

ABSTRACT
We assessed the tolerability and antitumor activity of solitomab, a bispecific T-cell engager (BiTE®) antibody construct targeting epithelial cell adhesion molecule (EpCAM). Patients with relapsed/refractory solid tumors not amenable to standard therapy received solitomab as continuous IV infusion in a phase 1 dose-escalation study with six different dosing schedules. The primary endpoint was frequency and severity of adverse events (AEs). Secondary endpoints included pharmacokinetics, pharmacodynamics, immunogenicity, and antitumor activity. Sixty-five patients received solitomab at doses between 1 and 96 µg/day for ≥28 days. Fifteen patients had dose-limiting toxicities (DLTs): eight had transient abnormal liver parameters shortly after infusion start or dose escalation (grade 3, n = 4; grade 4, n = 4), and one had supraventricular tachycardia (grade 3); all events resolved with solitomab discontinuation. Six patients had a DLT of diarrhea: four events resolved (grade 3, n = 3; grade 4, n = 1), one (grade 3) was ongoing at the time of treatment-unrelated death, and one (grade 3) progressed to grade 5 after solitomab discontinuation. The maximum tolerated dose was 24 µg/day. Overall, 95% of patients had grade ≥3 treatment-related AEs, primarily diarrhea, elevated liver parameters, and elevated lipase. Solitomab half-life was 4.5 hours; serum levels plateaued within 24 hours. One unconfirmed partial response was observed. In this study of a BiTE® antibody construct targeting solid tumors, treatment of relapsed/refractory EpCAM-positive solid tumors with solitomab was associated with DLTs, including severe diarrhea and increased liver enzymes, which precluded dose escalation to potentially therapeutic levels.
KEYWORDS: AMG 110; solitomab; MT110; immunotherapy; bispecific; BiTE®; EpCAM, phase 1; solid tumor; CD3
Introduction
The epithelial cell adhesion molecule (EpCAM, CD326) is a transmembrane, 40 kDa glycoprotein highly expressed in colon, gastric, prostate, ovarian, lung, and pancreatic cancer and often correlated with a poor prognosis, thus representing an attractive therapeutic target.1-4 Although EpCAM can be detected on the basolateral membrane of various normal epithelial tissues, including colon, small intestine, and hepatoblasts,5,6 it is believed to be less accessible to traditional antibody constructs due to sequestration within tight cellular junctions.7,8 The univalent EpCAM antibodies edrecolomab and adecatumumab have shown modest single-agent activity in the treatment of colon, prostate, and breast cancer.9-11
Bispecific T cell-engager (BiTE®) antibody constructs combine two single chain Fv domains from two different antibodies: one recognizes a tumor-associated antigen expressed on target cells while the other binds to the CD3ε epitope of the T-cell receptor, resulting in the formation of a cytotoxic synapse followed by tumor cell lysis. In comparison to checkpoint inhibitors, BiTE® antibody constructs12 induce an MHC-independent T-cell response, thereby circumventing specific immune escape mechanisms.13 Blinatumomab was the first BiTE® antibody construct to be clinically evaluated, focusing on hematologic malignancies. Blinatumomab has demonstrated efficacy in patients with Philadelphia chromosome-negative relapsed/refractory acute lymphoblastic leukemia (ALL)14 and antilymphoma activity in patients with relapsed/refractory diffuse large B-cell lymphoma.15
The number of EpCAM-positive cancers with limited therapeutic options at advanced stage formed the rationale for developing the EpCAM/CD3 BiTE® antibody construct immunotherapy solitomab (MT110, AMG 110). In a preclinical study, solitomab induced target cell lysis through the activation of cytolytic T-cell synapses.16
This report describes results from a first-in-human, phase 1 dose-escalation study that assessed the tolerability and evidence of antitumor activity of solitomab in patients with refractory, EpCAM-expressing solid tumors. The study represents one of the first investigations of a BiTE® antibody construct in patients with solid tumors.
Results
Patients
Between April 2008 and December 2013, 65 patients were enrolled at four study sites in Germany. Most patients were ≤65 years old and had stage IV disease, liver metastases, and/or abnormal liver parameters at baseline (Table 1). The most common diagnoses were colorectal, ovarian, and gastric cancer. EpCAM expression was detected in 80% (52 of 65) of archival tissue samples. Among these, 57% and 14% had high and low EpCAM expression, respectively. EpCAM expression was undetectable in 6% (4 of 65) of samples. Expression data were unavailable for the remaining samples. All patients received at least one dose of solitomab during cycle 1. Eighteen patients (28%) started two or more treatment cycles. Forty-nine patients had at least one treatment interruption (mean 0.3 days; range, 0.0–10.4 days) due to technical or logistical reasons; 10 patients had at least one interruption due to adverse events (AEs). Most patients discontinued the study because of disease progression (n = 25; 39%) or AEs (n = 25; 39%). Other reasons for study discontinuation included withdrawal of consent (n = 6), non-compliance (n = 1), death (n = 1), and other reasons (n = 7).
Table 1.
Baseline demographics and clinical characteristics.
| Characteristic | All Patients (N = 65) |
|---|---|
| Age, n (%) | |
| ≤65 years | 47 (72) |
| >65 years | 18 (28) |
| Sex, n (%) | |
| Female | 27 (42) |
| Male | 38 (59) |
| Primary Tumor Type, n (%) | |
| Colorectal | 35 (54) |
| Ovarian | 10 (15) |
| Gastric | 8 (12) |
| Non-small cell lung | 6 (9) |
| Small cell lung | 3 (5) |
| Hormone-refractory prostate | 3 (5) |
| Disease Stage, n (%) | |
| Stage III/IIIc | 2 (3) |
| Stage IV | 51 (78) |
| Unknown | 12 (18) |
| Liver, n (%) | |
| Metastases at screening | 41 (63) |
| Abnormal parameters at screening | 42 (65) |
| EpCAM expression, n (%) | |
| High | 41 (63) |
| Low | 11 (17) |
| Negative | 4 (6) |
| Missing | 9 (14) |
| ECOG performance status, n (%) | |
| 0 | 25 (39) |
| 1 | 35 (54) |
| 2 | 5 (8) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group.
Spizzo G, Went P, Dirnhofer S,et al: High EpCAM expression is associated with poor prognosis in node-positive breast cancer. Breast Can Res Treat. 86:207-213, 2004.
Dose-limiting toxicities
Patients received solitomab in 14 dose groups using six different dosing schedules and doses ranging from 1 to 96 µg/day (dosing schedule A1, n = 6; A2, n = 3; A3, n = 2; B1, n = 4; B2, n = 4; B3, n = 5; Bx1, n = 7; Bx2, n = 7; Bx3, n = 7; Bz1, n = 3; C1, n = 4; C2, n = 3; D1, n = 3; D2, n = 7; Figure 1). Across all dosing schedules, 15 patients had dose-limiting toxicities (DLTs). Two patients that did not present with clinical symptoms continued treatment per standard clinical practice and careful risk-benefit evaluation (Table 2). Most DLTs were transient grade 3 or 4 abnormal liver parameters that occurred shortly after infusion start or dose escalation and resolved under continued solitomab administration or, in some cases, after discontinuation of solitomab. Based on the observation that increases in liver parameters were less pronounced during cycle 2 compared with cycle 1 (Fig. S1), dosing schedules B, C, and D were explored in an effort to mitigate DLTs like diarrhea and liver toxicity (Figure 1). Additionally, administration of dexamethasone during the first three days of each dosing cycle was implemented. However, the maximum tolerated dose (MTD) could not be increased above 24 µg/day for cohorts A through D, although individual patients could tolerate higher doses. DLTs occurred in three of the seven patients receiving 48 µg/day and in two of the seven patients receiving 96 µg/day.
