

Summary
T cells infiltrate affected organs in chronic infections and malignancy, but they may fail to eradicate virus-infected cells or tumor because of exhaustion. This report describes a Yin Yang-1 (YY1)-centered mechanism for diverse components that have been correlated with exhaustion. Utilizing an in vitro reconstruction of chronic T cell activation, YY1 is shown to positively regulate the checkpoint receptors PD1, Lag3, and Tim3 and to negatively regulate the type I cytokines interleukin-2 (IL-2) (in collaboration with Ezh2 histone methyltransferase) and interferon gamma (IFN-γ). Other tests suggest that IL-2 failure drives a large component of cytotoxic functional decline rather than solely checkpoint receptor-ligand interactions that have been the focus of current anti-exhaustion therapies. Clinical evaluations confirm elevated YY1 and Ezh2 in melanoma tumor-infiltrating lymphocytes and in PD1+ T cells in patients with HIV. Exhaustion is revealed to be an active process as the culmination of repetitive two-signal stimulation in a feedback loop via CD3/CD28→p38MAPK/JNK→YY1→ exhaustion.
Subject Areas: Cancer Systems Biology, Immune Response, Immune System Disorder
Graphical Abstract
Highlights
-
•
Transcription factor YY1 is shown to serve as master regulator of T cell exhaustion
-
•
YY1 recruits Ezh2 histone methyltransferase to co-repress IL-2 cytokine production
-
•
Persistent signal 1+2 stimulation via p38MAPK/JNK pathway promotes YY1 transcription
-
•
In vivo melanoma and HIV exhausted PD1+ T cells are confirmed for elevated YY1 and Ezh2
Cancer Systems Biology; Immune Response; Immune System Disorder
Introduction
The T cells of patients affected by malignancy or chronic viral infections become exhausted because of persistent antigenic stimulation (Zajac et al., 1998, Baitsch et al., 2011, Balkhi et al., 2015, Wherry and Kurachi, 2015). Exhausted T cells have low proliferative capacity; express inhibitory checkpoint receptor molecules such as programmed death 1 (PD1), T cell immunoglobulin and mucin domain-containing protein 3 (Tim3), and lymphocyte-activation gene 3 (Lag3); and concurrently lose the ability to produce type I cytokines and kill antigenic targets (Baitsch et al., 2011, Balkhi et al., 2015).
Our group previously observed that activation of T cells in vitro with two signals leads to abundant interleukin-2 (IL-2) production on initial antigen exposure, which abruptly declines on repeated stimulation with concomitant slowing of T cell proliferation (Emtage et al., 2008). As a type I cytokine, IL-2 plays a pivotal role in clonal expansion and persistence of virus- and tumor-reactive T cells and in their effector activity (Rosenberg et al., 1985, Liao et al., 2013). The demonstrated therapeutic benefit of exogenously supplemented IL-2 in humans and in model systems of cancer and infections is one indicator of this impact of exhaustion that hampers T cells' ability to generate this same effector molecule (Rosenberg et al., 1985, Blattman et al., 2003, Emtage et al., 2008, Lo et al., 2010, Liao et al., 2013). Similarly, the effectiveness of antibodies against the checkpoint receptors to restore T cell function and generate clinical responses is additional testimony to the relevance of exhaustion to clinical disease (Barber et al., 2006, Nguyen and Ohashi, 2015). Lastly, there has been an appreciation that a therapeutic synergy may be derived by concurrently addressing the two axes of cytokines and checkpoint receptors (West et al., 2013).
Despite advances in the molecular and phenotypic characterization of exhausted T cells, the mechanisms underlying the initiation, progression, and maintenance of exhaustion are largely unknown. We exploited our in vitro observation of a depressed IL-2 response under repeated stimulation as a potential entrée to the exhaustion process to interrogate its molecular basis.
Results
In Vitro Exhaustion Model
Exhaustion is an in-vivo-defined phenomenon. To study exhaustion experimentally, we sought an in vitro system to recapitulate the process. Building on our prior observations (Emtage et al., 2008), we established a procedure whereby normal resting human T cells were continuously exposed to signal 1 + 2 with anti-CD3/CD28 beads, repeated at 2-day intervals, which was previously shown to stimulate and then lose the production of IL-2 (Figure 1A). This pattern of cytokine failure was confirmed in the current model for both IL-2 and interferon (IFN) γ (Figures 1B and S1). Upon repeated stimulation, CD4 and CD8 T cells also expressed markers of exhaustion, namely, checkpoint receptors PD1, Tim3, and Lag3, which progressively increased with each stimulation (Figure 1C). The cells maintained viability during these stimulations and sustained CD69 expression, a marker of T cell activation (Figure S2). Marker progression was independent of IL-2 depletion, as the same results were obtained with 330 IU/mL of exogenously supplemented IL-2 (Figure S3). Lastly, repeated stimulation of T cells with immobilized anti-CD3 or anti-CD28 antibody alone was insufficient to induce exhaustion (Figures S4A and S4B), confirming a key distinction from other processes such as anergy in which isolated signal 1 is effective (Appleman and Boussiotis, 2003, Balkhi et al., 2015). Taken together, this in vitro model mimics in vivo persistent stimulation of T cells entering into an antigen-rich environment and successfully recapitulates key features of the exhaustion phenotype. For economy of nomenclature/terminology, we provisionally denote such in vitro repeatedly stimulated T cells as “exhausted,” with the final judgment reserved for additional confirmations stated below.
Figure 1.

Persistent T Cell Activation In Vitro Induces IL-2 Shutdown and Checkpoint Receptor Elevation
(A) Schematic depicting repeat stimulation model. T cells were stimulated with anti-CD3/CD28 beads for 2 days; cells were counted at the end of 2 days, beads removed, and cells placed in new medium with fresh beads for the next stimulation. Control cells were cultured identically without beads.
(B) ELISA shows high secretion and then decline of IL-2 production after repeated stimulations.
(C) Flow cytometry of re-stimulated CD4 T cells analyzed at day 8 shows increasing expression of exhaustion markers, PD1, Lag3, and Tim3. CD8 T cells exhibited same pattern (data not shown). Similar results were obtained when the cells were analyzed right after first, second, third, and fourth stimulations.
(D) qRT-PCR and (E) IL-2 promoter luciferase assay in CD4 T cells shows fold change of IL-2 mRNA and promoter activity after re-stimulations. Four replicates performed per assay; data from one of three representative experiments. *p < 0.05.
YY1 Recruits Ezh2 to Repress IL-2
The IL-2 secretion pattern was paralleled in mRNA levels, starting high following activation and then declining (Figure 1D), and was also reflected in reporter assays using an IL-2 promoter construct in recurrently stimulated T cells (Figure 1E). To identify putative sites for transcription factor binding that could regulate IL-2 transcription during exhaustion, we performed an in silico analysis of the IL-2 promoter by searching the TRANSFAC database (Biobase). Several sites identified with high confidence were associated with transcriptional activation (Figure S5). In contrast, the presence of a single YY1-binding site attracted interest because of its known potential for negative as well as positive regulation. This site was in close proximity to sites for the high mobility group protein 1 (HMG1) and TATA-binding protein, marking YY1 a strong candidate for IL-2 gene repression.
Yin Yang 1 (YY1) is a ubiquitous and multifunctional zinc finger transcription factor that regulates diverse cellular functions, including B cell development, proliferation, differentiation, and tumorigenesis (Gordon et al., 2006, Liu et al., 2007). YY1 regulates both positively and negatively (whence the name yin-yang) after binding to the consensus sequence 5′-CCGCCATNTT-3′ in the promoters of these genes (Thomas and Seto, 1999). As a member of the polycomb group (PcG) proteins, YY1 may recruit and cooperate with other members of the complex that function as chromatin modifiers (Shi et al., 1997, Srinivasan and Atchison, 2004, Woo et al., 2013, Atchison, 2014). Enhancer of zeste homolog 2 (Ezh2) is a component of the Polycomb Repressor Complex 2 (PRC2), one of two classes of PcG, and possesses histone H3-K27 trimethylation (HKMT) activity that can directly impede gene transcription (Caretti et al., 2004), with prominent roles in B and T cell differentiation (Arvey et al., 2014, Gray et al., 2017).
