

Abstract
Cadmium (Cd) is a harmful heavy metal, which causes severe brain damage and neurotoxic effects. Polydeoxyribonucleotide (PDRN) stimulates adenosine A2A receptor, thus contrasting several deleterious mechanisms in course of tissue damages. We aimed to investigate the possible neuroprotective effect of PDRN in a murine model of Cd-induced brain toxicity. Male C57 BL/6J mice were treated as follows: vehicle (0.9% NaCl, 1 ml/kg/day), PDRN (8 mg/kg/day), CdCl2 (2 mg/kg/day), and CdCl2 + PDRN. Animals were tested with the Morris water maze test to assess spatial memory and learning. After 14 days of treatment, brains were processed to evaluate the presence of edema in the cerebral tissue, the expression of mammalian target of rapamycin kinase (mTOR) and brain-derived neurotrophic factor (BDNF), and the morphological behavior of the hippocampal structures. After CdCl2 administration, the escape latency was high, protein expression of BDNF was significantly decreased if compared to controls, mTOR levels were higher than normal controls, and brain edema and neuronal damages were evident. The coadministration of CdCl2 and PDRN significantly diminished the escape latency, increased BDNF levels, and decreased protein expression of mTOR. Furthermore, brain edema was reduced and the structural organization and the number of neurons, particularly in the CA1 and CA3 hippocampal areas, were improved. In conclusion, a functional, biochemical, and morphological protective effect of PDRN against Cd induced toxicity was demonstrated in mouse brain.
1. Introduction
Cadmium (Cd) is an extremely toxic metal with no known necessary function in the human body. It represents serious hazard to human health, as stated by the International Agency for Research on Cancer [1]. Major sources of Cd are food, cigarette smoke, and recharged nickel-cadmium batteries [2]. Foods as cereals, vegetables, nuts and pulses, starchy roots, potatoes, and meat products are the main source of Cd exposure for the nonsmoking population [3].
Several reports studied Cd toxicity in the brain as a whole or in its specific regions. In particular, Cd can experimentally induce neurotoxic effects either in vitro or in vivo. In fact, damages referred to Cd challenge were observed in cortical and trigeminal neurons [4, 5], in anterior pituitary cells [6], in glioma and neuroblastoma cells [7], and in nerve-glial cell cultures [8]. Neurotoxic effects were also described in neonatal mouse [9] and in adult rat brain [10] and in diabetic rat optic nerve experimentally exposed to Cd [11].
To date, the pathophysiological mechanism of Cd brain toxicity is not completely defined, even if reactive oxygen species (ROS) generation probably plays a crucial role in the detrimental neurotoxic cascade triggered by Cd [12]. Specifically, under these conditions ROS can promote an exaggerated inflammatory response characterized by increased cytokine expression and intercellular adhesion molecule-1 (ICAM-1) upregulation [13], particularly through the activation of nuclear factor-κB (NF-κB) [13]. Moreover, a peculiar role in neurotoxic damage following Cd exposure is played by mitogen-activated protein kinases (MAPKs) [12] able to promote apoptosis [14], as well as by Akt/mammalian target of rapamycin (mTOR) signaling pathway activation, which controls neuron proliferation, growth, and survival [15, 16].
An impaired neurogenesis, with strongly reduced neuronal differentiation and axonogenesis, was observed as a result of Cd-induced neurotoxicity; therefore, neuronal death occurred [17].
So far, in the brain of mammals, an intricate crosstalk underlying both neuroinflammation and neurogenesis provides many possible molecular targets; they might be harmfully impacted by Cd but also, on other side, by suitable therapeutic approaches to counteract Cd-induced neurotoxic effects [17, 18].
Among the neurotrophic factors that support differentiation [19], maturation [20], and survival of neurons [21], the brain-derived neurotrophic factor (BDNF) has neuroprotective effects under adverse conditions, such as glutamatergic stimulation, neuroinflammation, cerebral ischemia, hypoglycemia, and neurotoxicity [22].
Adenosine A2A receptor (ADORA2A) plays a crucial role in many physiological responses and pathological conditions [23]. However, it is still unclear if the role of ADORA2A in the control of neuroprotection is mostly due to the different homeostatic roles of these receptors related with the control of metabolism, of neuron-glial communication, of neuroinflammation, or of the control of action of growth factors [23].
ADORA2A is colocalized with BDNF in the brain, and the functional interaction between ADORA2A stimulation and BDNF action has been proposed [24]. Experimental data indicate that ADORA2A activation is a crucial requisite for the functioning of neurotrophic receptors at synapses. This has been shown for the facilitatory actions of BDNF on synaptic transmission [25, 26] typically on prolonged potentiation at the CA1 area of the hippocampus [27].
Interestingly, a high prevalence of brain function disorders, including cognitive and behavioral impairments, has been associated with mTOR signaling disturbances [28]; in particular, mTOR activation was related to two major signaling pathways, Ras-ERK and PI3K-Akt, that essentially control neuron survival, differentiation, and proliferation in response to extracellular signals [29]. So far, extracellular messengers linked to mTOR activation may involve the adenosine pathway through ADORA2A modulation in response to systemic inflammation [30].
Polydeoxyribonucleotide (PDRN) is the active fraction extracted from trout spermatozoa used for tissue repair [31]; acting through stimulation of ADORA2A, it can well contrast several harmful mechanisms observed in pathological conditions of low tissue perfusion [31–35].
A positive role of PDRN on Cd-induced structural changes of the blood-testis barrier was already demonstrated, suggesting that it may have a positive effect against Cd-induced structural lesions on gametogenesis [36].