Figure 1.
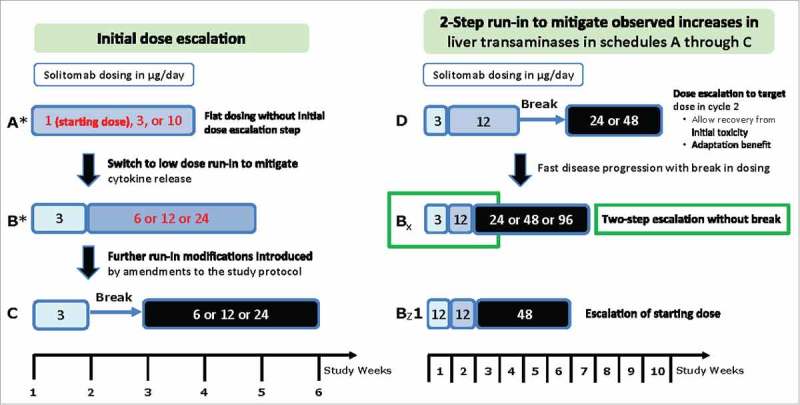
Treatment schema. Schedules A through C explored flat dosing and low-dose run-in schedules with and without a break in dosing. Treatment schedules D, Bx, and Bz1 explored various two-step run-in protocols with and without a break in dosing and optional extension weeks.
Table 2.
Dose-limiting toxicities.
| Patient | MedDRA Preferred Term | CTCAE Grade | Changes in Solitomab Infusion | Resolved | Dose at Time of DLT (μg/day) | Treatment Schedule |
|---|---|---|---|---|---|---|
| 1 | AST increased | 4 | None | Yes | 1 | A |
| ALT increased | 4 | None | Yes | |||
| Glutamate dehydrogenase increased | 4 | None | Yes | |||
| 2 | AST increased | 3 | Discontinued | Yes | 10 | A |
| 3 | Gamma-glutamyl transferase increased | 4 | Discontinued | Yes | 24 | B |
| 4 | Diarrhea | 3 | Discontinued | Yes | 24 | B |
| 5 | Abdominal pain | 3 | Discontinued | Yes | 24 | Bx |
| Diarrhea | 3 | Discontinued | Yes | |||
| 6 | Diarrhea | 3 | Discontinued | Yes | 48 | Bx |
| 7 | AST increased | 3 | Discontinued | Yes with sequelae | 12 | Bx |
| 8 | Blood bilirubin increased | 3 | Discontinued | Yes | 96 | Bx |
| 9 | Diarrhea | 4 | Discontinued | Yes | 96 | Bx |
| 10 | AST increased | 4 | Discontinued | Yes with sequelae | 12 | Bz1 |
| ALT increased | 4 | Discontinued | Yes | |||
| 11 | AST increased | 4 | None | Yes | 12 | Bz1 |
| 12 | Blood bilirubin increased | 3 | Discontinued | Yes | 24 | C |
| 13 | Diarrhea | 3 | Discontinued | Yes with sequelae | 24 | C |
| 14 | Diarrhea | 3 | Discontinued | Yes | 48 | D |
| 15 | Supraventricular tachycardia | 3 | Discontinued | Yes | 48 | D |
Abbreviations: AST, aspartate aminotransferase; ALT, alanine aminotransferase; CTCAE, Common Terminology Criteria for Adverse Events; MedDRA, Medical Dictionary for Regulatory Activities; DLT, dose-limiting toxicity.
Treatment schedules B, C, and D could be performed in parallel as independent groups.
Event was not considered a DLT by the investigator per protocol provision (see Methods) given the rapid normalization of liver abnormalities and the absence of clinical signs and symptoms. The patient had increased liver parameters at baseline following anticoagulation therapy, which was discontinued at study start. Liver parameters had normalized before solitomab administration but increased again once anticoagulation treatment was restarted. These events triggered specific protocol amendments that resulted in modifications of the dose and dosing schedules and the definition of DLT (Supplementary Data).
Resolution with sequale indicates that resolution of the DLT to grade 1 or baseline level did not occur until the end of the study. In patient 7, the AST increase resolved to grade 2 at the end of study. In patient 10, the AST increase resolved to grade 3 at the end of the study. In patient 13, diarrhea resolved to grade 2 at the end of the study and subsequently received other chemotherapy.
This DLT was not documented as an AE by the investigator due to the absence of clinical signs and symptoms.
Changes in liver parameters
Changes in liver parameters did not show a clear dose-dependency (Table 2), and they were not associated with demographic characteristics (data not shown). All liver toxicity was evaluated in the context of clinical signs/symptoms and an individual patient's recovery, medical history, and diagnosis. Additionally, ultrasound or CT hepatic imaging and liver biopsy were performed in four patients across dosing schedules B and C. Imaging did not detect pathological findings associated with abnormal liver parameters, and individual biopsy evaluation did not show liver damage typically associated with severe drug-induced liver injury, consistent with the normalization of liver enzymes over time (Fig. S1). Typically, the peak in elevation of transaminases occurred at day 2 with levels returning to baseline in most cases despite continuing treatment. Increases in transaminases were less pronounced during cycle 2 after a two-week treatment-free interval, suggesting that tachyphylaxis developed upon continued treatment with solitomab, possibly as a result of immunological feedback loops or upregulation of cytokine receptors.
Diarrhea
In four of the six patients with a DLT of diarrhea, the events resolved with treatment discontinuation. Diarrhea severity was dependent on dose and treatment duration. One grade 3 event of diarrhea was ongoing at the time the patient died of treatment-unrelated septic shock with pneumonia; the other was a grade 5 event considered probably related to solitomab treatment. The patient had grade 3 diarrhea at the time of infusion discontinuation; the fatal event occurred 60 days after solitomab discontinuation and followed a complicated clinical course, including Staphylococcus epidermidis infection and increased nausea and vomiting.
Patients with persistent gastrointestinal AEs after treatment had ended were evaluated further. One patient with lung adenocarcinoma had ongoing abdominal pain one week after solitomab infusion (3 µg/day for 8 days, 12 µg/day for 3 days) was stopped. Endoscopic examination of the duodenum revealed widespread mucosal atrophy. IHC staining of duodenal tissue from the same patient showed robust EpCAM expression on epithelial cells in duodenal crypts and along villi but not on other cells of the mucosa or Brunner's glands (Figure 2A). There was also strong infiltration of mononuclear cells, especially lymphocytes and single eosinophil and neutrophil granulocytes, but only few B cells, indicating localized inflammation associated with epithelial tissue damage. CD3- and TIA-positive lymphocytes were present on basolateral sites of the epithelium and between epithelial cells, suggesting potential cytotoxic activity (Figure 2B). Histologic examination indicated damage of duodenal crypt structure (Figure 2C) and ongoing regenerative processes in areas adjacent to the damaged tissue (data not shown). There was no damage to submucosa or muscularis.
Figure 2.