Western blotting confirmed marked increases in YY1 and Ezh2 protein with repeated stimulations in enriched CD4 (Figure 2A) and CD8 T cells (not shown). In contrast, the response from single stimulations did not recapitulate the pattern of protein increases over time, confirming the necessity of repeat stimulations for active product accumulation (Figure 2B; compare day 8 signals for 1–4 stimulations). Immunofluorescence confirmed high levels of YY1 and Ezh2 in repeatedly stimulated T cells compared with resting T cells (Figure 2C). No increase in expression of YY1 or Ezh2 was observed when T cells were repeatedly stimulated with immobilized anti-CD3 or anti-CD28 antibody alone (Figures S6A and S6B), confirming the necessity of two signals for these two nuclear proteins' expression, as also noted for the classic surface exhaustion markers above (Figures 1C and S4).
Figure 2.

Persistent T Cell Activation Upregulates YY1 and Ezh2 to Epigenetically Silence IL-2
(A) Western blots show increasing expression of YY1 and Ezh2 after re-stimulation of CD4 T cells, analyzed on day 8. Similar results obtained with CD8 T cells (not shown). β-Actin is the loading control.
(B) More YY1 and Ezh2 from persistent activation by comparing 1–4 stimulations with results assessed on day 8 days as in (A).
(C) Immunofluorescence (IF) performed on four times stimulated exhausted T cells and unstimulated PBMCs analyzed on day 8 showing YY1 and Ezh2 expression. Right panels show magnified single cell single positive for CD3 or double positive for CD3/YY1 and CD3/Ezh2. In unstimulated PBMCs, Ezh2 is seen to be expressed in non–T cells that correspond to the B cells that are present and known to express Ezh2 (van Galen et al., 2004).
(D) Western blot shows decreased levels of H3K27me3 with Ezh2 inhibitor. T cells were treated continuously with Ezh2 inhibitor or mock reagent before third stimulation and analyzed after four stimulations.
(E) qPCR for IL-2 promoter shows high H3K27me3 methylation with recurrent stimulation that is decreased with Ezh2 inhibitor or YY1 shRNA. Chromatin immunoprecipitation (ChIP) with anti-H3K27me3 antibody or IgG in twice-stimulated CD4 T cells, transduced before third stimulation with plasmid for control-shRNA (lane 2) or YY1 shRNA (lane 3), or maintained in the presence of Ezh2 inhibitor (lane 4).
(F) ELISA shows sustained IL-2 production from CD4 T cells stimulated repeatedly in vitro without or with Ezh2 inhibitor.
(G) ELISA assay shows recovered IL-2 production from exhausted T cells with YY1 shRNA. CD4 T cells stimulated twice produced high IL-2 but afterward these cells were exhausted. Twice-stimulated exhausted cells were transduced with plasmids for YY1-specific shRNA and control shRNA or no plasmid just before the third or fourth stimulations. Western blot shows effective YY1 knockdown by shRNA, analyzed 2 days after shRNA before the fourth stimulation.
(H) Co-immunoprecipitation (coIP) confirms YY1 interaction with Ezh2 in repeatedly stimulated T cells. Immunoprecipitation (IP) was with Ezh2 antibody, and the blot was probed with YY1 (upper panel) or Ezh2 antibody (bottom panel). No Ezh2 or complex was recovered by Ezh2 IP from unstimulated T cells.
(I) Model showing YY1 acting in association with Ezh2 to repress IL-2 transcription.
Graphs in (E)–(G) are from a minimum of three replicates; assays representative of at least three experiments. *p < 0.05.
If H3-K27 trimethylation and IL-2 failure result from obligatory cooperation of YY1 and Ezh2 during exhaustion, it follows that blocking YY1 or Ezh2 should be equally preventative. To examine this possibility, we maintained a small molecule Ezh2 inhibitor in the activation cultures or we transduced T cells with short hairpin RNA (shRNA) to knock down YY1 expression. As anticipated, both approaches suppressed H3-K27 trimethylation of the IL-2 promoter by western blotting (Figure 2D; Ezh2 inhibitor) and by chromatin immunoprecipitation (ChIP)/qPCR (Figure 2E; YY1 shRNA and Ezh2 inhibitor). Confirming the hypothesized suppressive effect of histone trimethylation, T cells continuously blocked for H3-K27 trimethylation with Ezh2 inhibitor sustained IL-2 at high levels throughout, exceeding that of control T cells by a factor of >40 after the fourth stimulation (Figure 2F).
In a separate test using YY1 antisense shRNA (Figure 2G), T cells underwent two control stimulations, releasing ∼1500 pg/mL IL-2 after each stimulation, and then on day 4, just before a third stimulation, were either transduced with shRNA plasmid or control plasmid or were left untransduced. In contrast to Ezh2 inhibition, which was continuous through the experiment, YY1 inhibition was applied as a transient system because the delivered plasmid that generates shRNA is not stably retained and expressed in the T cells. IL-2 secretion was fully exhausted after the second stimulation, now releasing only 35 pg/mL following a third stimulation. In contrast, the same twice-stimulated exhausted T cells treated with YY1 shRNA had 30-fold higher IL-2 levels after the third stimulation, in excess of 1,000 pg/mL. In a further repeat of the assay, using now the control thrice-stimulated exhausted T cells that previously released just 35 pg/mL IL-2, a fourth stimulation was performed. This yielded 15 pg/mL IL-2 for control but 350 pg/mL from the same exhausted T cells pretreated with YY1 shRNA, yielding a >20-fold IL-2 increase. YY1 was effectively suppressed when assayed 2 days after shRNA plasmid exposure (Figure 2G inset); shorter times were not evaluated.
These YY1 shRNA tests allow a key conclusion, i.e., IL-2 failure can be reversed by our interventions rather than merely prevented. This ability to reverse IL-2 exhaustion implies a need for continuously effective histone trimethylation for continued IL-2 suppression, marking “exhaustion” itself as an active, energy-requiring process rather than simply a depleted state.
The YY1-shRNA-rescued IL-2 levels after the fourth stimulation (350 pg/mL) were less complete than after the third stimulation (>1000 pg/mL) (Figure 2G). This outcome would be consistent with higher YY1 mRNA and protein levels at the fourth stimulation (Figures 2B and 4B) and higher degrees of histone modification that require longer times to reverse than under our 2-day test condition. This hypothesis was not further investigated.
Figure 4.

cJun/ATF2 Stimulates YY1 Transcription
(A) YY1 promoter sequence shows cAMP response element (CRE)-binding site for cJun/ATF2 at −136 relative to transcription start site (TSS).
(B) Western blot (upper) and qRT-PCR (lower) show increase of YY1 protein and mRNA in CD8 T cells with re-stimulation. Three replicates per assay.
(C) Luciferase reporter gene assay of YY1 promoter shows increased activity with re-stimulation (blue bars) and activity loss with mutation of CRE site (red bars).
(D) Western blot shows increased phospho-cJun and phospho-ATF2 in CD4 T cells after re-stimulation. Total cJun and ATF2 levels unchanged.
(E) Native gel analysis shows increased phospho-cJun dimer formation with restimulation and loss of monomer detected by specific antibodies.
(F) Electrophoretic mobility shift assay (EMSA) confirms binding of phospho-cJun and phospho-ATF2 to site on YY1 promoter. YY1 probe was mixed with nuclear extract from four times stimulated CD8 T cells and incubated with antibody to phospho-cJun or phospho-ATF2 before gel loading. Bottom panel: western blot developed after species-specific secondary antibody to detect primary antibodies bound to the probe.
The presumed physical association of YY1 and Ezh2 to mediate IL-2 suppression was confirmed by co-immunoprecipitation assays using extract from T cells under exhaustion conditions (Figures 2H and S7). These results thus support a model of T cell exhaustion in which the IL-2 promoter binds YY1, YY1 recruits Ezh2, Ezh2 trimethylates chromatin, and IL-2 transcription is repressed (Figure 2I).
As a key control in this effort, Ezh2 inhibition did not reverse the IFN-γ loss that follows repeated T cell stimulation (Figure S8). YY1 was previously shown to disrupt transcription of this type I cytokine by a distinct, non-Ezh2-dependent mechanism from what we newly describe for IL-2, i.e., by sterically blocking AP1 binding via an overlapping YY1-binding site, in which AP1 is a prime activator of IFN-γ transcription (Ye et al., 1996).