In light of this background, PDRN effects in the brain of mice exposed to Cd chloride (CdCl2) were investigated to better elucidate the role of this adenosine agonist.
2. Materials and Methods
2.1. Experimental Protocol
All procedures complied with the standards for care and use of animal subjects indicated by the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Academy of Sciences, Bethesda, MD, USA); they were carried out also in accordance with Directive 2010/63/EU on the protection of animals used for scientific experiments [37, 38]. Fifty-six male adult C57 BL/6J mice (25–30 g), obtained from Charles River Laboratories Italia srl (Calco, Italy), were provided a standard diet ad libitum with free access to tap water under a 12 h light/dark cycle. They were divided into four groups: (i) animals administered with a vehicle solution consisting in 0.9% NaCl (1 ml/kg, ip, daily), indicated as “control + vehicle animals,” (ii) animals administered with PDRN (8 mg/kg, ip daily), indicated as “control + PDRN animals,” (iii) animals challenged with CdCl2 plus with the vehicle as above (2 mg/kg, ip, daily), indicated as “CdCl2 + vehicle animals,” and (iv) animals challenged with CdCl2 (2 mg/kg, ip, daily) and treated with PDRN (8 mg/kg, ip, daily), immediately following CdCl2 administration, indicated as “CdCl2 + PDRN animals.”
2.2. Drugs
CdCl2 was purchased from Sigma-Aldrich Srl (Milan, Italy) and diluted to the requested concentration in 0.9% NaCl. PDRN was donated by Mastelli Srl (Sanremo, Italy). All chemicals and reagents were of commercially available reagent grades.
2.3. Assessment of Cognitive Performance
To assess spatial memory and learning, animals were tested with the Morris water maze (MWM) test [39]. The test was performed in a round white pool (diameter 80 cm and depth 55 cm). The pool was filled to a depth of 30 cm with water made opaque with white nontoxic water-based tempura paint. Pool temperature was maintained at 22 ± 0.5°C by adding warm water. The escape platform was a 25 cm2 plexiglas square, placed in the center of one quadrant of the pool, 15 cm from the pool's edge, and submerged 1 cm below the water surface. The mouse was gently placed in the water pool between the quadrants, facing the wall of the pool changing the order every day during each trial. The mice were given four trial sessions each day for five consecutive days, with an intertrial interval of 15 min. Escape latency time (ELT), that is, the time taken by the animal to move from the starting quadrant to find the hidden platform in the target quadrant, was recorded in each trial, and the average time, expressed in seconds (s), for each day was calculated. If the mouse failed to find the platform within 60 s, it was guided gently onto the platform and allowed to remain there for 20 s. Significant decrease in ELT from that of the first session was considered as successful learning. During all trials, the experimenter always stood in the same position. All trials were performed between 9.00 and 16.00 h in a sound dampened room.
2.4. Brain Collection
The experiments lasted 14 days, until the mice were sacrificed with an ip overdose of ketamine and xylazine (100/20 mg/kg, resp.) and then subjected to decapitation. Their skulls were quickly opened, and the brains were extracted on ice and washed with cold phosphate-buffered saline (PBS). The brains of 14 animals for each group were divided as follows: seven brains were used for histological study. From the other seven brains, one half was stored at −80°C for Western blot analysis, and one half was used for edema evaluation.
2.5. Evaluation of Brain Edema
To evaluate the extent of edema, brain sections from each group of animals were assayed for water content using wet weight/dry weight. Freshly dissected tissue samples of the hippocampus were weighed on aluminum foil, dried for 24 h at 105°C, and reweighed as previously described [40]. The percentage of water was calculated as follows: water content (%) = (wet weight − dry weight)/wet weight × 100.
2.6. Histological Evaluation
Brains were immediately fixed in 4% paraformaldehyde in 0.2 M phosphate buffer solution (PBS), dehydrated in graded ethanol, cleared in xylene, and embedded in paraffin (Paraplast, Supplies SPI, West Chester, PA, USA). 5 μm coronal sections, cut with a RM2125 RT microtome (Leica Instruments, Nußloch, Germany), were cleared with xylene, rehydrated in ethanol, and stained with hematoxylin and eosin (H&E). Histological identification of nervous structures was made according to the atlas of Franklin and Paxinos [41], and the slides were photographed with a Nikon Ci-L (Nikon Instruments, Tokyo, Japan) light microscope; the images were taken with a digital camera Nikon Ds-Ri2 and processed to the final magnification of 800x.
2.7. Morphometric Evaluation
Five not serial sections per animal were evaluated for each group. Two experienced investigators, blinded to the experimental group of each animal, independently performed cell counting. The results gave an intraobserver and interobserver variation less than 5%. For hippocampal neurons counts, a region of interest (unit area (UA)) of 0.1 mm2 (316 × 316 μm) in both the CA1 and CA3 regions was selected for each section; the cells overlapping the left and the bottom boundaries were counted, whereas the cells that touched the right and top boundaries were not included in the evaluation. Criteria for neurons to be counted were well-defined cytoplasm, clearly visible nucleus, and evident nucleolus.
2.8. Determination of Protein Content
Total cellular proteins were extracted in a lysis buffer composed of 25 mM Tris-HCl pH 7.4, 1.0 mM ethylene glycol tetraacetic acid (EGTA), 1.0 mM ethylenediaminetetraacetic acid (EDTA), and 0.5 mM phenyl methylsulphonyl fluoride, added with protease and phosphatase inhibitors (100 mM Na3VO4, aprotinin, leupeptin, and pepstatin (10 μg/ml each)). After centrifugation of the cell lysate for 15′ at 13000 rpm, the protein concentration was determined from the supernatant by Bio-Rad protein assay (Bio-Rad, Richmond, CA, USA).
