

Abstract
Well-nourished cells in a favorable environment (well supplied with growth factors, cytokines, and/or hormones and free from stresses, ionizing radiation, etc.) will grow, replicate their genome, and divide into two daughter cells, fully prepared to repeat the process. This cycle of DNA replication and division underlies all aspects of biological growth, reproduction, repair and development. As such, it is essential that the cell’s genome be guarded against damage during the replication/division process, lest the error(s) be irrevocably passed down to all future generations of progeny. Hence, cell cycle progression is closely guarded against major sources of errors, in particular DNA damage and misalignment of replicated chromosomes on the mitotic spindle. In this review article we examine closely the molecular mechanisms that maintain genomic integrity during the cell division cycle, and we find an unexpected and intriguing arrangement of concatenated and nested bistable toggle switches. The topology of the network seems to play crucial roles in maintaining the stability of the genome during cell proliferation.
Introduction
The fundamental purpose of the cell division cycle is two-fold: to replicate the DNA molecules contained in the cell’s chromosomes, and to segregate the replicated chromosomes to two daughter cells at division. In eukaryotic cells, these two processes occur in two distinct phases of the cell cycle: the chromosomes are replicated in S phase (for DNA ‘synthesis’) and segregated to daughter cells in M phase (for ‘mitosis’). In most eukaryotic cells, S and M phases are separated by two gaps, G1 (unreplicated chromosomes) and G2 (replicated chromosomes). By tradition, the three phases G1 + S + G2 are often referred to as ‘interphase’ [1].
Of utmost importance to the cell division cycle is maintaining the integrity of the genome while propagating it from one generation to the next; that is, ensuring that DNA is replicated only once and virtually without error and that replicated chromosomes are precisely partitioned at cell division, so that each daughter cell receives one and only one copy of each chromosome. The accuracy of these processes is maintained by two ‘quality-control’ devices, each of which consists of a ‘surveillance mechanism’ (that identifies errors and attempts to correct them) and a downstream ‘checkpoint’ (that halts further progress through the cell division cycle until the errors are resolved). One might think of the checkpoint as the gate at a border crossing and the surveillance mechanism as the hut where border agents examine travelers’ documents [2]. If all the documents are in order, a message is passed to lift the gate, and the travelers may continue on their way.
With regard to the cell division cycle, the documents that the border-crossing agents are checking are: (1) Is the cell’s DNA damaged in any way? If so, then the damaged DNA should not be replicated, so entry into S phase is blocked. (2) Are all the replicated chromosomes (i.e., the sister chromatid pairs) properly bioriented on the mitotic spindle? If not, then the cell should not attempt to separate the sister chromatids to opposite poles of the spindle, so nuclear division is blocked. Hence, these two crucial transitions in the cell cycle are guarded by checkpoints, and whether to lift the checkpoint or not is a binary decision that the cell must make on the basis of information it receives from the associated surveillance mechanisms. (DNA damage is checked not only at the G1/S transition but also at later transitions of the cell cycle [3], but we shall limit our attention in this review article to the DNA-damage checkpoint at the G1/S transition.)
These checkpoint decisions are implemented by networks of interacting genes and proteins, and, as we have emphasized repeatedly over the past 25 years, binary decisions are made possible by bistable (toggle) switches in the underlying macromolecular regulatory networks [4–6]. In the following sections we describe the molecular mechanisms and physiological characteristics of the toggle switches that ensure genomic integrity at the G1/S and metaphase/anaphase transitions of the cell cycle. Both transitions are governed, we find, by nested control elements, consisting of a pair of interacting toggle switches [7]. But, before we can reach this conclusion, we must consider the mechanistic details of the quality-control machineries in the cell cycle.
Switching between interphase and mitosis
As we have said, DNA synthesis occurs during interphase and sister chromatid separation during M phase. M phase is further decomposed into five sequential phases: prophase (chromosome condensation), prometaphase (nuclear envelope breakdown, spindle assembly, and chromosome capture by microtubules), metaphase (brief period when all the sister-chromatid pairs are bioriented on the mitotic spindle), anaphase (segregation of sister chromatids to opposite poles of the spindle), and telophase (re-formation of nuclear envelopes around the separate collections of chromosomes, followed by cell division). Progression into M phase requires massive phosphorylation of proteins on chromosomes, nuclear membranes, and cytoskeletal filaments. These phosphorylations are catalyzed by Cyclin B-dependent kinases that are activated as cells transition from interphase into mitosis [8]. To exit from mitosis (anaphase and telophase), these Cyclin B-dependent kinases must be inactivated by degradation of Cyclin B, and their activities must be replaced by counter-acting phosphatases that remove the M-phase phosphorylations and return the cell to the low-phosphoprotein conditions associated with interphase [9]. To be sure, there are crucial phosphorylation events during interphase, driven by Cyclin E- and Cyclin A-dependent kinases (e.g., the phosphorylation of replication complexes at origins of DNA replication). Nonetheless, we suggest that interphase is a low-phosphoprotein state compared to M phase and that switching back-and-forth between these two phases is governed by a fundamental antagonism between the activities of cyclin-dependent protein kinase (CDK) and its counter-acting protein phosphatase (CAP). In interphase CDK activity is low and CAP activity is high, whereas in M phase CDK activity is high and CAP activity is low. Switching back and forth between these two states is fundamental to cycling between interphase (when DNA is replicated) and M phase (when sister chromatids are partitioned to daughter cells). These switches must be ‘irreversible’ in the sense that, once a cell commits itself to a new round of DNA replication, it does not back up and initiate another round of DNA replication before it goes through mitosis. Worse yet would be to back up and make a second try at mitosis without replicating the DNA.
