

ABSTRACT
The recent breakthroughs in the understanding of tumor immune biology have given rise to a new generation of immunotherapies, harnessing the immune system to eliminate tumors. As the typology and frequency of encountered infections are susceptible to shape the immune system, it could also impact the efficiency of immunotherapy. In this review, we report evidences for an indirect link between personal history of infection and different strategies of immunotherapy. In the current context of interest rise for personalized medicine, we discuss the potential medical applications of considering personal history of infection to design immunotherapeutic strategies.
KEYWORDS: Immunotherapy, Infections, Personalized medicine, Cancer vaccine, Immune checkpoints, Adoptive cell transfer
Introduction
Genomic instability, common to all cancers,1 is a challenge for current cytotoxic therapies and represents the main cause of therapy failure and cancer relapse. Indeed, treatments generally target the dominant clones within the tumor and result in massive cell death. Nevertheless, it may paradoxically provide a selective pressure for the expansion of variant cells resisting to the drug used.2 Combinations of chemotherapies have been tested, and have shown encouraging results, but only delay emergence of resistance and rarely avoid it.3 This is why the research on cancer treatment has evolved from relatively nonspecific cytotoxic agents to selective, mechanism-based therapeutics. These ‘targeted therapies’ have been designed to interfere with specific molecular targets that are known to have a critical role in tumor growth or progression.4 For instance, treatments based on VEGF have increased the time before the cancer progresses as well as improving survival for patient with renal cell carcinoma.5
Another key point in the recent evolution of cancer therapies is the observation that cancerous patients can be stabilized over metronomic treatments (chemo-, radio- or targeted therapies) through the induction of a long-term anti-cancer immune response or vaccine-like effect.6 To this extent, the recent breakthroughs in the understanding of tumor immune biology have given rise to a new generation of treatments which attempts to harness the exquisite power and specificity of the immune system for the treatment of malignancy. This cancer immunotherapy could allow overcoming immune tolerance to tumors and the production of adaptive anti-cancer responses able to deal with genomic instability.7
Indeed, since the beginning of the 20th century, it has been hypothesized that immune system may play a role in cancer surveillance and prevention. The pioneering works of Dr William B. Coley have showed that stimulation of immune system by infections could be associated with tumor growth control.8 However, direct evidence for immunosurveillance of human cancers has been hard to prove experimentally and immunotherapy has mainly consisted in passive transfer of anti-cancer antibodies and T cells from donor to induce an immediate immune reaction against cancer.9 Recently, the clinical success of new immunotherapies, such as immune checkpoint inhibitors leading to complete tumor elimination in some patients with advanced cancers,10 supports the idea that immune system is a powerful weapon against cancer. Nevertheless, the successes of immunotherapy are extremely heterogeneous, sometimes associated with dramatic side-effects,11 and resistance has already been reported.12 Thus, identification of predictive biomarkers allowing selection of patients predisposed to respond to immunotherapy remains a challenge for immunologists and oncologists.
In addition to immunosurveillance of cancer, a prime function of immune system is the defense against infectious agents (i.e., viruses, bacteria, fungi, protozoans and metazoans that exploit other organisms, called hosts, to complete their life cycle). At an individual scale, humans are exposed to a high number of infections (through contact, ingestion or inhalation among other possibilities) which sometimes result in infraclinical symptoms.13 The community of organisms which have infected an individual during its life represents the personal history of infection. As cancer cells and infections interact with organisms' defense mechanisms, typology and frequency of encountered infections are susceptible to shape the functioning of immune system, and consequently the efficiency of immunotherapy. Here, we review the indirect evidences suggesting that personal history of infections may impact, in a non-negligible way, many mechanisms targeted by immunotherapeutic strategies and therefore may explain their contrasting successes as well as open new opportunities to improve them.
Strategies envisioned for immunotherapy
Today, several categories of immunotherapy exist which differ according to their target. First, we can distinguish immunotherapies designed to directly target tumor cells but which rarely result in a long-lasting anti-tumor immune response. It includes anti-tumor antibodies as well as cellular therapy, also known as adoptive cell transfer, aiming to select and/or to engineer tumor-specific immune cells that are thereafter administrated by local injection (i.e., intra-tumoral).14 A second category includes immunotherapies that stimulate the patient own immune system to elicit cancer cells elimination and an immune memory protective against tumor-associated antigens (TAAs). They are generally administrated by intravenous injection to generate a systemic immune response. They include antibodies directed against immune-modulating molecules present on immune cells such as CTLA-4, PD-1 and PDL-1, PDL-1 being also expressed by tumor cells themselves. At the interface between these two strategies, oncolytic therapies are based on viruses that show a tropism for cancer cells. This cytotoxic therapy stimulates both cancer cell destruction as well as anti-tumor immune response. Finally, immunotherapy also includes strategies based on immune-mechanisms, such as vaccines that either treat existing cancer or prevent development of a cancer.
Cellular therapy
CAR T cells
Adoptive transfer of T cells engineered to express chimeric antigen receptor (CAR) has emerged as a powerful immunotherapeutic strategy and is now the focus of numerous clinical trials. Especially, CAR-modified T cells targeting the B cell specific antigen CD19 have shown great promise in the treatment of chronic lymphocytic leukemia (CLL)15 and acute lymphocytic leukemia (ALL).16 The most notable toxicity of this treatment is the cytokine release syndrome (CRS), which is a systemic inflammatory process resulting from attack by CAR T cells of non-malignant cells that express targeted tumor antigen.17 In some patients, CD19-targeted T cells therapy has also been associated to neurological disturbances. A study has shown that CAR T cells are able to penetrate blood-brain barrier18 and they have been observed in the cerebrospinal fluid of the patient experiencing neurological toxicities.19 We can expect that neurological complications may be more frequent in patients with a high level of B cell infiltration in the brain. Indeed, the immune response produced by CAR T cells recognizing CD19 B cells could be associated with cytotoxicity to the surrounding tissues.
In addition to the natural variability between individuals, the level of cerebral infiltration by B cells could also be the result of past infections. Notably, acute malaria infection has been associated to a significant increase of CD19 B lymphocyte percentages in the brain of infected animals in a rodent malaria model.20 This increase is susceptible to protect against cerebral malaria, which is a frequent and deadly complication in children.21 It may be worth mentioning that certain viral brain infections (i.e., cytomegalovirus (CMV), West Nile Virus) may also be associated with an infiltration of immune cells and especially immune-regulatory B cells in the brain, as suggested by an experimental study on mice.22 However, to our knowledge, the persistence of B cells within the brain after recovery of infection has not been investigated. If this was confirmed, neurological complications with CD19-targeted CAR T cells could be more common in patients with an history of malaria or such viral infections.
Another approach to generate CAR T cells is to use natural virus-specific T cells that have been modified to express anti-tumor CAR. These techniques generally lay on the dual-specificity of CAR T cells for infected cells and for tumor cells. In this manner, clonal expansion resulting from response to infection may benefit to tumor cell control and elimination. For example, EBV-specific T cells have been transduced with tumor-specific chimeric receptors that allow the recognition of GD2, a TAA present on several tumors of neural crest origin.23 The authors have observed that EBV-specific CAR T cells recognize EBV-infected targets through their conventional T-cell receptors and tumor targets through their chimeric receptors.24 Consequently, they are maintained by the presence of EBV-infected B cells and induce a higher anti-tumor activity which has increased survival in two mice tumor models: neuroblastoma and Hodgkin diseases. Influenza and CMV have also been used to generate dual-specific CAR T cells25,26 but their efficiencies in presence of the virus have not been tested.