2.9. Malondialdehyde (MDA) and Glutathione (GSH) Determination
MDA content was determined in all experimental groups with a colorimetric commercial kit (Lipid Peroxidation Assay kit, cat number 437634; Calbiochem-Novabiochem Corp, Darmstadt, Germany), as previously described [42], and expressed in nmol/mg protein. GSH content was also determined in all experimental groups according to the method of Gong et al. [43].
2.10. Determination of BDNF and mTOR by Western Blot Analysis
The supernatant was diluted with Laemmli buffer. Protein samples, denatured in reducing buffer (62 mM Tris-HCl pH 6.8, 10% glycerol, 2% SDS, 5% beta-mercaptoethanol, and 0.003% bromophenol blue), were separated by electrophoresis on SDS polyacrylamide gel (6% or 10%), for approximately 1 h. The separated proteins were moved to a PVDF membrane in a transfer buffer (39 mM glycine, 48 mM Tris-HCl (pH 8.3), and 20% methanol) at 200 mA for 1 h. The reaction was blocked with 5% nonfat dry milk in TBS-0.1% Tween-20 for 1 h at room temperature. Membranes were washed three times for 10 min each in TBS-0.1% Tween-20 and incubated with primary antibodies for mTOR (1 : 500 in TBS-0.1% Tween-20; Cell Signaling, Beverly, MA, USA) and BDNF (1 : 1000 in TBS-0.1% Tween-20; Abcam, Cambridge, UK). The following day, the membranes were washed three times for 10 min in TBS-0.1% Tween-20 and were incubated with a specific peroxidase-conjugated secondary antibody (1 : 10.000; KPL, USA) for 1 h at room temperature. After further washings, the membranes were analyzed by enhanced chemiluminescence (KPL, USA). Protein signals were quantified by scanning densitometry with a Bio Image Analysis system (C-DiGit Blot Scanner with Image Studio software), and the results were expressed as relative integrated intensity compared to controls. α-Tubulin (Cell Signaling Technology, Beverly, MA, USA) was used to confirm equal protein loading and blotting.
2.11. Statistical Analysis
Primary outcome measures were assessment of cognitive performance and neuron morphology. The statistical significance of differences among groups was performed with the ANOVA comparison test, followed by the Bonferroni post hoc test. The MedCalc 12.2.1.0 statistical software (MedCalc Software, Ostend, Belgium) was used. A p value ≤0.05 was considered statistically significant. Values are provided as mean ± standard deviation (SD).
3. Results
3.1. Effects of PDRN on MDA and GSH Content
The levels of MDA were significantly increased in Cd-challenged mice. The coadministration of CdCl2 and PDRN significantly decreased the levels of MDA in brains (Table 1). On the contrary, a significant decrease in the activity of GSH was observed in Cd-challenged mice. The treatment with PDRN significantly increased GSH levels in brains of Cd-treated mice (Table 1).
Table 1.
Malondialdehyde (MDA) and glutathione (GSH) content in mice exposed to cadmium chloride (CdCl2; 2 mg/kg ip) plus vehicle, as compared to mice exposed to CdCl2 (2 mg/kg ip) plus PDRN (8 mg/kg/day ip) or to control mice treated with vehicle or PDRN alone.
| Group | MDA (nmol/mg protein) | GSH (μmol/g tissue) |
|---|---|---|
| Control + vehicle | 0.13 ± 0.04 | 66 ± 5 |
| Control + PDRN | 0.12 ± 0.06 | 69 ± 6 |
| CdCl2 + vehicle | 0.81 ± 0.31a | 47 ± 7a |
| CdCl2 + PDRN | 0.20 ± 0.09b | 64 ± 3b |
All the values are expressed as mean ± SD, n = 7 animals for each group. ap < 0.05 versus both controls; bp < 0.05 versus CdCl2 + vehicle.
3.2. Effects of PDRN on BDNF and mTOR Brain Expression
The expression of BDNF was observed in the brain of control mice treated with vehicle or PDRN (Figure 1(a)). CdCl2 caused a marked reduction on BDNF brain expression in mice (Figure 1(a)). Conversely, in mice treated with PDRN, the brain levels of BDNF were significantly higher than in the vehicle-treated CdCl2 group (Figure 1(a)).
Figure 1.

Representative Western blot analysis of BDNF (a) and mTOR (b) in brains of controls and CdCl2- (2 mg/kg ip) challenged mice treated with vehicle or PDRN (8 mg/kg ip), respectively. ∗p < 0.05 versus both controls; §p < 0.05 versus CdCl2 + vehicle. Bars indicate mean ± SD of 7 experiments.
Low expression of mTOR was observed in the brain of control mice treated with vehicle or PDRN (Figure 1(b)). A higher expression of mTOR was detected in CdCl2-treated animals (Figure 1(b)). mTOR expression was significantly reduced after PDRN administration if compared to mice treated with CdCl2 alone (Figure 1(b)).
3.3. Brain Edema Assessment
No differences in brain water content were observed in both controls of hippocampal tissue (Figure 2). CdCl2 challenge caused brain edema in the mouse hippocampus (Figure 2). PDRN treatment showed a significant reduction of brain edema when compared to CdCl2-treated animals (Figure 2).
Figure 2.