The molecular mechanism that governs this fundamental alternation of interphase and mitosis is presented as a regulatory motif in Figure 1A and as a detailed biochemical mechanism in Figure 1B. The motif consists of two positive feedback loops, one governing the activation of CDK (in particular, CycB:Cdk1) and the other governing the activation of CAP (in particular, B55:PP2A). These positive feedback loops underlie two bistable switches (Figure 1C), as has been abundantly proven by many experimental studies [10–13]. As stressed in Ref [12], these switches are tightly coupled by mutually inhibitory signals. For this reason, when a cell enters mitosis and the CDK switch flips on (Figure 1C left), the CAP switch flips off (Figure 1C right). Entry into mitosis is driven by the accumulation of Cyclin B in G2 phase (Figure 1C left). When the level of Cyclin B crosses a threshold, CycB:Cdk1 is abruptly activated by engaging the positive feedback loops through Wee1 and Cdc25 (Figure 1B left) [10,11] and B55:PP2A is abruptly inactivated through the action of Greatwall kinase and ENSA (Figure 1B right) [12]. Consequently, the excess of kinase activity over phosphatase activity leads to massive protein phosphorylation (Figure 1D left).
Figure 1. Switching between interphase and mitosis.
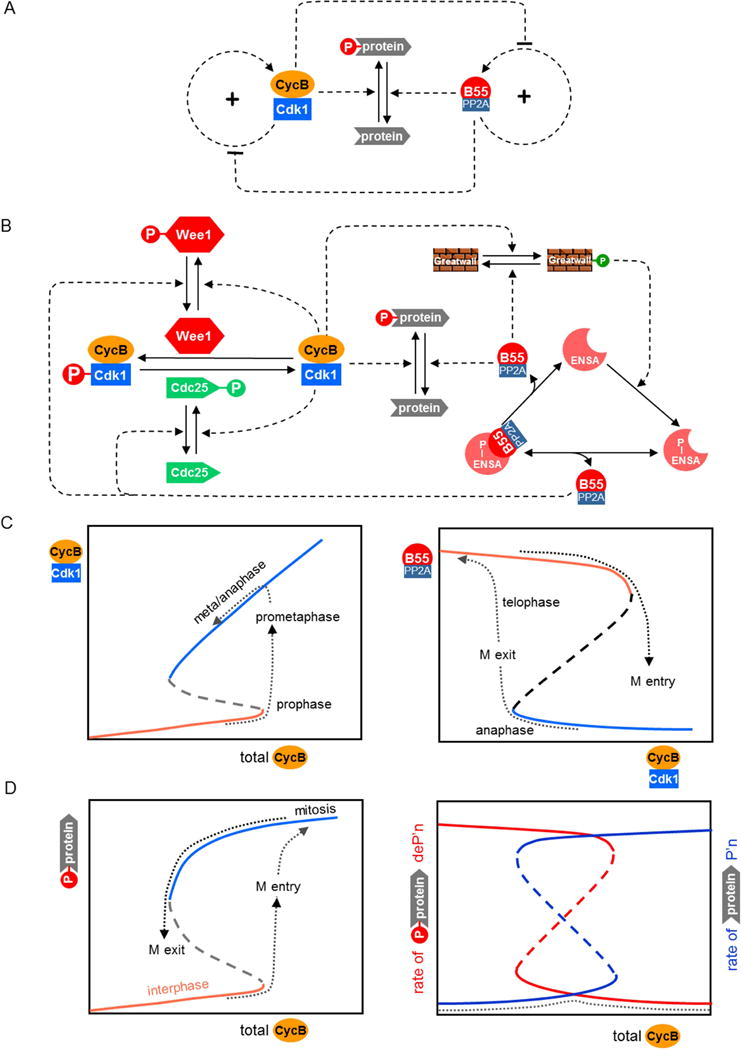
(A) Regulatory motif. Many cellular proteins are phosphorylated during mitosis by Cyclin B-dependent protein kinase-1 (CycB:Cdk1). During interphase most of these proteins are unphosphorylated, due to the action of protein phosphatases, such as B55:PP2A. Both CycB:Cdk1 and B55:PP2A are activated by positive feedback loops that create two separate bistable switches. The two switches are mutually inhibitory, so that, if the kinase switch is ON, then the phosphatase switch is OFF, and vice versa. (B) Detailed biochemical mechanism. CycB:Cdk1 is inhibited by phosphorylation of the kinase subunit by Wee1 and activated by dephosphorylation by Cdc25. Both Wee1 and Cdc25 are phosphorylated by CycB:Cdk1; an inhibitory phosphorylation in the first case and activatory in the second [39]. These redundant positive feedback loops create a bistable toggle switch for CycB:Cdk1 activity [4]. On the other side, B55:PP2A is inactivated by binding to phosphorylated ENSA [40,41]. B55:PP2A can activate itself by dephosphorylating P-ENSA [42], but this action is opposed by a kinase, Greatwall in its phosphorylated form. This motif, called a ‘feedback-amplified domineering substrate’, creates a bistable toggle switch for B55:PP2A activity [43]. The two toggle switches are mutually inhibitory, because CycB:Cdk1 is the kinase that phosphorylates Greatwall, and B55:PP2A is the phosphatase that dephosphorylates Wee1 and Cdc25 [7,12]. (C) Signal-response curves. (Left panel) Steady state activity of CycB:Cdk1 as a function of total Cyclin B in a cell entering mitosis and proceeding into anaphase. (Right panel) Steady state activity of B55:PP2A as a function of CycB:Cdk1 activity in a cell. B55:PP2A activity switches OFF as a cell enters mitosis and switches ON as a cell exits mitosis. (D) Substrate phosphorylation during interphase and mitosis. Total concentration of B-type Cyclins increases during interphase and early mitosis, and decreases as cells exit mitosis and return to interphase. (Left panel) Cdk-targeted substrates are heavily phosphorylated in mitosis and mostly unphosphorylated in interphase. (Right panel) During interphase the rate of substrate phosphorylation is low and dephosphorylation is high, and vice versa during mitosis. Consequently, the rate of futile cycling (dashed line) is low throughout the cell cycle, because CDK and CAP are never active simultaneously.
The cell leaves M-phase (as described below) by degrading CycB, which allows the bistable switches to flip to the high-CAP, low-CDK state (Figure 1C right) and mitotic proteins to be dephosphorylated (Figure 1D left).
This mechanism (Figure 1B) ensures that CDK and CAP are never active at the same time. Consequently, when the rate of protein phosphorylation is high, the rate of protein dephosphorylation is low, and vice versa (Figure 1D right). This antagonism between CDK and CAP is greatly beneficial to the cell, because if they were both active at the same time, then their target proteins would be continuously phosphorylated and dephosphorylated and the cell would suffer considerable “futile cycling” of ATP (ATP + H2O ➔ ADP + Pi). On the contrary, because CDK and CAP are not active at the same time, the rate of futile cycling (the dotted curve in Figure 1D, right) is always low.