We would like to propose here that the establishment of personal history of infection (especially by viruses and P. falciparum), before treatment administration, may help to select patients in order to prevent the apparition of dramatic adverse-effect, such as those observed with CD19-trageted CAR T cells. In addition, we can suspect that the use of virus-based CAR T cells in patients who have been already infected by the natural virus, could show a higher efficiency. For instance, EBV infects more than 80% of the adult population.27 In these patients, the use of EBV-specific CAR T cells without simultaneous introduction of the virus, or with lower viral dose to avoid side-effects, could improve anti-tumor responses (Fig. 1). The generation of a cocktail of dual-specific CAR T cells, specific to persisting pathogens (EBV, CMV, herpesvirus) and engineered to target a panel of common TAAs, could improve immunotherapy efficiency and raises new perspectives for prevention.
Figure 1.
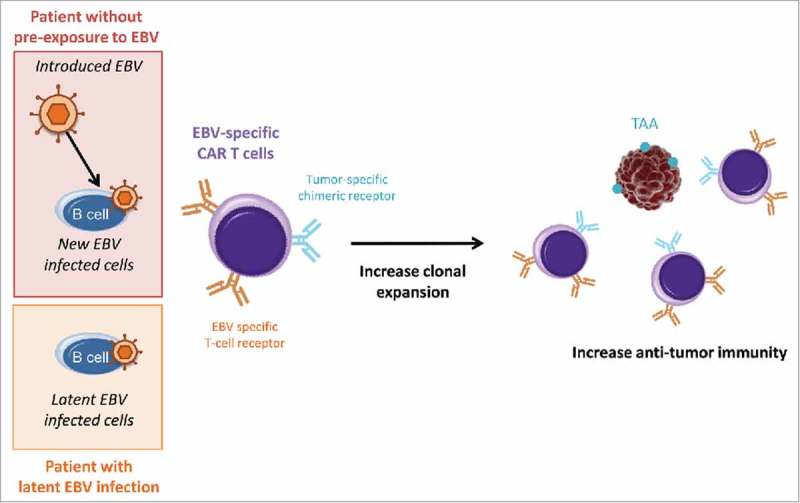
Influence of latent EBV infection on treatment with dual-specific CAR T cells. Latent and introduced virus could both be recognized by T-cell receptors and could increase CAR T cell clonal expansion. Thus, in patient with latent viral infection, the use of EBV-specific CAR T cells could result in stronger anti-tumor responses even without introduced EBV. TAA: tumor-associated antigens.
TILs
Given the lack of well-defined TAAs for most human tumors, the use of naturally occurring tumor infiltrating lymphocytes (TILs) represents a good alternative to CAR T cells. In fact, TILs are able to recognize multiple TAAs that increase their ability to keep tumor growth in check. Nevertheless, insufficient migration28 and decreased anti-tumor function in the immunosuppressive tumor microenvironment29 are significant limitations to treat cancer patients with adoptive transfer of ex-vivo expanded TILs. However, the identification of cytotoxic tumor-infiltrating T cells that recognize clonal mutations, shared by all tumor cells, hold promise for adoptive cellular therapy strategies in combination with the appropriate immunomodulatory agent.30
Recently, tumor-specific T cells from patients have been engineered to carry a second T cell receptor (TCR) recognizing antigens from the bacteria Listeria monocytogenes.31 These polyclonal dual-specific T cells show a greater anti-tumor effect in presence of specific bacterial antigens, which are simultaneously injected at the tumor site. Indeed, the presence of bacterial antigens allows a greater clonal expansion of dual-specific T cells and decreases immunosuppression in tumor micro-environment. However, patients who have already been exposed to the bacteria, or a closely related one, could show a stronger immune response against these antigens. This may result in a quick elimination of the bacterial antigens, neutralizing the positive impact on clonal expression and thus decreasing therapy efficiency. But, the consequences of this pre-exposition in term of treatment efficacy have not been investigated. Thus, within the context of personalized medicine rise, the choice of an additional infectious agent in adoptive TILs transfer should rely on the personal history of infections of the patient in order to maximize therapy efficiency.
Immune checkpoint inhibitors
Anti-CTLA-4
Cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) is a negative regulator of T cell activation. CTLA-4 acts by competing with the co-stimulatory molecule CD28 for binding to shared ligands CD80 and CD86 expressed by antigen-presenting cells. Antibodies that block CTLA-4, such as the ipilimumab, are known to increase T-cell activation and proliferation. Therefore, blocking CTLA-4 has the potential to enhance antitumor immune responses.32 Treatment with ipilimumab can cause significant tumor response in patients with metastatic melanoma.33 However, complete response has rarely been observed34 and immune-related adverse events, related to tolerance break and consequent auto-immunity, happen at high frequency.35
Regarding the interactions between infections and CTLA-4, a study has shown that the expression of CTLA-4 was depressed in peripheral blood mononuclear cells from tuberculosis patients,36 suggesting that people exposed to Mycobacterium tuberculosis could be more responsive to such immunotherapy. Nowadays, up to one-third of the world's population is infected with M. tuberculosis and the majority of infected persons have a clinically latent infection.37 Even if the prevalence of tuberculosis is expected to be low in developed countries, where such immunotherapeutic strategy is available, this mechanism could be mimicked by TB vaccination (Bacillus Calmette-Guérin (BCG) is routinely administrated in many countries) or applied to infection with other mycobacteria.
Interestingly, intravesical injection of BCG vaccine is approved for the treatment of non-invasive bladder cancer38 and its efficiency has been tested in combination with anti-CTLA-4 antibodies in a murine model. It has been observed that mice treated with BCG and anti-CTLA-4 show an improved survival than mice receiving BCG only.39 Therefore, the use of ipilimumab in patients with a history of mycobacterial infection, where the target of ipilimumab is already under-expressed, could block the immunosuppressive action of remaining molecules and could therefore result in stronger immune activation (Fig. 2A). Thus, it would be intriguing to evaluate in clinical trials whether tuberculosis/vaccination status might have an impact on response to anti-CTLA-4 but also whether it would allow diminishing the dose of administration to avoid immune-related adverse events (Fig. 2B).
Figure 2.