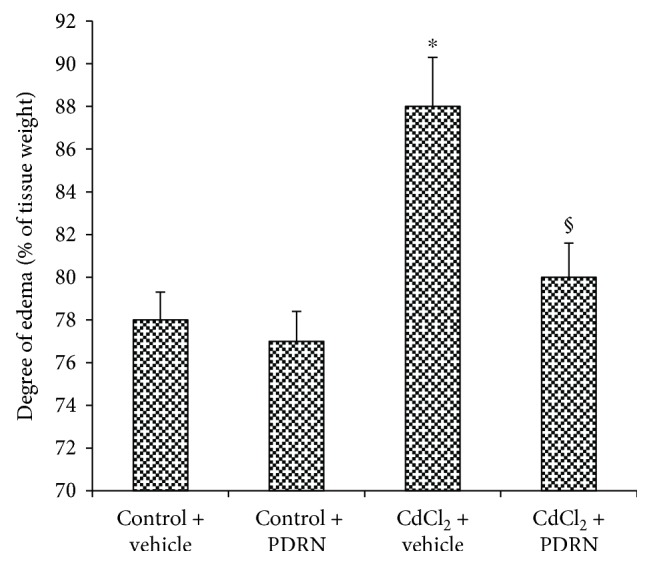
Brain edema evaluated through water content in the hippocampus of controls and CdCl2 (2 mg/kg ip) challenged mice treated with vehicle or PDRN (8 mg/kg ip), respectively. ∗p < 0.05 versus both controls; §p < 0.05 versus CdCl2 + vehicle. Bars indicate mean ± SD of 7 experiments.
3.4. Administration of PDRN Counteracts CdCl2-Induced Neuronal Changes
In both control groups of mice, CA1 and CA3 hippocampal regions showed normal organization (Figures 3(a), A1, A2, and 3b, B1, B2). In contrast, CA1 and CA3 regions of CdCl2-challenged mice showed evident neuronal loss with degenerating pyramidal cells and interstitial edema (Figure 3(c), C1, C2). PDRN administration significantly reduced neuronal morphological changes in both CA1 and CA3 regions (Figure 3(d), D1, D2). The morphometric analysis showed a significant reduction of neurons in both the CA1 and CA3 regions in CdCl2-challenged mice, which was normalized when PDRN was coadministered (Figure 3(e)).
Figure 3.

Structural organization of the hippocampus from mice of control plus vehicle (0.9% NaCl, 1 ml/kg/day ip), control plus PDRN (8 mg/kg/day ip), CdCl2 (2 mg/kg/day ip) plus vehicle, and CdCl2 plus PDRN (HE stain). (a, A1, A2, b, B1, B2) In both control plus vehicle and control plus PDRN-treated mice, the normal morphology of the nervous tissue of the hippocampus, particularly in CA1 (arrowhead) and CA3 (arrow) areas, is evident. (c, C1, C2) In CdCl2 plus vehicle-treated mice, neuronal loss and mild edema of the nervous tissue of the hippocampus, particularly in CA1 (arrowhead) and CA3 (arrow) areas, are evident. (d, D1, D2) In CdCl2 plus PDRN-treated mice, the hippocampus and CA1 (arrowhead) and CA3 (arrow) areas in particular show a well-preserved neuronal architecture. (e) Quantitative evaluation of neurons in both CA1 and CA3 regions in the different groups of mice. ∗p < 0.05 versus both controls; †p < 0.05 versus CdCl2 plus vehicle (scale bar: a, b, c, d = 500 μm; A1, A2, B1, B2, C1, C2, D1, D2 = 50 μm).
3.5. PDRN Enhances Cognitive Performance
In both controls, a gradual shortening in ELT was observed along the five-consecutive-day trials (Table 2). CdCl2 administration significantly increased ELT when compared with both control groups (Table 2). On the contrary, PDRN administration significantly reduced the time spent by mice to find the platform (Table 2).
Table 2.
Results obtained from the escape latency time (the time to reach the platform in seconds) evaluated with the Morris water maze test in mice exposed to cadmium chloride (CdCl2; 2 mg/kg/day ip) plus vehicle, as compared to control mice treated with vehicle or PDRN alone or to mice exposed to CdCl2 (2 mg/kg/day ip) plus PDRN (8 mg/kg/day ip).
| Group | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 |
|---|---|---|---|---|---|
| Control + vehicle | 36 ± 2 | 31 ± 3 (13.4%)a | 19 ± 2 (47.3%)a,b | 14 ± 3 (61.2%)a,b,c | 11 ± 2 (69.5%)a,b,c,d |
| Control + PDRN | 34 ± 3 | 32 ± 3 (5.9%) | 18 ± 4 (47.1%)b | 15 ± 2 (55.9%)a,b,c | 12 ± 3 (64.8%)a,b,c,d |
| CdCl2 + vehicle | 38 ± 3 | 34 ± 4 (10.5%)a | 27 ± 3 (28.9%) a,b,e | 22 ± 3 (42.1%) a,b,c,e | 20 ± 4 (47.3%)a,b,c,e |
| CdCl2 + PDRN | 33 ± 4 | 32 ± 4 (3.1%) | 20 ± 2 (39.4%) b,f | 14 ± 4 (57.6%)a,b,c,f | 12 ± 2 (63.7%)a,b,c,f |
All the values are expressed as mean ± SD, n = 14 animals for each group. ap < 0.05 versus day 1 of the same group; bp < 0.05 versus day 2 of the same group; cp < 0.05 versus day 3 of the same group; dp < 0.05 versus day 4 of the same group; ep < 0.05 versus both controls at the same day; fp < 0.05 versus CdCl2 + vehicle at the same day.