In the next two sections, we show that the checkpoint mechanisms for DNA damage at the G1/S transition and for chromosome alignment at the metaphase checkpoint are embedded within the two alternative states created by the antagonism between CDK and CAP.
Controlling the transition from G1 into S phase
A major threat to genome integrity is DNA damage. If damage occurs in G1 phase, it must be sensed and repairs must be initiated, and, in the meantime, progression into S phase must be blocked. The DNA-damage surveillance mechanism consists of proteins that, in response to single-strand and double-strand breaks in DNA, induce a rise in the activity of a transcription factor (p53) that subsequently upregulates the expression of repair enzymes and of a CDK-inhibitory protein called p21. By inhibiting Cyclin E- and Cyclin D-dependent kinases [14,15], p21 prevents passage through the ‘restriction point’ (RP) (see Figure 2A, B).
Figure 2. Controlling the transition from G1 into S phase.
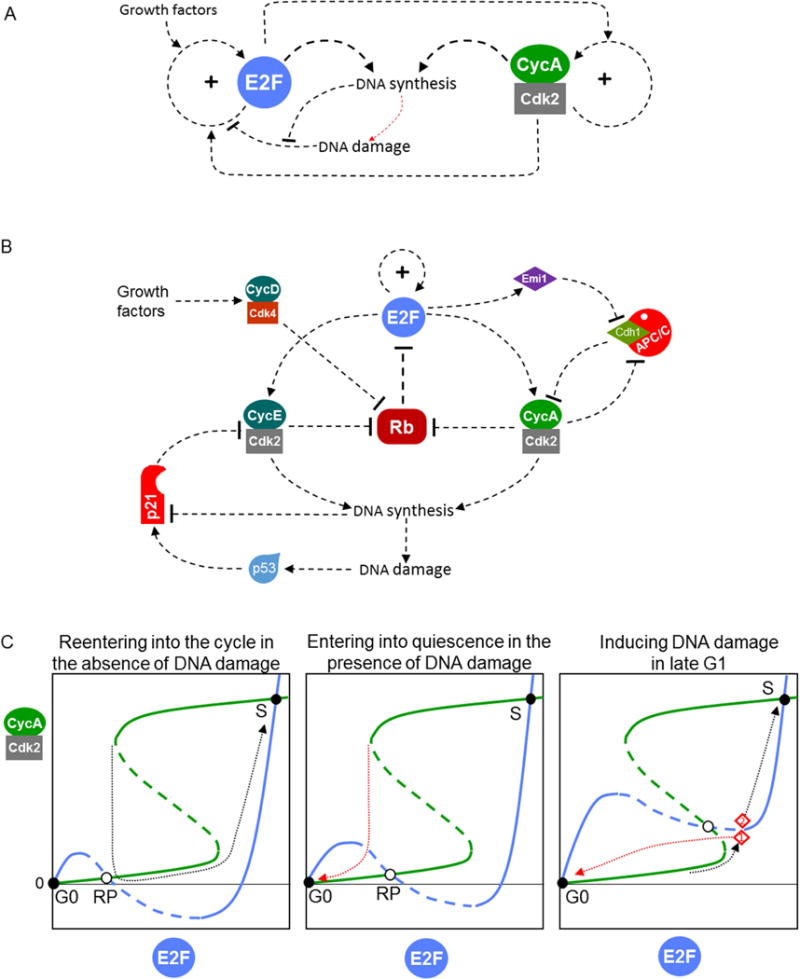
(A) Regulatory motif. The ‘restriction point’ in the mammalian cell cycle is based on a positive feedback loop controlling the activity of a family of E2F transcription factors. DNA-damage signals tend to inactivate E2F and impose a checkpoint on further progression through G1, whereas growth factor signals tend to activate E2F and lift the checkpoint. The G1/S transition is controlled by a positive feedback loop on Cyclin A-dependent kinase, which creates a second toggle switch. The two toggle switches are mutually activatory. The process of DNA synthesis dampens the ‘damage’ signal created by single- and double-strand DNA breaks. (B) Detailed influence diagram. E2F upregulates its own expression as well as the expression of Cyclin E [44]. RB (the retinoblastoma protein) binds to and silences E2F. Phosphorylation of RB by CycE:Cdk2 causes RB to detach from E2F. The two positive feedback loops (promoting E2F expression and activity, respectively) combine to form a robust bistable switch. Downstream of E2F is a second bistable switch caused by mutual antagonism of CycA:Cdk2 and Cdh1:APC/C (the kinase phosphorylates and inactivates Cdh1, whereas the ubiquitin ligase labels Cyclin A for degradation). E2F pushes this switch in favor of CycA:Cdk2 by upregulating the synthesis of both Cyclin A and Emi1 (an inhibitor of Cdh1:APC/C). CycA:Cdk2 returns the favor by phosphorylating RB. Growth-factor signaling is mediated by expression of Cyclin D, and DNA-damage signaling is mediated by p53 and p21 (a stoichiometric inhibitor of Cyclin E- and Cyclin A-dependent kinases). DNA synthesis creates damage signals (single- and double-strand breaks), but the signal doesn’t get through because DNA synthesis also promotes degradation of p21. (C) Dividing-of-the-ways. CycA:Cdk2 activity is a bistable function of E2F level (green curve), and E2F level is a bistable function of CycA:Cdk2 activity (blue curve). The curves are plotted for cells well supplied with growth factors. (Left panel) A cell that enters G1 with no residual DNA damage has enough activity associated with Cyclin D, Cyclin E and E2F to pass the restriction point and proceed directly to the G1/S transition (the black dotted trajectory). (Middle panel) For a cell that enters G1 with DNA damage left over from the previous cycle, the blue curve is raised up a little, and the cell cannot pass the restriction point. Instead, it is attracted (red dotted trajectory) to the quiescent state (E2F low, and both Cyclin E and Cyclin A low). (Right panel) For a cell that receives massive DNA damage in the previous cycle or in the present G1 phase, the blue curve is raised up significantly, and the quiescent state has a much larger ‘basin of attraction’. For example, a cell that is quite far along in G1 (point 1 in the panel) can still be recalled to the quiescent state (red dashed trajectory), whereas another cell that is a little deeper into the G1/S transition (point 2 in the panel) cannot be recalled (black dashed trajectory). Somewhere between these two trajectories is the ‘point-of-no-return’.