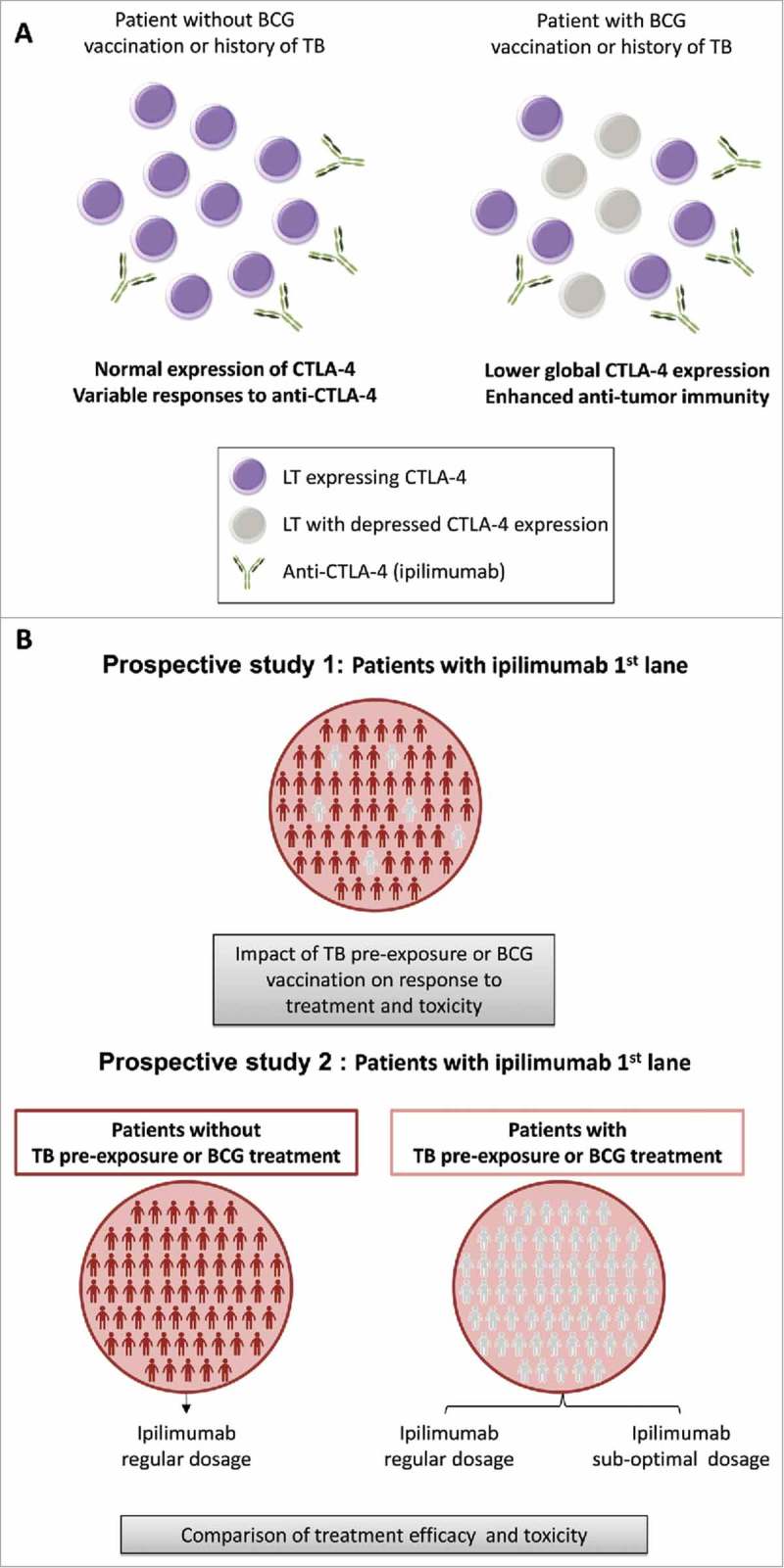
TB – CTLA-4 interaction hypothesis. (A) In patients with BCG vaccination or past history of tuberculosis (TB), CTLA-4 expression is down-regulated. For the same dose of ipilimumab (or other anti-CTLA-4 drugs), these patients will better respond to the treatment as more lymphocytes T (LT) will be targeted. (B) Proposition of clinical trial design to measure the impact of TB pre-exposure or BCG vaccination on treatment efficiency.
Anti-PD-1 & anti-PD-L1
Programmed death 1 (PD-1) is a key immune-checkpoint receptor expressed by activated T cells that mediates down-regulation of immune system. Expression of PD-1, signed the “exhausted phenotype” of effector T cells, has been first identified through studies of chronic viral infections in mice in which PD-1/PD-L1 was found to be an important negative regulatory feedback loop to ensure immune homeostasis.40 Cancer immunotherapies based on the administration of anti-PD-1 (such as nivolumab and pembrolizumab) or anti-PD-L1 (such as atezolizumab, avelumab or durmalumab) aim to inhibit the interaction between PD-L1, expressed by tumor cells but also by stromal cells, and PD-1, expressed by T cells, and thus restore T-cell antitumor activity.41 Many investigators have addressed the question of whether the expression of PD-1 or PD-L1 might be a prognostic marker of treatments with immune checkpoint inhibitors.42,43 Although it is still a matter of debate for PD-L1, PD-1 expression within the tumor reflecting the presence of specific anti-tumor lymphocytes has been associated with a positive prognostic feature in many cancers.44
Importantly, the basal level of PD-1 expression by T cells could be shaped by personal history of infection.45 As mentioned above, the increase of PD-1 expression on antigen-experienced effector T cells during chronic infections, is a physiological mechanism to constrain the adaptive immune response and prevent auto-immunity.46 For instance, HIV infection has been associated with an increase of PD-1 expression on NK cells.47
The alteration of PD-1 expression in the context of acute infections has also attracted increasing attention. It has been reported that acute infections with M. tuberculosis and influenza virus result in a significant increase of PD-1 on NK cells and CD8+ T cells respectively.48,49 Moreover, evidences suggest that PD-1 expression could also be modified at long-term through immunological memory. In fact, memory CD8+ T cells excessively up-regulate PD-1 expression following vaccinia virus reinfection.50 Hence patients who have experienced a relatively high number of infections may also express high level of PD-1, in activated as well as memory immune cells, reflecting the immunosuppression of pre-existing T cell immunity.
Contrarily to NK cells, CD8+ T cells are antigen-dependant and thus specific T cells could target cancer cells only if TAAs share similarities with infectious agents. This situation has already be suggested between influenza virus and lung cancer cells.51 In this context, we could expect that checkpoint inhibitors will present a lower efficacy in patients with a high basal level of PD-1. We also suggest that this effect may not be limited by clonal selection of cancer cells. In fact, cancer cells which harbor the ligand PD-L1 may have been favored because it allows them to escape immune destruction by the high number of immune cells expressing PD-1. If so, new strategies, aiming to increase immunotherapy efficiency and avoid dangerous side-effects, should consider adapting the dose of treatments with antagonists of the PD-1/PDL-1 interaction, according to the past history of infections of each patient.
Tumor-targeting monoclonal antibodies
Although developed to directly target malignant cells, it is now recognized that the therapeutic activity of tumor antigen-targeting monoclonal antibodies (mAb) is associated to their capacity to mobilize, through FcγR-dependent mechanisms, both innate and adaptive immune cells. However, tumor escape from mAb-induced tumor immune surveillance remains one of the main clinical issues.52,53 As an example, anti-HER2/ErbB2 mAb (Trastuzumab) aims to reduce cell proliferation by blocking a receptor (ErbB2) of a growth factor highly expressed by many cancer cells. This therapy has substantially improved outcome of HER2-positive breast cancer54 and, given the importance of immune responses in trastuzumab therapy,55 therapy combined with immune stimulating agents have been used with success.56
Recently, co-injection of polyinosinic-polycytidylic acid (PolyI:C) and CpG oligodeoxynucleotides (CpG) with trastuzumab shows higher efficiency than anti-ErbB2 therapy used alone.57 The authors suggest that these molecules may represent pertinent adjuvant stimulating innate responses including interferon expression which is required in trastuzumab therapy.58 PolyI:C is a synthetic viral-like double-stranded RNA (dsRNA) which bind to the innate immunity receptors TLR3, RIG-I and MDA-5. It has been used to mimic activation of dendritic cells by viral infections as most of viruses produce dsRNA at some point in their replication.59 CpG, a ligand for TLR9, mimics the presence of bacterial DNA and is also considered as a pathogen-associated molecular pattern that activates DC and B cells in particular.60
Thus, whether a history of viral and/or bacterial infections could mimic the action of these molecules, and especially the increase expression of interferons, remains an interesting hypothesis. In accordance with this idea, it has been shown that interferon (α and γ) levels were significantly higher in patients with chronic hepatitis B infection but also in patients who cleared the virus.61 Thus, one can suggest that personal history of hepatitis B infection might naturally boost anti-tumor responses in patients who received anti-HER2 therapy. In addition, over the last few years, an increasing number of works uncovered that innate immune cells (including monocytes, macrophages, and natural killer cells) display prolonged changes in their functionalities after infections or vaccination therapies, a phenomenon called “trained immunity”.62 According to this idea, innate immune cells could harbor an increased production of cytokines and other inflammatory mediators, including interferons, after recent infections. The increase in innate immune cells responsiveness may potentiate the adjuvant effect and thus the efficiency of anti-tumor targeting therapies.