4. Discussion
Oxidative stress is strongly related to neuroinflammatory mechanisms, so that it is particularly difficult to exert neuroprotective effects on the brain. In addition, glial activation involving astrocytes, microglial cells, and/or reactive mediators and/or growth factors are also hallmarks of inflammatory reaction [44]. This justifies the research strategies that rely on multiple mechanisms involving antiradical scavenging activity and antiapoptotic mechanisms, resulting in increased neuronal proliferation.
Neurotoxic effects may play a key role in the systemic toxic consequences of Cd exposure [45]. Therefore, the mechanism of Cd neurotoxicity should be better clarified, and measures should be taken to reduce Cd exposure in the general population to minimize the risk of adverse human health effects [46]. In this context, we previously demonstrated a positive effect of PDRN, an ADORA2A, which was demonstrated on Cd-induced damages of the blood-testis barrier, suggesting that it should also counteract the role of Cd as an endocrine disruptor [36].
Therefore, our data represent novel findings on the effects of PDRN on the brain, since few information on the molecular pathways involved are currently available.
Indeed, in our in vivo experimental model, we observed that mice challenged with CdCl2 alone showed a significant increase of MDA and a decrease of GSH; on the contrary, PDRN administration protected mice against oxidative stress, thus confirming the harmful role of Cd in triggering the lipid peroxidation pathways [36].
Furthermore, an increased expression of mTOR in CdCl2-challenged mice was demonstrated, whereas PDRN administration significantly reduced mTOR expression. This feature strongly suggests that Cd-induced neuronal toxicity is related to induction of ROS, which, in turn, leads to oxidative stress. In fact, it has been recently shown that Cd induces ROS generation in a time- and concentration-dependent manner in PC12 and SH-SY5Y cells [47], causing apoptosis of neuronal cells, particularly via activation of MAPKs and mTOR signaling pathways [12, 14–16, 47].
Accordingly, we observed that PDRN administration significantly increased BDNF levels in mice. It has been shown that BDNF in vivo can rescue different types of neurons from ischemic, traumatic, and toxic brain injury [48]. Recent evidences indicate that the protective effect of BDNF in hippocampal neurons against toxicity is mostly mediated by the PI3K and the Ras/MAPK signaling pathways and involves a long-term change in protein synthesis [29].
Moreover, the role of serine/threonine protein kinase mTOR was also considered [49]. In particular, it has been suggested that mTOR affects the translational control of proteins necessary for the formation and functional maturation of dendritic spines [50]. Moreover, it has been proposed that the neuroprotective effect of BDNF is mediated by autophagy through the PI3K/Akt/mTOR pathway [51]. Our results also show that PDRN administration resulted in a significant reduction of brain edema when compared to the water content of CdCl2-treated animals. These effects may be strictly linked with previous explored molecular pathways because it has been suggested that ROS, cytokine overproduction, and neurotrophin reduction are strongly related to brain edema formation in animals challenged with neurotoxic agents [40, 52, 53]. Furthermore, in humans, acute Cd toxicity led to brain intracellular accumulation of the metal with consequent cell dysfunction, blood-brain barrier disruption, and even lethal cerebral edema [54]. It is likely that PDRN can also reinforce the detoxification mechanisms, such as antioxidant systems through the induction of protective macromolecules (heat shock proteins, etc.), production of specific metal inclusion bodies or binding proteins, and biotransformation reactions (methylation, conjugation, etc.) localized in the choroid plexus [17]. Accordingly, ADORA2A was highly expressed in the choroid plexus [55].
The biochemical and molecular patterns correlated very well with the histological analysis. In fact, following CdCl2 administration, we observed a significant neuronal loss in both CA1 and CA3 areas, which are susceptible to Cd-induced neurotoxic injury [56]. In contrast, PDRN administration showed significant neuroprotective effects, with a normal number of neurons/UA and structural organization.
Finally, the positive effects of PDRN treatment were also supported in our model by the evidence of a good protection against the behavioral changes that accompanied Cd administration. In fact, PDRN injection significantly improved ELT in mice tested with MWM following CdCl2 challenge. This observation could have a strong translational impact considering that Cd causes learning disabilities and hyperactivity in environmentally exposed children [17] and neurological disorders, such as amyotrophic lateral sclerosis [57], Parkinsonism [58], and Parkinson's and Alzheimer's disease [59], in occupationally exposed subjects.
Taken together, our data suggest that adenosine receptor manipulation/modulation is a pertinent avenue of research for novel strategies in order to modulate neuroinflammatory signal into the brain in diseases characterized by impaired immune response induced by toxic agents.
Moreover, in light of our results, we feel that new environmental research on Cd should take aim at the role of neurotoxicity in causing the health effects following Cd exposure.
Both short- and long-duration epidemiological studies are required to determine the optimal doses of antioxidant products and dietary supplements alone and in combination, to provide safe and effective therapeutic strategies against Cd toxicity. In this context, PDRN, an agonist of ADORA2A, might offer a structural model for the production of new analog compounds (cosmeceuticals, nutraceuticals, and/or phytochemicals) that, properly combined with good agricultural practice to minimize Cd contamination in food crops and animals, could also provide a definite strategy to prevent and counteract severe damages in Cd-induced brain toxicity.
Acknowledgments
The investigation was granted by a departmental funding. The authors thank Mister Sebastiano Brunetto of the Department of Biomedical Sciences, University of Messina, for his dedicated technical expertise.