The RP is a bistable switch [16] whose function is to turn on E2F transcription factors that upregulate expression of proteins required for DNA replication, including Cyclins E and A (Figure 2B left). Consequently, the RP serves as a hub for signals that control entry into the DNA replication-division cycle [17]. In addition to DNA-damage signals, which (through p21) keep the RP checkpoint imposed, the RP machinery receives signals from growth factors (through Cyclin D) and (most likely) from overall cell growth to lift the checkpoint.
After cell division, proliferating cells in G1 phase exhibit a ‘dividing of the ways’ [18], a decision that depends on residual activity of Cyclin E (left over from the previous cell cycle) in association with Cdk2. Newborn cells with low activity of CycE:Cdk2 enter a ‘quiescent’ state, whereas newborn cells with slightly higher CycE:Cdk2 activity continue proliferating (see Figure 2C left and middle panels). This sort of behavior is indicative of a bistable switch, which can be very sensitive to CycE:Cdk2 activity at the start of a new cell cycle. Because CycE:Cdk2 activity is a function of the amounts of both Cyclin E and p21 in a newborn cell, this dividing-of-the-ways in G1 is apparently a maneuver to protect the genome from endogenous DNA damage sustained during S/G2/M phases of the previous cell cycle [15,19,20].
Downstream of RP is a second bistable switch, the G1/S checkpoint [21], whose function is to maintain degradation of Cyclin A, so that CycA:Cdk2 is kept low and it cannot initiate DNA replication (by phosphorylating proteins at origins of replication). The degradation of Cyclin A by proteasomes is controlled by polyubiquitination of Cyclin A by an E3 ubiquitin ligase called APC/C (Anaphase Promoting Factor/Cyclosome), in conjunction with a targeting subunit called Cdh1 [22]. These two components (Cyclin A and Cdh1) are locked in a mutually antagonistic ‘embrace’ (Figure 2B right) that creates alternative stable steady states [23,24]: (1) Cdh1:APC/C is active, and Cyclin A level is low (the pre-transition state; checkpoint imposed); and (2) CycA:Cdk2 is active, and Cdh1 is phosphorylated and inactive (the post-transition state; checkpoint lifted). The RP switch helps to lift the G1/S checkpoint by inducing the synthesis of Cyclin A and of Emi1 [21,24], an inhibitor of Cdh1:APC/C [25].
The chaining of two bistable switches (at RP and G1/S) creates a time-window between satisfying the requirements of the restriction point and turning on DNA replication. This time window may be useful, for example, for making deoxyribonucleotide precursors for DNA synthesis. If, during this time window, the DNA is damaged, the damage surveillance mechanism will generate a new pulse of p21. Because p21 inhibits both Cyclins E and A, DNA damage during this transient period is still able to block entry into S phase (Figure 2C right panel) [24].
After the cell enters S phase, the process of DNA replication creates single- and double-strand breaks in DNA (by production of Okazaki fragments and relief of supercoiling), and these breaks are recognized as DNA damage by the surveillance mechanism [3]. To prevent reactivation of the checkpoint, the process of DNA replication activates an E3 ubiquitin ligase (Crl4:Cdt2) that promotes rapid degradation of p21 during S phase [19,26]. If unrepaired DNA damage is incurred during S phase or G2 phase, it may prevent entry into M phase or it may be dealt with in G1 phase of the next cell cycle [19,20].
Controlling the transition from prometaphase into anaphase
The major threat to genome integrity during M phase is misorientation of replicated chromosomes on the mitotic spindle. This threat is detected by a surveillance mechanism that is sensitive to tensionless (unattached) kinetochores [27] and sends an inhibitory signal to the Anaphase Promoting Factor, APC/C (see Figure 3A). The two sister chromatids of a replicated chromosome are held together by cohesin rings at their centromeric regions [28]. Each sister chromatid has a kinetochore (KT), where spindle microtubules (MTs) can bind. If the KTs of sister chromatids are bound to MTs from opposite poles of the spindle, then, because of the constraints imposed by the cohesin rings, the KTs are subjected to tensile forces that stretch the entire centromeric assembly and create gaps between the inner and outer KT regions of each chromatid. The centromere surveillance mechanism can sense when the centromeric region is not under tension, i.e., when the KTs are not bioriented, and create a mitotic checkpoint complex (MCC) (see Figure 3B), which is a potent inhibitor of APC/C in association with Cdc20 [29]. (Cdc20 is a molecular relative of Cdh1.) The job of Cdc20:APC/C is to polyubiquitinate securin and Cyclin B and thereby promote their proteolysis [30]. Securin degradation releases separase, an endoprotease that cleaves cohesin rings and allows bioriented sister chromatids to be pulled to opposite poles of the mitotic spindle during anaphase [31]. Clearly, Cdc20:APC/C must be kept inactive by MCC until all replicated chromosomes are properly bioriented on the mitotic spindle.
Figure 3. Controlling the transition from prometaphase into anaphase.
