

Abstract
Four crystal structures are presented of Histone Deacetylase 6 (HDAC6) complexes with para-substituted phenylhydroxamate inhibitors, including bulky peptoids. These structures provide insight regarding the design of capping groups that confer selectivity for binding to HDAC6, specifically with regard to interactions in a pocket formed by the L1 loop. Capping group interactions may also influence hydroxamate-Zn2+ coordination with monodentate or bidentate geometry.
For TOC use only:
INTRODUCTION
Metal-dependent histone deacetylases (HDACs) catalyze the hydrolysis of acetyl-L-lysine to yield acetate and free L-lysine side chains in protein substrates, and reversible lysine acetylation rivals the reversible phosphorylation of serine, threonine, and tyrosine in the regulation of myriad cellular processes.1–4 Phylogenetic analysis indicates three isozyme subgroups: class I, (HDACs 1, 2, 3, and 8); class II, further subdivided into class IIa HDACs 4, 5, 7, and 9, and the class IIb HDACs 6 and 10; and the sole class IV enzyme, HDAC11.5 Structural and enzymological studies of metal-dependent HDAC isozymes indicate a common fold and chemical mechanism for catalysis.6–8 Class III HDACs are better known as sirtuins, but these enzymes are structurally and mechanistically unrelated to the metal-dependent HDACs.9
Dysregulation of HDAC function underlies diverse clinical conditions, and individual HDAC isozymes serve as validated targets for therapeutic intervention.10–13 For example, the class IIb isozyme HDAC614,15 is the cytosolic tubulin deacetylase;16,17 inhibition of HDAC6 suppresses microtubule dynamics and leads to cell cycle arrest and apoptosis, making HDAC6 a critical target for cancer chemotherapy as well as the treatment of neurodegenerative diseases and other disorders.18–21 However, the general conservation of active site structure and chemistry among class I, II, and IV HDACs presents significant challenges to the design of isozyme-selective inhibitors.
Recent crystal structure determinations of HDAC6 facilitate the analysis of inhibitor structure-affinity and structure-selectivity relationships.22–24 Notably, phenylhydroxamic acid exhibits 17–68-fold selectivity for inhibition of HDAC6 class I isozymes,25,26 and derivatization of phenylhydroxamic acid with bulky capping groups further enhances inhibitory selectivity. For example, N-hydroxy-4-(2-[(2-hydroxyethyl)(phenyl)amino]-2-oxoethyl)benzamide (HPOB) and N-hydroxy-4-[(N(2-hydroxyethyl)-2-phenylacetamido)methyl)-benzamide)] (HPB) exhibit 52-fold and 36-fold selectivity for the inhibition of HDAC6 over HDAC1, respectively.27,28 Crystal structures of these compounds in complex with HDAC6 reveal an unusual monodentate hydroxamate-Zn2+ binding mode that is only 0.5 kcal/mol less stable than the canonical bidentate hydroxamate-Zn2+ binding mode observed in other complexes.22,24 Curiously, many HDAC6-selective inhibitors with bulky and rigid capping groups bind with monodentate hydroxamate-Zn2+ coordination.
As part of our continuing series of studies probing structure-affinity and structure-selectivity relationships in HDAC6-inhibitor complexes, we now present X-ray crystal structures of catalytic domain 2 (CD2) of HDAC6 from Danio rerio (zebrafish) complexed with four different phenylhydroxamic acid-based inhibitors at 1.47–2.20 Å resolution. Our studies focus on the inhibition of CD2 and not catalytic domain 1 (CD1), since only CD2 exhibits broad-specificity catalytic activity;22 moreover, it is this domain that is established to be the tubulin deacetylase.29 We previously demonstrated that zebrafish HDAC6 CD2 (henceforth simply “HDAC6”) is an excellent and more readily-studied surrogate of the actual drug target, human HDAC6 CD2.22 Molecular structures of the phenylhydroxamate-based inhibitors are shown in Figure 1. Inhibitors 1 and 2 contain large peptoid capping groups and exhibit better than 200-fold selectivity in comparison with HDAC2,30 whereas structurally related inhibitor 3 is essentially non-selective. Bavarostat (4) exhibits better than 16000-fold selectivity in comparison with HDAC1, HDAC2, and HDAC3.31 Bavarostat is used as a brain-penetrant positron emission tomography (PET) probe for imaging HDAC6 in the central nervous system.31 Here, our structural studies show that peptoid inhibitors 1–3 bind to HDAC6 with primarily monodentate hydroxamate-Zn2+ coordination geometry; however, Bavarostat (4) binds with canonical bidentate hydroxamate-Zn2+ coordination geometry. Comparisons with available structures of HDAC6-inhibitor complexes22–24 suggest that the hybridization of the linker benzylic nitrogen and the steric bulk of the capping group influence the denticity of hydroxamate-Zn2+ coordination. Interactions of the capping group in a pocket defined by the L1 loop further contribute to selectivity for binding to HDAC6.
