

Abstract
Thoracic aortic aneurysm (TAA) is an asymptomatic and progressive dilatation of the thoracic aorta. Ascending aortic dissection (AAD) is an acute intraparietal tear, occurring or not on a pre-existing dilatation. AAD is a condition associated with a poor prognosis and a high mortality rate. TAA and AAD share common etiology as monogenic diseases linked to transforming growth factor β signaling pathway, extracellular matrix defect, or smooth muscle cell protein mutations. They feature a complex pathogenesis including loss of smooth muscle cells, altered phenotype, and extracellular matrix degradation in aortic media layer. A better knowledge of the mechanisms responsible for TAA progression and AAD occurrence is needed to improve healthcare, nowadays mainly consisting of aortic open surgery or endovascular replacement. Recent breakthrough discoveries allowed a deeper characterization of the mechanisms of gene regulation. Since alteration in gene expression has been linked to TAA and AAD, it is conceivable that a better knowledge of the causes of this alteration may lead to novel theranostic approaches. In this review article, the authors will focus on epigenetic regulation of gene expression, including the role of histone methylation and acetylation, deoxyribonucleic acid methylation, and noncoding ribonucleic acids in the pathogenesis of TAA and AAD. They will provide a translational perspective, presenting recent data that motivate the evaluation of the potential of epigenetics to diagnose TAA and prevent AAD.
Keywords: noncoding RNAs, histone acetylation, DNA methylation
Aortic progressive dilation (aneurysm) is a potentially life-threatening condition that puts patients at risk of aortic dissection or rupture, a devastating clinical event with high mortality. Most aneurysms are painless and cause no symptomatology to alert the patient or clinician to the associated risk. Unfortunately, the hidden nature of progressive extracellular matrix (ECM) defect (with or without dilation) means that often the diagnosis is only made at the time of an aortic dissection. Thousands of deaths annually can be attributed to aortic aneurysms and dissections. 1
Epigenetics can be defined as stable heritable traits independent of changes in deoxyribonucleic acid (DNA) sequence. In many cardiovascular disorders, significant epigenetic modifications have been shown to affect disease development or progression. Work has progressed most rapidly in cardiomyocytic lineages where intensive effort has gone into understanding transcriptional networks at the epigenetic level. 2 In cardiomyocytic lineages, multiple noncoding ribonucleic acids (RNAs) have been identified that coordinate protein complexes to direct promoter modification or other remote transcriptional enhancers. Despite significant progress in the identification of the genetic basis of aortic aneurysms, the full range of chromatin modifications that affect disease progression is just beginning to be investigated. This review will cover the known literature regarding epigenetic control of ascending thoracic aortic aneurysm (TAA) and ascending aortic dissection (AAD), and highlight opportunities for future research in this important topic.
Thoracic Aortic Aneurysm and Dissection: Pathophysiology
Dilating remodeling of arterial tissues is defined as the structural consequence of dysregulated biological activities controlling the ECM and cell homeostasis within the wall. Aneurysms, regardless of their localization, are characterized by breakdown of the ECM, and aortic smooth muscle cell (aSMC) loss, leading to progressive dilation of the arteries, thinning of the medial layer, and, eventually leading to rupture of the arterial wall. Risk factors for aortic aneurysm are dependent on the regional propensity of the affected segment. Descending thoracic aortic aneurysm (DTAA) demonstrates a pathology characterized by atheroma, aSMC disarray and apoptosis, intense phagocytosis, and intraluminal thrombus usually observed in abdominal aortic aneurysms (AAA). Enzymatic degradation of the aortic ECM leads to structural weakening, dilation, and eventually rupture if left untreated. 3 A large proportion of patients with DTAA also coexpress AAA. In contrast, the ascending thoracic aorta is relatively protected. Instead, TAAs have a pathologic appearance characterized by a leukocyte-independent “medial degeneration” in which medial aSMCs play the major role.
Thoracic aortic aneurysm, a progressive dilation of the thoracic ascending aorta, is a chronic pathology involving an imbalance between proteolytic degradation of the ECM and wall compensating resistance to proteolytic injury, 4 involving aSMC adaptive capacities (phenotypic switches, mainly from contractile to synthetic state). TAA associates with highly penetrant genetic conditions that segregate with single genes, 5 but can also have a degenerative etiology in elderly patients, or be associated with bicuspid aortic valves, in which a specific hemodynamic disorder could play an important role. 6 Whatever the etiology, hemodynamics, including high shear stress and arterial blood pressure, play a direct or indirect role in progressive dilation or acute tear.
The identity of genes mediating TAA often represents ECM components or regulators and genes required for the contractile function of aSMC. Increased expression of matrix-degrading enzymes, mucoid accumulation, and aSMC apoptosis are commonly noted in most forms of aneurysmal disease. Disruption of structural ECM components such as collagens and elastin is a commonly accepted mechanism of aneurysm pathogenesis. It comes as no surprise, therefore, that mutations that directly influence the ECM components can cause aneurysm. Examples include mutations in ELN, 7 encoding elastin, COL3A1, 8 encoding collagen type 3-α1, microfibril-associated protein 5, 9 or FBN1, encoding fibrilin1, and responsible for Marfan syndrome. 10 Additionally, mutations in genes encoding proteins required for elastogenesis and collagen metabolism such as EFEMP2 11 and LOX 12 promote TAA. In these disorders, altered assembly or decreased expression of matrix components results in weakness of the aortic media.
In addition to genetic changes that directly alter ECM components, human mutations have been discovered that interfere with vascular smooth muscle-mediated aortic homeostasis in genes encoding members of the smooth muscle contraction apparatus, such as MYH11, 13 encoding smooth muscle myosin and ACTA2, 14 encoding α-smooth muscle actin, and PRKG-1, 15 encoding type I cGMP-dependent protein kinase. Mutations are commonly encountered in genes encoding the various members of the canonical transforming growth factor β (TGF-β) signaling cascade such as TGF-β receptors TGFBR1 and TGFBR2, 16 their cognate ligands TGF-β2, 17 TGF-β3, 18 and SMAD3, an intracellular effector of TGF-β signaling. 19 Mutations in TGF-β receptors are responsible for the Loeys–Dietz syndrome. 16
Aortic dissection is an acute intramural rupture, which can occur in a normal or moderately dilated aorta, 20 without compensatory mechanisms. TAA and AAD correspond respectively to a progressive or acute loss of the ability of the arterial wall to withstand the arterial blood pressure.
TAA and AAD share common etiologies, including monogenic diseases in young patients. 13 21 They also feature common pathological signatures, including aSMC cell disappearance, areas of mucoid degeneration, 22 and degradation of collagen and elastic fibers. 23 Overall, TAA and AAD develop through interactions between numerous factors, including genetics—from mutations causing Mendelian traits to genetic susceptibility, environmental factors such as specific hemodynamic conditions, 24 and aSMC phenotypic changes, some of which are induced by epigenetic modifications.
Epigenetics: General Concepts
As displayed in Fig. 1 , epigenetic modifications encompass different mechanisms: modifications of DNA-associated histone proteins, DNA methylation, and noncoding RNAs-mediated modifications. 25 These mechanisms target either DNA molecules, transcriptional machinery, or transcription products, resulting in modulation of gene expression and consequent protein synthesis.
Fig. 1.