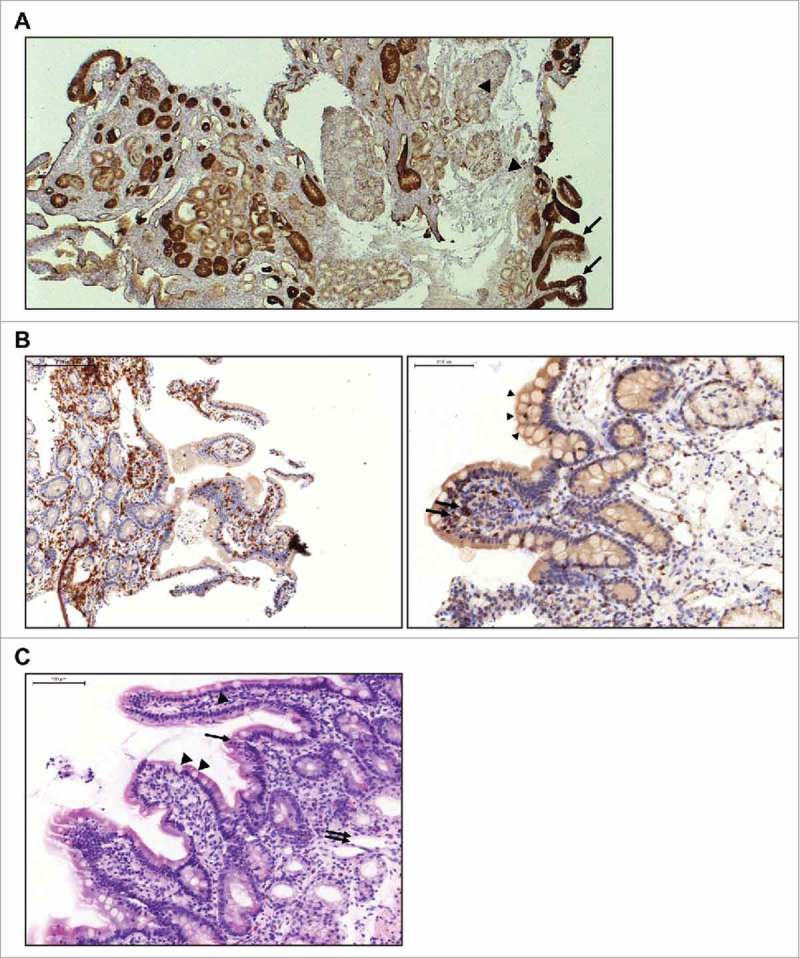
Immunohistochemical evaluation of duodenal biopsy tissue from a patient with lung adenocarcinoma treated with solitomab (3 µg/day for 8 days, 12 µg/day for 3 days). A, IHC staining of EpCAM expression shows EpCAM-positive epithelial cells in duodenal crypts and along villis (arrows) and EpCAM-negative cells of the mucosa and Brunner's cells (arrow heads). B, Infiltration of duodenal epithelium by CD3-positive (left) and T-cell restricted intracellular antigen (TIA)-positive (right; arrows) lymphocytes. Vacuolated tip enterocytes (arrow heads) are also present. C, HE staining showing damage to the crypt structure with villus collapse (arrow) and mucosal ulceration (double arrow). Vacuolated tip enterocytes (arrow heads) are visible along the villi.
Given the severity of the diarrhea, we conducted a series of controlled concurrent preclinical experiments to confirm and expand on the single biopsy findings described above. Treatment of mice with 0.05 mg/kg/day of muS110, a murine surrogate of solitomab, for two days caused body weight loss and hypoactivity in all animals and diarrhea in a subset. Enterocytes in the mouse small intestine showed robust expression of EpCAM along the villi in both treatment groups (Figure 3A). Similar to the biopsy findings, duodenal tissue from treated animals, but not from control, showed significant damage as evident in vacuolated enterocytes along the villi tips, villous collapse, and mucosal ulceration (Figure 3B) as well as signs of advanced repair including crypt elongation and enterocyte hyperplasia. Importantly, no tissue damage was detected in the colon of the same animals (data not shown), suggesting duodenal origin of the diarrhea. Duodenal tissue damage was accompanied by CD3+ lymphocyte infiltration, a subset of which were Granzyme B-positive, and increased cleaved caspase-3 immunoreactivity within enterocytes at villi tips (Figure 3C). Collectively, the duodenal lesions and immunohistochemical staining patterns imply acute enterocyte injury and concurrent repair associated with muS110 treatment. Cytokine levels were noticeably elevated in the serum of animals treated with muS110, compared with control, along with markers of epithelial barrier damage (Table S1).
Figure 3.
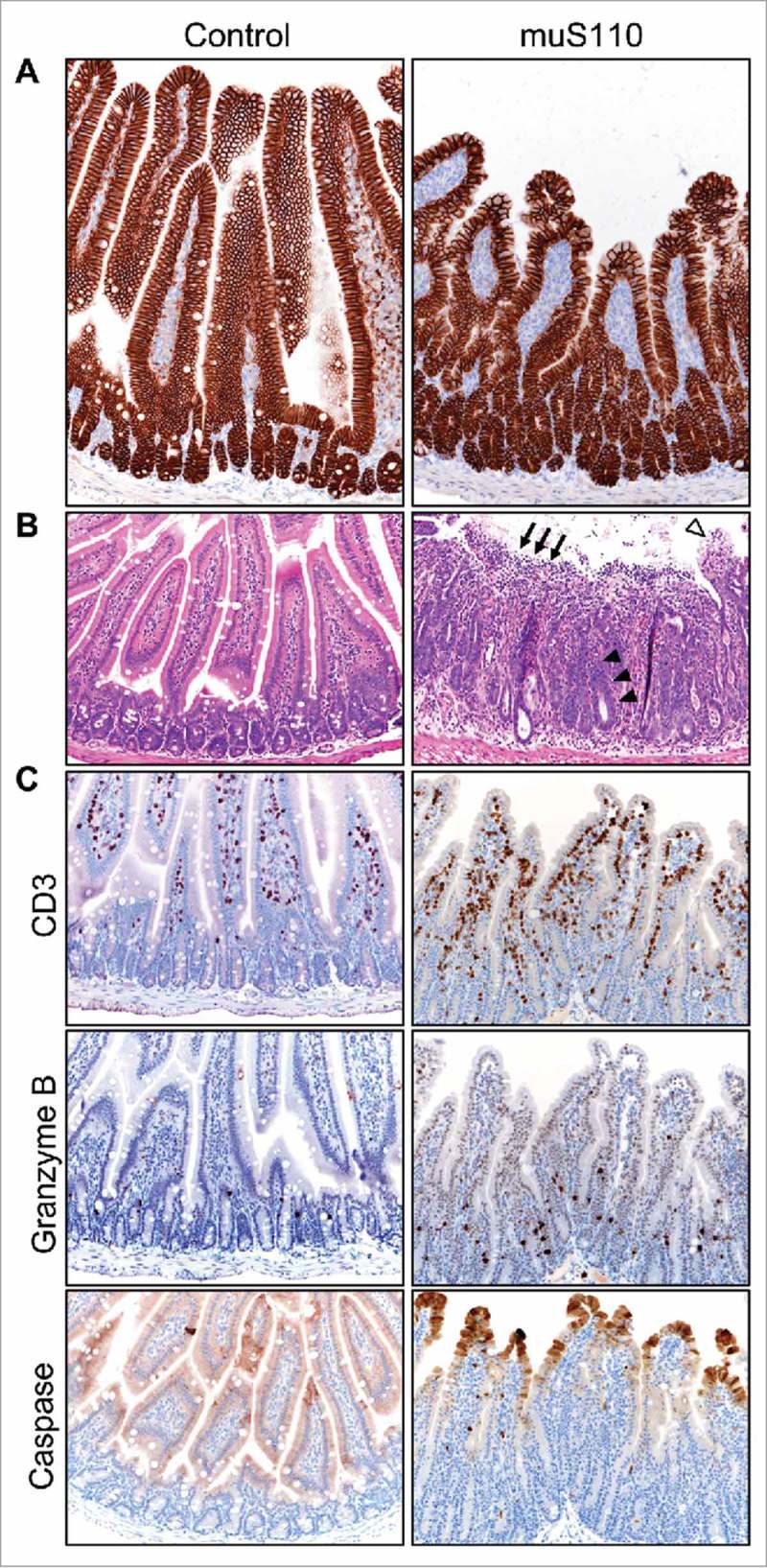
Effect of treatment with muS110 on mouse duodenal tissue. A, IHC showing similar expression of EpCAM in enterocytes from animals treated with vehicle control (left) or muS110 (right). B, HE stain of mouse duodenal tissue from vehicle control-treated animal (left) showing normal, healthy crypt epithelium and villi; and from muS110-treated animals showing crypt elongation with enterocyte hyperplasia (arrow head), villous collapse and mucosal ulceration (arrow) as well as vacuolated tip enterocytes (open arrow head). C, IHC illustrating presence of CD3-positive lymphocytes as well as Granzyme B- and caspase-positive cells in animals treated with vehicle control or muS110. Ten animals per group received 0.05 mg/kg/day of muS110 once every day for two days. Images from representative animals are shown (200 × ).