YY1 Upregulates Exhaustion Markers
The simultaneous rise of Ezh2 along with PD1, Lag3, and Tim3 suggests coordinate regulation of these genes by YY1 as well. Searching the TRANSFAC database (Biobase), PD1 and Lag3 promoters were found to contain consensus binding sites for YY1 (Figures S9A and S9B), whereas Tim3 and Ezh2 lacked such sites (data not shown).
We examined the PD1 and Lag3 genes for regulation by YY1. Gel shift assay confirmed that YY1 binds specifically to the consensus sites present in PD1 and Lag3 promoters (Figures 3A and 3B). Reporter gene assays showed increased transcription with repeat T cell stimulations that was abrogated by mutation of the YY1-binding sites, confirming that PD1 and Lag3 genes are positively regulated by YY1 (Figures 3C and 3D). Finally, knockdown of YY1 in repeatedly stimulated cells markedly reduced not only PD1 and Lag3, as predicted, but also Tim3 (Figures 3E and 3F), suggesting that YY1 regulates Tim3 as well but by indirect means. The Tim3 promoter carries a binding site for GATA3 (Figure S9C), which is known to bind YY1 to enhance transcriptional activity in the absence of a separate YY1-binding site (Hwang et al., 2013), again supporting the centrality of YY1 to broad elements of the exhaustion process as studied here in vitro. Knockdown of YY1 had no impact on Ezh2 levels (not shown), implying that Ezh2 regulation is through an independent mechanism that parallels rather than depends on YY1.
Figure 3.

YY1 Upregulates Checkpoint Receptors in Exhaustion
(A and B) Electrophoretic mobility shift assay (EMSA) shows YY1 binding to PD1 and Lag3 promoters. Recombinant YY1 protein was incubated with PD1 and Lag3 probes containing intact (lane 2) or mutated YY1-binding sites (lane 3). Epstein-Barr nuclear antigen (EBNA) extract was the control non-specific extract (lane 4).
(C and D) Luciferase reporter assay shows increased activity with PD1 and Lag3 reporter plasmids in re-stimulated CD8 T cells with intact YY1-binding sites and activity loss with YY1-mutated sites. Three replicates per assay; *p < 0.05.
(E) Flow cytometry shows suppression of increase of PD1, Lag3, and Tim3 in CD8 T cells expressing lentiviral YY1-shRNA. Samples analyzed after four stimulations.
(F) YY1 knockdown achieved with YY1 shRNA compared with control after four stimulations.
cJun/ATF2 Upregulates YY1
We next sought to understand the connection between T cell activation and exhaustion, specifically, what precedes YY1 upregulation. Examination of the YY1 promoter revealed an octameric palindrome cyclic adenosine monophosphate (cAMP) response element (CRE) (TGACGTCA) for binding the cJun/ATF2 transcription factor complex (van Dam and Castellazzi, 2001) (Figures 4A and S9D) (Figure S9E summarizes the relevant TF binding sites in this report). Repeated stimulation yielded increased YY1 mRNA and protein levels (Figure 4B) as above (Figures 2A–2C). A YY1 reporter assay showed progressively increased activity with repeat T cell stimulation that was abrogated upon mutating the CRE site (Figure 4C), thus confirming the essential role of cJun/ATF2 binding.
To stimulate transcription, non-phosphorylated cJun and ATF2 that predominate in resting state undergo conversion to their transcriptionally active phospho forms (Smeal et al., 1992) as seen on repeat stimulation in our system (Figure 4D). cJun and ATF2 exist as homodimers and as cJun-ATF2 heterodimers, where the latter may be more commonly recruited for promoter binding and activation (Hayakawa et al., 2004). With repeated stimulations in the T cells, the majority of phospho-cJun was confirmed as dimers on native gels (Figure 4E). Furthermore, dimer was seen to bind to YY1 probe on gel shift assay (Figure 4F, lanes 2–4), which was confirmed by antibody staining to contain phospho-cJun (lane 3) and phosph-ATF2 (lane 4). Taken together, these data confirm that phospho-cJun/ATF2 dimer physically binds to the CRE locus to upregulate YY1 expression in chronically stimulated T cells.
p38MAPK/JNK Pathway Phosphorylates cJun/ATF2
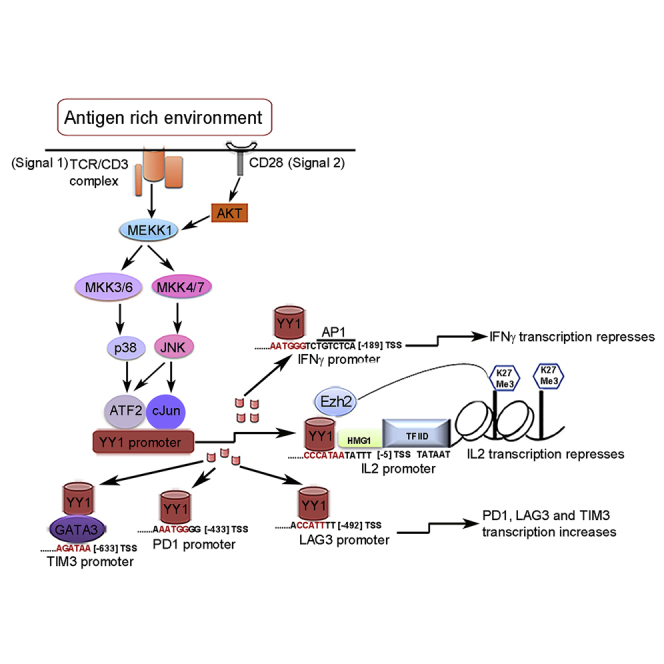
We then sought to define the upstream elements that precede cJun/ATF2 transcription factor phosphorylation and activation. The p38MAPK/JNK2 stress signaling pathway has a complex array of activities that are cell and context dependent, initiated in the current context by the combination of TCR/CD3 and CD28 signaling (Davis, 2000). Here, we hypothesize a model that this prominent pathway, upon persistent stimulation, leads to cJun and ATF2 phosphorylation, YY1 promoter activation, and ensuing T cell exhaustion (Figure 5A).
Figure 5.

p38MAPK/JNK Signaling Pathway Regulates YY1 Expression
(A) Model depicting p38MAPK/JNK signaling to cJun/ATF2 to regulate YY1 in T cells exposed to antigen-rich environment. T cell activation initiates phosphorylation of tyrosine residues within the ITAMs of the TCR/CD3 complex and CD28 co-stimulatory molecules by Src family tyrosine-protein kinases, Lck and Fyn (Arbour et al., 2002), to serve as docking site for ZAP70. Phospho-ZAP70(Tyr319/Tyr352) amplifies the downstream signaling cascade. CD28 co-stimulation complements TCR stimulation by activating and integrating serine-threonine kinase AKT with MEKK kinase (Kane et al., 2001) to phosphorylate MKK3/6 and MKK4/7. Phoshpho-MKK3(Ser189)/MKK6(Ser207) and phospho-MKK4(Ser257/Thr261) activate p38 MAPK and the cJun-N-terminal kinase (JNK) pathways, respectively (Kang et al., 2006, Raingeaud et al., 1995). Phospho-p38 MAPK(Thr180/Tyr182) in turn activates ATF2 and phospho-JNK2 (Thr183/Tyr185) activates the cJun transcription factor (Freshney et al., 1994, Ichijo, 1999). Phospho-cJun and phospho-ATF2 form heterodimers that bind to the YY1 promoter to initiate transcription.
(B) Western blots show increased phosphorylation of kinases in p38MAPK/JNK pathway with T cell re-stimulation.
(C) YY1 is suppressed by inhibitors to p38 and JNK by reporter assay (upper) and by western blot (lower). Three replicates per assay; representative of three experiments. *p < 0.05.
To test the explanatory value of our model (Figure 5A), we examined lysates from T cells persistently activated in vitro to analyze phospho-p38α(Tyr323) (alternative p38 activation pathway), phospho-p38α(Thr180/Tyr182), phospho-AKT(S473), phospho-JNK2(T183/Tyr185), phospho-MKK4(Ser257/Thr261), phospho-MKK3(Ser189/Thr193), and phospho-ZAP70(Tyr319). Upon repeated stimulation, these kinases underwent a progressive increase in phosphorylation (Figure 5B).