Figure 1.

Phenylhydroxamate-based HDAC6 inhibitors and the corresponding selectivity data over the class I enzyme HDAC1. aRef. 30. bRef. 31.
RESULTS AND DISCUSSION
The 2.10 Å resolution structure of the HDAC6–1 complex reveals monodentate hydroxamate-Zn2+ coordination in both monomers in the asymmetric unit (Figure 2a). Selected interatomic distances are recorded in Table 1. The phenyl linker is nestled in the aromatic crevice formed by F583 and F643 with average inter-ring separations of 3.5 Å each. The peptoid carbonyl is oriented away from “gatekeeper” residue S531 in the L2 loop22 and accepts a hydrogen bond from the guanidinium group of R601 in an adjacent monomer. This places the dimethylphenyl substituent in van der Waals contact with the side chains of L1 loop residues H463 and P464, which define a small pocket designated the “L1 loop pocket”. The cyclohexylamide carbonyl forms hydrogen bonds with two water molecules, one of which interacts with the backbone carbonyl of A641 while the other forms a hydrogen bond with another water molecule that interacts with Zn2+ ligand H614. The cyclohexyl moiety is oriented away from the enzyme surface, packing against the side chain of R736 in the adjacent monomer in the crystal lattice.
Figure 2.

Polder omit maps (green mesh) contoured at 3.0 sigma for compounds (a) 1 (orange), (b) 2 (orange/purple), (c) 3 (yellow), and (d) Bavarostat (blue) complexed with HDAC6 (white). The catalytic Zn2+ ion appears as a grey sphere; metal coordination and hydrogen bond interactions are shown as solid and dashed black lines, respectively. Atoms from a symmetry-related molecule in the crystal lattice are shown with dark grey carbon atoms. In (b), the monodentate hydroxamate conformation of 2 is shown in orange and the bidentate conformation is shown in purple. The binding of 1 (a) and 3 (c) illustrates exclusive monodentate hydroxamate-Zn2+ coordination; the binding of 4 (Bavarostat) (d) illustrates exclusive bidentate hydroxamate-Zn2+ coordination.
Table 1.
Average intermolecular interactions in monoden-tate HDAC6–inhibitor complexes (Å)
| Inhibitor | 1 | 2 | 3 |
|---|---|---|---|
| N–O−---Zn2+ | 2.1 | 1.8 | 2.0 |
| H2O---Zn2+ | 2.1 | 2.0 | 2.2 |
| C=O---OH2 | 2.8 | 2.7 | 2.7 |
| O−---O (Y745) | 2.5 | 2.8 | 2.8 |
| H2O---N (H573) | 2.6 | 2.6 | 2.4 |
| H2O---N (H574) | 2.9 | 2.9 | 2.9 |
The crystal structure of the HDAC6–2 complex determined at 1.47 Å resolution reveals a monodentate hydroxamate-Zn2+ binding mode in monomers A, C, and D in the asymmetric unit generally similar to that of compound 1; selected interatomic distances are recorded in Table 1. In monomer B, electron density is consistent with a mixture of monodentate and bidentate hydroxamate-Zn2+ binding modes with refined occupancies of 0.63 and 0.37, respectively (Figure 2b). The lower occupancy bidentate conformation exhibits Zn2+---O distances of 2.5 Å for the hydroxamate C=O and 2.2 Å for the hydroxamate N–O− group.