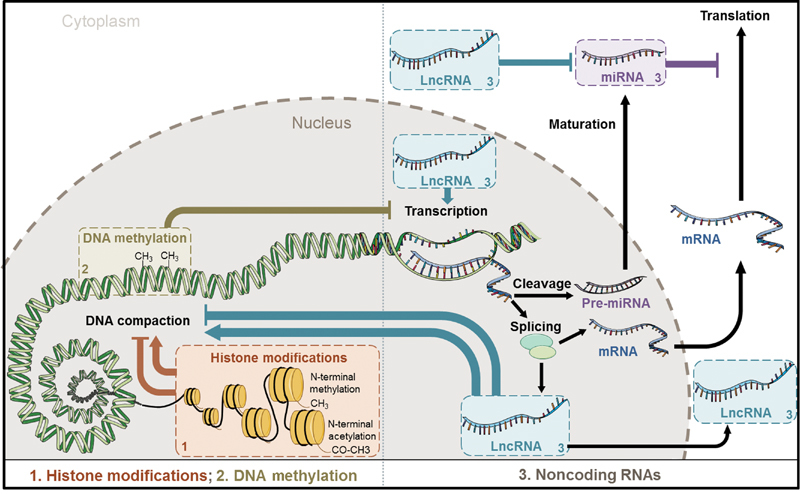
Overview of epigenetic mechanisms. Epigenetic modifications include (1) modifications of DNA-associated histone proteins, (2) DNA methylation, and (3) noncoding RNAs. Abbreviations: DNA, deoxyribonucleic acid; LncRNA, long noncoding RNA; mRNA, messenger RNA; miRNA, microRNA; RNA, ribonucleic acid.
Histones are DNA-associated proteins responsible for chromatin compaction. They are prone to modifications such as acetylation, methylation, phosphorylation, adenosine diphosphate-ribosylation, and sumoylation that generally occur on specific amino acid residues (arginine and lysine) of their N-extremity. 26 Histone acetylation is associated with chromatin loosening and activates transcription, while histone methylation can either activate or repress transcription. 27
Performed by DNA methyltransferases, 28 DNA methylation forms a covalent but reversible bond between a methyl group and the DNA base cytosine, resulting in the production of 5-methylcytosine, sometimes considered as the “fifth base” of DNA. DNA methylation is commonly associated with repression of transcription. Methylated DNA can interact with other epigenetics changes as histone-code modifications. 29
Noncoding RNAs are defined as RNA molecules lacking protein-coding potential. They are generally classified according to their size: small noncoding RNAs contain less than 200 nucleotides, while long noncoding RNAs (lncRNAs) contain at least 200 base pairs. Different types of small noncoding RNAs have been characterized, among which 20-nucleotides-long microRNAs (miRNAs) predominate. 30 MiRNAs possess a seed sequence (6 to 8 nucleotides in length), homologous with the 3′ untranslated region of their target genes. The binding of miRNAs to their target gene through base complementarity results in repression of the expression of the target gene. 31 This repression occurs through messenger RNA (mRNA) degradation when the complementarity between miRNAs and target genes is complete and through inhibition of translation when this complementarity is partial. 32 Importantly, several miRNAs can bind the same mRNA and a single miRNA can bind different mRNAs, attesting for the complexity of gene regulation by miRNAs.
The mechanisms of gene regulation by lncRNAs are more complex and diverse than miRNAs. LncRNAs can act as signal, decoy, guide, scaffold, or enhancer. 33 Some lncRNAs mediate their effects by guiding or recruiting proteinaceous complexes to initiate transcription. 34 The lncRNA Xist modulates gene expression by regulation of imprinting. 35 The lncRNA named CDR1-AS is circular and acts as a miRNA-sponge, preventing the binding of miRNAs to target genes, hence favoring their expression. 36 LncRNAs can also compete with other genes or RNA transcripts, acting as competitive endogenous RNA. 37
The presence of miRNAs in the circulation was revealed almost a decade ago 38 and a plethora of studies on the potential of miRNAs for use as biomarkers of cardiovascular disease rapidly emerged. 39 The biomarker potential of circulating lncRNAs has been more recently evidenced. 33 40 41 Circular RNAs, which are more stable than their linear counterparts due to resistance to exonuclease degradation, also appear to possess an interesting biomarker potential. 42 43
Epigenetics in Thoracic Aortic Aneurysm Disease
Histone Modifications
Experimental studies reported an activation of TGF-β in aneurysms related to Marfan syndrome. 44 TGF-β signaling in syndromic and nonsyndromic aneurysmal diseases was investigated in different types of aneurysmal ascending aortic specimens. The TGF-β1 mRNA was not significantly changed. In contrast, the amounts of TGF-β1 protein retained within and released by an aneurysmal media layer were greater than for a healthy aortic media layer. This observation fitted with the observed increase in LTBP-1 mRNA and protein, 45 associated with the increase in ECM protein turnover reported in TAA. 46 This was associated with higher levels of Smad2 mRNA, phosphorylated Smad2 protein, and nucleus translocation in the ascending aortic wall from all types of aneurysm. Activation of TGF-β pathway was correlated with the degree of elastic fiber fragmentation. Surprisingly, there was no consistent colocalization between the nuclear location of phospho-Smad2 and extracellular TGF-β staining. This first study highlights independent dysregulations of TGF-β retention and Smad2 signaling in TAA whatever their etiologies, suggesting a non-TGF-β dependent activation and nuclear translocation of Smad2 in TAA. 45
In a second study performed on human aortic samples, including TAAs of different etiologies and normal aortas from which tissue extracts and aSMCs and fibroblasts were obtained in primary cultures, it was observed that all types of TAA share a complex dysregulation of Smad2 signaling, independent of TGF-β1 in TAA-derived aSMCs. 47 The Smad2 dysregulation was characterized by an aSMC-specific (not seen in fibroblasts) heritable activation and overexpression of Smad2, compared with normal aortas. The cell specificity and heritability of this overexpression of Smad2 suggested the implication of an epigenetic control. Using chromatin immunoprecipitation assay, the authors showed that Smad2 overexpression in TAA involves increases in H3K9/14 acetylation and H3K4 methylation. 47 These results demonstrated the heritability, cell specificity, and independence with regard to TGF-β1 and genetic backgrounds of the Smad2 dysregulation in thoracic aneurysms and the involvement of epigenetic mechanisms regulating histone marks in this process.
In a third study, the histone-modifying enzymes, transcription factors, and cofactors responsible for Smad2 promoter activation in aSMC from TAA patients were explored to understand the mechanisms regulating Smad2 overexpression. It was shown that Smad2 promoter activation is driven by the recruitment of a multipartner complex, including the transcription factor p53 and histone acetyltransferases. 48 Remarkably, the transcriptional regulatory network of the Smad2 promoter was dramatically altered in human aneurysmal aSMCs in vitro and in situ with a switch from Myc-dependent repression of Smad2 in normal vessel to a p53-dependent constitutive activation of Smad2 in aneurysms. Furthermore, histone acetyltransferases p300 and P300/CBP-associated proteins played a major role in Smad2 promoter activation by acting on histone acetylation and p53 recruitment. 48 These results provided evidence for a major role of p53 and the complex composed of histone acetyltransferases p300 and p300/CBP-associated proteins in Smad2 activation in human aneurysmal aSMCs.
In a fourth study, protease nexin-1 (PN-1) and plasminogen activator inhibitor-1 (PAI-1), both inhibitors of serine-protease were overexpressed in medial tissue extracts and primary aSMCs from TAA compared with acute dissections of ascending aortic and healthy aorta. 49 Furthermore, TGF-β increased PN-1 expression in control but not in aneurysmal aSMCs. PN-1 and PAI-1 overexpression by aneurysmal aSMCs was associated with increased Smad2 binding on their respective gene promoters. This phenomenon was restricted to aneurysms and not observed in acute dissections. 49 Thus, epigenetically regulated PN-1 overexpression promotes development of an antiproteolytic aSMC phenotype which might favor progressive aneurysmal dilation. Absence of this counter regulation may lead to acute wall rupture (dissection, Fig. 2 ).
Fig. 2.