Abbreviations
- CdCl2:
Cadmium chloride
- ROS:
Reactive oxygen species
- HE:
Hematoxylin and eosin
- MAPK:
Mitogen-activated protein kinase
- MDA:
Malondialdehyde
- GSH:
Glutathione
- mTOR:
Mammalian target of rapamycin kinase
- BDNF:
Brain-derived neurotrophic factor
- ADORA2A:
Adenosine receptor A2A
- PDRN:
Polydeoxyribonucleotide.
Data Availability
The data used to support the findings of this study are included within the article.
Disclosure
The paper was published as an abstract in the Proceedings of the Italian Society of Pharmacology, 38th National Meeting, 2017.
Conflicts of Interest
The authors declare no actual or potential competing financial interests.
References
- 1.ATSDR. Agency for Toxic Substance and Disease Registry. Atlanta: U.S. Toxicological Profile for Cadmium, Department of Health and Humans Services, Public Health Service, Centers for Disease Control; 2012. [Google Scholar]
- 2.Järup L., Akesson A. Current status of cadmium as an environmental health problem. Toxicology and Applied Pharmacology. 2009;238(3):201–208. doi: 10.1016/j.taap.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 3.Chunhabundit R. Cadmium exposure and potential health risk from foods in contaminated area, Thailand. Toxicological Research. 2016;32(1):65–72. doi: 10.5487/TR.2016.32.1.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.López E., Figueroa S., Oset-Gasque M. J., González M. P. Apoptosis and necrosis: two distinct events induced by cadmium in cortical neurons in culture. British Journal of Pharmacology. 2003;138(5):901–911. doi: 10.1038/sj.bjp.0705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Habeebu S. S., Liu Y., Park J. D., Klaassen C. D. Strain differences in the toxicity of cadmium to trigeminal ganglia in mice. Toxicology and Applied Pharmacology. 2001;177(3):200–207. doi: 10.1006/taap.2001.9311. [DOI] [PubMed] [Google Scholar]
- 6.Poliandri A. H., Cabilla J. P., Velardez M. O., Bodo C. C., Duvilanski B. H. Cadmium induces apoptosis in anterior pituitary cells that can be reversed by treatment with antioxidants. Toxicology and Applied Pharmacology. 2003;190(1):17–24. doi: 10.1016/S0041-008X(03)00191-1. [DOI] [PubMed] [Google Scholar]
- 7.Wätjen W., Beyersmann D. Cadmium-induced apoptosis in C6 glioma cells: influence of oxidative stress. BioMetals. 2004;17(1):65–78. doi: 10.1023/A:1024405119018. [DOI] [PubMed] [Google Scholar]
- 8.Sugawara N., Aoshima K., Kasuya M. Effect of cadmium chloride and Cd-metallothionein on the nervous tissue culture. Toxicology Letters. 1983;16(1-2):95–101. doi: 10.1016/0378-4274(83)90016-4. [DOI] [PubMed] [Google Scholar]
- 9.Webster W. S., Valois A. A. The toxic effects of cadmium on the neonatal mouse CNS. Journal of Neuropathology and Experimental Neurology. 1981;40(3):247–257. doi: 10.1097/00005072-198105000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Carageorgiou H., Tzotzes V., Pantos C., Mourouzis C., Zarros A., Tsakiris S. In vivo and in vitro effects of cadmium on adult rat brain total antioxidant status, acetylcholinesterase, (Na+, K+)-ATPase and Mg2+-ATPase activities: protection by L-cysteine. Basic & Clinical Pharmacology & Toxicology. 2004;94(3):112–118. doi: 10.1111/j.1742-7843.2004.pto940303.x. [DOI] [PubMed] [Google Scholar]
- 11.Demir N., Akkoyunlu G., Yargicoglu P., Agar A., Tanriöver G., Demir R. Fiber structure of optic nerve in cadmium-exposed diabetic rats: an ultrastructural study. The International Journal of Neuroscience. 2009;113(3):323–337. doi: 10.1080/00207450390162128. [DOI] [PubMed] [Google Scholar]
- 12.Liu J., Qu W., Kadiiska M. B. Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicology and Applied Pharmacology. 2009;238(3):209–214. doi: 10.1016/j.taap.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeong E. M., Moon C. H., Kim C. S., et al. Cadmium stimulates the expression of ICAM-1 via NF-κB activation in cerebrovascular endothelial cells. Biochemical and Biophysical Research Communications. 2004;320(3):887–892. doi: 10.1016/j.bbrc.2004.05.218. [DOI] [PubMed] [Google Scholar]
- 14.Qu W., Ke H., Pi J., et al. Acquisition of apoptotic resistance in cadmium-transformed human prostate epithelial cells: Bcl-2 overexpression blocks the activation of JNK signal transduction pathway. Environmental Health Perspectives. 2007;115(7):1094–1100. doi: 10.1289/ehp.10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan Y., Wang Y., Hu F. F., et al. Cadmium activates reactive oxygen species-dependent AKT/mTOR and mitochondrial apoptotic pathways in neuronal cells. Biomedical and Environmental Sciences. 2016;29(2):117–126. doi: 10.3967/bes2016.013. [DOI] [PubMed] [Google Scholar]
- 16.Chen L., Xu B., Liu L., et al. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radical Biology & Medicine. 2011;50(5):624–632. doi: 10.1016/j.freeradbiomed.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang B., Du Y. Cadmium and its neurotoxic effects. Oxidative Medicine and Cellular Longevity. 2013;2013:12. doi: 10.1155/2013/898034.898034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al Omairi N. E., Radwan O. K., Alzahrani Y. A., Kassab R. B. Neuroprotective efficiency of Mangifera indica leaves extract on cadmium-induced cortical damage in rats. Metabolic Brain Disease. 2018;33(4):1121–1130. doi: 10.1007/s11011-018-0222-6. [DOI] [PubMed] [Google Scholar]
