suppressor cells (Liang et al., 2017). In another study, chronic interferon exposure has been shown to promote the derepression and transcription of anti-sense endogenous retrovirus-coding sequences at the STAT1 and EZH2 promoters. The resulting double-stranded RNA, and dsDNA produced through reverse-transcription, sustains MAVS and cGAS-STING signaling and promotes mesenchymal cell traits, treatment resistance, and an immune suppressive microenvironment (Cañadas et al., 2018). These findings have important implications on therapeutic design and lead us to question the role of long-term use of cytotoxic therapies in intact or large residual tumors. Indeed, lingering viable tumor cells can acquire migratory and mesenchymal phenotypes after prolonged neoadjuavnt treatment with cytotoxic therapies, which, in some pre-clinical breast cancer models, are associated with increased metastasis to the liver (Karagiannis et al., 2017; Li et al., 2016). It is tempting to postulate that these effects are secondary to chronic inflammatory stimulation, such that if the therapy were unsuccessful in eliminating all tumor cells, the residual disease acquires an inducible aggressive phenotype.
Concluding remarks: CIN as a therapeutic vulnerability
The importance of CIN in tumor evolution is becoming increasingly appreciated and an understanding of the dual role of CIN as an activator of innate immune signaling as well as a vehicle for tumor adaptation is critical to our ability to target it for a therapeutic benefit. Despite recent advances in our understanding of CIN in biological systems, clinical adoption of CIN-directed therapies remains at its infancy and there are currently no drugs in the clinic that can be used to suppress chromosome segregation errors. Successful clinical implementation will hinge on interdisciplinary integration of cell biology, computational modeling, single-cell analyses, genetically engineered mouse models, as well as the use of human tumor samples (Figure 6). Careful patient selection will undoubtedly be central to our ability to determine when it is beneficial to modulate CIN and when its effects might be deleterious, as is the case with chronic inflammation. Furthermore, identifying means to prevent tumor cell adaptation to cytosolic DNA will be key to our ability to target an otherwise lethal feature of cancer for a therapeutic benefit (Figure 6). Given the widespread nature of CIN in human cancer, CIN-directed therapies have the potential to profoundly impact clinical outcomes including minimizing the onset of therapeutic resistance, treating advanced and metastatic disease, and augmenting systemic anti-tumor immunity.
Figure 6. An integrated approach to exploit CIN for a therapeutic benefit.
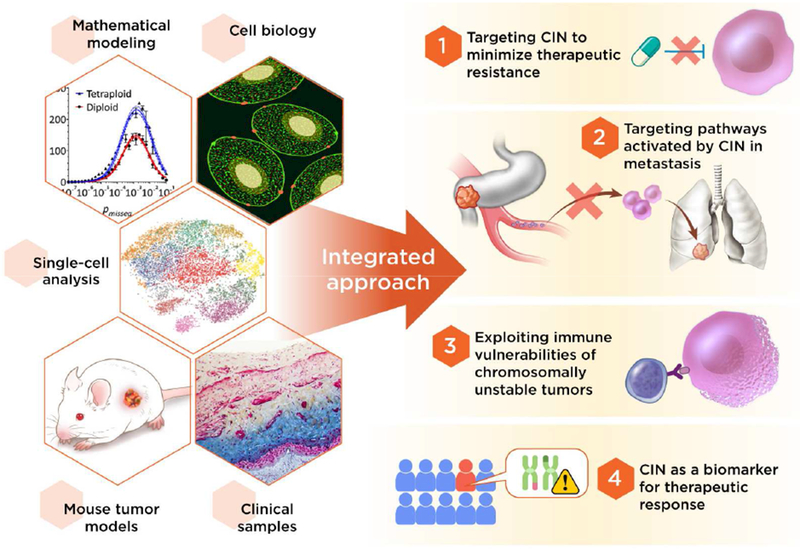
A viable adoption of CIN-directed therapies in the clinic must integrate a multi-disciplinary approach as well as careful patient selection. The widespread prevalence of CIN in advanced tumors offers an opportunity to devise therapeutic strategies that aim to target multi-drug resistance and metastatic progression. Furthermore, a better understanding of the mechanisms of CIN-induced inflammation might enable the development of novel approaches to augment anti-tumor immunity and synergize with existing immunotherapeutic agents used in the context of advanced metastatic disease.