To probe the relation of this pathway to YY1 expression, we employed selective inhibitors that block phosphorylation of JNK, p38MAPK, and MEK1 (Figure S10A), with the last of these included as a non-pathway control. YY1 luciferase reporter showed high transcriptional activity after four stimulations, which was suppressed by inhibitors to p38MAPK or JNK, confirming their roles in YY1 transcription, with no impact from MEK1 inhibitor, confirming that the MEK1 pathway is not involved (Figure 5C upper). These inhibitor tests also showed markedly reduced YY1 protein in the same cells (Figure 5C lower).
These data are consistent with a pathway beginning at the membrane with two-signal T cell stimulation to p38MAPK/JNK activation to phosphorylation of ATF2/cJun in the cytoplasm with nuclear translocation and then activation of YY1 transcription, which in turns directs the exhaustion phenotype that ensues. The total YY1-centered circuit from T cell activation to exhaustion as revealed by these studies is depicted graphically in Figure S11.
As noted, Ezh2 exhibits similar expression kinetics to YY1 with T cell restimulation, but its promoter lacks YY1-binding sites. However, a cJun-binding site was identified (TGAATCA; Figure S9E). We speculate that the JNK pathway through cJun homodimerization or heterodimerization with other molecules could be regulating Ezh2 expression. This would put YY1 and Ezh2 regulation under the same signaling pathway. This was not investigated further.
Restimulation Induces Functional Exhaustion
Conventional markers of T cell exhaustion (e.g., checkpoint receptor expression, cytokine loss) are indicators or mediators: the key functional outcome of exhaustion is its adverse impact on killing of pathogenic targets, i.e., tumor or virus-infected cells. Our in vitro model successfully recapitulates the markers of in vivo exhausted T cells, but it cannot directly address cytotoxic activities without antigenic targeting. In this section, we extend our analysis via a related functional model that allows antigen selection as well as control of the number of signals involved. We then further apply this model to dissect the contributions of the separate cytokine and checkpoint axes of exhaustion.
We previously created chimeric antigen receptor (CAR)-modified T cells with one or two signals that enable a direct assay of killing against cells expressing carcinoembryonic antigen (CEA) (Figure 6A) (Emtage et al., 2008). We incubated the CAR-T cells with an excess of CEA+ tumor cells and followed the T cells for exhaustion marker responses. PD1 and Lag3 progressively increased in the CAR-T fraction (∼50%), but only with the construct generating two signals (Figures 6B and 6C), paralleling our results in restimulated normal unmodified T cells (Figures 1C and S4). (All subsequent data pertain to signal 1 + 2 [second generation] CAR-T.) As previously determined (Emtage et al., 2008) and as also reproduced in normal unmodified T cells above, there was a high initial secretion of IL-2 via signal 1 + 2 by CAR-T on CEA+ targets that declined with subsequent tumor cell exposures (Figure 6D). Finally, referencing cytotoxicity as the ultimate functional outcome, killing was robust with the first CAR-T exposure to tumor, but then declined with subsequent exposures, compatible with T cell functional exhaustion (Figure 6E). Our in vitro restimulation model is therefore confirmed by this final functional criterion as replicating in vivo T cell exhaustion responses.
Figure 6.

Functional Assessment of T Cell Exhaustion
(A) Design of CAR-T cells. An anti-CEA sFv-antibody-binding domain is fused with CD3 zeta (signal 1) or CD3 zeta plus CD28 (signal 1 + 2) signaling chains in first- and second-generation CAR-T cells.
(B and C) Increased expression of checkpoint receptors with restimulation only on second-generation CAR-T cells. CAR-T cells were created with approximately 50% modification and then co-cultured three times for two days with MIPCEA or MIP101 cell lines and analyzed by fluorescence-activated cell sorting (FACS) for PD1 and Lag3. (All remaining assays are with second-generation CAR-T cells).
(D) IL-2 ELISA shows high initial IL-2 and then decline in MIPCEA/CAR-T cell co-cultures. Antigen-negative MIP101 cells do not stimulate IL-2 production.
(E) Functional exhaustion shown by live images of MIPCEA target cells before and after co-culture with CAR-T cells. T cells from first co-culture were recovered and re-exposed to second and third batches of target tumor cells. High clearing of plates in first co-cultures indicates high initial killing that shows less clearing (less killing) in subsequent co-cultures.
(F) IL-2 ELISA as in (D) except co-culture performed in the presence of Ezh2 inhibitor, now showing sustained IL-2 for MIPCEA + CAR-T.
(G) Reversal of functional exhaustion shown by live images of MIPCEA and control MIP101 target cells after co-culture with CAR-T cells in the presence of Ezh2 inhibitor without or with anti-IL-2 antibody that blocks exhaustion reversal.
(H) Cell counts obtained from experiment in (E) and (G). Three replicates per assay; representative of three experiments.
To assess selectively the contributions of IL-2 failure versus checkpoint receptor action in the functional decline of exhausted T cells, we asked if blocking Ezh2 or checkpoint receptor PD1 could restore the cytotoxic potency of the exhausted T cells. Because Ezh2 inhibition appears only to reverse IL-2 suppression with no impact on other aspects of the exhaustion phenotype, any activity of the checkpoint receptors to mediate this exhaustion could continue uninterrupted. Specifically, the MIPCEA cell line is strongly PDL1+ (Figure S12) with the potential to continue to drive exhaustion if the PD1-PDL1 axis were the prime regulator of exhaustion.
As expected from the normal T cell results (Figure 2F), CAR-T cells treated with Ezh2 inhibitor sustained their antigen-specific IL-2 production under exhaustion-promoting conditions (Figure 6F). The CAR-T cells in the presence or absence of Ezh2 inhibitor performed robust killing against antigenic targets equally during the first co-culture with tumor cells (compare Figure 6G with Figure 6E), whereas only Ezh2-inhibitor-treated CAR-T cells maintained efficient killing with each subsequent exposure for a net 5-fold benefit by the third co-culture (Figure 6H). An anti-IL-2-neutralizing antibody applied to the Ezh2 inhibitor group successfully blocked 60%–70% of the restored killing activity, confirming that the benefit of Ezh2 inhibitor was from sustained IL-2 (Figures 6G and 6H).
A trivial reason for loss of cytotoxic function would be death of T cells in the IL-2-deficient cultures. Accordingly, we monitored the viability of the T cells in Figure 6H by trypan blue and propidium iodide staining in flow cytometry. Values after the third co-culture with MIPCEA revealed preserved CAR-T cell viability (74%–79%) across all tests: (1) control, exhausted cells (IL-2 suppressed); (2) experimental, Ezh2 inhibitor (abundant IL-2); and (3) control, Ezh2 inhibitor + anti-IL-2 Ab (IL-2 blocked). Stimulation with two signals is known to render T cells resistant to short-term IL-2 withdrawal via Bcl-xl upregulation (Boise et al., 1995), and this is the likely reason for preserved viability in the IL-2-deficient cultures (1 and 3). Although the presence or absence of IL-2 did not affect the viability in these tests, the absence of IL-2 with repeated two-signal stimulation appeared to engender cytotoxic exhaustion. Although IL-2 has been considered important for survival of activated T cells (Emtage et al., 2008), this result emphasizes a second, non-survival action of IL-2 to support T cell cytotoxic functions.
In other tests, we performed blocking of the PD1-PDL1 axis with anti-PD1 antibody, nivolumab. In line with nivolumab's action to oppose exhaustion in human cancer therapies with major anti-tumor benefits (Rajan et al., 2016), nivolumab partially rescued the loss of T cell cytotoxic potency during repeated exposures of CAR-T cells to tumor, but to a lesser degree than Ezh2 inhibition (30% rescue by nivolumab versus 100% rescue by Ezh2 inhibitor in third co-culture; Figures S13A and S13D). This result could suggest that IL-2 failure may be of even greater importance than PD1-PDL1 signaling to functional suppression under some exhaustion conditions. Nivolumab interrupts the action of PD1 to upregulate phosphatases that inhibit TCR activation (Freeman et al., 2000) by a non-IL-2-dependent mechanism. Nivolumab-treated CAR-T cells experienced the same IL-2 loss as control T cells, indicating that restoring IL-2 is not part of how nivolumab acts (Figure S13E).