- 19.Binder D. K., Scharfman H. E. Brain-derived neurotrophic factor. Growth Factors. 2009;22(3):123–131. doi: 10.1080/08977190410001723308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Acheson A., Conover J. C., Fandl J. P., et al. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374(6521):450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- 21.Huang E. J., Reichardt L. F. Neurotrophins: roles in neuronal development and function. Annual Review of Neuroscience. 2001;24(1):677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maisonpierre P. C., le Beau M. M., Espinosa R., III, et al. Human and rat brain-derived neurotrophic factor and neurotrophin-3: gene structures, distributions, and chromosomal localizations. Genomics. 1991;10(3):558–568. doi: 10.1016/0888-7543(91)90436-I. [DOI] [PubMed] [Google Scholar]
- 23.Gomes C. V., Kaster M. P., Tomé A. R., Agostinho P. M., Cunha R. A. Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2011;1808(5):1380–1399. doi: 10.1016/j.bbamem.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Jeon S. J., Rhee S. Y., Ryu J. H., et al. Activation of adenosine A2A receptor up-regulates BDNF expression in rat primary cortical neurons. Neurochemical Research. 2011;36(12):2259–2269. doi: 10.1007/s11064-011-0550-y. [DOI] [PubMed] [Google Scholar]
- 25.Diógenes M. J., Fernandes C. C., Sebastião A. M., Ribeiro J. A. Activation of adenosine A2A receptor facilitates brain-derived neurotrophic factor modulation of synaptic transmission in hippocampal slices. Journal of Neuroscience. 2004;24(12):2905–2913. doi: 10.1523/JNEUROSCI.4454-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tebano M. T., Martire A., Potenza R. L., et al. Adenosine A(2A) receptors are required for normal BDNF levels and BDNF-induced potentiation of synaptic transmission in the mouse hippocampus. Journal of Neurochemistry. 2008;104(1):279–286. doi: 10.1111/j.1471-4159.2007.05046.x. [DOI] [PubMed] [Google Scholar]
- 27.Fontinha B. M., Diógenes M. J., Ribeiro J. A., Sebastião A. M. Enhancement of long-term potentiation by brain-derived neurotrophic factor requires adenosine A2A receptor activation by endogenous adenosine. Neuropharmacology. 2008;54(6):924–933. doi: 10.1016/j.neuropharm.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 28.Borrie S. C., Brems H., Legius E., Bagni C. Cognitive dysfunctions in intellectual disabilities: the contributions of the Ras-MAPK and PI3K-AKT-mTOR pathways. Annual Review of Genomics and Human Genetics. 2017;18(1):115–142. doi: 10.1146/annurev-genom-091416-035332. [DOI] [PubMed] [Google Scholar]
- 29.Shaw R. J., Cantley L. C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y. W., Yang T., Zhao L., et al. Activation of adenosine 2A receptor inhibits neutrophil apoptosis in an autophagy-dependent manner in mice with systemic inflammatory response syndrome. Scientific Reports. 2016;6(1, article 33614) doi: 10.1038/srep33614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altavilla D., Bitto A., Polito F., et al. Polydeoxyribonucleotide (PDRN): a safe approach to induce therapeutic angiogenesis in peripheral artery occlusive disease and in diabetic foot ulcers. Cardiovascular & Hematological Agents in Medicinal Chemistry. 2009;7(4):313–321. doi: 10.2174/187152509789541909. [DOI] [PubMed] [Google Scholar]
- 32.Minutoli L., Arena S., Bonvissuto G., et al. Activation of adenosine A2A receptors by polydeoxyribonucleotide increases vascular endothelial growth factor and protects against testicular damage induced by experimental varicocele in rats. Fertility and Sterility. 2011;95(4):1510–1513. doi: 10.1016/j.fertnstert.2010.07.1047. [DOI] [PubMed] [Google Scholar]
- 33.Altavilla D., Squadrito F., Polito F., et al. Activation of adenosine A2A receptors restores the altered cell-cycle machinery during impaired wound healing in genetically diabetic mice. Surgery. 2011;149(2):253–261. doi: 10.1016/j.surg.2010.04.024. [DOI] [PubMed] [Google Scholar]
- 34.Minutoli L., Antonuccio P., Squadrito F., et al. Effects of polydeoxyribonucleotide on the histological damage and the altered spermatogenesis induced by testicular ischaemia and reperfusion in rats. International Journal of Andrology. 2012;35(2):133–144. doi: 10.1111/j.1365-2605.2011.01194.x. [DOI] [PubMed] [Google Scholar]
- 35.Minutoli L., Arena S., Antonuccio P., et al. Role of inhibitors of apoptosis proteins in testicular function and male fertility: effects of polydeoxyribonucleotide administration in experimental varicocele. BioMed Research International. 2015;2015:9. doi: 10.1155/2015/248976.248976 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Squadrito F., Micali A., Rinaldi M., et al. Polydeoxyribonucleotide, an adenosine-A2A receptor agonist, preserves blood testis barrier from cadmium-induced injury. Frontiers in Pharmacology. 2017;7 doi: 10.3389/fphar.2016.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. https://grants.nih.gov/grants/olaw/guide-for-the-care-and-use-of-laboratory-animals.pdf.