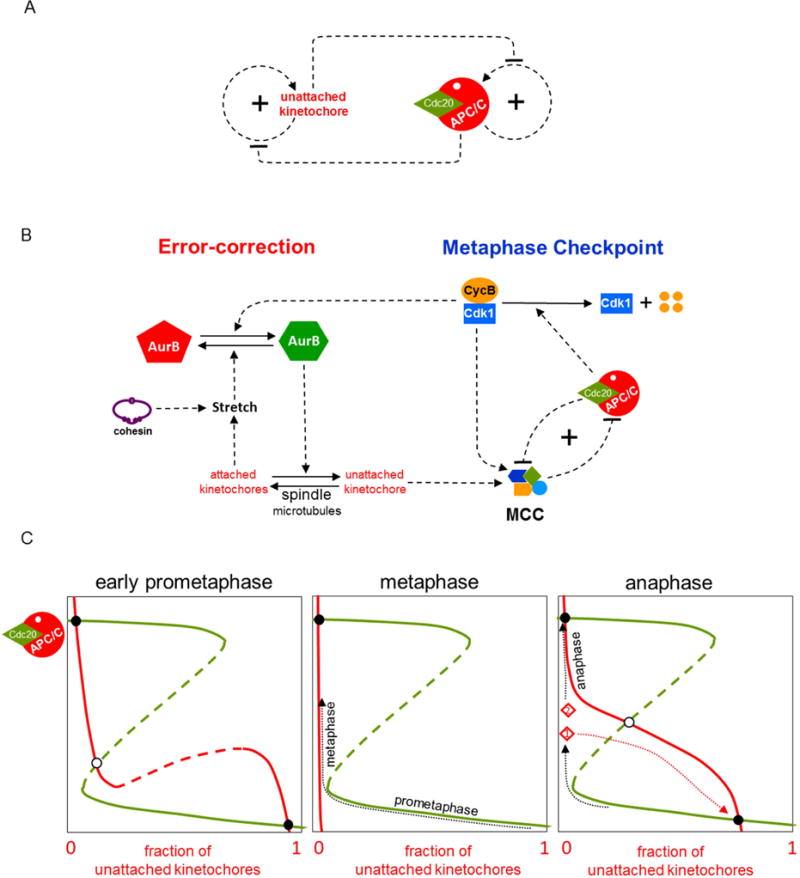
(A) Regulatory motif. Mitotic cells have a surveillance mechanism for detecting and correcting unattached kinetochores (uKTs). Positive feedback in this mechanism creates a bistable switch that allows replicated chromosomes to accumulate in a bioriented state (with sister KTs stretched by their attachment to opposite poles of the mitotic spindle). Meanwhile, uKTs send an inhibitory signal to the ‘anaphase promoting complex/cyclosome’ (APC/C), whose activity is also governed by a bistable switch. When the APC/C is activated, it silences a component in the uKT positive feedback loop [7,45]. (B) Detailed influence diagram. (Left side) The error-correction module. If a pair of sister chromatids is properly attached to the mitotic spindle at both kinetochores (i.e., bioriented), then the attached kinetochores (aKT) become stretched, which blocks the action of Aurora B (AurB) kinase. If a pair of uKTs is improperly attached by only a single kinetochore, then active AurB at that centromere induces detachment of microtubules (MTs) from the aKT. This error-correcting action of AurB allows the replicated chromosome to try again to achieve biorientation on the mitotic spindle. The mutual antagonism between aKT and AurB creates a bistable switch that maintains a population of uKTs until all the cell’s chromosomes are properly aligned on the spindle. (Right side) The metaphase checkpoint. uKTs promote assembly of a mitotic checkpoint complex (MCC), which binds to and inhibits the APC/C, thereby blocking progression of the cell into anaphase. Active APC/C, on the other hand, promotes disassembly of the MCC; but this double-negative feedback loop is insufficient, in its own right, to create a bistable switch. Bistability requires that active APC/C promotes the degradation of CycB; thereby destroying CycB:Cdk1 activity, which is necessary for assembling the MCC [46]. The entire motif (on the right side of the diagram) is called a ‘feedback-amplified domineering substrate’, and the conditions for bistability are described in Ref [43]. Experimental evidence indicates that the metaphase checkpoint is indeed a bistable toggle switch [45,47]. In addition, CycB:Cdk1 activity feeds back to the error-correction module as a co-activator of AurB. (C) The dynamics of mitotic progression. (Left panel) Green curve: steady state activity of APC/C as a function of the fraction of uKTs; red curve: steady state fraction of uKTs as a function of APC/C activity. During prometaphase, the control system persists at the stable steady state with low APC/C activity (the black circle in the lower-right corner), with many chromosomes in the 00-state of KT attachment and the APC/C inactive. As chromosomes become bioriented (11-state), the red curve moves steadily to the left. (middle panel) When all the chromosomes are properly attached to the mitotic spindle (metaphase), the red curve is pressed against the vertical axis, and the cell lifts the metaphase checkpoint by activating APC/C. (There is now a single stable steady state, the black circle in the upper-left corner.) (Right panel) As the cell proceeds through metaphase, if some aKTs become unattached (either spontaneously or by experimental manipulation), an error-signal recreates the prometaphase steady state (the APC/C inactive state in the lower-right corner) and a cell that has not yet passed the ‘point-of-no-return’ will return to prometaphase (the red trajectory from point 1). Cleavage of cohesins during anaphase eliminates kinetochore stretch and recreates the APC/C inactive steady state, but anaphase cells (point 2 in the panel) are beyond the ‘point-of-no-return’ and so they maintain their progression toward the upper-left steady state, where APC/C is fully active.
In this case, the error-correction module is sensitive to the attachment states of the centromeric region of each replicated chromosome. If both KTs are unattached (00), there is nothing to be done until, by chance, a spindle MT binds to a KT and starts pulling. In this sense, the 00-state is a stable steady state of the error-correction module. The half-attached states (01 or 10) must be transient states: some time is allowed for the other KT to get attached to the opposite pole; but if this happy state (11) is not achieved quickly, then the single KT-MT attachment must be dissolved (the error-correction mechanism) so that the chromosome can start again from 00 and try to reach 11 [32]. The bioriented state (11) must be a stable steady state of the error-correction module, so that the cell can steadily build up a collection of bioriented chromosomes.
These considerations led us to postulate [33,34] that the surveillance-mechanism/error-correction module of the ‘spindle assembly checkpoint’ is itself a bistable switch that relays a message (the MCC) to the downstream ‘metaphase checkpoint’ that governs whether cells are blocked in prometaphase or allowed to proceed into anaphase; as diagrammed in Figure 3B). The metaphase checkpoint is imposed (gate down) as long as the error-correction module is unhappily producing MCC and inhibiting Cdc20:APC/C, as in Figure 3C left panel. Once the error-correction module confirms that all chromosomes are bioriented, MCC production is cut off. But it takes some time for the cell to rid itself of MCC and activate the Cdc20:APC/C, thereby lifting the gate into anaphase. This lag phase is what we see as ‘metaphase’, a transient state between prometaphase and anaphase (Figure 3C middle panel). If MT-KT attachments are lost during metaphase (either spontaneously or by laser ablation in the laboratory), the error-correction module can snap back into action (as in Figure 3C right panel) [35]. The transition from prometaphase to anaphase is still reversible at this stage.