Other adverse events
Throughout the study the most frequently reported (≥20% of patients) treatment-related grade ≥3 AEs were diarrhea, elevations in liver function tests, and increases in lipase. The most common treatment-related AEs of any grade were pyrexia, peripheral edema, nausea, vomiting, and abdominal pain. Serious AEs were restricted to diarrhea and abdominal pain and did not include AEs, such as hepatic encephalopathy or hepatobiliary obstruction (Table 3), or laboratory abnormalities, such as decreased Prothrombin Time and International Normalized Ratio (Table S2), typically associated with clinically relevant liver toxicity. No cases of CNS toxicity were observed. A total of four (6%) patients had fatal AEs on study, including septic shock with pneumonia, ileus, sepsis, and the diarrhea DLT noted earlier. None of these fatal events, except the diarrhea DLT, were considered related to solitomab by the investigator.
Table 3.
Patient Incidence of adverse events.
| All Patients (N = 65) |
|||
|---|---|---|---|
| AEs Any Grade | Treatment-related AEs Grade ≥3 | AEs Serious | |
| AEs Occurring in >5% of Patients, n (%) | |||
| Diarrhea | 30 (46) | 11 (17) | 8 (12) |
| Pyrexia | 28 (43) | 0 (0) | 1 (2) |
| Peripheral edema | 26 (40) | 0 (0) | 0 (0) |
| Nausea | 25 (39) | 1(2) | 3 (5) |
| Vomiting | 22 (34) | 1 (2) | 2 (3) |
| Abdominal pain | 21 (32) | 3 (5) | 4 (6) |
| Fatigue | 19 (29) | 3 (5) | 1 (2) |
| Dysgeusia | 15 (23) | 1 (2) | 0 (0) |
| Cough | 11 (17) | 0 (0) | 0 (0) |
| Insomnia | 11 (17) | 1 (2) | 0 (0) |
| Anorexia | 10 (15) | 1 (2) | 0 (0) |
| Dyspnea | 10 (15) | 0 (0) | 2 (3) |
| Flatulence | 8 (12) | 0 (0) | 0 (0) |
| Headache | 8 (12) | 0 (0) | 0 (0) |
| Infection | 8 (12) | 1 (2) | 4 (6) |
| Sleep disorder | 8 (12) | 0 (0) | 0 (0) |
| General physical health deterioration | 7 (11) | 0 (0) | 2 (3) |
| Upper abdominal pain | 7 (11) | 0 (0) | 0 (0) |
| Hypertension | 7 (11) | 1 (2) | 0 (0) |
| Constipation | 6 (9) | 0 (0) | 0 (0) |
| Nasopharyngitis | 6 (9) | 0 (0) | 0 (0) |
| Weight decreased | 6 (9) | 2 (3) | 0 (0) |
| Candidiasis | 5 (8) | 0 (0) | 0 (0) |
| Dyspepsia | 5 (8) | 0 (0) | 0 (0) |
| Jaundice | 5 (8) | 1 (2) | 0 (0) |
| Overdose | 5 (8) | 2 (3) | 5 (8) |
| Ascites | 4 (6) | 1 (2) | 0 (0) |
| Back pain | 4 (6) | 0 (0) | 0 (0) |
| Device related infection | 4 (6) | 0 (0) | 3 (5) |
| Dizziness | 4 (6) | 0 (0) | 0 (0) |
| Dry skin | 4 (6) | 0 (0) | 0 (0) |
| Nocturia | 4 (6) | 0 (0) | 0 (0) |
| Urinary tract infection | 4 (6) | 0 (0) | 1 (2) |
Abbreviations: AE, adverse event.
Among 63 patients with evaluable samples for antibody testing, seven (11%) developed anti-solitomab antibodies during treatment. Five of these patients had neutralizing antibodies at the end of the study, and two patients receiving 96 μg/day developed high-titer neutralizing antibodies that resulted in a reduction of measurable solitomab levels. One of the patients with neutralizing antibodies received a second treatment cycle without clinical signs or symptoms of anaphylactic reactions.
Pharmacokinetics and pharmacodynamics
The increase in solitomab steady-state concentration (Css) was dose-proportional, although solitomab was not detectable at doses below 3 µg/day (Fig. S2). Css was reached within 24 hours after solitomab infusion start. The elimination half-life was 4.5 hours. At doses ≥48 µg/day, solitomab concentrations were comparable to published in vitro EC90 values from colorectal cancer cell lines.17
After infusion start or dose escalation, a swift decrease of peripheral T-cell counts was observed (Fig. S3A). In most patients, recovery of T-cell counts was delayed, remaining low for more than two weeks. T cells increased again after treatment stop. This lymphocyte redistribution was not dose-dependent. Peripheral CD4+ and CD8+ T cells showed similar, redistribution patterns typical for T cells (Fig. S3B). There was also transient expression of the T-cell activation markers CD69 and LFA-1 (data not shown). Measured serum cytokines, most notably IL-6, IL-8, and IFN-γ, increased from baseline following infusion (Fig. S3C and S3D). Cytokine concentrations peaked mainly during cycle 1 after escalation to target dose and decreased quickly within 24 to 48 hours. There was no true dose-dependency, likely because of dexamethasone pretreatment (Fig. S3D).
Antitumor activity
Fifty-four of 65 enrolled patients had a response assessment according to RECIST. Eight patients (15%) without measureable target lesions were not evaluable; 11 patients (17%) had no follow-up tumor assessment. Confirmed stable disease (SD) was the best overall response. A total of 18 patients had at least SD as best response, with a median duration of 84 days (range, 21‒355 days). Of those, 17 patients (31%) had a best response of SD and one had an unconfirmed partial response; 28 (52%) patients had progressive disease. The unconfirmed partial response was observed in a patient with ovarian cancer who received solitomab 48 µg/day. The patient, who was 76 years old, had progressive disease and new hepatic lesions after anthracycline-based chemotherapy with pegylated doxorubicin. After three months of solitomab therapy, treatment was stopped because of diarrhea. The patient showed a 39% reduction from baseline tumor dimensions (per serial CT scans) 22 weeks after the end of treatment. Another patient with metastatic ovarian cancer, who received one cycle at 96 µg/day, showed an almost complete disappearance of ascites, accompanied by marked clinical improvement (based on the investigator's assessment). However, the patient developed rapid disease progression after cycle 2 and discontinued treatment.