Oncolytic viruses
Oncolytic viruses have emerged as a novel class of immune-based cancer therapy through their natural preference for cancerous cells and their capacity to trigger lysis of tumor cells as well as activation of anti-tumor immunity.63 Advances in molecular biology have also allowed the modification of other viruses to make them specific to neoplastic tissues and/or to combine them with immunogenic chemotherapy or immune checkpoint blockers to break tumor-induced immune tolerance.64,65 For instance, recombinant measles viruses have been intravenously injected to treat human patients with bone-marrow cancer.66
As for any non-oncolytic infections, the strength of the immune response to oncolytic viruses may be dependent of previous exposition. For instance, treatment based on the recombinant measles viruses led to a significant resolution of tumor only in the two patients who were measles-seronegative,66 suggesting that absence of prior exposure to measles virus has allowed mounting an immune response strong enough to eliminate cancerous cells. This highlights that personal infection history could be crucial to identify which oncolytic agents can be envisioned in these strategies.
In addition, a genetically engineered herpes simplex virus, called T-VEC, has been recently approved to treat advanced melanoma.67 The treatment is administrated by intra-tumoral injection to seronegative patients.68 However, 67% of the population (0–49 years) had current herpes simplex virus-1 infection in 2012 (WHO). Thus, the proportion of patients who could receive the treatment could be lower than excepted. Such considerations could be applied to other oncolytic therapy such as poxvirus vaccine in the treatment of prostate cancer69 or BCG therapy for bladder cancer.38 Thus, it suggests the necessity to investigate the potential of less common virus to design new oncolytic therapies.
Prevention strategies
Prevention with infectious vaccines
Several evidences show that vaccination against specific infectious agents could be used to prevent cancer. Protection against melanoma, lymphoma or leukemia has been reported 10 years after BCG, vaccinia or yellow fever vaccination.70,71 These findings might be explained by non-specific effects of these vaccines through the shifting of the immune response towards a Th1 profile or through cross reactivity.72 Indeed, vaccines may contain pathogen antigens with amino-acid sequences that are homologous with those of certain TAAs.73 By this cross-reactive effect, vaccination allows eliminating malignant cells as soon as they expressed the shared-antigens. For instance, a prior immunization with BCG vaccine, which has antigenic similarity with human endogenous retroviruses (HERV-K-MEL) expressed in 95% of malignant melanocytes,74 has been associated with a better survival in patients with melanoma.75 Such considerations highlight the importance to consider vaccination as a component of personal history of infection and to study their potential for immunotherapeutic prevention. However, such studies may appear hardly conceivable because of methodological constrains such as cohort size and follow-up duration. Meta-analyses of several cohort studies and systematic reviews should nevertheless be considered to circumvent these limitations.
Preventive cancer vaccines
The potential of TAAs as targets for immunotherapy has already been largely investigated. Recently, it has been suggested that these antigens could be also candidates for preventive vaccines, which could hamper initiation of cancer cell proliferation.76 However, as it seems to be the case for therapy, the response to cancer vaccines could be conditioned by past history of infection. Here, we report examples of interaction between immune responses to infections and to cancer associated antigens.
Mumps and MUC1 vaccine
MUC1 is a normal epithelial cell antigen that was characterized as a TAA due to its abnormal expression in most epithelial adenocarcinomas, and the presence of anti-MUC1 antibodies in patients with cancer has already been associated with a more favorable prognosis.77 This suggests that pre-existing anti-MUC1 humoral response could help to control tumor growth or even prevent cancer proliferation. Interestingly, it has been described that mumps parotitis causes abnormal MUC1 expression on salivary gland and a negative correlation has early been observed between ovarian cancer and past history of mumps infection.78,79 Taken together, it has suggested that generation of anti-MUC1 immunity during mumps infection might protect against cancer development. Reinforcing such hypothesis, it has been reported that women who have experienced childhood mumps show a higher proportion of anti-MUC1 antibodies which could increase humoral responses and immune recognition of MUC1.80 Accordingly, MUC1 has been studied as a target for antibody-based immunotherapies, some of which have been able to induce the elimination of ovarian cancer cells which express MUC1.81
Multiple MUC1 vaccines for treatment of ovarian cancer are now in development and have given significant results.82 The elaboration of prophylactic vaccines has also been proposed and tested in a murine model of colon adenoma where it elicits a long term memory.83 However, mumps is a very common childhood disease suggesting that a large proportion of the population could have been exposed. We proposed that the consequences of a pre-existing immune memory developed against the infectious antigens should be investigated in the case of vaccines based on similar antigens. Indeed, it may help to better adapt the number of vaccine boosts which are necessary to reestablish a potent anti-tumor immune environment.84
Varicella and vaccine against cyclin B1
Several tumors express high level of cyclin B1 such as lung, breast and prostate cancers,76 and it has been demonstrated that cyclin B1-specific immune responses may be important in cancer immunosurveillance. Indeed, a prophylactic vaccine against cyclin B1 can delay spontaneous cyclin B1+ tumor growth in a murine lymphoma model and increases median survival.85
Interestingly, aberrant expression of cyclin B1-specific antibodies has also been reported in healthy individuals86 and infections seem to be a reasonably good candidate to explain the presence of cyclin B1-specific responses in healthy individuals. Indeed, a study has shown that cyclins are highly induced in varicella-zoster virus (VZV) infected cells.87 After the primary infection, Varicella-Zoster virus is able to establish a latent infection in the nervous system.88 The immune memory against the virus can persist for more than 20 years after the primo-infection89 and immune responses can be subsequently boosted either by endogenous re-exposure (silent reactivation of latent virus) or exogenous re-exposure (environmental).90
Thus, one can speculate that immune memory against VZV infection could boost immunosurveillance of cancer cells but could also interfere with the efficiency of cycline B1-based vaccines. In this context, the number of vaccine boosts should be determined in accordance with past history of VZV infection.84
Personal history of infection and vaccine elaboration
There is a growing body of evidence showing that infections, both in childhood and adulthood, can provide a protection against the risk of developing malignancies.91 For example, exposures to childhood diseases have been observed to reduce the risk of developing multiple cancers, including melanoma.92
Among childhood diseases, a particular attention has been paid to measles virus. It has been observed that specific MHC class I self peptide (Hsp90β570–578) are expressed both on melanoma cells93 and on measles-infected cells.94 Thus, the auto-reactive CD8+T cells generated after measles infection could persist and enhance immunosurveillance of cancer cells, giving another example of cross-reactivity. During adulthood, history of acute infections (i.e., common colds and influenza) has been associated with a decreased of cancer risk in control-case study.95 This has been experimentally confirmed by a study showing that influenza infected mice harbor smaller lung tumor than naïve mice. They have also reported the overexpression of 5 TAAs (GAPDH, H4, MDH2, A2) both in infected cells and in tumor cells.51
These evidences suggest that immune memory for antigens produced during influenza or measles infection has allowed the production of long-lasting antibodies against these specific TAAs which could be involved in cancer cell elimination. From these two examples, it seems that therapeutic potential of molecules, such as Hsp90β570–578 and GAPDH, should be investigated. However, studies should not under-estimate the effects of previous natural infections, especially in the case of recurrent infections of fast-evolving virus (i.e., influenza virus), on vaccine efficiency.