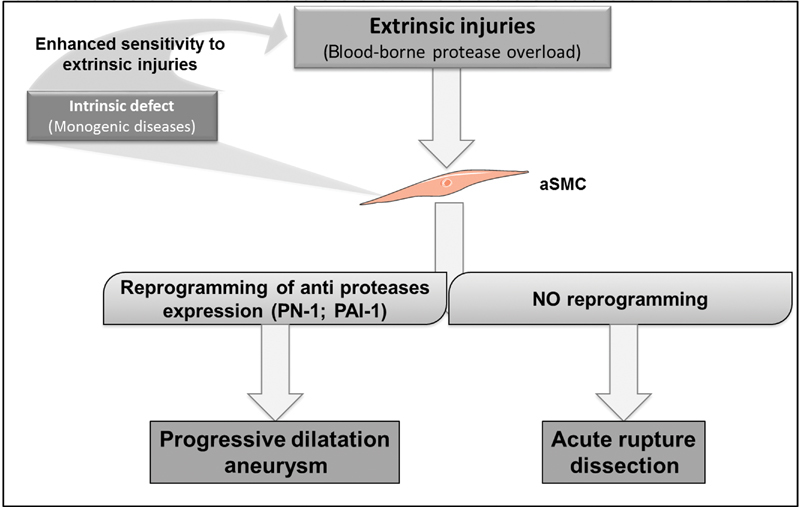
Schematic representation of the differential biology between chronic dilation of the aorta (aneurysm) and acute intraparietal rupture (dissection). The progressive development of chromatin remodeling in aortic smooth muscle cells (aSMC) in response to small dilation or matrix proteolytic injury could reduce the risk of acute rupture. Abbreviations: PN-1, protease nexin-1, PAI-1, plasminogen activator inhibitor-1.
DNA Methylation
A genome-wide DNA methylation study comparing aortic tissues from patients with two different etiologies of TAA, bicuspid and tricuspid aortic valves, revealed that several genes involved in cardiovascular development were differentially methylated, 50 suggesting that DNA methylation may be involved in TAA development and dissection.
MicroRNAs
Although the functional contribution of miRNAs to the development of TAA and progression toward dissection is still poorly characterized, some associations between miRNAs expression levels, TAA and AAD, have been reported ( Table 1 ).
Table 1. Differentially expressed miRNAs in aortic tissues from patients with TAA or AAD compared with controls.
| AAD aortic tissue | TAA aortic tissue | |||||
|---|---|---|---|---|---|---|
| ID | Expression versus control | Technology | Reference | Expression versus control | Technology | Reference |
| miR-1 | ↓ | Microarray, PCR | 52 54 | |||
| miR-15a | ↓ | Microarray | 51 | ↑ | Microarray | 53 |
| miR-21 | ↑ ; ↓ | Microarray; PCR | 52 53 54 | |||
| miR-21 a | ↑ | Microarray | 53 54 | |||
| miR-22 | ↓ | Microarray, PCR | 51 | ↓ | Microarray | 53 a |
| miR-25 | ↑ | Microarray | 53 | |||
| miR-29a | ↓ | Microarray | 51 | ↓ | Microarray, PCR | 52 54 |
| miR-29b | ↑ | Microarray; PCR | 53 57 | |||
| miR-30c a | ↓ | PCR | 54 | |||
| miR-125a-3p | ↓ | Microarray | 53 | |||
| miR-126–3p | ↑ | Microarray; PCR | 53 a 54 | |||
| miR-128 | ↑ | Microarray | 53 | |||
| miR-133a | ↓ | Microarray | 51 | ↓ | Microarray, PCR | 52 53 54 |
| miR-133b | ↓ | Microarray | 51 | ↓ | Microarray | 53 |
| miR-138–1 a | ↑ | Microarray | 51 | ↑ | Microarray | 53 |
| miR-142–5p | ↑ | Microarray | 53 | |||
| miR-143 | ↓ | Microarray, PCR | 51 | ↓ | PCR | 67 |
| miR-145 | ↓ | Microarray, PCR | 51 | ↓ ; ↑ | Microarray; PCR | 53 54 66 67 |
| miR-146b-5p | ↑ | Microarray | 53 54 | |||
| miR-155 | ↓ | PCR | 54 | |||
| miR-183 a | ↑ | Microarray, PCR | 51 | ↑ | Microarray | 53 |
| miR-204 | ↓ | PCR | 54 | |||
| miR-422a | ↑ | Microarray | 53 | |||
| miR-433 | ↑ | Microarray, PCR | 51 | |||
| miR-486–5p | ↓ ; ↑ | Microarray; PCR | 53 54 | |||
| miR-487b | ↑ | Microarray | 53 | |||
| miR-491–3p | ↑ | Microarray, PCR | 51 | ↑ | Microarray | 53 b |
| miR-553 | ↑ | Microarray, PCR | 51 | |||
| miR-638 | ↓ | Microarray | 53 b 54 | |||
| miR-940 | ↓ | Microarray | 53 | |||
|
miR-193a-3p;
miR-768–5p; miR-886–5p; miR-30e; miR-195; miR-101; miR-140–5p; miR-744 |
↓ | Microarray | 51 | ↑ | Microarray | 53 a |
| miR-193a-5p | ↓ | Microarray | 51 | ↓ | Microarray | 53 b |
Abbreviations: AAD, ascending aortic dissection; PCR, polymerase chain reaction; TAA, thoracic aortic aneurysm
Only in males.
Only in females.
↑: upregulated in disease versus control.
↓: downregulated in disease versus control.
Underlined: miRNAs differentially expressed in TAA and AAD.
MicroRNA Profiles
Technological developments of high-throughput approaches such as microarray facilitated the characterization of disease-associated miRNA profiles.
Using microarray and quantitative polymerase chain reaction (PCR), a panel of four up-regulated and three down-regulated miRNAs was identified in six aortic tissues from AAD patients compared with six control subjects 51 ( Table 1 ). In another study, four miRNAs were down-regulated in aortic tissues from TAA patients compared with control subjects, and their expression levels were inversely correlated with aortic diameter. 52 Also with microarray, a set of 16 miRNAs was found to be regulated in 10 nonfamilial and nonsyndromic aneurysmal ascending aorta compared with 10 control aorta. 53 Interestingly, gender-associated differences in miRNA profiles were observed in this study. Yet, no validation by PCR was reported. More recently, 6 up-regulated and 2 down-regulated miRNAs were observed in 11 aortic tissues from TAA patients compared with 8 control tissues, using quantitative PCR. 54 Noteworthy, even though two different diseases (AAD and TAA) were investigated in these four studies, 51 52 53 54 at least nine miRNAs (miR-22/-29a/-133a/-133b/-138–1*/-145/-183*/-193a-5p/-491–3p) showed similar expression patterns ( Table 1 ). Considering that TAA and AAD display similar features, these studies support the involvement of miRNAs in TAA and AAD pathogenesis. However, miR-1/-30c-2/-145/-204/-331–3p were up-regulated in human aortic tissue samples from patients with TAA compared with DTAA. 55 Since TAA and DTAA possess different clinical features and etiologies, such differences of miRNA profiles strengthen the concept that miRNAs may play specific roles in TAA and DTAA pathogenesis.
Investigation of the functional role of miRNAs in TAA and AAD remains difficult in human aortic tissues in situ. Gender-associated differences need to be considered. The use of primary human aSMC and experimental models in mice allowed a better characterization of the mechanisms of action of miRNAs, as detailed hereafter.
MiR-29 Family
MiR-29 family contains three members, miR-29a/-29b/-29c, all involved in fibrosis. 56 MiR-29b was highly expressed in human aSMC compared with endothelial cells and monocytes. 57 Its expression was higher in aorta from TAA patients compared with healthy controls, whereas expression of miR-29a and miR-29c was comparable between the two groups. 57 In humans, however, miR-29a expression was decreased during TAA formation. 52 54 In mice, miR-29 family members were overexpressed in aorta from fibulin-4 deficient animals. 57 MiR-29b was also overexpressed in angiotensin II-infused mice and its silencing with locked nucleic acid (LNA)-modified antisense oligonucleotides inhibited angiotensin II-triggered aortic dilation. 57 MiR-29b was up-regulated in the ascending aorta of Marfan Fbn1 (C1039G/ + ) mice and its inhibition with LNA-modified antisense oligonucleotides prevented aneurysm development, reduced apoptosis in aortic wall, and increased ECM components synthesis. 58
In vitro, miR-29b induced aSMC migration by inhibiting DNA methyltransferase 3 that negatively regulates matrix metalloproteinases 2 and 9 expression. 59 MiR-29b directly targets the genes encoding the antiapoptotic proteins MCL1 (a member of the BCL2 family of apoptosis regulators) 60 and KLF4 (Kruppel-like factor 4). 61 KLF4 is a transcription factor involved in aSMC proliferation and is activated by Sp1. 62 MiR-29 family members also down-regulate the expression of several genes coding for ECM proteins such as collagens 1a1, 1a2, 3a1, eln1, fbn1, 63 and Adamts7. 64 TGF-β1 stimulation increased miR-29b expression in aSMC from Fbn C1039G/+ mice but not from wild-type mice. 58 This stimulation reduced nuclear factor kappa B (NFκB) activity and, since miR-29b expression is inhibited by NFκB in ascending aorta of young Fbn C1039G/+ mice, the authors proposed that up-regulation of miR-29b expression by TGF-β1 may involve the NFκB pathway. 58
These studies support a therapeutic potential for miR-29b for the prevention of TAA expansion, acting through modulation of aSMC synthetic phenotypic switch and ECM composition. The cross-talk between miR-29b and TGF-β is consistent with a role for miR-29b in the pathogenesis of conditions due to TGF-β pathway alteration, not limited to TAA and AAD. The observation that TGF-β impacts miR-29b expression suggests that TAA and AAD pathogenesis is controlled by complex epigenetic mechanisms that remain to be fully deciphered.