Cohesin cleavage at the onset of anaphase presents a conundrum (the ‘anaphase problem’ [36]), because tension at the KTs is immediately lost, which might be recognized by the centromere surveillance mechanism as misaligned chromosomes, causing MCC to be produced and the APC/C to be inhibited. To prevent this, the surveillance mechanism is dependent on CycB:Cdk1 activity (see Figure 3B), and Cyclin B is degraded by Cdc20:APC/C along with securin. For this mechanism to work properly, the timing of Cyclin B and securin degradation must be precisely ordered [30]. If this timing is disturbed, the surveillance-mechanism/error-correction module is reactivated, and anaphase segregation of sister chromatids is aborted [37,38].
Conclusion
Putting together Figures 1–3, we come to an intriguingly symmetric picture (Figure 4) of eukaryotic cell cycle regulation:
A matched pair of mutually inhibitory, bistable switches (for CycB:Cdk1 and B55:PP2A) control switching between a kinase-dominated phase of the cell cycle (mitosis) and a phosphatase-dominated phase of the cell cycle (interphase).
In interphase, a matched pair of mutually activating, bistable switches (for E2F and CycA:Cdk2) control switching from G1 phase (unreplicated chromosomes) into S phase (chromosome replication). These switches impose a DNA-damage checkpoint on cell cycle progression in late G1.
In mitosis, a matched pair of mutually inhibitory, bistable switches (for unattached KTs and APC/C), control switching from prometaphase to anaphase (i.e., segregation of replicated chromosomes). These switches impose the spindle-assembly checkpoint on cell cycle progression.
Figure 4. Overview of the molecular mechanisms controlling cell cycle progression and genome stability.
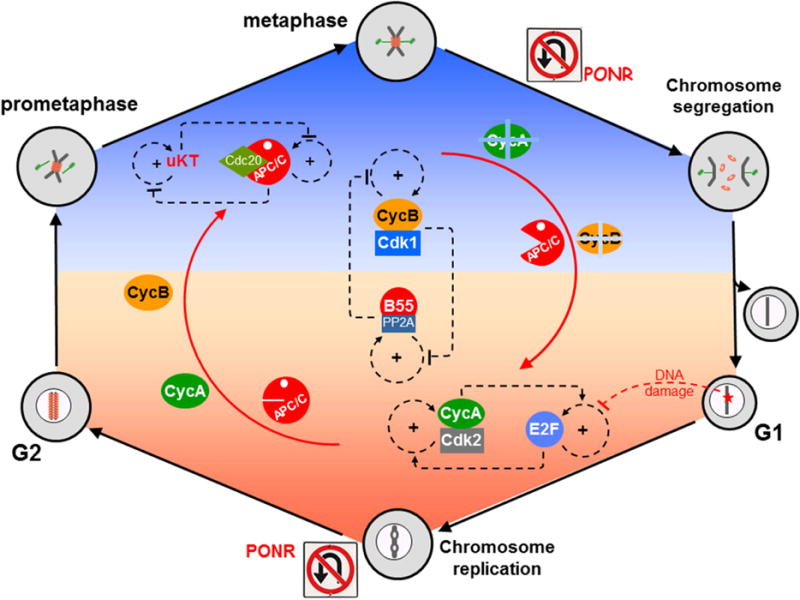
The basic alternation between interphase (orange) and mitosis (blue) is governed by the interplay between Cyclin B-dependent kinase (CycB:Cdk1) and B55-dependent protein phosphatase 2A (B55:PP2A). Each of these components is the output of a bistable switch, and their mutual antagonism ensures that if CycB:Cdk1 is active then B55:PP2A is inactive (the blue phase), and vice versa in the orange phase. Within the orange phase (interphase), the cell’s commitment to a new round of DNA replication is governed by another pair of bistable switches, based on activation of E2F (a transcription factor) at the restriction point (RP) and on the downstream activation of CycA:Cdk2, a kinase that initiates S phase by phosphorylating proteins at origins of replication on the DNA. These two bistable switches are linked by mutual activation. DNA damage imposes a checkpoint at RP by preventing the activation and accumulation of E2F, which in turn prevents accumulation of Cyclin A and entry into S phase. If and when the damage is repaired, the checkpoint is lifted, E2F is turned ON, Cyclin A accumulates, and the cell enters S phase. CycA:Cdk2 locks the E2F switch in the E2Fhigh position, preventing DNA damage signals from blocking progression through S phase. Within the blue phase (mitosis), a third pair of bistable switches implements the ‘spindle assembly checkpoint’ and progression from prometaphase to anaphase. The first element of this pair controls signaling from unattached kinetochores (uKT). It is a surveillance-mechanism/error-correction module that maintains uKT signaling as long as some of the replicated chromosomes are not bioriented on the mitotic spindle. uKTs promote assembly of a mitotic checkpoint complex, which binds to the APC/C and blocks the second bistable switch in the APC/Clow position. When all chromosomes are properly aligned on the spindle, the APC switch can flip to its APC/Chigh position and promote entry into anaphase. In its APC/Chigh position, the second switch locks down the uKT switch in its uKT = 0 state, thereby preventing the cell from slipping back from anaphase into prometaphase. PONR = point-of-no-return.
In Figures 2A and 4 we have drawn the regulatory motif in G1 phase as a pair of mutually activating, bistable switches for E2F and CycA:Cdk2 because it is conventional to view the motif from E2F’s perspective. It is important to realize however that E2F and RB are locked in a mutually inhibitory loop (Figure 2B), so we could just as well replace E2F in Figure 2A and 4 by RB, in which case RB and CycA:Cdk2 would compose a pair of mutually inhibitory, bistable switches controlling the G1/S transition. The crucial feature in all three cases is that the pair of bistable switches are themselves coupled in a bistable loop of mutual inhibition (or activation).
The bistable properties of these switches are essential for maintaining unidirectional progression through the cell division cycle, while allowing damage-sensing signals to halt progression and permissive signals (growth factors, cytokines) to push quiescent cells into the cycle. The processing of DNA-damage signals and chromosome-alignment signals is especially important for ensuring the integrity of the cell’s genome from one generation to the next. The decisions that a cell must make in response to these signals are intimately tied to the nature of ‘irreversible’ transitions governed by (bistable) toggle switches.