Discussion
The development of BiTE® antibody constructs against cancer cells has opened a new pathway in immunotherapy. Proof of concept was shown early with blinatumomab in the setting of hematologic malignancies;12,14,15 however, information on the applicability of BiTE® antibody constructs to the solid tumor setting remains very limited.18 We report here the final results of the first-in-human phase 1 study of solitomab, a BiTE® antibody construct immunotherapy against EpCAM, in solid tumors.
Sixty-five patients were treated in a dose escalation design with prolonged solitomab infusions on the basis that therapeutic T-cell activation and proliferation may be augmented by sustained stimulation. Due to its short half-life (4.5 hours), solitomab was administered by continuous intravenous infusion (cIV) over at least 4 weeks, allowing for continuous exposure. Ambulatory treatment was feasible in all patients.
The DLTs consisted of clinically significant but transient increases in liver parameters and severe diarrhea accompanied by abdominal pain. Most patients with increased liver parameters did not present with clinical symptoms and none had severe pathological liver findings. Several different dosing regimens were explored in an effort to mitigate liver enzyme increases. Steroid premedication was recommended to mitigate diarrhea, which was clearly dependent on dose and treatment duration, and steroid treatment at first diarrhea onset to prevent symptoms from worsening. Similar to experience with blinatumomab,15,19 most of the clinically significant AEs emerged within the first days after infusion start, which guided the definition of the DLT period. Based on preclinical experiments with the murine solitomab surrogate muS110 showing that a one-week low-dose adaptation period permitted prolonged therapy at higher doses thereafter,20 we incorporated step-dosing, which proved generally more tolerable. Although increases in liver parameters did not show a clear dose-dependency, they were much less pronounced with low-dose run-in and at the start of the second cycle compared with the first cycle. Despite dose modifications and high-dose steroid administration during the first three days of solitomab infusion, DLTs continued to occur. The MTD was 24 µg/day, although individual patients could tolerate higher doses (up to 96 µg/day). Although two patients with ovarian cancer who received 96 µg/day showed antitumor effects of short duration, the nature and severity of AEs did not allow dose escalation to potentially therapeutic levels.
Adverse events and DLTs of diarrhea were not entirely unexpected given EpCAM expression throughout the gastrointestinal tract. Early preclinical experiments in mice using muS110 showed dose-dependent diarrhea as a side effect.20 However, the severity and duration of the diarrhea in the clinical study and its limited responsiveness to management were unforeseen. Immunohistochemical staining of duodenal biopsy samples showed damage to EpCAM-expressing tissue along with some regenerative processes and evidence of T cell infiltration and apoptotic activity. The findings were confirmed in concomitant, vehicle-controlled animal studies with muS110, which also revealed that the tissue damage was limited to the duodenum and did not involve the colon. Diarrhea of duodenal origin is a far more severe and less manageable form, which is consistent with the clinical study observations. Moreover, the data from the animal experiments, which were collected 48 hours post dosing, showed clear signs of diminishing damage and beginning regeneration, indicating that more severe tissue destruction may have been detectable at earlier time points. Based on these data, we hypothesize that solitomab localized in the duodenal mucosa recruits and activates intraepithelial T cells colocalized among EpCAM-expressing enterocytes, leading to T cell-mediated epithelial apoptosis, villus collapse, and acute lesions causing severe diarrhea; however, this hypothesis requires further investigation. The presence of granzyme B and caspase at the tip of villi in muS110-treated mice is in line with the expected BiTE® mode of action, indicating muS110 activity in healthy duodenal tissue. Although some nontumor activity of solitomab was expected given the expression of EpCAM in the gastrointestinal tract, the severity of the tissue damage was surprising and may be linked to duodenal regeneration and associated increased accessibility of EpCAM molecules. The duodenum has the highest regeneration rate of any tissue, and even more so after injury. The duodenum of muS110-treated mice showed crypt elongation and enterocyte hyperplasia, suggesting robust regeneration and possibly high EpCAM expression in many newly formed cells. Regeneration starts in the villi troughs where cells are not entirely organized and tight junctions not fully formed, possibly making EpCAM molecules more accessible to muS110 or solitomab. Given the advanced disease state of the study population, patients may have entered the study with preexisting injury to the duodenum from previous cancer treatments and thus, easily accessible EpCAM molecules. This hypothesis is supported by the observation that the severity of diarrhea varied among patients, which would be in line with different degrees of preexisting tissue damage.
The AEs associated with solitomab treatment likely represent a direct targeting effect, since EpCAM is expressed on normal liver bile ducts and GI tract epithelia.5 Similar AEs, including liver toxicities and diarrhea, have been reported in a phase 1 study of the trifunctional antibody catumaxomab, which targets EpCAM, CD3, and Fcγ receptors. However, catumaxomab appears to mediate a target-independent interaction between Fcγ and Kupffer cells in the liver.21 The exact mechanism leading to changes in liver parameters after the start of solitomab infusion or after dose escalation remains unclear. A current hypothesis proposes that solitomab administration may lead to local lysis of EpCAM-positive cells in the liver, accompanied by transient and localized release of proinflammatory cytokines by involved cytotoxic T cells. Locally released cytokines may affect target-negative hepatocytes, resulting in bile acid transport impairment and accumulation of bile acids. The ensuing transient increase in liver enzymes and bilirubin in serum may reflect a reversible and self-limiting damage to hepatocytes and bile duct epithelial cells. Despite continued dosing with solitomab in most patients, increases in liver parameters did not indicate chronic or severe drug-induced liver injury. Step-wise intrapatient dose escalation and concomitant dexamethasone partly mitigated the initial elevation in liver parameters. However, the potential for liver injury must be carefully balanced against anticipated benefit with solitomab, which, in most patients, was limited by the occurrence of sometimes severe diarrhea with longer infusions at higher doses.
Only patients treated at or above 24 µg/day achieved a solitomab Css of at least 1 ng/mL required for efficient tumor cell lysis in vitro,22 which likely contributed to the limited antitumor activity. The observed lymphocyte redistribution (and transient expression of T cell activation markers) was similar to what has been reported for the BiTE® antibody construct blinatumomab.12,23 In contrast with blinatumomab, which targets CD19, recurrence of peripheral T cells during solitomab treatment was delayed or absent in most patients during the first 50 hours of treatment in the first patients enrolled, possibly indicating T-cell migration from the peripheral blood to the solid tumor; however, this hypothesis require further investigation. Prolonged therapy with dexamethasone was necessary in patients with impacted lymphocyte levels. Similarly, the transient increase in cytokines following solitomab treatment is in line with the BiTE® mode of action. Of note, the pharmacodynamic data were primarily collected in the early study cohorts, with limited data available from later dosing schedules.
As indicated above, BiTE®-directed cytolysis, in addition to lymphocyte- and cytokine-mediated effects, may have been responsible for the observed increases in liver parameters and diarrhea, underlining the potency of this therapeutic approach. Continued development of BiTE® antibody constructs in the solid tumor setting is desirable given that the BiTE® mechanism of action may circumvent common tumor escape mechanisms, such as the loss of MHC molecules or impaired antigen processing and/or transport. However, harnessing its potency in the solid tumor setting will require identification of targets that are restricted or significantly overexpressed in tumor tissue. Accordingly, clinical studies with BiTE® antibody constructs directed against CEA (NCT02291614) and PSMA (NCT01723475) were underway.