Intriguingly, in aorta from young Fbn C1039G/+ mice, losartan administration reduced aneurysm formation and miR-29b expression. 58 Furthermore, expression of miR-29 family members was increased in aorta of aged mice compared with young mice. 57 These observations may explain, at least in part, the heterogeneity of results obtained from human studies, mostly negative. Medication and age are certainly confounding factors that need to be taken into account in future clinical studies.
MiR-143/-145 Family
MicroRNA-143 and miR-145 are expressed from the same bicistronic precursor. 65 Both miRNAs were down-regulated in aorta from AAD patients compared with controls 51 and miR-145 was up-regulated in TAA compared with DTAA. 55 In another study, miR-145 expression was higher in TAA tissues compared with control tissues and was positively correlated with collagen III levels and aortic diameter. 66 Elsewhere, miR-143/-145 expression was deficient in aortic tissues from TAA patients compared with controls. 67 These discrepancies may be attributed to technical bias (use of microarray or PCR to measure miRNAs) or differences between patient cohorts and disease etiology.
In mice, miR-143 and miR-145 were enriched in aorta compared with other organs. 67 Homozygous miR-143/-145 knocked-out mice showed altered aortic structure compared with the wild-types, as characterized by diminution of medial thickness, and aSMC dedifferentiation and migration toward the intima. 67
In vitro, miR-145 has been proposed as a phenotypic marker of vascular smooth muscle cells (vSMC). 68 MiR-143/-145 appear to play an important role in vSMC phenotypic switch since their inhibition increased cell migration and decreased proliferation. 67 Furthermore, miR-143/-145 up-regulation increased the expression of vSMC differentiation markers. 68 The effects of miR-145 on vSMC are mediated by modulation of the TGF-β pathway 66 and targeting of KLF4 and KLF5. 65 Using cocultured vascular endothelial cells and vSMC, a communication involving exosomes containing miR-143/-145 and transferring from endothelial cells to vSMC could be demonstrated. 69
Despite some heterogeneity observed in human studies, miR-143/-145 family is apparently deeply involved in the regulation of aSMC biology and ECM composition. Since aSMC phenotypic switch and ECM alterations are critical in aortic media dysfunction, miR-143 and miR-145 may play important roles in TAA and AAD pathogenesis that could be interesting for therapeutic purposes.
MicroRNAs in aSMC Phenotypic Switch
Due to the importance of aSMC phenotypic switch in the pathogenesis of TAA and AAD, the following chapter focuses on miRNAs ( Fig. 3 ) and their target genes ( Table 2 ) known to regulate this switch.
Fig. 3.
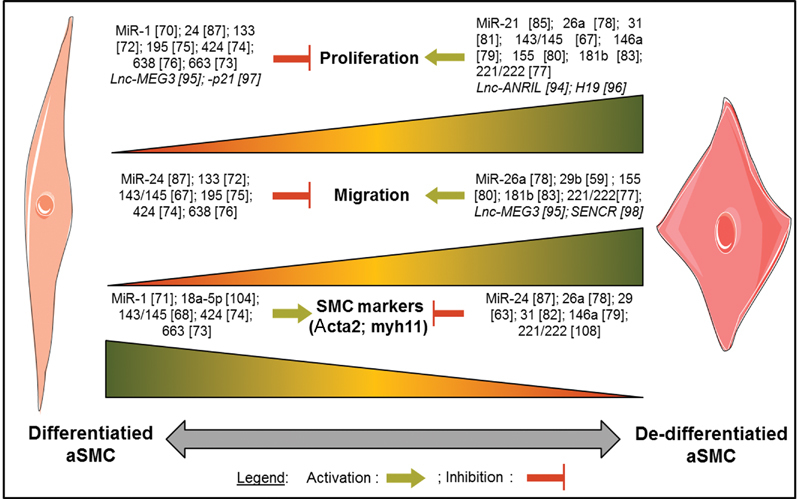
Involvement of microribonucleic acids (miRNAs) and long noncoding RNAs (lncRNAs) in aortic smooth muscle cell (aSMC) phenotypic switch.
Table 2. MicroRNAs target genes modulating aSMC phenotypic switch.
| ID | Target genes | Effect in aSMC | Reference |
|---|---|---|---|
| miR-1 | Klf4 | Induces aSMC markers expression: acta2, tagln | 71 |
| miR-133 | Sp1 | Inhibits cnn1; tagln2; acta2 and sfr; induces myh11 expression | 72 |
| miR-143/145 | Klf4, CamkII-δ, Klf5 | Induces acta2; cnn1; myh11; col3 expression and TGF-B | 65 66 68 |
| miR-146a | Klf4 | Inhibits acta2; induces NFKB phosphorylation | 79 102 |
| miR-181b | Eln | Inhibits p21/p27; induces CDK4 and cyclin D1. Induces erk1/2 and jnk/jnk phosphorylation | 83 103 |
| miR-18a-5p | Induces cnn1; acta2 expression | 104 | |
| miR-21 | Sp1; c-Ski | Inhibits myh11 expression | 105 106 |
| miR-221/222 | p27(Kip1), p57(Kip2), and c-Kit | Inhibits aSMC markers: acta2, cnn, and tagln expression | 77 107 108 |
| miR-24 | HMOX-1; PDGFRB and c-Myc | Inhibits HMGB1; erk, and akt activation and inhibits aSMC markers acta2 and ccn1 expression | 86 87 109 |
| miR-26a | Inhibits myh11; acta2 ; smad1 and smad 4 | 78 | |
| miR-29b | Mcl-1 ; Klf4 ; Col1a1 ; col1a2 ; col3a1 ; eln ; fbn1 ; adamts7 | Inhibits col1a2 ; col3a1 ; eln ; adamts7 expression | 57 60 61 63 64 |
| miR-31 | CREG; lats2 | Inhibits acta2 expression | 81 82 |
| miR-424 | Induces acta2, myh11 and cnn1 expression | 74 | |
| miR-663 | JunB and Myl9 | Induces acta2; myh11, tagln, ccn1 | 73 |
Abbreviations: aSMC, aortic smooth muscle cell; CREG, cellular repressor of E1A-stimulated gene; MicroRNA, microribonucleic acid.