In Vivo Markers of Exhaustion in Cancer and HIV
T cells reactive to melanoma antigen Melan-A/MART1 commonly infiltrate tumor sites (i.e., tumor-infiltrating lymphocytes [TILs]) but are functionally exhausted, as evidenced by loss of type I cytokines; expression of genes associated with exhaustion, including PD1; and lack of tumor cell killing (Baitsch et al., 2011). We examined 15 human melanoma samples and 10 normal skin biopsies and confirmed that the majority of TILs were exhausted per PD1 positivity, whereas normal skin T cells were negative (Figure 7A). We then assayed these tissues for the novel features revealed in our in vitro exhaustion system. Staining confirmed elevated phospho-cJun(Ser63/73) protein in the TILs, indicating activation of the p38MAPK/JNK pathway and a broad YY1 positivity (Figures 7B and 7C). YY1-cofactor Ezh2 was also elevated (Figure 7D). Resident CD3+ T cells in normal skin were negative for all exhaustion markers. These in vivo data on exhausted T cells thus replicate and validate the key novel findings derived from our in vitro modeling: activation of p38MAPK/JNK pathway in TILs drives YY1 expression, which in turn upregulates PD1 and mediates the other diverse aspects of the exhaustion phenotype (Figure S11).
Figure 7.

New Exhaustion Markers Confirmed in Melanoma TILs
(A and B) Human melanoma sections underwent immunofluorescence (IF) staining, showing most infiltrating T cells are PD1+ (CD3 and PD1 double staining) and activated phospho-cJun(ser 63/73) positive (CD3/phospho-cJun(ser 63/73) double staining). The tumor cells themselves are positive for phospho-cJun. Normal skin T cells were negative for PD1 and phospho-cJun (arrows point to CD3 cells negative for PD1 in A and CD3 cells negative for phospho-cJun in B). Magnified panels shown for double staining.
(C and D) IHC showing expression of YY1 and Ezh2 in TILs, confirmed on double staining (CD3 is brown, YY1 and Ezh2 are red; merged IHC dark red for double positive). Normal skin T cells were negative for YY1 and Ezh2, bottom panels (arrows).
Scale bar represents 20 μm. These stainings were performed five times with a minimum of two melanoma and two skin samples in each test with concordant results.
As a further test of the breadth of the model for exhaustion regulation, we examined T cells from an infectious disease setting. T cell exhaustion was first recognized in infections rather than cancer, specifically in experimental lymphocytic choriomeningitis virus (LCMV) infections in mice, and later in chronic infections in humans, including HIV (Barber et al., 2006, Wherry, 2011). Similar to melanoma, PD1 expression on CD4 and CD8 T cells in untreated HIV infection has been associated with functional exhaustion, with higher fractions of PD1+ CD4 cells correlating with higher viral load and low CD4 counts (Day et al., 2006, Grabmeier-Pfistershammer et al., 2011). Furthermore, the PD1+ CD4 T cell subset, which is restrained from activation and proliferation by exhaustion, has been favored as a reservoir for latent HIV in patients on antiretroviral therapy (Hatano et al., 2013). We analyzed CD4 T cells by flow cytometry from n = 10 chronically infected and untreated patients with HIV and an equal number of healthy donors (Figures 8A and 8B). These tests confirm that the PD1+ populations are shifted toward YY1 expression and Ezh2 expression in HIV+ subjects (Figure 8C), in parallel with our findings with the in vitro exhaustion model (Figures 2A–2C) and the in vivo melanoma tests (Figure 7) and also in the smaller fraction of PD1+ CD4 T cells in normals (Figure 8C).
Figure 8.

New Exhaustion Markers Confirmed in HIV T Cells
(A and B) CD4 T cells from (A) control subjects and (B) patients with HIV were stained and analyzed by flow cytometry for PD1 and YY1 or PD1 and Ezh2 as in Transparent Methods.
(C) Summary of MFI values for YY1 and Ezh2 in PD1- and PD1+ CD4 T cells. MFI for YY1 and Ezh2 is higher in PD1+ than PD1- cells at p<0.02 (paired two-tailed t-test).
Discussion
In the recent decade, T cell exhaustion has come to be recognized as critically important both in the resistance to virus elimination in chronic infections and in the failure of immune surveillance in cancer. To understand the molecular underpinnings of exhaustion and its regulation, we required an in vitro model that we could manipulate to dissect the underlying elements. The restimulation model we employed was shown to recapitulate key defining elements that distinguish exhausted T cells from differentiated effector and memory T cells (Wherry, 2011): a requirement for chronic two-signal stimulation; upregulation of checkpoint receptors PD1, Lag3, and Tim3; downregulation of type 1 cytokines IL-2 and IFN-γ; loss of proliferative potential; and cytotoxic failure.
Still, our data are derived from an in vitro model, whereas exhaustion is fundamentally an in-vivo-defined phenomenon. Corroboration was sought in the presence of YY1, Ezh2, and cJun in in vivo exhausted T cells in melanoma and HIV, which is reassuring for the relevance of our findings to human biology. However, more work will be required to firmly link these discoveries to in vivo exhaustion. These in vitro results have provided a vital roadmap to suggest where to look in in vivo systems and how to perturb those systems for this ultimate corroboration, as discussed below.
By the studies in our in vitro T cell exhaustion model, the functionally disparate axes of checkpoint receptor action and cytokine deprivation are unified for the first time under a common mechanistic origin. Our results reveal the previously unknown role of transcription factor Yin Yang-1 to orchestrate diverse elements of exhaustion progression in our in vitro model, displaying both positive and negative regulations as embodied in its name. Persistent two-signal stimulation was shown to be required for the upregulation of YY1, yet the process leading to exhaustion in this model is shown to begin already with the very first interval of T cell activation.
Components of both TCR signal 1 and CD28 signal 2 merge into the p38MAPK/JNK pathway (Figure S11), which leads to phosphorylation of cJun and ATF2 and conversion to their transcriptionally active forms. Phospho-cJun/ATF2 dimers bind to the CRE locus in the YY1 promoter, activating transcription for YY1 accumulation. We noted in every setting that Ezh2 increases in parallel with YY1 but Ezh2 is not regulated by YY1. However, Ezh2, like YY1, contains a cJun site in its promoter that could also be regulated through the same p38MAPK/JNK pathway. This was not investigated further. Increasing YY1 then exerts its dual roles to upregulate checkpoint receptors PD1, Lag3, and Tim3 and to downregulate cytokines IL-2—recruiting Ezh2 for histone trimethylation—and IFN-γ, and functional cytotoxic exhaustion ensues.
The novel elements predicted in this study were corroborated in in vivo sample examinations of tumor tissues and blood from normal subjects and patients with HIV. Melanoma TILs were confirmed for PD1 positivity, marking them as exhausted as previously defined (Ahmadzadeh et al., 2009). Staining showed elevated phospho-cJun that drives transcription of YY1, which in turn drives PD1 expression from our in vitro model. Ezh2 was highly expressed in melanoma TILs in parallel with YY1, potentially also driven by phospho-cJun, as stated above. PD1 positivity in T cells of patients with HIV, also associated with exhaustion (Wherry et al., 2003, Day et al., 2006, Wherry and Kurachi, 2015), was similarly shown to correlate with YY1 and Ezh2 expression, consistent with our in vitro model and the in vivo findings in melanoma. Exhausted T cells exist in normal settings as a self-protective mechanism and in disease, where it can have a negative impact to hamper effective immune responses. Seemingly, the clinical setting (normal, infection, or cancer) does not differentially affect how exhausted T cells are generated: all work through a continued T cell activation-driven transcription of YY1.
Our studies are solely in humans with uncertain correspondence to mouse systems, which can exhibit important differences (Mestas and Hughes, 2004). However, clues exist to analogous processes in mice. Search of an RNA-seq database from LCMV chronic infection (http://rstats.immgen.org/Skyline/skyline.html) showed increased T cell YY1 mRNA (61 versus 36 in control) and Ezh2 mRNA (190 versus 36 in control) (Yu et al., 2017). Inasmuch as this analysis was in bulk CD8 T cells, it would underestimate predicted changes in YY1 and Ezh2 in the PD1+ exhausted virus-specific fractions. The significance of the co-expression of YY1 and Ezh2 and the potential for collaboration was not part of their investigation. Similarly, in a tumor model, upregulation of Ezh2 mRNA was observed in exhausted ova-specific T cells infiltrating B16-ova melanoma tumors (OT-1 TILs) when compared with non-exhausted non-specific T cells (P14 TILs) (Mognol et al., 2017). Both tumor and control T cells were pre-activated before infusion, and therefore differences in these markers could be underrepresented. No linkage of Ezh2 to a transcription factor was investigated that would direct the Ezh2 action (e.g., YY1).