In conclusion, in this study of a BiTE® antibody construct in solid tumors, significant target-related AEs, consistent with the solitomab mechanism of action, prevented dose escalation to therapeutic levels. Solitomab showed preliminary signs of antitumor activity. While solid tumor therapy with BiTE® antibody constructs remains an attractive treatment approach, selection of appropriate targets is crucial.
Methods
Patients
Eligible patients were ≥18 years old with locally advanced, recurrent or metastatic solid tumors known to express EpCAM: adenocarcinoma of the lung or gastroesophageal junction, small cell lung, colorectal, gastric, endometrial, ovarian, breast or hormone-refractory prostate cancer. Key inclusion criteria were Eastern Cooperative Oncology Group (ECOG) performance status ≤2, recovery from any previous anticancer chemotherapy, nonmeasureable disease or at least one measurable tumor lesion per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0.24 Key exclusion criteria were evidence of CNS metastases at baseline per computed tomography (CT) or contrast-enhanced magnetic resonance imaging (MRI) scan; inadequate hematologic, renal or hepatic function; chronic systemic corticosteroid therapy for >2 months or any other immunosuppressive therapies; presence of human anti-murine antibodies. The study protocol was approved by the independent ethics committee at each research center, and patients provided written informed consent before any study-specific procedures. The study was conducted in accordance with the Declaration of Helsinki and consistent with Good Clinical Practice (ClinicalTrials.gov: NCT00635596).
Study design and treatment
This was a phase 1 dose-escalation study. Patients received solitomab by continuous intravenous infusion (cIV) via a portable pump. The starting dose was 1 µg/day during weeks 1 to 4 based on the in vitro minimal anticipated biological effect level. The initial study protocol included one intrapatient dose escalation, based on preclinical observations, in the event of early, non-cumulative toxicity.20,25
Solitomab was administered in six-week dosing cycles (four weeks of solitomab cIV followed by a two-week treatment-free period), until disease progression, withdrawal of informed consent, or unacceptable side effects. Dose escalation followed a 3+3 design with six different schedules and doses ranging from 1 to 96 µg/day (Figure 1). Treatment schedule A followed a first-in-human flat dosing regimen. Schedules B, C, and D were performed independently and in parallel and included a run-in phase with lower doses to mitigate cytokine release, followed by higher doses to allow for clinical experience at increased doses, but only after a Data Review Committee (DRC) evaluated DLTs and all clinically relevant safety data at the patient, dose group, and study level. The starting dose for the first treatment week in schedules B and C was the maximum-tolerated dose (MTD) from schedule A, or a lower dose decided by the DRC. Schedule B was only performed if schedule A resulted in an MTD of 10 μg/day or lower. Schedule D included a two-step run-in phase, followed by a break, then dosing at higher levels. Additional treatment schedules (Bx and Bz1, with two optional extension weeks at the same dose level) were introduced to mitigate initial increases in liver transaminases and to achieve a potentially therapeutic solitomab dose. Additional details of the dose escalation are included in the Supplementary Data.
A DLT was defined as any grade 3 or 4 AE, with noted exceptions (Table S3), that was considered at least possibly related to solitomab per investigator review. Three to six patients were enrolled in sequential dose groups with increasing doses if no DLT occurred during the DLT period, defined as the first week of treatment at the target dose (in schedule Bz it also included the first week of treatment). Selected liver-specific laboratory values were excluded from the definition of DLT (see footnotes of Table S3) via protocol amendments early in the study conduct. Liver biopsy was not required, but was performed in patients with elevated liver parameters who provided informed consent for the procedure. Solitomab dosing was stopped immediately in case of a DLT, or a life-threatening or potentially life-threatening clinical event. If more than one DLT occurred in a given dose group, dose escalation was stopped. The MTD was the highest dose level in a treatment schedule at which no more than one in six patients experienced a DLT. All patients could receive H1 and H2 blockers on day 1 of cycle 1. Dexamethasone (8 mg every 12 hours) was administered on days 1 through 3 of cycle 1 (investigator discretion) to mitigate cytokine release.
The severity of AEs was graded using the Common Terminology Criteria for Adverse Events (CTCAE) version 3.0.26 Due to high EpCAM expression in the pancreas and pancreatic toxicity observed with a similar compound,27,28 serum amylase and lipase were monitored throughout the study. Tumor dimensions were assessed using CT or contrast-enhanced MRI every second cycle and at the end of study. Response to solitomab was evaluated according to modified RECIST version 1.0 at each study center.24
Immunohistochemistry of human duodenal tissue
Endoscopic duodenal tissue biopsies were acquired from one consenting patient. Biopsies were not protocol-specified but were performed as medically needed per investigator decision. Tissue was fixed in formalin and embedded in paraffin following standard procedures. Freshly cut sections (4 to 7 µm) were pretreated in a PT Link Pretreatment Module and then processed in an Autostainer Link 48 Module (Dako, Glostrup, Denmark) using mouse anti-T‒cell intracellular antigen 1 (TIA-1; clone 4i389; Zytomed Systems, Germany ), anti-EpCAM (clone VU-1D9; Leica Biosystems, Wetzlar, Germany), mouse anti-CD8 (clone C8/144B; Dako), rabbit anti-CD3 (Dako), and mouse anti-CD4 (clone 4B12; Dako) primary antibodies. Bound antibody was visualized using the EnVision Kit (Dako) following the manufacturer's directions. Sections were also stained with hematoxylin and eosin (H&E) following standard procedures. Images were acquired using a Zeiss Axioscop (Zeiss, Jena, Germany).
In vivo murine studies
All animal studies were performed according to the German Animal Welfare Law with permission from the responsible local authorities. Female BALB/c mice (Janvier, Le Genest-Saint-Isle, France) 7 weeks old were injected IV with muS110 or vehicle control at 0.05 mg/kg/day (10 animals per group) for 2 days. On day 3, animals were euthanized and the small intestinal tissue was fixed in formalin and embedded in paraffin following standard procedures. Freshly cut sections (5 µm) were deparaffinized and rehydrated in graded alcohol solutions. Sections were heated in DIVA Decloaker antigen retrieval buffer (Biocare Medical, Pacheco, CA), then subsequently rinsed, endogenous peroxidase activity was quenched (Peroxidazed 1, Biocare Medical), and a protein blockade was applied (Background Sniper, Biocare Medical). After another buffer rinse, sections were incubated overnight with rabbit anti-EpCAM (Abcam, Cambridge, MA; catalog #ab71916), rat anti-mouse CD3 (clone CD3-12; BioRad, Hercules, CA; catalog #MCA1477), rabbit anti-Granzyme B (Abcam; catalog #ab4059), or rabbit anti-cleaved caspase 3 (Cell Signaling, Danvers, MA; catalog #9664) primary antibodies. After a final rinse, sections were incubated with a relevant peroxidase-conjugated secondary antibody reagent (various) and developed with DAB (Biocare Medical) and counterstained with hematoxylin. Sections were also subjected to H&E staining following standard procedures. Images were acquired using a Nikon Eclipse Ci microscope and Nikon NIS Elements software.
Immunogenicity
Blood samples were collected at screening, at the end of each treatment cycle, and at end of study. In patients with a positive immunogenicity test, further samples were collected in post-study follow-up to determine anti-solitomab antibody titers. Anti-solitomab antibodies were detected using a sandwich electrochemiluminescence (ECL) assay using the Sector Imager 2400 (Meso Scale Diagnostics, Rockville, MD, USA). Neutralizing capacity of detected anti-solitomab antibodies was subsequently tested in a cell-based CD69 activation assay using FACSCanto™ II (Becton Dickinson, Franklin Lakes, NJ).