MicroRNAs promoting aSMC differentiation . MiR-1 expression was induced by myocardin in aSMC, 70 targeted KLF4, and induced aSMC differentiation from embryonic stem cells. 71 MiR-133 repressed Sp1 (which activates KLF4 62 ), resulting in maintenance of aSMC in a differentiated status. 72 MiR-133 also inhibited aSMC migration, acta2 and srf expression, and was associated with an increase in myh11 expression. 72 MiR-663 and miR-424 induced the expression of aSMC differentiation markers (acta2; myh11) and decreased aSMC migration. 73 74 MiR-195 repressed proliferation, migration, and protein secretion of aSMC. 75 MiR-638 inhibited aSMC proliferation and migration. 76
MicroRNAs promoting aSMC dedifferentiation . MiR-221 and miR-222 are two close homologous miRNAs sharing the same seed sequence. They are highly expressed in aSMC, inducing not only their proliferation but also their migration, and inhibiting their apoptosis, presumably through modulation of their target genes p27 (Kip1), p57 (Kip2), and c-Kit. 77 Interestingly, miR-221/-222 had opposite effects on endothelial cells compared with aSMC. 77 MiR-26a targets the TGF-β/BMP (bone morphogenic protein) pathway, thereby inhibiting aSMC apoptosis and differentiation, and activating proliferation and migration. 78 MiR-146a and miR-155 induced aSMC migration and repressed apoptosis. 79 80 MiR-31 was strongly expressed in aSMC and induced their proliferation, 81 probably through modulation of cellular repressor of E1A-stimulated genes and acta2. 82 MiR-181b induced proliferation, and migration of aSMC through the PI3K and MAPK pathways. 83
MicroRNAs promoting phenotype switch . The role of miR-21 in aSMC phenotype switch is less clear since it appears to be able to promote aSMC differentiation 84 as well as proliferation 85 which is considered as a feature of dedifferentiation. Likewise, miR-24 inhibited aSMC proliferation and migration 86 but repressed the expression of contractile gene markers. 87
Overall, the differential expression of multiple miRNAs in aneurysmal aortic tissues, as well as their functional involvement in aSMC biology and ECM composition, strongly supports their role in the pathogenesis of TAA and AAD. The challenge resides in the differentiation between simple bystanders and miRNAs that actively contribute to the disease, the latter category containing the most interesting therapeutic candidates. MiR-29b appears to be such a good candidate.
To our knowledge, the biomarker potential of circulating miRNAs has not been studied in the context of TAA. The potential use of circulating miRNAs as markers of AAA and aneurysm growth has only recently been reported. 88 Being able to predict dissection with a simple blood test, based on miRNA measurements, for instance, would represent a major achievement.
Long Noncoding RNAs
Contrarily to miRNAs, a few studies have addressed the association between lncRNAs and TAA. The lncRNA AK056155 was up-regulated in serum and aortic tissue samples from patients with Loeys–Dietz syndrome compared with controls. 89 Since these patients generally develop TAA, it is conceivable that AK056155 is also associated with TAA. However, all patients with Loeys–Dietz syndrome enrolled in this study had AAA, precluding evaluation of the association between AK056155 and TAA. Another lncRNA, named HIF1α-AS1 for its localization on the antisense strand of the gene coding the hypoxia inducible factor 1 α, was strongly up-regulated in serum of patients with thoraco-AAA. 90 HIF1α-AS1 down-regulation in palmitic acid-treated aSMC inhibited apoptosis and this effect was associated with a decrease in caspase-3, -8, and bcl 2 expression. 90 91
Mice lacking the lncRNA GAS5 display a thickening of the aortic media. 92 GAS5 is expressed in aortic wall, endothelial cells, and aSMC. In aSMC, its down-regulation increased viability and proliferation and lowered expression of aSMC markers. 92 Interestingly, GAS5 may act as paracrine mediator since conditioned medium obtained from endothelial cells overexpressing GAS5 induced a decrease in proliferation and migration of aSMC. 92 Knocking out the lncRNA RNCR3 in mice lowered aSMC proliferation, an effect attributed to a competition for miR-185–5p binding. 93
In vitro experiments revealed that, like miRNAs, lncRNAs affect aSMC proliferation and migration ( Fig. 3 ). The lncRNA ANRIL (CDKN2B antisense RNA 1) induced aSMC proliferation and up-regulated some genes involved in TAA such as eln and col3a1. 94 MEG3 reduced proliferation and increased migration of aSMC, effects accompanied by p53 and mmp2 overexpression. 95 H19 induced aSMC proliferation, an effect mediated by miR-675. 96 LincRNA-p21 inhibits aSMC proliferation and apoptosis by regulating p53 activity. 97 SENCR is highly expressed in aSMC and its down-regulation repressed the expression of aSMC markers and increased cell migration. 98
The role of lncRNAs in TAA is only emerging. The observation that some lncRNAs are present in blood supports a potential use as biomarkers. A deeper characterization of the functional involvement and biomarker potential of lncRNAs in TAA and AAD is warranted.
Conclusion and Future Directions
Genetic alterations critically account for the development of TAA and the progression toward dissection. However, other etiologies of TAA or AAD such as degenerative TAA or AAD occurring on medium-sized aorta are unrelated to any known DNA polymorphism. In this context, epigenetic modifications, which do not change DNA sequence, may also trigger, or at least contribute to, TAA and AAD. Several lines of evidence strongly support a causative role of epigenetic modifications and noncoding RNAs in the pathogenesis of TAA and AAD.
Since epigenetic modifications may occur under the influence of environmental changes such as hemodynamic modifications, it would be interesting to address whether chromatin remodeling mediated by histone modifications is related to the hemodynamic modifications featured in TAA. Indeed, the dilation (increased radius, r ) observed in TAA is associated with a decreased media thickness ( h ), leading to a dramatic increase in wall tension ( T ) at a constant blood pressure ( P ), according to the Laplace's law ( T = P . r /2 h ). The nuclear envelope is mechanically coupled to mechanotransduction through aSMC adhesion to matrix, coupling of integrins to actin, intermediate filaments (tensegrity 99 ) and linkers of nucleoskeleton to cytoskeleton. 100 101 It has been proposed that the mechanical environment impacts the chromatin status of aSMC, thereby controlling vascular gene expression and function. 87 It is, therefore, likely that the increased wall tension in TAA leading to altered mechanotransduction signaling between the matrix and the nucleus will impact chromatin remodeling in aSMC, an issue that has not been addressed. In addition, since chromatin remodeling is associated with chronic progressive dilation but not with acute intraparietal rupture (dissection without important dilation), chromatin remodeling in aSMC could be a hallmark of TAA as compared with dissecting aortas.
The impact of noncoding RNAs on TAA and AAD pathogenesis is far from being well characterized. Increasing evidences show that both short and lncRNAs interact to regulate chromatin remodeling, aSMC phenotype, and ECM composition, sometimes through mediation of cell–cell communication.
Since miRNAs and lncRNAs are present in the blood, it is conceivable that they may emanate, actively or passively, from the diseased vascular wall and inform about its stability or propensity to rupture. The possibility that noncoding RNAs could be used as biomarkers of TAA and dissection is attractive and remains to be tested.
Additional research is warranted to reach a better knowledge of the epigenetic mechanisms gearing TAA development and aneurysm rupture. Several studies reported here showed the involvement of noncoding RNAs in TAA and dissection. However, their role in the pathogenesis of these diseases is only partly understood. Increasing interest is observed toward DNA methylation and histone modification. New insights into their role in TAA and dissection pathogenesis are expected to emerge in the upcoming years. Since epigenetic changes can be modulated, this may lead to novel tools for better diagnosis and treatment of these severe conditions.
Acknowledgements
None.
Funding Statement
Funding A.B. is funded by the National Research Fund (grant # AFR 8832104). Y.D. is supported by the National Research Fund and the Ministry of Higher Education and Research of Luxembourg. M.E.L. is supported by the National Institutes of Health (grant #HL130113), and the Toomey Fund for Aortic Dissection Research. J.B.M. is funded by INSERM and Agence Nationale pour la Recherche GDPMs, NONAGES, and EU FP7: Fighting Aneurysmal Diseases.
Conflicts of Interest The authors declare no conflict of interest related to this article.
On behalf of the Cardiolinc™ network (www.cardiolinc.org) .