Finally, Blimp-1 has been proposed to have a role in exhaustion and IL-2 shutdown (Wherry, 2011) based on a Blimp-1-binding site 2 kb from the IL-2 promoter and knockout studies in mice (Martins et al., 2008). However, this IL-2-repressive role for Blimp-1 was not evaluated by promoter mutation studies as we do here, and the site for Blimp-1 binding is also not conserved in the human IL-2 promoter and therefore cannot be a direct mediator of human IL-2 decline as we show for YY1. Furthermore, any residual effect of Blimp-1 to repress IL-2 in our system was inapparent given that blocking YY1 activity yielded sustained IL-2 production, whatever Blimp-1's contribution. Formally, one cannot exclude the possibility that Blimp-1 has a role in mouse that is distinct from humans and that exhaustion is mediated by different mechanisms between the species. On the other hand, all promoters in which we have shown functional YY1-binding sites in humans are also present in mice, including GATA-3 in mouse Tim3, strongly suggesting that a YY1-centered mechanism will be shown to pertain in mouse T cell exhaustion as well.
A survey of promoters for all 24,135 expressed genes identified 765 or ∼3% with YY1 binding (Xi et al., 2007). Notably, of the five genes that we selected in an unbiased fashion a priori for study based on their association with exhaustion (PD1, Lag3, Tim3, IL-2, INF-γ), all (5/5) have sites for YY1 binding (or GATA-3 in Tim3 that can mediate YY1 binding). The probability of such an association by chance across all 5 genes is (0.03)5 = 2.4 × 10−8, and if not by chance, the “purpose” of the YY1 association with these genes would be compatible with a role for YY1 to coordinately regulate all of these components to mediate T cell exhaustion. The same statistical argument applies to the mouse setting above.
Because we prove that signal 1 + 2 is required for YY1 expression in our exhaustion model, it begs the question as to the source of signal 2 in these settings. In HIV infection, signal 2 could derive from B7.1 (CD80) on monocytes and macrophages that harbor HIV viruses and that also scavenge HIV proteins from plasma (Giguere et al., 2004). In contrast, B7.1 is typically absent from solid tumors. However, its homolog B7.2 (CD86), also a CD28 ligand, is expressed more widely and was found to be positive on 11/12 specimens of melanoma in the human protein atlas database (http://www.proteinatlas.org/ENSG00000114013-CD86/cancer); melanoma is the tumor in which human T cell exhaustion is most prominent and well characterized among human cancers (Baitsch et al., 2011).
We previously showed that repeat two-signal stimulation resulted in IL-2 shutdown and a severely restricted T cell proliferation (Emtage et al., 2008) as anticipated with exhaustion, but the impact on functional killing, the ultimate consequential outcome, was not directly addressed until now. An unanticipated study result was the degree to which IL-2 failure appeared to underlie cytotoxic failure. To ameliorate the adverse functional effects of exhaustion, most current efforts have focused on checkpoint receptor interactions and their interruption. From our analyses, however, it appears possible that IL-2 failure may in some settings contribute even more to functional cytotoxic exhaustion than checkpoint receptors.
If our in vitro results can be confirmed for relevance to human in vivo clinical settings, a number of interventions of therapeutic potential are enabled by these results. Three types of interventions are demonstrated: (1) those that interrupt Ezh2 activity, with the principle impact on rescuing IL-2 production; (2) those that interrupt checkpoint receptor and ligand interactions, undampening TCR activation; and (3) those that interrupt YY1 action, disrupting both axes for what could be a more complete block of exhaustion. Of these, only checkpoint receptor-ligand interruption was previously known and exploited with important clinical benefits.
YY1 is readily inactivated in therapeutic T cells (TIL and CAR-T) with RNAi, as used here (Figure 2G), but not directly by small molecules at this point in time. However, small molecule inhibitors can act at points along the p38MAPK/JNK2 pathway to block YY1 transcription (Figure 5C). This rationale is bolstered by the observation that JNK2 knockout mice have heightened proliferation of CD8 T cells in vivo and increased IL-2 production with improved virus control (Dong et al., 2000, Arbour et al., 2002, Conze et al., 2002). Our data for the first time provide the means to understand this result: by our model (Figure S11), JNK2 knockout would prevent cJun phosphorylation and block YY1 transcription with T cell restimulation, escaping cytokine failure and checkpoint receptor upregulation and thereby avoiding exhaustion for an improved therapeutic outcome.
Ezh2, on the other hand, can be directly inhibited systemically with small molecules that have shown favorable safety profiles in early-stage clinical trials primarily in Ezh2-expressing lymphomas (Ribrag et al., 2014). Ezh2 could likewise be interrupted with RNAi in engineered therapeutic T cells to avoid the need for continuous drug exposures. Ezh2 has attracted interest as a cancer target because of its overexpression and mutational hyperactivity in some tumors (Kim and Roberts, 2016). However, such rationales for the directed treatment of malignant tumor cells with Ezh2 inhibitors are conceptually wholly distinct from the rationales emerging from these new discoveries surrounding Ezh2's role as a critical co-factor in immune regulation. Rather, targeting of Ezh2 or YY1 in the current context is solely for the directed treatment of exhausted T cells, irrespective of tumor Ezh2 gene expression, with potentially broad implications for infections as well as cancer.
Exhaustion with IL-2 failure is widely observed in oncology and infectious diseases, prompting the use of systemic IL-2 in melanoma and renal cell cancers (Atkins et al., 2000, Klapper et al., 2008) and in infection models (Blattman et al., 2003), and as a critical adjunct in TIL and CAR-T therapies (Rosenberg et al., 1988, Lo et al., 2010, Katz et al., 2015, Junghans, 2017). Avoiding IL-2 failure by inhibiting Ezh2 or YY1 could alleviate the need for systemic IL-2 and its associated toxicity and cost. On the other hand, where checkpoint inhibitor therapies have a proven utility, the co-application of Ezh2 inhibitor to maintain endogenous IL-2 would be rational: synergy of exogenous IL-2 with checkpoint blockade was previously shown in infections (West et al., 2013) with high probability of utility for their co-application in cancer as well. Ezh2 inhibitor co-applied with checkpoint antibodies to combat IL-2 failure and checkpoint suppression could be equivalent to blockade of YY1, which coordinates both.
Exhaustion, as revealed by these in vitro studies, is seen to be an integral feature of the T cell immune response that initiates lead elements with the very first T cell activation. Exhaustion is an active, energy-requiring process that depends critically on recurrent stimulation for continued YY1 transcription to achieve and maintain exhaustion's full expression. CD4 and CD8 T cells were highly concordant in their expression of conventional exhaustion markers and in their functional declines of cytokine and cytotoxicity, emphasizing that both T cell arms are restrained by exhaustion for a more complete suppression. Similar to anergy, exhaustion likely evolved to limit anti-self reactions that escaped TCR editing in the thymus. Anergy is conceived via signal-1-only stimulation, whereas exhaustion as defined here is distinguished by its suppressive activity in settings with co-stimulation (signal 1 + 2) that could otherwise elicit robust autoimmune responses (Leavy, 2015). In pathologic states of malignancy and chronic infection, however, exhaustion is a limiting factor that restrains the full potential of T cells to cure.