Pharmacokinetics
Solitomab concentrations in serum were determined using a proprietary electrochemiluminescence (ECL)-based sandwich assay (lower limit of quantification: 0.20 ng/mL) developed and validated by Amgen Research (Munich) GmbH (Munich, Germany). Briefly, solitomab in serum samples from patients was captured on a microtiter plate (Meso Scale Diagnostics, LLC; Rockville, MD, USA) coated with anti-solitomab monoclonal antibody, 6E12 (Bioventix, Farnham, United Kingdom). Bound solitomab was then detected via a biotin-labeled antibody (Qiagen, Hilden, Germany) directed against the histidine tag encoded within solitomab, followed by SULFO-TAG™-labeled streptavidin (Meso Scale Diagnostics). Test plates were analyzed using a Sektor Imager 2400 (Meso Scale Diagnostics).
Pharmacodynamics
Lymphocyte subpopulations were measured using density gradient-separated peripheral blood mononuclear cells prepared as previously described,23 or whole peripheral blood collected at screening (for a subset of patients), at baseline (within 1 hour before infusion start), at various timepoints during the first two treatment cycles, and at the end of the treatment period. Lymphocyte subpopulations were analyzed by flow cytometric determination of cell surface markers to determine the relative cellular composition of blood samples using an eight-color FACSCanto™ II instrument (Becton Dickinson, Franklin Lakes, NJ, USA), a five-color FC500 instrument (Beckman Coulter, Brea, CA, USA), or a ten-color FACS NAVIOS instrument (Beckman Coulter). Monoclonal antibodies labeled with different fluorescent dyes were used to detect CD45, CD19, CD3, CD4, CD8, CD28, CD45RA, CCR7, and CD69. By combining percentage values of certain lymphocyte subpopulations with the absolute lymphocyte count determined via hemogram, absolute numbers for the respective subpopulations were calculated. To explore the immunogenic mechanism of action of solitomab, serum concentrations of the pro- and anti-inflammatory cytokines IL-6, IL-10, and IFN-γ were determined from peripheral blood using a FACS-based cytometric bead array system (BD Biosciences, San Jose, CA, USA; Catalog Number: 551809). In addition, serum concentrations of IL-8 were determined using ELISA (Diaclone; Catalog number: 850.050).
Study endpoints and statistical analysis
The primary endpoint was the overall frequency and severity of AEs, including DLTs, clinical symptoms, laboratory abnormalities, and serious AEs. Secondary endpoints included pharmacokinetics of solitomab, T-cell counts, T-cell kinetics, serum cytokine concentrations, immunogenicity, and antitumor activity. Primary and secondary endpoints were evaluated in an exploratory fashion using descriptive statistics.
Supplementary Material
Conflict of interest
Maria-Elisabeth Goebeler: consultant or advisory role with Roche, GEMoAb, and Novartis; travel, accommodations, and/or expenses paid by Amgen Inc., Novartis, Roche, and Janssen-Cilag. Annette Hasenburg: leadership role and stock or other ownership in Theraclion; honoraria from Amgen Research (Munich) GmbH, Novartis, Roche Pharma AG, BVF, Georg Thieme Verlag, Celgene GmbH, GlaxoSmithKline, FBA GmbH, TEVA GmbH, Med Update GmbH Wiesbaden, and Urban & Fischer Verlag Elsevier GmbH; consultant or advisory role with Theraclion, Pharma Mar, and Roche; research funding from Celgene. Ruth Seggewiss-Bernhardt: consultant or advisory role with AstraZeneca and Novartis; honoraria, travel, accommodations, and/or expenses paid by Astellas, Bristol-Myers Squibb, Novartis, Roche Pharma GmbH, and Celgene. Beate Rautenberg: honoraria from Roche and Eisai; travel, accommodations, and/or expenses paid by Celgene. Djordje Atanackovic: Speakers' Bureau for Celgene, Takeda, Onyx, and Bristol-Myers Squibb. Andrea Kratzer, Stefanie Elm, Ingrid Patzak, Sabine Stienen: employee of Amgen Research (Munich) GmbH and share holder in Amgen Inc. James B. Rottman: employee of Amgen Cambridge, MA at the time the research was conducted and shareholder in Amgen Inc. Matthias Friedrich: employee of Amgen Research (Munich) GmbH and share holder in Amgen Inc.; Amgen patent proprietor. Eva Vieser: employee of and share holder in Amgen Inc.; recipient of payments associated with patents, royalties, or other intellectual property from Amgen Inc. Dorothea Wessiepe: employee of Metronomia Clinical Research GmbH whose work is funded by Amgen Inc. Walter Fiedler: consultant or advisory role with Amgen Inc., Ariad, and Novartis; research funding from Kolltan Pharmaceuticals and Amgen Inc.; recipient of patents, royalties, or other intellectual property from Amgen Inc.; travel, accommodations, and/or expenses paid by Teva GmbH, Gilead Sciences, Jazz Pharmaceuticals, and Amgen Inc. Maxim Kebenko, Martin Wolf, Barbara Ritter: no conflicts of interest to disclose.
Research support
This study was supported by Amgen Inc.
Previous presentation
The results of this study have not been previously published or submitted for publication elsewhere. The results were presented in part as an oral presentation at the American Society of Clinical Oncology 48th Annual Meeting, Chicago, Illinois, USA, June 1 to 5, 2012.
Acknowledgment
The authors thank Till Krech, M.D. (Dept. of Pathology, University Medical Center Hamburg-Eppendorf) for contributing the human immunohistochemistry data; Pamela Bogner (Amgen Research [Munich] GmbH) for assistance with analysis of the pharmacokinetic data; and James Ziobro (funded by Amgen Inc.), and Beate D. Quednau, PhD (Amgen Inc.) for editorial assistance in the preparation of this manuscript.