Conclusions
Even if accumulating evidence suggests that infections may play a role in cancer risk,96,97 their influence in the responses to immunotherapy has been rarely investigated. Here, we report the evidences for indirect links between personal history of infections and different immunotherapeutic strategies. First, infections could be associated with a modified expression of the therapeutic target, such as increase of PD-1 following acute infections. Second, pre-exposition to additional infectious agents (as latent EBV infection in the case of EBV-specific CAR T cells) could increase cancer therapy efficiency. Finally, non-oncogenic pathogens may induce long-lasting immunity against antigens shared with cancerous cells (i.e., mumps and antigens associated with ovarian cancer) and thus modify immunotherapy efficiency. Some immunotherapeutic strategies already mimic or rely on the effect of infection on immune system.98 It confirms that approaches, linking immunotherapy and personal history of infection, should be developed in a greater extent to consider the huge diversity of infectious agents, chronic but also acute, that may play a role.
While the potential interactions of infectious agents with immunotherapies cited here need to be experimentally tested and clarified, identifying the links between personal history of infection and immunotherapy could help clinicians to personalize their immunotherapeutic strategies, improving them or even envisioning new ones. First, it may help to adapt therapies through the determination of an optimal dose of administration. In addition, it may orient the clinicians' choice toward the most adapted strategy, in accordance with the patient's immune profile. Second, it could bring crucial insights to understand the diversity of responses to immunotherapy in cancer patients but also to forecast the apparition of adverse effects which could be crucial to design safe treatments. Third, considering personal history of infection could help to design new oncolytic therapies based on less common viruses as it is expected that they will generate stronger immune responses. Finally, it may allow the elaboration of new preventive measures based on the knowledge of immune mechanisms involved in response to infections. It is worth mentioning that the role of vaccines to induce some of the immune mechanisms described in the context natural infections should be investigated as they represent a good opportunity of prevention. Retrospective analyses of data coming from immunotherapy trials could already give crucial clues to figure out the contribution of personal history of infection and vaccination on the heterogeneity of outcomes observed after the treatments.
Funding Statement
Funding with Agence Nationale pour la Recherche, EVOCAN and STORY.
Competiting interest
The authors declare no competing interests.
Acknowledgments
The authors thank CREEC sponsors CNRS, Labex MabImprove, and Andre HOFFAMNN (Fondation Mava).
References
- 1.Hanahan D, Weinberg RA. Review hallmarks of cancer : The next generation. Cell [Internet]. 2011;144:646–74. Available from: doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Greaves M, Maley CC. Clonal evolution in cancer. Nature [Internet]. 2012. [cited 2014July9];481:306–13. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3367003&tool=pmcentrez&rendertype=abstract doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bozic I, Reiter JG, Allen B, Antal T, Chatterjee K, Shah P, Moon YS, Yaqubie A, Kelly N, Le DT, et al.. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife [Internet]. 2013. [cited 2016February23];2:e00747 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3691570&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sawyers C. Targeted cancer therapy. Nature [Internet]. 2004. [cited 2017March24];432:294–7. Available from: http://www.nature.com/doifinder/10.1038/nature03095 doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- 5.Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, Jassem J, Zolnierek J, Maroto JP, Mellado B, et al.. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol [Internet]. 2015. [cited 2018April5];16:1473–82. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26482279 doi: 10.1016/S1470-2045(15)00290-9. [DOI] [PubMed] [Google Scholar]
- 6.Pasquier E, Kavallaris M, André N. Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol [Internet]. 2010. [cited 2018January2];7:455–65. Available from: http://www.nature.com/doifinder/10.1038/nrclinonc.2010.82 doi: 10.1038/nrclinonc.2010.82. [DOI] [PubMed] [Google Scholar]
- 7.Yaghmour G, Pandey M, Ireland C, Patel K, Nunnery S, Powell D, Baum S, Wiedower E, Schwartzberg LS, Martin MG. Role of genomic instability in immunotherapy with checkpoint inhibitors. Anticancer Res [Internet]. 2016. [cited 2017July24];36:4033–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27466509. [PubMed] [Google Scholar]
- 8.Coley WB. The treatment of inoperable sarcoma by bacterial toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc R Soc Med. 1910;3:1–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dougan M, Dranoff G. Immune therapy for cancer. AnnuRevImmunol. 2009;27:83–117. [DOI] [PubMed] [Google Scholar]
- 10.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, et al.. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–30. doi: 10.1200/JCO.2013.53.0105. PMID:24590637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Couzin-Frankel J. Cancer immunotherapy. Science (80-). 2013;342:1432–3. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 12.Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer [Internet]. 2016. [cited 2016February4];16:121–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26822578 doi: 10.1038/nrc.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnes E. Diseases and human evolution. Albuquerque: Univ New Mex Press; 2005. [Google Scholar]
- 14.Shi H, Sun M, Liu L, Wang Z. Chimeric antigen receptor for adoptive immunotherapy of cancer: latest research and future prospects. Mol Cancer [Internet]. 2014. [cited 2017July24];13:219 Available from: http://www.ncbi.nlm.nih.gov/pubmed/25241075 doi: 10.1186/1476-4598-13-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med [Internet]. 2011;3:95ra73 Available from: http://stm.sciencemag.org/content/3/95/95ra73.full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al.. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med [Internet]. 2013;368:1509–18. Available from: http://www.nejm.org/doi/pdf/10.1056/NEJMoa1215134 doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL. How I Treat: Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–96. doi: 10.1182/blood-2014-05-552729. PMID:24876563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu Y, Sun J, Wu Z, Yu J, Cui Q, Pu C, Liang B, Luo Y, Shi J, Jin A, et al.. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J Hematol Oncol [Internet]. 2016;9:70 Available from: http://jhoonline.biomedcentral.com/articles/10.1186/s13045-016-0299-5 doi: 10.1186/s13045-016-0299-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davila ML, Riviere I, Wang X, Bartido S, Park J, Chung SS, Stefanski J, Borquez-ojeda O, Qu J, Wasielewska T, et al.. Efficacy and toxicity management of 19–28z CAR T Cell therapy in B Cell acute lymphoblastic leukemia. Sci Transl Med. 2015;6:224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lacerda-Queiroz N, Lima OCO, Carneiro CM, Vilela MC, Teixeira AL, Carvalho AT, Araújo MSS, Martins-Filho OA, Braga ÉM, Carvalho-Tavares J. Plasmodium berghei NK65 induces cerebral leukocyte recruitment in vivo: An intravital microscopic study. Acta Trop [Internet]. 2011;120:31–9. Available from: doi: 10.1016/j.actatropica.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 21.Bao LQ, Huy NT, Kikuchi M, Yanagi T, Senba M, Shuaibu MN, Honma K, Yui K, Hirayama K. CD19(+) B Cells confer protection against experimental cerebral malaria in semi-immune rodent model. PLoS One. 2013;8:1–13. doi: 10.1371/journal.pone.0064836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mutnal MB, Hu S, Schachtele SJ, Lokensgard JR. Infiltrating regulatory B cells control neuroinflammation following viral brain infection. J Immunol [Internet]. 2014. [cited 2017July24];193:6070–80. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25385825 doi: 10.4049/jimmunol.1400654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mujoo K, Kipps TJ, Yang HM, Cheresh DA, Wargalla U, Sander DJ, Reisfeld RA. Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res [Internet]. 1989. [cited 2018April6];49:2857–61. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2720646. [PubMed] [Google Scholar]