Nonetheless, the ‘irreversibility’ of these switches is tempered by the mutually antagonistic coupling of the dual switches (the surveillance mechanism and the checkpoint mechanism) at the G1/S and metaphase/anaphase transitions. This mode of coupling creates ‘transient states’ between the restriction point and the G1/S transition (sometimes called ‘late G1’) and between chromosome biorientation and anaphase onset (i.e., ‘metaphase’). During these transient states, the error-detection systems are still re-imposable (reversible). Only after the second switch is thrown (initiation of DNA synthesis and cohesin cleavage, respectively) are the switches locked down irreversibly, and these lock-downs are identifiable as ‘points-of-no-return’ in the cell cycle. These interplays between reversibility and irreversibility of cell cycle transitions, due to the complex topologies of the regulatory networks underlying cell cycle progression, seem to play crucial roles in the information processing by cells to maintain genome stability during cell proliferation.
Highlights.
Cell cycle transitions share common motif comprised of two nested bistable switches.
Interphase/mitosis transitions are governed by coupled kinase-phosphatase switches.
Futile cycling (pointless ATP hydrolysis) is kept to a minimum during the cycle.
In interphase, the G1/S switch stabilizes passage through the restriction point.
In mitosis, a pair of bistable switches implements the spindle assembly checkpoint.
Acknowledgments
This review article was written while JJT was on sabbatical in the Department of Biochemistry at the University of Oxford, made possible by generous support from Merton College and Virginia Tech. JJT also acknowledges financial support from the US National Institutes of Health (5R01-GM078989-11) administered through Colorado State University (PI: Jean Peccoud). FSH and BN are funded by the BBSRC Strategic LoLa grant (BB/M00354X/1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morgan DO. The Cell Cycle: Principles of Control. London: New Science Press; 2007. [Google Scholar]
- 2.Nasmyth K. Viewpoint: putting the cell cycle in order. Science. 1996;274:1643–1645. doi: 10.1126/science.274.5293.1643. [DOI] [PubMed] [Google Scholar]
- 3.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 4.Novak B, Tyson JJ. Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos. J Cell Sci. 1993;106:1153–1168. doi: 10.1242/jcs.106.4.1153. [DOI] [PubMed] [Google Scholar]
- 5.Novak B, Tyson JJ, Gyorffy B, Csikasz-Nagy A. Irreversible cell-cycle transitions are due to systems-level feedback. Nat Cell Biol. 2007;9:724–728. doi: 10.1038/ncb0707-724. [DOI] [PubMed] [Google Scholar]
- 6.Tyson JJ, Novak B. Temporal organization of the cell cycle. Curr Biol. 2008;18:R759–R768. doi: 10.1016/j.cub.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutter LH, Rata S, Hochegger H, Novak B. Interlinked bistable mechanisms generate robust mitotic transitions. Cell Cycle. 2017:1–8. doi: 10.1080/15384101.2017.1371885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nurse P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344:503–508. doi: 10.1038/344503a0. [DOI] [PubMed] [Google Scholar]
- 9.Barr FA, Elliott PR, Gruneberg U. Protein phosphatases and the regulation of mitosis. J Cell Sci. 2011;124:2323–2334. doi: 10.1242/jcs.087106. [DOI] [PubMed] [Google Scholar]
- 10.Pomerening JR, Sontag ED, Ferrell JE., Jr Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat Cell Biol. 2003;5:346–351. doi: 10.1038/ncb954. [DOI] [PubMed] [Google Scholar]
- 11.Sha W, Moore J, Chen K, Lassaletta AD, Yi CS, Tyson JJ, Sible JC. Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts. Proc Natl Acad Sci U S A. 2003;100:975–980. doi: 10.1073/pnas.0235349100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12•.Mochida S, Rata S, Hino H, Nagai T, Novak B. Two Bistable Switches Govern M Phase Entry. Curr Biol. 2016 doi: 10.1016/j.cub.2016.10.022. The hysteresis characteristic of the PP2A:B55 regulatory network was demonstrated with a reconstituted system and mathematical modelling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13•.Araujo AR, Gelens L, Sheriff RS, Santos SD. Positive Feedback Keeps Duration of Mitosis Temporally Insulated from Upstream Cell-Cycle Events. Mol Cell. 2016;64:362–375. doi: 10.1016/j.molcel.2016.09.018. Robust progression through mitosis is provided by feedback-amplified Cdk1 activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell-Crowley L, Swindell E, et al. Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang HW, Chung M, Kudo T, Meyer T. Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature. 2017;549:404–408. doi: 10.1038/nature23880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao G, Lee TJ, Mori S, Nevins JR, You L. A bistable Rb-E2F switch underlies the restriction point. Nat Cell Biol. 2008;10:476–482. doi: 10.1038/ncb1711. [DOI] [PubMed] [Google Scholar]
- 17.Yao G. Modelling mammalian cellular quiescence. Interface Focus. 2014;4:20130074. doi: 10.1098/rsfs.2013.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, Meyer T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell. 2013;155:369–383. doi: 10.1016/j.cell.2013.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19•.Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, Novak B, Bakal C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat Commun. 2017;8:14728. doi: 10.1038/ncomms14728. This paper and ref [20] show that DNA damage inherited by a daughter cell from its mother activates p21 and causes G1 arrest in a proliferating population. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20•.Arora M, Moser J, Phadke H, Basha AA, Spencer SL. Endogenous Replication Stress in Mother Cells Leads to Quiescence of Daughter Cells. Cell Rep. 2017;19:1351–1364. doi: 10.1016/j.celrep.2017.04.055. See ref [19]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barr AR, Heldt FS, Zhang T, Bakal C, Novak B. A Dynamical Framework for the All-or-None G1/S Transition. Cell Syst. 2016;2:27–37. doi: 10.1016/j.cels.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Primorac I, Musacchio A. Panta rhei: the APC/C at steady state. J Cell Biol. 2013;201:177–189. doi: 10.1083/jcb.201301130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pines J. Cubism and the cell cycle: the many faces of the APC/C. Nat Rev Mol Cell Biol. 2011;12:427–438. doi: 10.1038/nrm3132. [DOI] [PubMed] [Google Scholar]