Inter-ring separations between the phenyl linker of 2 and the side chains of F583 and F643 are 3.3 Å and 3.9 Å, respectively, and the capping group adopts an essentially identical conformation for both metal-binding modes. The peptoid carbonyl is oriented away from the enzyme surface toward bulk solvent. The dimethylaniline group resides in the L1 loop pocket, and its nitrogen atom makes a van der Waals contact with P464. Interestingly, crystal packing places the dimethylaniline moieties of separate inhibitor molecules adjacent to each other, such that they form a staggered π-stacking interaction with a ring separation distance of 3.9 Å. The tolylamide carbonyl interacts with Zn2+ ligand H614 through two hydrogen bonded water molecules, as observed for compound 1. The tolyl moiety is accordingly oriented away from the enzyme surface and makes a van der Waals contact with the dimethylaniline methyl group of an inhibitor bound to an adjacent monomer in the crystal lattice.
The crystal structure of the HDAC6–3 complex determined at 2.20 Å resolution largely resembles that of the HDAC6–2 complex. All four monomers in the asymmetric unit uniformly adopt monodentate hydroxamate-Zn2+ coordination geometry (Figure 2c, Table 1). The phenyl linker resides in the aromatic crevice and is 3.3 Å and 4.0 Å away from the phenyl groups of F583 and F643, respectively. The peptoid carbonyl is oriented away from the protein surface and out toward bulk solvent. As in the HDAC6–2 complex, the dimethylaniline group resides in the L1 loop pocket. The dimethylaniline groups in adjacent monomers form staggered π-stacking interactions with a ring separation of 3.8 Å. The benzylamide carbonyl engages in the same solvent-mediated interaction with Zn2+ ligand H614 as observed for the binding of compound 2. The benzyl moiety of compound 3 also makes a van der Waals contact with the dimethylaniline group of an inhibitor bound to an adjacent monomer in the crystal lattice.
The crystal structure of the HDAC6–4 (Bavarostat) complex determined at 1.98 Å resolution, containing only one monomer in the asymmetric unit, clearly reveals canonical bidentate hydroxamate-Zn2+ coordination geometry with Zn2+---O separations of 2.2 and 2.0 Å, respectively, for the hydroxamate C=O and N–O− groups (Figure 2d). The catalytic histidine residues, H573 and H574, form hydrogen bonds with the hydroxamate N–O− and NH groups (N573---O separation = 2.5 Å and N574---N separation = 2.8 Å). The Y745 phenol oxygen hydrogen bonds with the hydroxamate carbonyl (O---O separation = 2.4 Å). The 2-fluorophenyl linker is situated in the aromatic crevice such that the fluorine atom is 3.3 Å from the side-chain methylene group of S531, 3.1 Å from the Ca atom of G582, 3.6 Å from the side chain of F583, and 3.1 Å from the side chain of F643. The benzylic tertiary amine is pyramidalized such that the lone pair on the nitrogen is oriented away from the side chain of gatekeeper residue S531, which revises a prior prediction.31 Instead, this nitrogen is oriented toward bulk solvent. The adamantyl capping group is nestled in the L1 loop pocket.
Bulky capping groups bind in the L1 loop pocket.