References
- 1.Benjamin E J, Blaha M J, Chiuve S E et al. Heart Disease and Stroke Statistics-2017 Update: a report from the American Heart Association. Circulation. 2017;135(10):e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruneau B G. Epigenetic regulation of the cardiovascular system: introduction to a review series. Circ Res. 2010;107(03):324–326. doi: 10.1161/RES.0b013e3181f17dfe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim J B, Kim K, Lindsay M E et al. Risk of rupture or dissection in descending thoracic aortic aneurysm. Circulation. 2015;132(17):1620–1629. doi: 10.1161/CIRCULATIONAHA.114.015177. [DOI] [PubMed] [Google Scholar]
- 4.Jondeau G, Michel J B, Boileau C. The translational science of Marfan syndrome. Heart. 2011;97(15):1206–1214. doi: 10.1136/hrt.2010.212100. [DOI] [PubMed] [Google Scholar]
- 5.Vapnik J S, Kim J B, Isselbacher E M et al. Characteristics and outcomes of ascending versus descending thoracic aortic aneurysms. Am J Cardiol. 2016;117(10):1683–1690. doi: 10.1016/j.amjcard.2016.02.048. [DOI] [PubMed] [Google Scholar]
- 6.Michelena H I, Della Corte A, Prakash S K, Milewicz D M, Evangelista A, Enriquez-Sarano M. Bicuspid aortic valve aortopathy in adults: incidence, etiology, and clinical significance. Int J Cardiol. 2015;201:400–407. doi: 10.1016/j.ijcard.2015.08.106. [DOI] [PubMed] [Google Scholar]
- 7.Szabo Z, Crepeau M W, Mitchell A L et al. Aortic aneurysmal disease and cutis laxa caused by defects in the elastin gene. J Med Genet. 2006;43(03):255–258. doi: 10.1136/jmg.2005.034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Superti-Furga A, Gugler E, Gitzelmann R, Steinmann B. Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J Biol Chem. 1988;263(13):6226–6232. [PubMed] [Google Scholar]
- 9.Barbier M, Gross M S, Aubart M et al. MFAP5 loss-of-function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am J Hum Genet. 2014;95(06):736–743. doi: 10.1016/j.ajhg.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Magenis R E, Maslen C L, Smith L, Allen L, Sakai L Y. Localization of the fibrillin (FBN) gene to chromosome 15, band q21.1. Genomics. 1991;11(02):346–351. doi: 10.1016/0888-7543(91)90142-2. [DOI] [PubMed] [Google Scholar]
- 11.Dasouki M, Markova D, Garola R et al. Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am J Med Genet A. 2007;143A(22):2635–2641. doi: 10.1002/ajmg.a.31980. [DOI] [PubMed] [Google Scholar]
- 12.Guo D C, Regalado E S, Gong L et al. LOX mutations predispose to thoracic aortic aneurysms and dissections. Circ Res. 2016;118(06):928–934. doi: 10.1161/CIRCRESAHA.115.307130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu L, Vranckx R, Khau Van Kien P et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38(03):343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 14.Guo D C, Pannu H, Tran-Fadulu V et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39(12):1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 15.Guo D C, Regalado E, Casteel D E et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. 2013;93(02):398–404. doi: 10.1016/j.ajhg.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loeys B L, Chen J, Neptune E R et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(03):275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 17.Lindsay M E, Schepers D, Bolar N A et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44(08):922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertoli-Avella A M, Gillis E, Morisaki H et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015;65(13):1324–1336. doi: 10.1016/j.jacc.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van de Laar I M, Oldenburg R A, Pals G et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43(02):121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 20.LeMaire S A, Russell L. Epidemiology of thoracic aortic dissection. Nat Rev Cardiol. 2011;8(02):103–113. doi: 10.1038/nrcardio.2010.187. [DOI] [PubMed] [Google Scholar]
- 21.Boileau C, Guo D C, Hanna N et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44(08):916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Figueiredo Borges L, Martelli H, Fabre M, Touat Z, Jondeau G, Michel J B. Histopathology of an iliac aneurysm in a case of Menkes disease. Pediatr Dev Pathol. 2010;13(03):247–251. doi: 10.2350/08-08-0516.1. [DOI] [PubMed] [Google Scholar]
- 23.de Figueiredo Borges L, Jaldin R G, Dias R R, Stolf N A, Michel J B, Gutierrez P S. Collagen is reduced and disrupted in human aneurysms and dissections of ascending aorta. Hum Pathol. 2008;39(03):437–443. doi: 10.1016/j.humpath.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Bäck M, Gasser T C, Michel J B, Caligiuri G. Biomechanical factors in the biology of aortic wall and aortic valve diseases. Cardiovasc Res. 2013;99(02):232–241. doi: 10.1093/cvr/cvt040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Egger G, Liang G, Aparicio A, Jones P A.Epigenetics in human disease and prospects for epigenetic therapy Nature 2004429(6990):457–463. [DOI] [PubMed] [Google Scholar]
- 26.Lee K K, Workman J L. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8(04):284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 27.Geiman T M, Robertson K D. Chromatin remodeling, histone modifications, and DNA methylation-how does it all fit together? J Cell Biochem. 2002;87(02):117–125. doi: 10.1002/jcb.10286. [DOI] [PubMed] [Google Scholar]
- 28.Bestor T H. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9(16):2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 29.Jin B, Li Y, Robertson K D. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer. 2011;2(06):607–617. doi: 10.1177/1947601910393957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Devaux Y. Transcriptome of blood cells as a reservoir of cardiovascular biomarkers. Biochim Biophys Acta. 2017;1864(01):209–216. doi: 10.1016/j.bbamcr.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 31.He L, Hannon G J. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(07):522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 32.Goretti E, Wagner D R, Devaux Y. Regulation of endothelial progenitor cell function by micrornas. Minerva Cardioangiol. 2013;61(06):591–604. [PubMed] [Google Scholar]
- 33.Devaux Y, Zangrando J, Schroen B et al. Long noncoding RNAs in cardiac development and ageing. Nat Rev Cardiol. 2015;12(07):415–425. doi: 10.1038/nrcardio.2015.55. [DOI] [PubMed] [Google Scholar]
- 34.Uchida S, Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ Res. 2015;116(04):737–750. doi: 10.1161/CIRCRESAHA.116.302521. [DOI] [PubMed] [Google Scholar]
- 35.Engreitz J M, Pandya-Jones A, McDonel Pet al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome Science 2013341(6147):1.237973E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansen T B, Jensen T I, Clausen B Het al. Natural RNA circles function as efficient microRNA sponges Nature 2013495(7441):384–388. [DOI] [PubMed] [Google Scholar]
- 37.Guo G, Kang Q, Zhu X et al. A long noncoding RNA critically regulates Bcr-Abl-mediated cellular transformation by acting as a competitive endogenous RNA. Oncogene. 2015;34(14):1768–1779. doi: 10.1038/onc.2014.131. [DOI] [PubMed] [Google Scholar]
- 38.Mitchell P S, Parkin R K, Kroh E M et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goretti E, Wagner D R, Devaux Y. miRNAs as biomarkers of myocardial infarction: a step forward towards personalized medicine? Trends Mol Med. 2014;20(12):716–725. doi: 10.1016/j.molmed.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Kumarswamy R, Bauters C, Volkmann I et al. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ Res. 2014;114(10):1569–1575. doi: 10.1161/CIRCRESAHA.114.303915. [DOI] [PubMed] [Google Scholar]
- 41.Vausort M, Wagner D R, Devaux Y. Long noncoding RNAs in patients with acute myocardial infarction. Circ Res. 2014;115(07):668–677. doi: 10.1161/CIRCRESAHA.115.303836. [DOI] [PubMed] [Google Scholar]