ACKNOWLEDGEMENTS
We would like to thank Ashley Laughney, Nadeem Riaz (MSKCC) for constructive feedback and Wenjing Wu and Terry Helms (MSKCC) for assistance with illustrations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
S.F.B. declares no competing interests. L.C.C. owns equity in, receives compensation from, and serves on the board of directors and scientific advisory board of Agios Pharmaceuticals. He is also a founder of and receives laboratory support from Petra Pharmaceuticals.
REFERENCES
- Abe T, and Barber GN (2014). Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 88, 5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Xia T, Konno H, Konno K, Ruiz P, and Barber GN (2014). Inflammation-driven carcinogenesisis mediated through STING. Nat Commun 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ame JC, Cimini D, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, and Schreiber V (2008). Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim. Biophys. Acta 1786, 32–40. [DOI] [PubMed] [Google Scholar]
- Baird JR, Friedman D, Cottam B, Dubensky TW, Kanne DB, Bambina S, Bahjat K, Crittenden MR, and Gough MJ (2016). Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res 76, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Danilova OV, Kaur P, Levy NB, and Compton DA (2011). Chromosomal instability substantiates poor prognosis in patients with diffuse large B-cell lymphoma. Clin. Cancer Res 17, 7704–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Kabeche L, Murnane JP, Zaki BI, and Compton DA (2014). DNA-Damage Response during Mitosis Induces Whole-Chromosome Missegregation. Cancer Discovery 4, 1281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Kabeche L, Wood MD, Laucius CD, Qu D, Laughney AM, Reynolds GE, Louie RJ, Phillips J, Chan DA, et al. (2015). Numerical chromosomal instability mediates susceptibility to radiation treatment. Nat Commun 6, 5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker B, Bakker B, Taudt A, Taudt A, Belderbos ME, Belderbos ME, Porubsky D, Porubsky D, Spierings DCJ, Spierings DCJ, et al. (2016). Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol 17, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielski CM, Zehir A, Penson AV, Donoghue MTA, Chatila W, Armenia J, Chang MT, Schram AM, Jonsson P, Bandlamudi C, et al. (2018). Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkbak NJ, Eklund AC, Li Q, McClelland SE, Endesfelder D, Tan P, Tan IB, Richardson AL, Szallasi Z, and Swanton C (2011). Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res 71, 3447–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri T (1914). Zur Frage der Entstehung maligner Tumoren.