- 24.Rossig C, Bollard CM, Nuchtern JG, Rooney CM, Brenner MK. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: Potential for improved immunotherapy. Blood. 2002;99:2009–16. doi: 10.1182/blood.V99.6.2009. PMID:11877273. [DOI] [PubMed] [Google Scholar]
- 25.Murphy A, Westwood JA, Brown LE, Teng MWL, Moeller M, Xu Y, Smyth MJ, Hwu P, Darcy PK, Kershaw MH. Antitumor activity of dual-specific T cells and influenza virus. Cancer Gene Ther [Internet]. 2007;14:499–508. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17332777 doi: 10.1038/sj.cgt.7701034. [DOI] [PubMed] [Google Scholar]
- 26.Heemskerk M, Hoogeboom M, Hagedoorn R, Kester M, Willemze R, Falkenburg J. Reprogramming of virus-specific T cells into leukemia-reactive T cells using T cell receptor gene transfer. J Exp Med [Internet]. 2004;199:885–94. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15051765%5Cnpapers3://publication/uuid/DB3D1BCB-2CA5-4F15-A871-226C5E66766B doi: 10.1084/jem.20031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balfour HH, Sifakis F, Sliman JA, Knight JA, Schmeling DO, Thomas W. Age-specific prevalence of epstein-barr virus infection among individuals aged 6–19 Years in the united states and factors affecting its acquisition. J Infect Dis [Internet]. 2013. [cited 2017June22];208:1286–93. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23868878 doi: 10.1093/infdis/jit321. [DOI] [PubMed] [Google Scholar]
- 28.Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (80-). 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- 29.Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol [Internet]. 2013;14:1014–22. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24048123 doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al.. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (80-) [Internet]. 2016. [cited 2017December23];351:1463–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26940869 doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xin G, Schauder DM, Jing W, Jiang A, Joshi NS, Johnson B, Cui W. Pathogen boosted adoptive cell transfer immunotherapy to treat solid tumors. Proc Natl Acad Sci [Internet]. 2017:201614315 Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1614315114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (80-). 1996;271:1734–6. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 33.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med [Internet]. 2010. [cited 2017July25];363:711–23. Available from: http://www.nejm.org/doi/abs/10.1056/NEJMoa1003466 doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, White DE, Levy CL, Rosenberg SA, Phan GQ. CTLA-4 blockade with ipilimumab: Long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012;18:2039–47. doi: 10.1158/1078-0432.CCR-11-1823. PMID:22271879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bertrand A, Kostine M, Barnetche T, Truchetet M-E, Schaeverbeke T. Immune related adverse events associated with anti-CTLA-4 antibodies: Systematic review and meta-analysis. BMC Biol [Internet]. 2013;11:EE Available from: 10.1186/s12916-015-0455-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gong JH, Zhang M, Modlin RL, Linsley PS, Iyer D, Lin Y, Barnes PF. Interleukin-10 downregulates Mycobacterium tuberculosis-induced Th1 responses and CTLA-4 expression. Infect Immun. 1996;64:913–8. PMID:8641800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flynn JL, Chan J. Tuberculosis : Latency and reactivation. Infect Immun. 2001;69:4195–201. doi: 10.1128/IAI.69.7.4195-4201.2001. PMID:11401954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gontero P, Bohle A, Malmstrom P-U, O'Donnell MA, Oderda M, Sylvester R, Witjes F. The role of bacillus Calmette-Guérin in the treatment of non-muscle-invasive bladder cancer. Eur Urol [Internet]. 2010. [cited 2015October2];57:410–29. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19969411 doi: 10.1016/j.eururo.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 39.Mangsbo SM, Sandin LC, Anger K, Korman AJ, Loskog A, Tötterman TH. Enhanced tumor eradication by combining CTLA-4 or PD-1 Blockade with CpG Therapy. J Immunother [Internet]. 2010. [cited 2017August13];33:225–35. Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00002371-201004000-00001 doi: 10.1097/CJI.0b013e3181c01fcb. [DOI] [PubMed] [Google Scholar]
- 40.Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol [Internet]. 2011. [cited 2018January2];12:478–84. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21739670 doi: 10.1038/ni.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Topalian SL, Hodi FS, Brahmer JR, Al E. Safety, activity, and immune correlates of Anti–PD-1 antibody in cancer. N Engl J Med. 2012;366:339–54. doi: 10.1056/NEJMoa1200690. PMID:22276823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Filskov Sorensen S, Zhou W, Dolled-Filhart M, Baehr Georgsen J, Wang Z, Emancipator K, Wu D, Busch-Sørensen M, Meldgaard P, Hager H. PD-L1 Expression and survival among patients with advanced Non–small cell lung cancer treated with chemotherapy 1. Transl Oncol [Internet]. 2016. [cited 2018January19];9:64–9. Available from: https://ac.els-cdn.com/S1936523315300528/1-s2.0-S1936523315300528-main.pdf?_tid=5d9c545e-fcf4-11e7-927e-00000aacb35d&acdnat=1516351393_1a215b344e196e485a99260ba0f5d24e doi: 10.1016/j.tranon.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma K, Wei X, Dong D, Wu Y, Geng Q, Li E. PD-L1 and PD-1 expression correlate with prognosis in extrahepatic cholangiocarcinoma. Oncol Lett [Internet]. 2017. [cited 2018January19];14:250–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28693161 doi: 10.3892/ol.2017.6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao T, Li C, Wu Y, Li B, Zhang B. Prognostic value of PD-L1 expression in tumor infiltrating immune cells in cancers: A meta-analysis. PLoS One [Internet]. 2017. [cited 2018January2];12:e0176822 Available from: http://www.ncbi.nlm.nih.gov/pubmed/28453554 doi: 10.1371/journal.pone.0176822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salazar M d'A, Manuel ER, Tsai W, D'Apuzzo M, Goldstein L, Blazar BR, Diamond DJ. Evaluation of innate and adaptive immunity contributing to the antitumor effects of PD1 blockade in an orthotopic murine model of pancreatic cancer. Oncoimmunology. 2016;5:e1160184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol [Internet]. 2007. [cited 2017August13];8:239–45. Available from: http://www.nature.com/doifinder/10.1038/ni1443 doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 47.Norris S, Coleman A, Kuri-Cervantes L, Bower M, Nelson M, Goodier MR. PD-1 expression on natural killer cells and CD8 + T cells during chronic HIV-1 infection. Viral Immunol [Internet]. 2012;25:329–32. Available from: http://online.liebertpub.com/doi/abs/10.1089/vim.2011.0096 doi: 10.1089/vim.2011.0096. [DOI] [PubMed] [Google Scholar]
- 48.Alvarez IB, Pasquinelli V, Jurado JO, Abbate E, Musella RM, de la Barrera SS, García VE. Role played by the programmed death‐1–Programmed death ligand pathway during innate immunity against mycobacterium tuberculosis. J Infect Dis [Internet]. 2010;202:524–32. Available from: http://jid.oxfordjournals.org/lookup/doi/10.1086/654932 doi: 10.1086/654932. [DOI] [PubMed] [Google Scholar]