- 42.Devaux Y, Creemers E E, Boon R A et al. Circular RNAs in heart failure. Eur J Heart Fail. 2017;19(06):701–709. doi: 10.1002/ejhf.801. [DOI] [PubMed] [Google Scholar]
- 43.Vausort M, Salgado-Somoza A, Zhang L et al. Myocardial infarction-associated circular RNA predicting left ventricular dysfunction. J Am Coll Cardiol. 2016;68(11):1247–1248. doi: 10.1016/j.jacc.2016.06.040. [DOI] [PubMed] [Google Scholar]
- 44.Lindsay M E, Dietz H C.Lessons on the pathogenesis of aneurysm from heritable conditions Nature 2011473(7347):308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomez D, Al Haj Zen A, Borges L F et al. Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol. 2009;218(01):131–142. doi: 10.1002/path.2516. [DOI] [PubMed] [Google Scholar]
- 46.Borges L F, Gomez D, Quintana M et al. Fibrinolytic activity is associated with presence of cystic medial degeneration in aneurysms of the ascending aorta. Histopathology. 2010;57(06):917–932. doi: 10.1111/j.1365-2559.2010.03719.x. [DOI] [PubMed] [Google Scholar]
- 47.Gomez D, Coyet A, Ollivier V et al. Epigenetic control of vascular smooth muscle cells in Marfan and non-Marfan thoracic aortic aneurysms. Cardiovasc Res. 2011;89(02):446–456. doi: 10.1093/cvr/cvq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomez D, Kessler K, Michel J B, Vranckx R. Modifications of chromatin dynamics control Smad2 pathway activation in aneurysmal smooth muscle cells. Circ Res. 2013;113(07):881–890. doi: 10.1161/CIRCRESAHA.113.301989. [DOI] [PubMed] [Google Scholar]
- 49.Gomez D, Kessler K, Borges L F et al. Smad2-dependent protease nexin-1 overexpression differentiates chronic aneurysms from acute dissections of human ascending aorta. Arterioscler Thromb Vasc Biol. 2013;33(09):2222–2232. doi: 10.1161/ATVBAHA.113.301327. [DOI] [PubMed] [Google Scholar]
- 50.Shah A A, Gregory S G, Krupp D et al. Epigenetic profiling identifies novel genes for ascending aortic aneurysm formation with bicuspid aortic valves. Heart Surg Forum. 2015;18(04):E134–E139. doi: 10.1532/hsf.1247. [DOI] [PubMed] [Google Scholar]
- 51.Liao M, Zou S, Weng J et al. A microRNA profile comparison between thoracic aortic dissection and normal thoracic aorta indicates the potential role of microRNAs in contributing to thoracic aortic dissection pathogenesis. J Vasc Surg. 2011;53(05):1341–1.349E6. doi: 10.1016/j.jvs.2010.11.113. [DOI] [PubMed] [Google Scholar]
- 52.Jones J A, Stroud R E, O'Quinn E C et al. Selective microRNA suppression in human thoracic aneurysms: relationship of miR-29a to aortic size and proteolytic induction. Circ Cardiovasc Genet. 2011;4(06):605–613. doi: 10.1161/CIRCGENETICS.111.960419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patuzzo C, Pasquali A, Malerba Get al. A preliminary microRNA analysis of non syndromic thoracic aortic aneurysms Balkan J Med Genet 201215(15, Suppl):51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venkatesh P, Phillippi J, Chukkapalli S et al. Aneurysm-specific miR-221 and miR-146a participates in human thoracic and abdominal aortic aneurysms. Int J Mol Sci. 2017;18(04):184. doi: 10.3390/ijms18040875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Premakumari V, Chukkapalli S, Rivera M et al. Microrna expression signature in human thoracic and abdominal aortic aneurysms. Atherosclerosis. 2014;235(02):e130. [Google Scholar]
- 56.Boon R A, Dimmeler S. MicroRNAs and aneurysm formation. Trends Cardiovasc Med. 2011;21(06):172–177. doi: 10.1016/j.tcm.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 57.Boon R A, Seeger T, Heydt S et al. MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res. 2011;109(10):1115–1119. doi: 10.1161/CIRCRESAHA.111.255737. [DOI] [PubMed] [Google Scholar]
- 58.Merk D R, Chin J T, Dake B A et al. miR-29b participates in early aneurysm development in Marfan syndrome. Circ Res. 2012;110(02):312–324. doi: 10.1161/CIRCRESAHA.111.253740. [DOI] [PubMed] [Google Scholar]
- 59.Chen K C, Wang Y S, Hu C Y et al. OxLDL up-regulates microRNA-29b, leading to epigenetic modifications of MMP-2/MMP-9 genes: a novel mechanism for cardiovascular diseases. FASEB J. 2011;25(05):1718–1728. doi: 10.1096/fj.10-174904. [DOI] [PubMed] [Google Scholar]
- 60.Mott J L, Kobayashi S, Bronk S F, Gores G J. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26(42):6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cushing L, Costinean S, Xu W et al. Disruption of miR-29 leads to aberrant differentiation of smooth muscle cells selectively associated with distal lung vasculature. PLoS Genet. 2015;11(05):e1005238. doi: 10.1371/journal.pgen.1005238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deaton R A, Gan Q, Owens G K. Sp1-dependent activation of KLF4 is required for PDGF-BB-induced phenotypic modulation of smooth muscle. Am J Physiol Heart Circ Physiol. 2009;296(04):H1027–H1037. doi: 10.1152/ajpheart.01230.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Rooij E, Sutherland L B, Thatcher J E et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Du Y, Gao C, Liu Z et al. Upregulation of a disintegrin and metalloproteinase with thrombospondin motifs-7 by miR-29 repression mediates vascular smooth muscle calcification. Arterioscler Thromb Vasc Biol. 2012;32(11):2580–2588. doi: 10.1161/ATVBAHA.112.300206. [DOI] [PubMed] [Google Scholar]
- 65.Cordes K R, Sheehy N T, White M Pet al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity Nature 2009460(7256):705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pei H, Tian C, Sun X et al. Overexpression of microRNA-145 promotes ascending aortic aneurysm media remodeling through TGF-β1. Eur J Vasc Endovasc Surg. 2015;49(01):52–59. doi: 10.1016/j.ejvs.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 67.Elia L, Quintavalle M, Zhang J et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009;16(12):1590–1598. doi: 10.1038/cdd.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng Y, Liu X, Yang J et al. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105(02):158–166. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hergenreider E, Heydt S, Tréguer K et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14(03):249–256. doi: 10.1038/ncb2441. [DOI] [PubMed] [Google Scholar]
- 70.Chen J, Yin H, Jiang Y et al. Induction of microRNA-1 by myocardin in smooth muscle cells inhibits cell proliferation. Arterioscler Thromb Vasc Biol. 2011;31(02):368–375. doi: 10.1161/ATVBAHA.110.218149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xie C, Huang H, Sun X et al. MicroRNA-1 regulates smooth muscle cell differentiation by repressing Kruppel-like factor 4. Stem Cells Dev. 2011;20(02):205–210. doi: 10.1089/scd.2010.0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Torella D, Iaconetti C, Catalucci D et al. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res. 2011;109(08):880–893. doi: 10.1161/CIRCRESAHA.111.240150. [DOI] [PubMed] [Google Scholar]
- 73.Li P, Zhu N, Yi B et al. MicroRNA-663 regulates human vascular smooth muscle cell phenotypic switch and vascular neointimal formation. Circ Res. 2013;113(10):1117–1127. doi: 10.1161/CIRCRESAHA.113.301306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Merlet E, Atassi F, Motiani R K et al. miR-424/322 regulates vascular smooth muscle cell phenotype and neointimal formation in the rat. Cardiovasc Res. 2013;98(03):458–468. doi: 10.1093/cvr/cvt045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Y S, Wang H Y, Liao Y C et al. MicroRNA-195 regulates vascular smooth muscle cell phenotype and prevents neointimal formation. Cardiovasc Res. 2012;95(04):517–526. doi: 10.1093/cvr/cvs223. [DOI] [PubMed] [Google Scholar]