- 38. http://ec.europa.eu/environment/chemicals/lab_animals/legislation_en.htm.
- 39.Vorhees C. V., Williams M. T. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nature Protocols. 2006;1(2):848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Minutoli L., Marini H., Rinaldi M., et al. A dual inhibitor of cyclooxygenase and 5-lipoxygenase protects against kainic acid-induced brain injury. Neuromolecular Medicine. 2015;17(2):192–201. doi: 10.1007/s12017-015-8351-0. [DOI] [PubMed] [Google Scholar]
- 41.Franklin K. B. J., Paxinos G. The Mouse Brain in Stereotaxic Coordinates. 3rd. Amsterdam: Elsevier; 2007. [Google Scholar]
- 42.Minutoli L., Micali A., Pisani A., et al. Research article flavocoxid protects against cadmium-induced disruption of the blood-testis barrier and improves testicular damage and germ cell impairment in mice. Toxicological Sciences. 2015;148(1):311–329. doi: 10.1093/toxsci/kfv185. [DOI] [PubMed] [Google Scholar]
- 43.Gong P., Chen F., Liu X., Gong X., Wang J., Ma Y. Protective effect of caffeic acid phenethyl ester against cadmium-induced renal damage in mice. The Journal of Toxicological Sciences. 2012;37(2):415–425. doi: 10.2131/jts.37.415. [DOI] [PubMed] [Google Scholar]
- 44.Ientile R., Currò M., Caccamo D. Transglutaminase 2 and neuroinflammation. Amino Acids. 2015;47(1):19–26. doi: 10.1007/s00726-014-1864-2. [DOI] [PubMed] [Google Scholar]
- 45.Rinaldi M., Micali A., Marini H., et al. Cadmium, organ toxicity and therapeutic approaches: a review on brain, kidney and testis damage. Current Medicinal Chemistry. 2017;24(35):3879–3893. doi: 10.2174/0929867324666170801101448. [DOI] [PubMed] [Google Scholar]
- 46.Rizwan M., Ali S., Adrees M., et al. A critical review on effects, tolerance mechanisms and management of cadmium in vegetables. Chemosphere. 2017;182:90–105. doi: 10.1016/j.chemosphere.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 47.Chen L., Liu L., Luo Y., Huang S. MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. Journal of Neurochemistry. 2008;105(1):251–261. doi: 10.1111/j.1471-4159.2007.05133.x. [DOI] [PubMed] [Google Scholar]
- 48.Lykissas M. G., Batistatou A. K., Charalabopoulos K. A., Beris A. E. The role of neurotrophins in axonal growth, guidance, and regeneration. Current Neurovascular Research. 2007;4(2):143–151. doi: 10.2174/156720207780637216. [DOI] [PubMed] [Google Scholar]
- 49.Laplante M., Sabatini D. M. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takei N., Nawa H. mTOR signaling and its roles in normal and abnormal brain development. Frontiers in Molecular Neuroscience. 2014;7 doi: 10.3389/fnmol.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen S.-D., Wu C.-L., Hwang W.-C., Yang D.-I. More insight into BDNF against neurodegeneration: anti-apoptosis, anti-oxidation, and suppression of autophagy. International Journal of Molecular Sciences. 2017;18(3) doi: 10.3390/ijms18030545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marini H., Altavilla D., Bellomo M., et al. Modulation of IL-1 β gene expression by lipid peroxidation inhibition after kainic acid-induced rat brain injury. Experimental Neurology. 2004;188(1):178–186. doi: 10.1016/j.expneurol.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 53.Marini H., Costa C., Passaniti M., et al. Levetiracetam protects against kainic acid-induced toxicity. Life Sciences. 2004;74(10):1253–1264. doi: 10.1016/j.lfs.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 54.Provias J. P., Ackerley C. A., Smith C., Becker L. E. Cadmium encephalopathy: a report with elemental analysis and pathological findings. Acta Neuropathologica. 1994;88(6):583–586. doi: 10.1007/BF00296497. [DOI] [PubMed] [Google Scholar]
- 55.Mills J. H., Kim D. G., Krenz A., Chen J. F., Bynoe M. S. A2A adenosine receptor signaling in lymphocytes and the central nervous system regulates inflammation during experimental autoimmune encephalomyelitis. Journal of Immunology. 2012;188(11):5713–5722. doi: 10.4049/jimmunol.1200545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.WANG S., HU P., WANG H., et al. Effects of Cd(2+) on AMPA receptor-mediated synaptic transmission in rat hippocampal CA1 area. Toxicology Letters. 2008;176(3):215–222. doi: 10.1016/j.toxlet.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 57.Bar-Sela S., Reingold S., Richter E. D. Amyotrophic lateral sclerosis in a battery-factory worker exposed to cadmium. International Journal of Occupational and Environmental Health. 2001;7(2):109–112. doi: 10.1179/oeh.2001.7.2.109. [DOI] [PubMed] [Google Scholar]
- 58.Okuda B., Iwamoto Y., Tachibana H., Sugita M. Parkinsonism after acute cadmium poisoning. Clinical Neurology and Neurosurgery. 1997;99(4):263–265. doi: 10.1016/S0303-8467(97)00090-5. [DOI] [PubMed] [Google Scholar]
- 59.Chin-Chan M., Navarro-Yepes J., Quintanilla-Vega B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Frontiers in Cellular Neuroscience. 2015;9 doi: 10.3389/fncel.2015.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are included within the article.