- 49.Kohlhapp FJ, Huelsmann EJ, Lacek AT, Schenkel JM, Lusciks J, Broucek JR, Goldufsky JW, Hughes T, Zayas JP, Dolubizno H, et al.. Non-oncogenic acute viral infections disrupt anti-cancer responses and lead to accelerated cancer-specific host death. Cell Rep. 2016;17:957–65. doi: 10.1016/j.celrep.2016.09.068. PMID:27760326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuse S, Tsai C-Y, Molloy MJ, Allie SR, Zhang W, Yagita H, Usherwood EJ. Recall responses by helpless memory CD8+ T cells are restricted by the up-regulation of PD-1. J Immunol [Internet]. 2009;182:4244–54. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2713929&tool=pmcentrez&rendertype=abstract doi: 10.4049/jimmunol.0802041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iheagwara UK, Beatty PL, Van PT, Ross TM, Minden JS, Finn OJ. Influenza virus infection elicits protective antibodies and T cells specific for host cell antigens also expressed as tumor associated antigens : A new view of cancer immunosurveillance. Cancer Immunol Res. 2015;2:263–73. doi: 10.1158/2326-6066.CIR-13-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michaud H-A, Eliaou J-F, Lafont V, Bonnefoy N, Gros L. Tumor antigen-targeting monoclonal antibody-based immunotherapy: Orchestrating combined strategies for the development of long-term antitumor immunity. Oncoimmunology [Internet]. 2014. [cited 2018January2];3:e955684 Available from: http://www.ncbi.nlm.nih.gov/pubmed/25941618 doi: 10.4161/21624011.2014.955684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.They L, Michaud H-A, Becquart O, Lafont V, Guillot B, Boissière-Michot F, Jarlier M, Mollevi C, Eliaou J-F, Bonnefoy N, et al.. PD-1 blockade at the time of tumor escape potentiates the immune-mediated antitumor effects of a melanoma-targeting monoclonal antibody. Oncoimmunology [Internet]. 2017. [cited 2018January2];6:e1353857 Available from: http://www.ncbi.nlm.nih.gov/pubmed/29123966 doi: 10.1080/2162402X.2017.1353857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swain SM, Baselga J, Kim S-B, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero J-M, Schneeweiss A, Heeson S, et al.. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med [Internet]. 2015;372:724–34. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25693012 doi: 10.1056/NEJMoa1413513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loi S, Michiels S, Salgado R, Sirtaine N, Jose V, Fumagalli D, Kellokumpu-Lehtinen PL, Bono P, Kataja V, Desmedt C, et al.. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann Oncol. 2014;25:1544–50. doi: 10.1093/annonc/mdu112. PMID:24608200. [DOI] [PubMed] [Google Scholar]
- 56.Nava-Parada P, Forni G, Knutson K. Peptide vaccine given with a Toll-like receptor agonist is effective for the treatment and prevention of spontaneous breast tumors. Cancer Res [Internet]. 2007;67:1326–34. Available from: http://cancerres.aacrjournals.org/content/67/3/1326.short doi: 10.1158/0008-5472.CAN-06-3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Charlebois R, Allard B, Allard D, Buisseret L, Turcotte M, Pommey S, Chrobak P, Stagg J. PolyI:C and CpG synergize with anti-ErbB2 mAb for treatment of breast tumors resistant to immune checkpoint inhibitors. Cancer Res. 2016;77:312–9. PMID:27872096. [DOI] [PubMed] [Google Scholar]
- 58.Stagg J, Loi S, Divisekera U, Ngiow SF, Duret H, Yagita H, Teng MW, Smyth MJ. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci [Internet]. 2011. [cited 2017August13];108:7142–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21482773 doi: 10.1073/pnas.1016569108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jacobs BL, Langland JO. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology [Internet]. 1996;219:339–49. Available from: http://www.sciencedirect.com/science/article/pii/S0042682296902597 doi: 10.1006/viro.1996.0259. [DOI] [PubMed] [Google Scholar]
- 60.Chaung HC. CpG oligodeoxynucleotides as DNA adjuvants in vertebrates and their applications in immunotherapy. Int Immunopharmacol. 2006;6:1586–96. doi: 10.1016/j.intimp.2006.06.001. PMID:16919831. [DOI] [PubMed] [Google Scholar]
- 61.Chu CM, Sheen IS, Yeh CT, Hsieh SY, Tsai SL, Liaw YF. Serum levels of interferon-alpha and -gamma in acute and chronic hepatitis B virus infection. Dig Dis Sci [Internet]. 1995. [cited 2017August13];40:2107–12. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7587774 doi: 10.1007/BF02208991. [DOI] [PubMed] [Google Scholar]
- 62.Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, ONeill LAJ, Xavier RJ. Trained immunity: A program of innate immune memory in health and disease. Science (80-) [Internet]. 2016. [cited 2018January25];352:aaf1098-aaf1098 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27102489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Russell SJ, Peng K-W, Bell JC. Oncolytic virotherapy. Nat Biotechnol [Internet]. 2012. [cited 2014July9];30:658–70. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3888062&tool=pmcentrez&rendertype=abstract doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bell J, McFadden G. Viruses for tumor therapy. Cell Host Microbe [Internet]. 2014. [cited 2014October29];15:260–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24629333 doi: 10.1016/j.chom.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fend L, Yamazaki T, Remy C, Fahrner C, Gantzer M, Nourtier V, Préville X, Quéméneur E, Kepp O, Adam J, et al.. Immune checkpoint blockade, immunogenic chemotherapy or IFN-α Blockade boost the local and abscopal effects of oncolytic virotherapy. Cancer Res [Internet]. 2017. [cited 2018January2];77:4146–57. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28536278 doi: 10.1158/0008-5472.CAN-16-2165. [DOI] [PubMed] [Google Scholar]
- 66.Russell SJ, Federspiel MJ, Peng KW, Tong C, Dingli D, Morice WG, Lowe V, O'Connor MK, Kyle RA, Leung N, et al.. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc [Internet]. 2014. [cited 2014October8];89:926–33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24835528 doi: 10.1016/j.mayocp.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ledford H. Cancer-fighting viruses near market. Nature. 2015:526. [DOI] [PubMed] [Google Scholar]
- 68.Rehman H, Silk AW, Kane MP, Kaufman HL. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J Immunother cancer [Internet]. 2016;4:53 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27660707%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC5029010 doi: 10.1186/s40425-016-0158-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaufman HL, Wang W, Manola J, DiPaola RS, Ko Y-J, Sweeney C, Whiteside TL, Schlom J, Wilding G, Weiner LM. Phase II randomized study of vaccine treatment of advanced prostate cancer (E7897): A trial of the eastern cooperative oncology group. J Clin Oncol. 2004;22:2122–32. doi: 10.1200/JCO.2004.08.083. PMID:15169798. [DOI] [PubMed] [Google Scholar]
- 70.Villumsen M, Sørup S, Jess T, Ravn H, Relander T, Baker JL, Benn CS, Sørensen TIa, Aaby P, Roth A. Risk of lymphoma and leukaemia after bacille Calmette-Guérin and smallpox vaccination: a Danish case-cohort study. Vaccine [Internet]. 2009. [cited 2015April21];27:6950–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19747577 doi: 10.1016/j.vaccine.2009.08.103. [DOI] [PubMed] [Google Scholar]
- 71.Mastrangelo G, Krone B, Fadda E, Buja A, Grange JM, Rausa G, de Vries E, Koelmel KF. Does yellow fever 17D vaccine protect against melanoma? Vaccine [Internet]. 2009. [cited 2015April21];27:588–91. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19010368 doi: 10.1016/j.vaccine.2008.10.076. [DOI] [PubMed] [Google Scholar]