- Burkard ME, and Weaver BA (2017). Tuning Chromosomal Instability to Optimize Tumor Fitness. Cancer Discovery 7, 134–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu Y-H, and Chen ZJ (2014). The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296. [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin M-L, et al. (2010). The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cañadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, Campisi M, Kuang Y, Zhang Y, Gjini E, et al. (2018). Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat. Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SLS, Carter SLS, Cibulskis KK, Cibulskis KK, Helman EE, Helman EE, McKenna AA, McKenna AA, Shen HH, Shen HH, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Eklund AC, Kohane IS, Harris LN, and Szallasi Z (2006). A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet 38, 1043–1048. [DOI] [PubMed] [Google Scholar]
- Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg M-C, Picard C, Neven B, Fischer A, Amigorena S, et al. (2017). Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J. Exp. Med 214, 1769–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, Zhou J, Hayakawa Y, Karaolis DKR, and Gravekamp C (2014). STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res 2, 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Chen G, Bradford WD, Bradford WD, Seidel CW, Seidel CW, Li R, and Li R (2012). Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 482, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang L, Jin J, Luan Y, Chen C, Li Y, Chu H, Wang X, Liao G, Yu Y, et al. (2017). p38 inhibition provides anti-DNA virus immunity by regulation of USP21 phosphorylation and STING activation. J. Exp. Med 214, 991–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, Kuo F, Kendall SM, Requena D, Riaz N, et al. (2017). Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, and Salmon ED (2001). Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol 153, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comaills V, Kabeche L, Morris R, Buisson R, Yu M, Madden MW, LiCausi JA, Boukhali M, Tajima K, Pan S, et al. (2016). Genomic Instability Is Induced by Persistent Proliferation of Cells Undergoing Epithelial-to-Mesenchymal Transition. Cell Rep 17, 2632–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquel F, Silva M-J, Techer H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz A-L, et al. (2018). SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61. [DOI] [PubMed] [Google Scholar]
- Coussens LM, and Werb Z (2002). Inflammation and cancer. Nature 420, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, and Pellman D (2012). DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, et al. (2006). Mutations in the gene encoding the 3”−5” DNA exonuclease TREX1 cause Aicardi-Goutièeres syndrome at the AGS1 locus. Nat. Genet 38, 917–920. [DOI] [PubMed] [Google Scholar]
- Davoli T, Uno H, Wooten EC, and Elledge SJ (2017). Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, and Elledge SJ (2013). Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denais CM, Denais CM, Gilbert RM, Gilbert RM, Isermann P, Isermann P, McGregor AL, McGregor AL, Lindert, te M, Lindert, te M, et al. (2016). Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vanpouille-Box C, Bakhoum SF, and Demaria S (2018). SnapShot: CGAS-STING Signaling. Cell 173, 276–276.e1. [DOI] [PubMed] [Google Scholar]
- Gaston J, Cheradame L, Yvonnet V, Deas O, Poupon M-F, Judde J-G, Cairo S, and Goffin V (2016). Intracellular STING inactivation sensitizes breast cancer cells to genotoxic agents. Oncotarget 7, 77205–77224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glück S, Guey B, Gulen MF, Wolter K, Kang T-W, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, and Ablasser A (2017). Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh JY, Feng M, Wang W, Oguz G, Yatim SMJM, Lee PL, Bao Y, Lim TH, Wang P, Tam WL, et al. (2017). Chromosome 1q21.3 amplification is a trackable biomarker and actionable target for breast cancer recurrence. Nat. Med 23, 1319–1330. [DOI] [PubMed] [Google Scholar]
- Graham NA, Minasyan A, Lomova A, Cass A, Balanis NG, Friedman M, Chan S, Zhao S, Delgado A, Go J, et al. (2017). Recurrent patterns of DNA copy number alterations in tumors reflect metabolic selection pressures. Mol. Syst. Biol 13, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, and Ablasser A (2017). Signalling strength determines proapoptotic functions of STING. Nat Commun 8, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenberg A, Gerdes JS, Jung K, Sander B, Gunawan B, Bock HC, Liersch T, Bruük W, Rohde V, and Füzesi L (2010). High chromosomal instability in brain metastases of colorectal carcinoma. Cancer Genet. Cytogenet. 198, 47–51. [DOI] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, and Hetzer MW (2013). Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrtwich L, Nanda I, Evangelou K, Nikolova T, Horn V, Sagar, Erny D, Stefanowski J, Rogell L, Klein C, et al. (2016). DNA Damage Signaling Instructs Polyploid Macrophage Fate in Granulomas. Cell 167, 1264–1280.e18. [DOI] [PubMed] [Google Scholar]