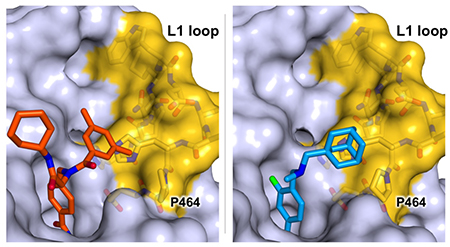
As the structures of HDAC6 complexed with compounds 1, 2, and 3 are compared, common features are evident for the binding of inhibitor capping groups. First, capping groups are similarly oriented, in that the smaller peptoid substituent (dime-thylphenyl for 1, dimethylaniline for 2 and 3) sits in the L1 loop pocket while the larger peptoid substituent is oriented away from the protein surface. Curiously, the crystal structure of isolated compound 1 (CSD ID: GADYOC)30 exhibits a cis peptoid conformation, whereas it exhibits a trans-peptoid conformation in its complex with HDAC6 (Figure 3a). The lower free energy difference between peptoid conformers presumably enables cis-trans isomerism to optimize the fit of the enzyme-inhibitor complex. The association of the smaller peptoid substituent with the L1 loop pocket thus appears to direct the peptoid conformation (Figure 3b). Notably, the adamantyl group of Bavarostat is similarly positioned in the L1 loop pocket (Figure 3c), as are cap substituents of other HDAC6-selective inhibitors.23,24,28 Taken together, these data indicate that the L1 loop pocket of HDAC6 provides a binding site for hydrophobic capping groups. Key residues defining this pocket (H463, P464, F583, and L712) are strictly conserved between human HDAC6, the actual drug target, and zebrafish HDAC6, the ortholog used for X-ray crystallographic studies. Notably, the L1 loop of HDAC6 is relatively rigid. Accordingly, the rigid, pre-formed nature of the L1 loop pocket presumably contributes to the selectivity of inhibitor binding to HDAC6 by minimizing the entropic cost of inhibitor binding site organization. Indeed, the binding of selective inhibitors to HDAC6 is generally characterized by entropy gain.26
Figure 3.

(a) Stereo superposition of crystal structures of free (light blue) and HDAC6-complexed (orange) conformations of compound 1. The phenyl linkers of these models have been aligned, highlighting conformational differences in the capping groups. (b) Binding of compound 1 to HDAC6 (light blue) with the dimethylphenyl group of the inhibitor cap packing against the L1 loop (H455–E465; yellow). (c) Binding of 4 (Bavarostat, blue) to HDAC6 viewed from a similar orientation to that shown in (b).
These results further support the hypothesis that interactions with the L1 loop are important for HDAC6–inhibitor selectivity. In the class I HDACs 1, 2, and 3, the L1 loop is shifted by approximately 1 Å relative to HDAC6, constricting the substrate binding pocket, and the L1 loop is buttressed when the enzyme is activated through the binding of corepressor and inositol tetraphosphate.32–34 This conformational difference would perturb the binding of a sterically bulky inhibitor capping group, thus rendering the inhibitor ineffective. We suggest that this effect accounts for the exceptional selectivity of Bavarostat for inhibition of HDAC6 relative to the class I HDACs 1, 2, and 3 (Figure 1).31
Surprisingly, inhibitor 3 binds with comparable affinity to HDAC6 compared with inhibitor 2, but 3 is much less selective than 2 with respect to inhibition of class I HDACs. We attribute this to the additional flexibility conferred by the benzylic substituent of 3 compared with the more rigid tolyl substituent of 2. The additional bulk and flexibility of 3 presumably enables binding to the more constricted active sites of class I HDACs, as exemplified for HDAC3 in Figure 4.
Figure 4.

Active site surfaces of (a) HDAC6 in its complex with inhibitor 1, and (b) HDAC3 (PDB 4A69) with inhibitor 1 modeled in the active site based on structural alignment with the HDAC6–1 complex. Zn2+ ions appear as grey spheres and metal coordination interactions are represented by solid yellow lines. The active site of HDAC6 is wider and more readily accommodates phenylhydroxamate inhibitors with bulky para-substituted substituents.
Capping group influence on hydroxamate-Zn2+ denticity.
Compared to other HDACs, the substrate binding groove of HDAC6 is wider (Figure 4). As a result, sterically bulky phenylhydroxamates can readily access the catalytic Zn2+ ion of HDAC6 but they cannot interact as easily with the catalytic Zn2+ ion of class I HDAC isozymes. However, the molecular features that distinguish monodentate versus bidentate hydroxamate-Zn2+ coordination in the HDAC6 active site are less clear.