- 72.Grange JM, Stanford JL, Stanford CA, Ko KF. Vaccination strategies to reduce the risk of leukemia and melanoma. J R Soc Med. 2003;96:389–92. doi: 10.1177/014107680309600806. PMID:12893854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Krone B, Kölmel KF, Henz BM, Grange JM. Protection against melanoma by vaccination with Bacille Calmette-Guerin (BCG) and/or vaccinia: An epidemiology-based hypothesis on the nature of a melanoma risk factor and its immunological control. Eur J Cancer [Internet]. 2005. [cited 2015April21];41:104–17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15617995 doi: 10.1016/j.ejca.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 74.Schiavetti F, Thonnard J, Colau D, Boon T, Coulie PG. A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Cancer Res [Internet]. 2002. [cited 2017July24];62:5510–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12359761. [PubMed] [Google Scholar]
- 75.Kölmel KF, Grange JM, Krone B, Mastrangelo G, Rossi CR, Henz BM, Seebacher C, Botev IN, Niin M, Lambert D, et al.. Prior immunisation of patients with malignant melanoma with vaccinia or BCG is associated with better survival. An European organization for research and treatment of cancer cohort study on 542 patients. Eur J Cancer [Internet]. 2005. [cited 2015April21];41:118–25. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15617996 doi: 10.1016/j.ejca.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 76.Finn OJ, Beatty PL. Cancer immunoprevention. Curr Opin Immunol [Internet]. 2016;39:52–8. Available from: 10.1016/j.coi.2016.01.002 doi: 10.1016/j.coi.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hamanakai Y, Suehiro Y, Fukui M, Shikichi K, Imai K, Hinoda Y. Circulating anti-MUC1 IGG antibodies as a favorable prognostic factor for pancreatic cancer. Int J Cancer. 2003;103:97–100. doi: 10.1002/ijc.10801. PMID:12455059. [DOI] [PubMed] [Google Scholar]
- 78.West RO. Epidemiologic study of malignancies of the ovaries. Cancer. 1966;19:1001–7. doi: 10.1002/1097-0142(196607)19:7%3c1001::AID-CNCR2820190714%3e3.0.CO;2-S. PMID:5939299. [DOI] [PubMed] [Google Scholar]
- 79.Menczer J, Modan M, Ranon L, Golan A. Possible role of mumps virus in the etiology of ovarian cancer. Cancer [Internet]. 1979. [cited 2018January19];43:1375–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/445337 doi: 10.1002/1097-0142(197904)43:4%3c1375::AID-CNCR2820430427%3e3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 80.Cramer DW, Vitonis AF, Pinheiro SP, McKolanis JR, Fichorova RN, Brown KE, Hatchette TF, Finn OJ. Mumps and ovarian cancer: moder interpretation of an historic association. Cancer Causes Control. 2011;21:1193–201. doi: 10.1007/s10552-010-9546-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deng J, Wang L, Chen H, Li L, Ma Y, Ni J, Li Y. The role of tumour-associated MUC1 in epithelial ovarian cancer metastasis and progression. Cancer Metastasis Rev. 2013;32:535–51. doi: 10.1007/s10555-013-9423-y. PMID:23609751. [DOI] [PubMed] [Google Scholar]
- 82.Gray HJ, Benigno B, Berek J, Chang J, Mason J, Mileshkin L, Mitchell P, Moradi M, Recio FO, Michener CM, et al.. Progression-free and overall survival in ovarian cancer patients treated with CVac, a mucin 1 dendritic cell therapy in a randomized phase 2 trial. J Immunother Cancer [Internet]. 2016;4:34 Available from: http://jitc.biomedcentral.com/articles/10.1186/s40425-016-0137-x doi: 10.1186/s40425-016-0137-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kimura T, McKolanis JR, Dzubinski LA, Islam K, Potter MD, Salazar AM, Schoen RE, Finn OJ. MUC1 vaccine for individuals with advanced adenoma of the colon: A cancer immunoprevention feasibility study. Cancer Prev Res (Phila). 2012;100:130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Finn OJ. Vaccines for cancer prevention: a practical and feasible approach to the cancer epidemic. Cancer Immunol Res [Internet]. 2014;2:708–13. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4163937&tool=pmcentrez&rendertype=abstract doi: 10.1158/2326-6066.CIR-14-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vella L, Mina Y, Amya P, Finn OJ. Cyclin B1 vaccine delays spontaneous tumors. Ann N Y Acad Sci. 2009;144:724–32. [Google Scholar]
- 86.Vella LA, Yu M, Fuhrmann SR, El-Amine M, Epperson DE, Finn OJ. Healthy individuals have T-cell and antibody responses to the tumor antigen cyclin B1 that when elicited in mice protect from cancer. Proc Natl Acad Sci U S A [Internet]. 2009;106:14010–5. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2720407&tool=pmcentrez&rendertype=abstract doi: 10.1073/pnas.0903225106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leisenfelder SA, Moffat JF. Varicella-zoster virus infection of human foreskin fibroblast cells results in atypical cyclin expression and cyclin-dependent kinase activity. J Virol [Internet]. 2006;80:5577–87. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1472175&tool=pmcentrez&rendertype=abstract doi: 10.1128/JVI.00163-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Croen KD, Ostrove JM, Dragovic LJ, Straus SE. Patterns of gene expression and sites of latency in human nerve ganglia are different for varicella-zoster and herpes simplex viruses. Proc Natl Acad Sci U S A [Internet]. 1988. [cited 2017July24];85:9773–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2849116 doi: 10.1073/pnas.85.24.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arvin AM. Humoral and cellular immunity to varicella‐zoster virus: An Overview. J Infect Dis [Internet]. 2008. [cited 2017July24];197:S58–60. Available from: https://academic.oup.com/jid/article-lookup/doi/10.1086/522123 doi: 10.1086/522123. [DOI] [PubMed] [Google Scholar]
- 90.Weinberg A, Levin MJ. VZV T Cell-Mediated Immunity [Internet]. In: Current topics in microbiology and immunology. 2010. [cited 2018January19]. page 341–57. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20473790. [DOI] [PubMed] [Google Scholar]
- 91.Kleef R, Jonas WB, Knogler W, Stenzinger W. Fever, cancer incidence and spontaneous remissions. Neuroimmunomodulation. 2001;9:55–64. doi: 10.1159/000049008. PMID:11549887. [DOI] [PubMed] [Google Scholar]
- 92.Hoption Cann SA, van Netten JP, van Netten C. Acute infections as a means of cancer prevention: opposing effects to chronic infections? Cancer Detect Prev [Internet]. 2006. [cited 2016April14];30:83–93. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16490323 doi: 10.1016/j.cdp.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 93.Jarmalavicius S, Welte Y, Walden P. High immunogenicity of the human leukocyte antigen peptidomes of melanoma tumor cells. J Biol Chem. 2012;287:33401–11. doi: 10.1074/jbc.M112.358903. PMID:22869377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Herberts CA, Van Gaans-van den Brink J, Van der Heeft E, Van Wijk M, Hoekman J, Jaye A, Poelen MCM, Boog CJP, Roholl PJM, Whittle H, et al.. Autoreactivity against induced or upregulated abundant self-peptides in HLA-A*0201 following measles virus infection. Hum Immunol. 2003;64:44–55. doi: 10.1016/S0198-8859(02)00707-3. PMID:12507814. [DOI] [PubMed] [Google Scholar]
- 95.Abel U, Becker N, Angerer R, Frentzel-beyme R, Kaufmann M, Wysoeki S, Schulz G. Common infections in the history of cancer patients and controls. J Cancer Res Clin Oncol. 1991;117:339–44. doi: 10.1007/BF01630717. PMID:2066354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jacqueline C, Bourfia Y, Hbid H, Sorci G, Thomas F, Roche B. Interactions between immune challenges and cancer cells proliferation: timing does matter! Evol Med Public Heal. 2016;in press:299–311. doi: 10.1093/emph/eow025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jacqueline C, Tasiemski A, Sorci G, Ujvari B, Maachi F, Missé D, Renaud F, Ewald P, Thomas F, Roche B. Infections and cancer: the “fifty shades of immunity” hypothesis. BMC Cancer. 2017;17:257. PMID:28049525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer [Internet]. 2014. [cited 2014July10];14:559–67. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24990523 doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]