- 24••.Cappell SD, Chung M, Jaimovich A, Spencer SL, Meyer T. Irreversible APC(Cdh1) Inactivation Underlies the Point of No Return for Cell-Cycle Entry. Cell. 2016;166:167–180. doi: 10.1016/j.cell.2016.05.077. The authors show that DNA damage or exposure to stress in G1 can revert cells back to a quiescent state until APC/CCdh1 is inactivated, constituting a point-of-no-return for cell cycle entry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller JJ, Summers MK, Hansen DV, Nachury MV, Lehman NL, Loktev A, Jackson PK. Emi1 stably binds and inhibits the anaphase-promoting complex/cyclosome as a pseudosubstrate inhibitor. Genes Dev. 2006;20:2410–2420. doi: 10.1101/gad.1454006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Havens CG, Walter JC. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011;25:1568–1582. doi: 10.1101/gad.2068611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Musacchio A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr Biol. 2015;25:R1002–1018. doi: 10.1016/j.cub.2015.08.051. [DOI] [PubMed] [Google Scholar]
- 28.Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet. 2009;43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- 29•.Izawa D, Pines J. The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature. 2015;517:631–634. doi: 10.1038/nature13911. By its two Cdc20 binding sites, MCC can both titrate away Cdc20 from APC/C and inhibit the activity of preformed APC/C-Cdc20 complexes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30••.Kamenz J, Mihaljev T, Kubis A, Legewie S, Hauf S. Robust Ordering of Anaphase Events by Adaptive Thresholds and Competing Degradation Pathways. Mol Cell. 2015;60:446–459. doi: 10.1016/j.molcel.2015.09.022. The competition of securin and CycB for APC/CCdc20 provides a robust mechanism for temporal ordering of anaphase and exit from mitosis. [DOI] [PubMed] [Google Scholar]
- 31.Uhlmann F, Lottspeich F, Nasmyth K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature. 1999;400:37–42. doi: 10.1038/21831. [DOI] [PubMed] [Google Scholar]
- 32.Tubman ES, Biggins S, Odde DJ. Stochastic Modeling Yields a Mechanistic Framework for Spindle Attachment Error Correction in Budding Yeast Mitosis. Cell Syst. 2017;4:645–650 e645. doi: 10.1016/j.cels.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang T, Oliveira RA, Schmierer B, Novak B. Dynamical scenarios for chromosome biorientation. Biophys J. 2013;104:2595–2606. doi: 10.1016/j.bpj.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalantzaki M, Kitamura E, Zhang T, Mino A, Novak B, Tanaka TU. Kinetochore-microtubule error correction is driven by differentially regulated interaction modes. Nat Cell Biol. 2015;17:421–433. doi: 10.1038/ncb3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dick AE, Gerlich DW. Kinetic framework of spindle assembly checkpoint signalling. Nat Cell Biol. 2013;15:1370–1377. doi: 10.1038/ncb2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vazquez-Novelle MD, Mirchenko L, Uhlmann F, Petronczki M. The ‘anaphase problem’: how to disable the mitotic checkpoint when sisters split. Biochem Soc Trans. 2010;38:1660–1666. doi: 10.1042/BST0381660. [DOI] [PubMed] [Google Scholar]
- 37.Oliveira RA, Hamilton RS, Pauli A, Davis I, Nasmyth K. Cohesin cleavage and Cdk inhibition trigger formation of daughter nuclei. Nat Cell Biol. 2010;12:185–192. doi: 10.1038/ncb2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rattani A, Vinod PK, Godwin J, Tachibana-Konwalski K, Wolna M, Malumbres M, Novak B, Nasmyth K. Dependency of the spindle assembly checkpoint on Cdk1 renders the anaphase transition irreversible. Curr Biol. 2014;24:630–637. doi: 10.1016/j.cub.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrell JE, Jr, Tsai TY, Yang Q. Modeling the cell cycle: why do certain circuits oscillate? Cell. 2011;144:874–885. doi: 10.1016/j.cell.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 40.Gharbi-Ayachi A, Labbe JC, Burgess A, Vigneron S, Strub JM, Brioudes E, Van-Dorsselaer A, Castro A, Lorca T. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science. 2010;330:1673–1677. doi: 10.1126/science.1197048. [DOI] [PubMed] [Google Scholar]
- 41.Mochida S, Maslen SL, Skehel M, Hunt T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science. 2010;330:1670–1673. doi: 10.1126/science.1195689. [DOI] [PubMed] [Google Scholar]
- 42.Williams BC, Filter JJ, Blake-Hodek KA, Wadzinski BE, Fuda NJ, Shalloway D, Goldberg ML. Greatwall-phosphorylated Endosulfine is both an inhibitor and a substrate of PP2A-B55 heterotrimers. Elife. 2014;3:e01695. doi: 10.7554/eLife.01695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hopkins M, Tyson JJ, Novak B. Cell cycle transitions: a common role for stoichiometric inhibitors. Mol Biol Cell. 2017 doi: 10.1091/mbc.E17-06-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trends Biochem Sci. 2004;29:409–417. doi: 10.1016/j.tibs.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 45.Mirkovic M, Hutter LH, Novak B, Oliveira RA. Premature Sister Chromatid Separation Is Poorly Detected by the Spindle Assembly Checkpoint as a Result of System-Level Feedback. Cell Rep. 2015;13:470–478. doi: 10.1016/j.celrep.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 46.Vazquez-Novelle MD, Sansregret L, Dick AE, Smith CA, McAinsh AD, Gerlich DW, Petronczki M. Cdk1 inactivation terminates mitotic checkpoint surveillance and stabilizes kinetochore attachments in anaphase. Curr Biol. 2014;24:638–645. doi: 10.1016/j.cub.2014.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He E, Kapuy O, Oliveira RA, Uhlmann F, Tyson JJ, Novak B. System-level feedbacks make the anaphase switch irreversible. Proc Natl Acad Sci U S A. 2011;108:10016–10021. doi: 10.1073/pnas.1102106108. [DOI] [PMC free article] [PubMed] [Google Scholar]