The data in this report advance our understanding of the relevance of T cell activation via a p38MAPK/JNK/YY1 feedback loop to IL-2 shutdown, checkpoint receptor upregulation, and T cell exhaustion, revealing novel points of intervention with potential to improve immunotherapies against chronic infections and metastatic cancers alike. Because the elucidated pathways were developed with in vitro exhausted T cells, additional studies can be considered to reinforce and validate the inferences derived here for in vivo settings. With experimental confirmation that the conserved YY1 sites in mouse also reflect conserved functions to upregulate checkpoint receptors and downregulate type 1 cytokines, animal testing may ensue. Conditional knockdown of YY1 in T cells in LCMV and YY1 antisense engineered into CAR-T in tumor models should show better control of infections and cancer if YY1 is a direct mediator of exhaustion. Finally, if this model is applicable in vivo, the use of checkpoint receptor antibodies to restore function of exhausted T cells in human cancer therapies should be further enhanced with the co-application of Ezh2 inhibitor in human trials, with a net increase in the potency and effectiveness of the intervention, providing both the ultimate validation and clinical benefit of these discoveries.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Gongxian Liao for providing CAR-T cells and target cell lines as well as know-how for their evaluation. We thank Dr. Liao and Dr. Steven Bunnell for their expert advice with FACS analysis. We thank Dr. Guangchao Sui of Wake Forest Health Sciences and Dr. Denis C. Guttridge of Ohio State University for providing the YY1-promoter luciferase construct and YY1 expression plasmid, respectively. We acknowledge Drs. Satori Iwamoto, Zhengke Wang, and Ting Zhao of Roger Williams Medical Center for providing materials and guidance in experimental procedures. We would like to acknowledge the contribution of Dr. Monika Pilichowska and Ms. Karen Krajewski for their help with the cytospin experiments. We thank Dr. Laura Kogelman for access to HIV samples and Dr. Nordine Bakouche for his help with HIV work. We acknowledge Nathan Li, Division of Laboratory Medicine, Tufts University, for his assistance with IHC. Finally, we would like acknowledge Dr. Seema Naheed, Dr. Sylvester Homsy, Stacey Bjorgaard, and Christopher Robertson for expert technical assistance. Grant support was in part from the U.S. Department of Defense (DAMD17-98-1-8284).
Author Contributions
M.Y.B. conducted all experimentation except immunostainings, which were performed by G.W. and F.X. M.Y.B. and R.P.J. oversaw the experimental design and data interpretation; and M.Y.B. and R.P.J. wrote the manuscript.
Declaration of Interests
Competing Interests: None.
Published: April 27, 2018
Footnotes
Supplemental Information includes Transparent Methods and 13 figures and can be found with this article online at https://doi.org/10.1016/j.isci.2018.03.009.
Supplemental Information
References
- Ahmadzadeh M., Johnson L.A., Heemskerk B., Wunderlich J.R., Dudley M.E., White D.E., Rosenberg S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleman L.J., Boussiotis V.A. T cell anergy and costimulation. Immunol. Rev. 2003;192:161–180. doi: 10.1034/j.1600-065x.2003.00009.x. [DOI] [PubMed] [Google Scholar]
- Arbour N., Naniche D., Homann D., Davis R.J., Flavell R.A., Oldstone M.B. c-Jun NH(2)-terminal kinase (JNK)1 and JNK2 signaling pathways have divergent roles in CD8(+) T cell-mediated antiviral immunity. J. Exp. Med. 2002;195:801–810. doi: 10.1084/jem.20011481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvey A., van der Veeken J., Samstein R.M., Feng Y., Stamatoyannopoulos J.A., Rudensky A.Y. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat. Immunol. 2014;15:580–587. doi: 10.1038/ni.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchison M.L. Function of YY1 in long-distance DNA interactions. Front. Immunol. 2014;5:45. doi: 10.3389/fimmu.2014.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins M.B., Kunkel L., Sznol M., Rosenberg S.A. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J. Sci. Am. 2000;6(Suppl 1):S11–S14. [PubMed] [Google Scholar]
- Baitsch L., Baumgaertner P., Devevre E., Raghav S.K., Legat A., Barba L., Wieckowski S., Bouzourene H., Deplancke B., Romero P. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J. Clin. Invest. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkhi M.Y., Ma Q., Ahmad S., Junghans R.P. T cell exhaustion and Interleukin 2 downregulation. Cytokine. 2015;71:339–347. doi: 10.1016/j.cyto.2014.11.024. [DOI] [PubMed] [Google Scholar]
- Barber D.L., Wherry E.J., Masopust D., Zhu B., Allison J.P., Sharpe A.H., Freeman G.J., Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Blattman J.N., Grayson J.M., Wherry E.J., Kaech S.M., Smith K.A., Ahmed R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat. Med. 2003;9:540–547. doi: 10.1038/nm866. [DOI] [PubMed] [Google Scholar]
- Boise L.H., Minn A.J., Noel P.J., June C.H., Accavitti M.A., Lindsten T., Thompson C.B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- Caretti G., Di Padova M., Micales B., Lyons G.E., Sartorelli V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004;18:2627–2638. doi: 10.1101/gad.1241904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conze D., Krahl T., Kennedy N., Weiss L., Lumsden J., Hess P., Flavell R.A., Le Gros G., Davis R.J., Rincon M. c-Jun NH(2)-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8(+) T cell activation. J. Exp. Med. 2002;195:811–823. doi: 10.1084/jem.20011508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R.J. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Day C.L., Kaufmann D.E., Kiepiela P., Brown J.A., Moodley E.S., Reddy S., Mackey E.W., Miller J.D., Leslie A.J., DePierres C. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- Dong C., Yang D.D., Tournier C., Whitmarsh A.J., Xu J., Davis R.J., Flavell R.A. JNK is required for effector T-cell function but not for T-cell activation. Nature. 2000;405:91–94. doi: 10.1038/35011091. [DOI] [PubMed] [Google Scholar]
- Emtage P.C., Lo A.S., Gomes E.M., Liu D.L., Gonzalo-Daganzo R.M., Junghans R.P. Second-generation anti-carcinoembryonic antigen designer T cells resist activation-induced cell death, proliferate on tumor contact, secrete cytokines, and exhibit superior antitumor activity in vivo: a preclinical evaluation. Clin. Cancer Res. 2008;14:8112–8122. doi: 10.1158/1078-0432.CCR-07-4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman G.J., Long A.J., Iwai Y., Bourque K., Chernova T., Nishimura H., Fitz L.J., Malenkovich N., Okazaki T., Byrne M.C. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freshney N.W., Rawlinson L., Guesdon F., Jones E., Cowley S., Hsuan J., Saklatvala J. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell. 1994;78:1039–1049. doi: 10.1016/0092-8674(94)90278-x. [DOI] [PubMed] [Google Scholar]
- Giguere J.F., Bounou S., Paquette J.S., Madrenas J., Tremblay M.J. Insertion of host-derived costimulatory molecules CD80 (B7.1) and CD86 (B7.2) into human immunodeficiency virus type 1 affects the virus life cycle. J. Virol. 2004;78:6222–6232. doi: 10.1128/JVI.78.12.6222-6232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S., Akopyan G., Garban H., Bonavida B. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene. 2006;25:1125–1142. doi: 10.1038/sj.onc.1209080. [DOI] [PubMed] [Google Scholar]
- Grabmeier-Pfistershammer K., Steinberger P., Rieger A., Leitner J., Kohrgruber N. Identification of PD-1 as a unique marker for failing immune reconstitution in HIV-1-infected patients on treatment. J. Acquir. Immune Defic. Syndr. 2011;56:118–124. doi: 10.1097/QAI.0b013e3181fbab9f. [DOI] [PubMed] [Google Scholar]