References
- 1.Fong D, Moser P, Kasal A, Seeber A, Gastl G, Martowicz A, Wurm M, Mian C, Obrist P, Mazzoleni G. Loss of membranous expression of the intracellular domain of EpCAM is a frequent event and predicts poor survival in patients with pancreatic cancer. Histopathology. 2014;64:683–92. doi: 10.1111/his.12307. PMID:24117877. [DOI] [PubMed] [Google Scholar]
- 2.Gastl G, Spizzo G, Obrist P, Dunser M, Mikuz G. Ep-CAM overexpression in breast cancer as a predictor of survival. Lancet. 2000;356:1981–2. doi: 10.1016/S0140-6736(00)03312-2. PMID:11130529. [DOI] [PubMed] [Google Scholar]
- 3.Spizzo G, Obrist P, Ensinger C, Theurl I, Dunser M, Ramoni A, Gunsilius E, Eibl G, Mikuz G, Gastl G. Prognostic significance of Ep-CAM AND Her-2/neu overexpression in invasive breast cancer. Int J Cancer. 2002;98:883–8. doi: 10.1002/ijc.10270. PMID:11948467. [DOI] [PubMed] [Google Scholar]
- 4.Spizzo G, Went P, Dirnhofer S, Obrist P, Moch H, Baeuerle PA, Mueller-Holzner E, Marth C, Gastl G, Zeimet AG. Overexpression of epithelial cell adhesion molecule (Ep-CAM) is an independent prognostic marker for reduced survival of patients with epithelial ovarian cancer. Gynecol Oncol. 2006;103:483–8. doi: 10.1016/j.ygyno.2006.03.035. PMID:16678891. [DOI] [PubMed] [Google Scholar]
- 5.Schmelzer E, Reid LM. EpCAM expression in normal, non-pathological tissues. Front Biosci. 2008;13:3096–100. doi: 10.2741/2911. PMID:17981779. [DOI] [PubMed] [Google Scholar]
- 6.Went PT, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G, Dirnhofer S. Frequent EpCam protein expression in human carcinomas. Hum Pathol. 2004;35:122–8. doi: 10.1016/j.humpath.2003.08.026. PMID:14745734. [DOI] [PubMed] [Google Scholar]
- 7.Ladwein M, Pape UF, Schmidt DS, Schnolzer M, Fiedler S, Langbein L, Franke WW, Moldenhauer G, Zöller M. The cell-cell adhesion molecule EpCAM interacts directly with the tight junction protein claudin-7. Exp Cell Res. 2005;309:345–57. doi: 10.1016/j.yexcr.2005.06.013. PMID:16054130. [DOI] [PubMed] [Google Scholar]
- 8.Nubel T, Preobraschenski J, Tuncay H, Weiss T, Kuhn S, Ladwein M, Langbein L, Zöller M. Claudin-7 regulates EpCAM-mediated functions in tumor progression. Mol Cancer Res. 2009;7:285–99. doi: 10.1158/1541-7786.MCR-08-0200. PMID:19276185. [DOI] [PubMed] [Google Scholar]
- 9.Schwartzberg LS. Clinical experience with edrecolomab: a monoclonal antibody therapy for colorectal carcinoma. Crit Rev Oncol Hematol. 2001;40:17–24. doi: 10.1016/S1040-8428(01)00131-7. PMID:11578913. [DOI] [PubMed] [Google Scholar]
- 10.Marschner N, Ruttinger D, Zugmaier G, Nemere G, Lehmann J, Obrist P, Baeuerle PA, Wolf A, Schmidt M, Abrahamsson PA. Phase II study of the human anti-epithelial cell adhesion molecule antibody adecatumumab in prostate cancer patients with increasing serum levels of prostate-specific antigen after radical prostatectomy. Urol Int. 2010;85:386–95. doi: 10.1159/000318055. PMID:20606402. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt M, Scheulen ME, Dittrich C, Obrist P, Marschner N, Dirix L, Schmidt M, Rüttinger D, Schuler M, Reinhardt C. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann Oncol. 2010;21:275–82. doi: 10.1093/annonc/mdp314. PMID:19633042. [DOI] [PubMed] [Google Scholar]
- 12.Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–7. doi: 10.1126/science.1158545. PMID:18703743. [DOI] [PubMed] [Google Scholar]
- 13.Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93:290–6. doi: 10.1038/icb.2014.93. PMID:25367186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Topp MS, Gokbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC, Dombret H, Fielding AK, Heffner L, Larson RA. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16:57–66. doi: 10.1016/S1470-2045(14)71170-2. PMID:25524800. [DOI] [PubMed] [Google Scholar]
- 15.Viardot A, Goebeler ME, Hess G, Neumann S, Pfreundschuh M, Adrian N, Zettl F, Libicher M, Sayehli C, Stieglmaier J. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood. 2016;127:1410–6. doi: 10.1182/blood-2015-06-651380. PMID:26755709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol. 2006;43:763–71. doi: 10.1016/j.molimm.2005.03.007. PMID:16360021. [DOI] [PubMed] [Google Scholar]
- 17.Schlereth B, Fichtner I, Lorenczewski G, Kleindienst P, Brischwein K, da Silva A, Kufer P, Lutterbuese R, Junghahn I, Kasimir-Bauer S. Eradication of tumors from a human colon cancer cell line and from ovarian cancer metastases in immunodeficient mice by a single-chain Ep-CAM-/CD3-bispecific antibody construct. Cancer Res. 2005;65:2882–9. doi: 10.1158/0008-5472.CAN-04-2637. PMID:15805290. [DOI] [PubMed] [Google Scholar]
- 18.Pishvaian M, Morse MA, McDevitt J, Norton JD, Ren S, Robbie GJ, Ryan PC, Soukharev S, Bao H, Denlinger CS. Phase 1 Dose escalation study of MEDI-565, a Bispecific T-Cell Engager that targets human Carcinoembryonic Antigen, in Patients with advanced Gastrointestinal Adenocarcinomas. Clin Colorectal Cancer. 2016;15:345–51. doi: 10.1016/j.clcc.2016.07.009. PMID:27591895. [DOI] [PubMed] [Google Scholar]
- 19.Topp MS, Gokbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32:4134–40. doi: 10.1200/JCO.2014.56.3247. PMID:25385737. [DOI] [PubMed] [Google Scholar]
- 20.Amann M, Friedrich M, Lutterbuese P, Vieser E, Lorenczewski G, Petersen L, Brischwein K, Kufer P, Kischel R, Baeuerle PA. Therapeutic window of an EpCAM/CD3-specific BiTE antibody in mice is determined by a subpopulation of EpCAM-expressing lymphocytes that is absent in humans. Cancer Immunol Immunother. 2009;58:95–109. doi: 10.1007/s00262-008-0529-y. PMID:18594818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mau-Sorensen M, Dittrich C, Dienstmann R, Lassen U, Buchler W, Martinius H, et al. A phase I trial of intravenous catumaxomab: a bispecific monoclonal antibody targeting EpCAM and the T cell coreceptor CD3. Cancer Chemother Pharmacol. 2015;75:1065–73. doi: 10.1007/s00280-015-2728-5. PMID:25814216. [DOI] [PubMed] [Google Scholar]
- 22.Brischwein K, Schlereth B, Guller B, Steiger C, Wolf A, Lutterbuese R, Offner S, Locher M, Urbig T, Raum T. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol. 2006;43:1129–43. doi: 10.1016/j.molimm.2005.07.034. PMID:16139892. [DOI] [PubMed] [Google Scholar]
- 23.Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119:6226–33. doi: 10.1182/blood-2012-01-400515. PMID:22592608. [DOI] [PubMed] [Google Scholar]
- 24.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC. New guidelines to evaluate the response to treatment in solid tumors. European organization for Research and treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. PMID:10655437. [DOI] [PubMed] [Google Scholar]
- 25.Amann M, Brischwein K, Lutterbuese P, Parr L, Petersen L, Lorenczewski G, Krinner E, Bruckmeier S, Lippold S, Kischel R. Therapeutic window of MuS110, a single-chain antibody construct bispecific for murine EpCAM and murine CD3. Cancer Res. 2008;68:143–51. doi: 10.1158/0008-5472.CAN-07-2182. PMID:18172306. [DOI] [PubMed] [Google Scholar]
- 26.Cancer Therapy Evaluation Program, Common Terminology Criteria for Adverse Events, Version 3.0, DCTD, NCI, NIH, DHHS Available at: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed 16March2016.
- 27.de Bono JS, Tolcher AW, Forero A, Vanhove GF, Takimoto C, Bauer RJ, Hammond LA, Patnaik A, White ML, Shen S. ING-1, a monoclonal antibody targeting Ep-CAM in patients with advanced adenocarcinomas. Clin Cancer Res. 2004;10:7555–65. doi: 10.1158/1078-0432.CCR-04-0729. PMID:15569986. [DOI] [PubMed] [Google Scholar]
- 28.Goel S, Bauer RJ, Desai K, Bulgaru A, Iqbal T, Strachan BK, Kim G, Kaubisch A, Vanhove GF, Goldberg G. Pharmacokinetic and safety study of subcutaneously administered weekly ING-1, a human engineere monoclonal antibody targeting human EpCAM, in patients with advanced solid tumors. Ann Oncol. 2007;18:1704–7. doi: 10.1093/annonc/mdm280. PMID:17693421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.