- Heyman M, Grandéer D, Bröndum-Nielsen K, Liu Y, Söderhäll S, and Einhorn S (1993). Deletions of the short arm of chromosome 9, including the interferon-alpha/-beta genes, in acute lymphocytic leukemia. Studies on loss of heterozygosity, parental origin of deleted genes and prognosis. Int. J. Cancer 54, 748–753. [DOI] [PubMed] [Google Scholar]
- Ho SSW, Zhang WYL, Tan NYJ, Khatoo M, Suter MA, Tripathi S, Cheung FSG, Lim WK, Tan PH, Ngeow J, et al. (2016). The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 44, 1177–1189. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, and Barber GN (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal-Hanjani M, A’Hern R, Birkbak NJ, Gorman P, Gronroos E, Ngang S, Nicola P, Rahman L, Thanopoulou E, Kelly G, et al. (2015). Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: a prospective validation cohort study from the TACT trial. Annals of Oncology 26, 1340–1346. [DOI] [PubMed] [Google Scholar]
- Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, et al. (2017). Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med 376, 2109–2121. [DOI] [PubMed] [Google Scholar]
- Karagiannis GS, Pastoriza JM, Wang Y, Harney AS, Entenberg D, Pignatelli J, Sharma VP, Xue EA, Cheng E, D’Alfonso TM, et al. (2017). Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, and Sandberg AA (1968). Chromosome pulverization in human cells with micronuclei. J. Natl. Cancer Inst 40, 165–179. [PubMed] [Google Scholar]
- Kis-Toth K, Szanto A, Thai T-H, and Tsokos GC (2011). Cytosolic DNA-activated human dendritic cells are potent activators of the adaptive immune response. J. Immunol 187, 1222–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Yamauchi S, Berglund A, Putney RM, Mule JJ, and Barber GN (2018). Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AR, Bert NL, Ho SS, Shen YJ, Tang LF, Xiong GM, Croxford JL, Koo CX, Ishii KJ, Akira S, et al. (2014). RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res 74, 2193–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau L, Gray EE, Brunette RL, and Stetson DB (2015). DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughney AM, Elizalde S, Genovese G, and Bakhoum SF (2015). Dynamics of Tumor Heterogeneity Derived from Clonal Karyotypic Evolution. Cell Rep 12, 809–820. [DOI] [PubMed] [Google Scholar]
- Lee H-S, Lee NCO, Kouprina N, Kim J-H, Kagansky A, Bates S, Trepel JB, Pommier Y, Sackett D, and Larionov V (2016). Effects of Anticancer Drugs on Chromosome Instability and New Clinical Implications for Tumor-Suppressing Therapies. Cancer Res 76, 902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee Y-A, de Silva U, Bailey SL, Witte T, Vyse TJ, et al. (2007). Mutations in the gene encoding the 3”−5” DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet 39, 1065–1067. [DOI] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, and Vogelstein B (1998). Genetic instabilities in human cancers. Nature 396, 643–649. [DOI] [PubMed] [Google Scholar]
- Lengauer C, Lengauer C, Kinzler KW, Kinzler KW, Vogelstein B, and Vogelstein B (1997). Genetic instability in colorectal cancers. Nature 386, 623–627. [DOI] [PubMed] [Google Scholar]
- Li Q, Ma Z, Liu Y, Kan X, Wang C, Su B, Li Y, Zhang Y, Wang P, Luo Y, et al. (2016). Low doses of paclitaxel enhance liver metastasis of breast cancer cells in the mouse model. Febs J 283, 2836–2852. [DOI] [PubMed] [Google Scholar]
- Liang D, Xiao-Feng H, Guan-Jun D, Er-Ling H, Sheng C, Ting-Ting W, Qin-Gang H, Yan-Hong N, and Ya-Yi H (2015). Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-jun/CCL22 signal. Biochim. Biophys. Acta 1852, 2494–2503. [DOI] [PubMed] [Google Scholar]
- Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, Zheng W, Mauceri H, Mack M, Xu M, et al. (2017). Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun 8, 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsley PS, Speake C, Whalen E, and Chaussabel D (2014). Copy number loss of the interferon gene cluster in melanomas is linked to reduced T cell infiltrate and poor patient prognosis. PLoS ONE 9, e109760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvin O, Schwartz S, Wan Z, Schild T, Rocco M, Oh NL, Chen B-J, Goddard N, Pratilas C, and Pe’er D (2015). Interferon α/β Enhances the Cytotoxic Response of MEK Inhibition in Melanoma. Mol. Cell 57, 784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P-R, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF, Birmann BM, Talkowski ME, Bakhoum SF, McCarroll SA, et al. (2018). Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 559, 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, Fachinetti D, Page DC, and Cleveland DW (2017). Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol 19, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, and de Lange T (2015). Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163, 1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J, et al. (2017). Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 171, 1259–1271.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S, Engleitner T, Maresch R, Zukowska M, Lange S, Kaltenbacher T, Konukiewitz B, Ollinger R, Zwiebel M, Strong A, et al. (2018). Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 554, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, Denroche RE, Liang S-B, Brown AMK, Kim JC, et al. (2016). A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 538, 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pailler E, Auger N, Lindsay CR, Vielh P, Islas-Morris-Hernandez A, Borget I, Ngo-Camus M, Planchard D, Soria J-C, Besse B, et al. (2015). High level of chromosomal instability in circulating tumor cells of ROS1-rearranged non-small-cell lung cancer. Annals of Oncology 26, 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsson K, and Johansson B (2007). Trisomy 8 as the sole chromosomal aberration in acute myeloid leukemia and myelodysplastic syndromes. Pathol. Biol 55, 37–48. [DOI] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, and Li R (2010). Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova TA, Zhu J, and Li R (2013). Aneuploidy and chromosomal instability: a vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev 32, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowald K, Mantovan M, Passos J, Buccitelli C, Mardin BR, Korbel JO, Jechlinger M, and Sotillo R (2016). Negative Selection and Chromosome Instability Induced by Mad2 Overexpression Delay Breast Cancer but Facilitate Oncogene-Independent Outgrowth. Cell Rep [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roylance R, Endesfelder D, Gorman P, Burrell RA, Sander J, Tomlinson I, Hanby AM, Speirs V, Richardson AL, Birkbak NJ, et al. (2011). Relationship of extreme chromosomal instability with long-term survival in a retrospective analysis of primary breast cancer. Cancer Epidemiol. Biomarkers Prev 20, 2183–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack LM, Davoli T, Li MZ, Li Y, Xu Q, Naxerova K, Wooten EC, Bernardi RJ, Martin TD, Chen T, et al. (2018). Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Richardson A, Iyer DR, M’Saad O, Zasadil L, Knouse KA, Wong YL, Rhind N, Desai A, and Amon A (2017). Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev. Cell 41, 638–651.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonen PM, Talens F, Stok C, Gogola E, Heijink AM, Bouwman P, Foijer F, Tarsounas M, Blatter S, Jonkers J, et al. (2017). Progression through mitosis promotes PARP inhibitor-induced cytotoxicity in homologous recombination-deficient cancer cells. Nat Commun 8, 15981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senovilla L, Senovilla L, Vitale I, Vitale I, Martins I, Martins I, Tailler M, Tailler M, Pailleret C, Pailleret C, et al. (2012). An immunosurveillance mechanism controls cancer cell ploidy. Science 337, 1678–1684. [DOI] [PubMed] [Google Scholar]
- Sheltzer JM, Ko JH, Replogle JM, Habibe Burgos NC, Chung ES, Meehl CM, Sayles NM, Passerini V, Storchova Z, and Amon A (2017). Single-chromosome Gains Commonly Function as Tumor Suppressors. Cancer Cell 31, 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J (2018). PARPi triggers STING-dependent immune response and enhances therapeutic efficacy of immune checkpoint blockade independent of BRCAness. BioRxiv 318980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Peng P, Tang Z, Zhao J, Wu W, Li H, Shao M, Li L, Yang C, Duan F, et al. (2017). Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci Rep 7, 39858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Schvartzman J-M, Socci ND, and Benezra R (2010). Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 464, 436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Stephens PJ, Greenman CD, Greenman CD, Fu B, Fu B, Yang F, Yang F, Bignell GR, Bignell GR, et al. (2011). Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, and Medzhitov R (2008). Trex1 Prevents Cell-Intrinsic Initiation of Autoimmunity. Cell 134, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Carmona M, Lesage J, Cataldo D, and Gilles C (2017). EMT and inflammation: inseparable actors of cancer progression. Mol Oncol 11, 805–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Sun L, Wu J, Wu J, Du F, Du F, Chen X, Chen X, Chen ZJ, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S-C (2011). Non-canonical NF-κB signaling pathway. Cell Res 21, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton C, Nicke B, Schuett M, Eklund AC, Ng C, Li Q, Hardcastle T, Lee A, Roy R, East P, et al. (2009). Chromosomal instability determines taxane response. Proc. Natl. Acad. Sci. U.S.a 106, 8671–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, Watanabe S, Kawamoto S, Miyata K, Barber GN, et al. (2018). Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 9, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima K, Takeda Y, Oshiumi H, Shime H, Okabe M, Ikawa M, Matsumoto M, and Seya T (2016). STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochemical and Biophysical Research Communications 478, 1764–1771. [DOI] [PubMed] [Google Scholar]
- Tang C-HA, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, and Hu C-CA (2016). Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res 76, 2137–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoli M, Ruhrberg C, Beaumont V, Reynolds K, Kirkham N, Collins WP, and Farzaneh F (1993). Whole chromosome 17 loss in ovarian cancer. Genes Chromosomes Cancer 8, 195–198. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, et al. (2018). Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 33, 676–689.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Bakhoum SF, and Compton DA (2010a). Mechanisms of chromosomal instability. Curr. Biol 20, R285–R295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Thompson SL, Compton DA, and Compton DA (2010b). Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 188, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T-H, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. (2018). The Immune Landscape of Cancer. Immunity 48, 812–830.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Torres EM, Sokolsky T, Sokolsky T, Tucker CM, Tucker CM, Chan LY, Chan LY, Boselli M, Boselli M, et al. (2007). Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317, 916–924. [DOI] [PubMed] [Google Scholar]
- Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, Nicol D, O’Brien T, Larkin J, Horswell S, et al. (2018). Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, et al. (2017). Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, and Demaria S (2017). DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 8, 15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CFA, Wu D, Goth SR, Baek J, Lollies A, Domhardt R, Grindel A, and Pessah IN (2013). Aryl hydrocarbon receptor signaling regulates NF-κB RelB activation during dendritic-cell differentiation. Immunol. Cell Biol 91, 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warth A, Herpel E, Krysa S, Hoffmann H, Schnabel PA, Schirmacher P, Mechtersheimer G, and Blaker H (2009). Chromosomal instability is more frequent in metastasized than in non-metastasized pulmonary carcinoids but is not a reliable predictor of metastatic potential. Exp. Mol. Med 41, 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Konno H, and Barber GN (2016a). Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res 76, 6747–6759. [DOI] [PubMed] [Google Scholar]
- Xia T, Xia T, Konno H, Konno H, Ahn J, Ahn J, Barber GN, and Barber GN (2016b). Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep 14, 282–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, and Chen ZJ (2017). cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. U.S.a 114, E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LR, Yates LR, Campbell PJ, and Campbell PJ (2012). Evolution of the cancer genome. Nat Rev Genet 13, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki BI, Suriawinata AA, Eastman AR, Garner KM, and Bakhoum SF (2014). Chromosomal instability portends superior response of rectal adenocarcinoma to chemoradiation therapy. Cancer 120, 1733–1742. [DOI] [PubMed] [Google Scholar]
- Zhang AW, McPherson A, Milne K, Kroeger DR, Hamilton PT, Miranda A, Funnell T, Little N, de Souza CPE, Laan S, et al. (2018). Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 173, 1755–1769.e22. [DOI] [PubMed] [Google Scholar]
- Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, and Pellman D (2015). Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut C, and Funabiki H (2017). The cytoplasmic DNA sensor cGAS promotes mitotic cell death. BioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]