- Gray S.M., Amezquita R.A., Guan T., Kleinstein S.H., Kaech S.M. Polycomb repressive complex 2-mediated chromatin repression guides effector CD8+ T cell terminal differentiation and loss of multipotency. Immunity. 2017;46:596–608. doi: 10.1016/j.immuni.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano H., Jain V., Hunt P.W., Lee T.H., Sinclair E., Do T.D., Hoh R., Martin J.N., McCune J.M., Hecht F. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J. Infect. Dis. 2013;208:50–56. doi: 10.1093/infdis/jis630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa J., Mittal S., Wang Y., Korkmaz K.S., Adamson E., English C., Ohmichi M., McClelland M., Mercola D. Identification of promoters bound by c-Jun/ATF2 during rapid large-scale gene activation following genotoxic stress. Mol. Cell. 2004;16:521–535. doi: 10.1016/j.molcel.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Hwang S.S., Kim Y.U., Lee S., Jang S.W., Kim M.K., Koh B.H., Lee W., Kim J., Souabni A., Busslinger M., Lee G.R. Transcription factor YY1 is essential for regulation of the Th2 cytokine locus and for Th2 cell differentiation. Proc. Natl. Acad. Sci. USA. 2013;110:276–281. doi: 10.1073/pnas.1214682110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18:6087–6093. doi: 10.1038/sj.onc.1203129. [DOI] [PubMed] [Google Scholar]
- Junghans R.P. The challenges of solid tumor for designer CAR-T therapies: a 25-year perspective. Cancer Gene Ther. 2017;24:89–99. doi: 10.1038/cgt.2016.82. [DOI] [PubMed] [Google Scholar]
- Kane L.P., Andres P.G., Howland K.C., Abbas A.K., Weiss A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-gamma but not TH2 cytokines. Nat. Immunol. 2001;2:37–44. doi: 10.1038/83144. [DOI] [PubMed] [Google Scholar]
- Kang Y.J., Seit-Nebi A., Davis R.J., Han J. Multiple activation mechanisms of p38alpha mitogen-activated protein kinase. J. Biol. Chem. 2006;281:26225–26234. doi: 10.1074/jbc.M606800200. [DOI] [PubMed] [Google Scholar]
- Katz S.C., Burga R.A., McCormack E., Wang L.J., Mooring W., Point G.R., Khare P.D., Thorn M., Ma Q., Stainken B.F. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified t-cell therapy for CEA+ liver metastases. Clin. Cancer Res. 2015;21:3149–3159. doi: 10.1158/1078-0432.CCR-14-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.H., Roberts C.W. Targeting EZH2 in cancer. Nat. Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapper J.A., Downey S.G., Smith F.O., Yang J.C., Hughes M.S., Kammula U.S., Sherry R.M., Royal R.E., Steinberg S.M., Rosenberg S. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma : a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer. 2008;113:293–301. doi: 10.1002/cncr.23552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavy O. Autoimmunity: benefits of exhaustion. Nat. Rev. Immunol. 2015;15:468. doi: 10.1038/nri3890. [DOI] [PubMed] [Google Scholar]
- Liao W., Lin J.X., Leonard W.J. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38:13–25. doi: 10.1016/j.immuni.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Schmidt-Supprian M., Shi Y., Hobeika E., Barteneva N., Jumaa H., Pelanda R., Reth M., Skok J., Rajewsky K. Yin Yang 1 is a critical regulator of B-cell development. Genes Dev. 2007;21:1179–1189. doi: 10.1101/gad.1529307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo A.S., Ma Q., Liu D.L., Junghans R.P. Anti-GD3 chimeric sFv-CD28/T-cell receptor zeta designer T cells for treatment of metastatic melanoma and other neuroectodermal tumors. Clin. Cancer Res. 2010;16:2769–2780. doi: 10.1158/1078-0432.CCR-10-0043. [DOI] [PubMed] [Google Scholar]
- Martins G.A., Cimmino L., Liao J., Magnusdottir E., Calame K. Blimp-1 directly represses Il2 and the Il2 activator Fos, attenuating T cell proliferation and survival. J. Exp. Med. 2008;205:1959–1965. doi: 10.1084/jem.20080526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestas J., Hughes C.C. Of mice and not men: differences between mouse and human immunology. J. Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- Mognol G.P., Spreafico R., Wong V., Scott-Browne J.P., Togher S., Hoffmann A., Hogan P.G., Rao A., Trifari S. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc. Natl. Acad. Sci. USA. 2017;114:E2776–E2785. doi: 10.1073/pnas.1620498114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.T., Ohashi P.S. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat. Rev. Immunol. 2015;15:45–56. doi: 10.1038/nri3790. [DOI] [PubMed] [Google Scholar]
- Raingeaud J., Gupta S., Rogers J.S., Dickens M., Han J., Ulevitch R.J., Davis R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- Rajan A., Kim C., Heery C.R., Guha U., Gulley J.L. Nivolumab, anti-programmed death-1 (PD-1) monoclonal antibody immunotherapy: role in advanced cancers. Hum. Vaccin. Immunother. 2016;12:2219–2231. doi: 10.1080/21645515.2016.1175694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribrag V., Soria J.C., Reyderman L., Chen R., Salazar P., Kumar N., Kuznetsov G., Keilhack H., Ottesen L.H., Italiano A. Phase 1 first-in-human study of the enhancer of zeste-homolog 2 (EZH2) histone methyl transferase inhibitor E7438 as a single agent in patients with advanced solid tumors or B cell lymphoma. Eur. J. Cancer. 2014;50:197. [Google Scholar]
- Rosenberg S.A., Mule J.J., Spiess P.J., Reichert C.M., Schwarz S.L. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. J. Exp. Med. 1985;161:1169–1188. doi: 10.1084/jem.161.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg S.A., Packard B.S., Aebersold P.M., Solomon D., Topalian S.L., Toy S.T., Simon P., Lotze M.T., Yang J.C., Seipp C.A. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988;319:1676–1680. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- Shi Y., Lee J.S., Galvin K.M. Everything you have ever wanted to know about Yin Yang 1. Biochim. Biophys. Acta. 1997;1332:F49–F66. doi: 10.1016/s0304-419x(96)00044-3. [DOI] [PubMed] [Google Scholar]
- Smeal T., Binetruy B., Mercola D., Grover-Bardwick A., Heidecker G., Rapp U.R., Karin M. Oncoprotein-mediated signalling cascade stimulates c-Jun activity by phosphorylation of serines 63 and 73. Mol. Cell. Biol. 1992;12:3507–3513. doi: 10.1128/mcb.12.8.3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan L., Atchison M.L. YY1 DNA binding and PcG recruitment requires CtBP. Genes Dev. 2004;18:2596–2601. doi: 10.1101/gad.1228204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M.J., Seto E. Unlocking the mechanisms of transcription factor YY1: are chromatin modifying enzymes the key? Gene. 1999;236:197–208. doi: 10.1016/s0378-1119(99)00261-9. [DOI] [PubMed] [Google Scholar]
- van Dam H., Castellazzi M. Distinct roles of Jun : Fos and Jun : ATF dimers in oncogenesis. Oncogene. 2001;20:2453–2464. doi: 10.1038/sj.onc.1204239. [DOI] [PubMed] [Google Scholar]
- van Galen J.C., Dukers D.F., Giroth C., Sewalt R.G., Otte A.P., Meijer C.J., Raaphorst F.M. Distinct expression patterns of polycomb oncoproteins and their binding partners during the germinal center reaction. Eur. J. Immunol. 2004;34:1870–1881. doi: 10.1002/eji.200424985. [DOI] [PubMed] [Google Scholar]
- West E.E., Jin H.T., Rasheed A.U., Penaloza-Macmaster P., Ha S.J., Tan W.G., Youngblood B., Freeman G.J., Smith K.A., Ahmed R. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J. Clin. Invest. 2013;123:2604–2615. doi: 10.1172/JCI67008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry E.J. T cell exhaustion. Nat. Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- Wherry E.J., Blattman J.N., Murali-Krishna K., van der Most R., Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry E.J., Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C.J., Kharchenko P.V., Daheron L., Park P.J., Kingston R.E. Variable requirements for DNA-binding proteins at polycomb-dependent repressive regions in human HOX clusters. Mol. Cell. Biol. 2013;33:3274–3285. doi: 10.1128/MCB.00275-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H., Yu Y., Fu Y., Foley J., Halees A., Weng Z. Analysis of overrepresented motifs in human core promoters reveals dual regulatory roles of YY1. Genome Res. 2007;17:798–806. doi: 10.1101/gr.5754707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Cippitelli M., Dorman L., Ortaldo J.R., Young H.A. The nuclear factor YY1 suppresses the human gamma interferon promoter through two mechanisms: inhibition of AP1 binding and activation of a silencer element. Mol. Cell. Biol. 1996;16:4744–4753. doi: 10.1128/mcb.16.9.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B., Zhang K., Milner J.J., Toma C., Chen R., Scott-Browne J.P., Pereira R.M., Crotty S., Chang J.T., Pipkin M.E. Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation. Nat. Immunol. 2017;18:573–582. doi: 10.1038/ni.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac A.J., Blattman J.N., Murali-Krishna K., Sourdive D.J., Suresh M., Altman J.D., Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.