- 76.Li P, Liu Y, Yi B et al. MicroRNA-638 is highly expressed in human vascular smooth muscle cells and inhibits PDGF-BB-induced cell proliferation and migration through targeting orphan nuclear receptor NOR1. Cardiovasc Res. 2013;99(01):185–193. doi: 10.1093/cvr/cvt082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu X, Cheng Y, Yang J, Xu L, Zhang C. Cell-specific effects of miR-221/222 in vessels: molecular mechanism and therapeutic application. J Mol Cell Cardiol. 2012;52(01):245–255. doi: 10.1016/j.yjmcc.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leeper N J, Raiesdana A, Kojima Y et al. MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J Cell Physiol. 2011;226(04):1035–1043. doi: 10.1002/jcp.22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dong S, Xiong W, Yuan J, Li J, Liu J, Xu X. MiRNA-146a regulates the maturation and differentiation of vascular smooth muscle cells by targeting NF-κB expression. Mol Med Rep. 2013;8(02):407–412. doi: 10.3892/mmr.2013.1538. [DOI] [PubMed] [Google Scholar]
- 80.Zhang J, Zhao F, Yu X, Lu X, Zheng G. MicroRNA-155 modulates the proliferation of vascular smooth muscle cells by targeting endothelial nitric oxide synthase. Int J Mol Med. 2015;35(06):1708–1714. doi: 10.3892/ijmm.2015.2181. [DOI] [PubMed] [Google Scholar]
- 81.Liu X, Cheng Y, Chen X, Yang J, Xu L, Zhang C. MicroRNA-31 regulated by the extracellular regulated kinase is involved in vascular smooth muscle cell growth via large tumor suppressor homolog 2. J Biol Chem. 2011;286(49):42371–42380. doi: 10.1074/jbc.M111.261065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang J, Yan C H, Li Y et al. MicroRNA-31 controls phenotypic modulation of human vascular smooth muscle cells by regulating its target gene cellular repressor of E1A-stimulated genes. Exp Cell Res. 2013;319(08):1165–1175. doi: 10.1016/j.yexcr.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 83.Li T J, Chen Y L, Gua C J, Xue S J, Ma S M, Li X D. MicroRNA 181b promotes vascular smooth muscle cells proliferation through activation of PI3K and MAPK pathways. Int J Clin Exp Pathol. 2015;8(09):10375–10384. [PMC free article] [PubMed] [Google Scholar]
- 84.Davis B N, Hilyard A C, Lagna G, Hata A.SMAD proteins control DROSHA-mediated microRNA maturation Nature 2008454(7200):56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ji R, Cheng Y, Yue J et al. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of microRNA in vascular neointimal lesion formation. Circ Res. 2007;100(11):1579–1588. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 86.Yang J, Chen L, Ding J et al. MicroRNA-24 inhibits high glucose-induced vascular smooth muscle cell proliferation and migration by targeting HMGB1. Gene. 2016;586(02):268–273. doi: 10.1016/j.gene.2016.04.027. [DOI] [PubMed] [Google Scholar]
- 87.Fiedler J, Stöhr A, Gupta S K et al. Functional microRNA library screening identifies the hypoxamir miR-24 as a potent regulator of smooth muscle cell proliferation and vascularization. Antioxid Redox Signal. 2014;21(08):1167–1176. doi: 10.1089/ars.2013.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wanhainen A, Mani K, Vorkapic E et al. Screening of circulating microRNA biomarkers for prevalence of abdominal aortic aneurysm and aneurysm growth. Atherosclerosis. 2017;256:82–88. doi: 10.1016/j.atherosclerosis.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 89.Yu B, Liu L, Sun H, Chen Y. Long noncoding RNA AK056155 involved in the development of Loeys-Dietz syndrome through AKT/PI3K signaling pathway. Int J Clin Exp Pathol. 2015;8(09):10768–10775. [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao Y, Feng G, Wang Y, Yue Y, Zhao W. Regulation of apoptosis by long non-coding RNA HIF1A-AS1 in VSMCs: implications for TAA pathogenesis. Int J Clin Exp Pathol. 2014;7(11):7643–7652. [PMC free article] [PubMed] [Google Scholar]
- 91.Wang S, Zhang X, Yuan Y et al. BRG1 expression is increased in thoracic aortic aneurysms and regulates proliferation and apoptosis of vascular smooth muscle cells through the long non-coding RNA HIF1A-AS1 in vitro. Eur J Cardiothorac Surg. 2015;47(03):439–446. doi: 10.1093/ejcts/ezu215. [DOI] [PubMed] [Google Scholar]
- 92.Wang Y N, Shan K, Yao M D et al. Long noncoding RNA-GAS5: a novel regulator of hypertension-induced vascular remodeling. Hypertension. 2016;68(03):736–748. doi: 10.1161/HYPERTENSIONAHA.116.07259. [DOI] [PubMed] [Google Scholar]
- 93.Shan K, Jiang Q, Wang X Q et al. Role of long non-coding RNA-RNCR3 in atherosclerosis-related vascular dysfunction. Cell Death Dis. 2016;7(06):e2248. doi: 10.1038/cddis.2016.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Congrains A, Kamide K, Katsuya T et al. CVD-associated non-coding RNA, ANRIL, modulates expression of atherogenic pathways in VSMC. Biochem Biophys Res Commun. 2012;419(04):612–616. doi: 10.1016/j.bbrc.2012.02.050. [DOI] [PubMed] [Google Scholar]
- 95.Liu W, Liu X, Luo M et al. dNK derived IFN-γ mediates VSMC migration and apoptosis via the induction of LncRNA MEG3: A role in uterovascular transformation. Placenta. 2017;50:32–39. doi: 10.1016/j.placenta.2016.12.023. [DOI] [PubMed] [Google Scholar]
- 96.Lv J, Wang L, Zhang J et al. Long noncoding RNA H19-derived miR-675 aggravates restenosis by targeting PTEN. Biochem Biophys Res Commun. 2017:S0006-291X(17)30011-6. doi: 10.1016/j.bbrc.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 97.Wu G, Cai J, Han Y et al. LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation. 2014;130(17):1452–1465. doi: 10.1161/CIRCULATIONAHA.114.011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bell R D, Long X, Lin M et al. Identification and initial functional characterization of a human vascular cell-enriched long noncoding RNA. Arterioscler Thromb Vasc Biol. 2014;34(06):1249–1259. doi: 10.1161/ATVBAHA.114.303240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ingber D E.Tensegrity II. How structural networks influence cellular information processing networks J Cell Sci 2003116(Pt 8, 116Pt 8):1397–1408. [DOI] [PubMed] [Google Scholar]
- 100.Wang N, Tytell J D, Ingber D E. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol. 2009;10(01):75–82. doi: 10.1038/nrm2594. [DOI] [PubMed] [Google Scholar]
- 101.Isermann P, Lammerding J. Nuclear mechanics and mechanotransduction in health and disease. Curr Biol. 2013;23(24):R1113–R1121. doi: 10.1016/j.cub.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sun S G, Zheng B, Han M et al. miR-146a and Krüppel-like factor 4 form a feedback loop to participate in vascular smooth muscle cell proliferation. EMBO Rep. 2011;12(01):56–62. doi: 10.1038/embor.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Di Gregoli K, Mohamad Anuar N N, Bianco R et al. MicroRNA-181b controls atherosclerosis and aneurysms through regulation of TIMP-3 and elastin. Circ Res. 2017;120(01):49–65. doi: 10.1161/CIRCRESAHA.116.309321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kee H J, Kim G R, Cho S N et al. miR-18a-5p microRNA increases vascular smooth muscle cell differentiation by downregulating Syndecan4. Korean Circ J. 2014;44(04):255–263. doi: 10.4070/kcj.2014.44.4.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li J, Zhao L, He X, Yang T, Yang K. MiR-21 inhibits c-Ski signaling to promote the proliferation of rat vascular smooth muscle cells. Cell Signal. 2014;26(04):724–729. doi: 10.1016/j.cellsig.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 106.Yang G, Pei Y, Cao Q, Wang R. MicroRNA-21 represses human cystathionine gamma-lyase expression by targeting at specificity protein-1 in smooth muscle cells. J Cell Physiol. 2012;227(09):3192–3200. doi: 10.1002/jcp.24006. [DOI] [PubMed] [Google Scholar]
- 107.le Sage C, Nagel R, Egan D A et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26(15):3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Davis B N, Hilyard A C, Nguyen P H, Lagna G, Hata A. Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem. 2009;284(06):3728–3738. doi: 10.1074/jbc.M808788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhu X F, Shan Z, Ma J Y et al. Investigating the role of the posttranscriptional gene regulator MiR-24- 3p in the proliferation, migration and apoptosis of human arterial smooth muscle cells in arteriosclerosis obliterans. Cell Physiol Biochem. 2015;36(04):1359–1370. doi: 10.1159/000430302. [DOI] [PubMed] [Google Scholar]