Insight regarding this phenomenon can be acquired from studying the 20 hydroxamate based crystal structures of HDAC6–inhibitor complexes determined to date, including the 4 structures reported herein.22,23,24,26,35 Of these, 10 display canonical bidentate binding, 8 display monodentate binding, and 2 exhibit fractional occupancy of each conformation in one crystallographic monomer. Bidentate metal coordination is generally observed for inhibitors that possess either flexible aliphatic linkers or aromatic linkers lacking a capping group. Addition of bulky and/or rigid capping groups to a phenylhydroxamate inhibitor generally leads to monodentate hydroxamate-Zn2+ coordination, but the steric bulk must be located close to the phenylhydroxamate moiety. Specifically, the capping group must branch at the second atom away from the phenyl ring, and both substituents at the branch must be bulky. While Bavarostat 4 contains a bulky adamantyl cap as one substituent at the amino branch of the capping group, the second substituent is only a methyl group, which thus enables the phenylhydrox-amate to more closely approach the catalytic Zn2+ ion to achieve bidentate coordination geometry. Another feature that may enable binding flexibility for Bavarostat is the sp3-hybridized nitrogen atom of the tertiary amino group itself. In contrast with the planar sp2-hybridized peptoid nitrogen atoms of inhibitors 1–3, the tertiary amino nitrogen of Bavarostat can rapidly invert between two pyramidal stereoisomers as needed to optimize enzyme-inhibitor interactions.
Compounds 1–3 contain relatively rigid peptoid moieties with two bulky substituents branching at the second atom away from the para-substituted phenylhydroxamate. Apart from one of four monomers in the HDAC6–2 complex, these inhibitors bind with monodentate hydroxamate-Zn2+ coordination geometry. Taking into account multiple independent copies of the enzyme-inhibitor complex in the asymmetric units of all three crystal structures, the overall monodentate:bidentate ratio is 3.6:0.4. This suggests that, for compound 2, monodentate coordination is ~1.3 kcal/mol more stable as observed in the crystal structure. In comparison, the mixture of monodentate and bidentate conformers in the 1.05 Å-resolution structure of the HDAC6–TSA complex suggested that bidentate coordination was 0.5 kcal/mol more stable.24 Therefore, it appears that the steric bulk and rigidity of inhibitor capping groups modulate the equilibrium between bidentate and monodentate hydroxamate-Zn2+ coordination and thereby direct the metal ion coordination mode.
CONCLUSIONS
The high-resolution crystal structures of HDAC6 complexes with bulky peptoid inhibitors 1–3 as well as Bavarostat (4) highlight the importance of the L1 loop pocket in accommodating large hydrophobic groups. This pocket is largely defined by H463, P464, F583, and L712, and these residues are conserved between human HDAC6, the actual human drug target, and zebrafish HDAC6, the ortholog used for X-ray crystal structure determinations. Each inhibitor studied is a phenylhydroxamate containing a capping group that branches at the second atom away from the aromatic ring. Analysis of HDAC6-inhibitor complexes suggests that if both branching substituents are sterically bulky, then the inhibitor will bind with monodentate hydroxamate-Zn2+ coordination geometry; if only one substituent is bulky, or if there is no substituent at all (i.e., a simple phenylhydroxamate26), then the inhibitor will bind with bidentate hydroxamate-Zn2+ coordination geometry. Future studies will allow us to further clarify structure-affinity and selectivity relationships for inhibitor binding to HDAC6.
EXPERIMENTAL SECTION
Reagents.
Chemicals used in buffer and crystallization conditions were obtained from Fisher, Millipore Sigma, or Hampton Research and used without further purification. Inhibitors 1 and 2 were synthesized as reported.30 Bavarostat was synthesized as described.31 Inhibitor 3 was synthesized through a similar approach as used for the synthesis of inhibitors 1 and 2, except that benzyl isocyanide was used as the isocyanide component and 4-(dimethylamino)benzoic acid was used as the carboxylic acid component in the Ugi four-component reaction. Product purity was confirmed to be greater than 95% based on RP-HPLC analysis. The compound characterization data of compound 3 are summarized below.
N-(2-(Benzylamino)-2-oxoethyl)-4-(dimethylamino)-N-(4-(hydroxycarbamoyl)benzyl)benzamide (3).
Colorless solid; mp: 193 °C; purity: 98.2 %. 1H NMR (300 MHz, DMSO-d6): δ = 11.20 (s, 1H), 9.03 (s, 1H), 8.60–8.32 (m, 1H), 7.83–7.66 (m, 2H), 7.46–7.14 (m, 9H), 6.75–6.55 (m, 2H), 4.67 (s, 2H), 4.30 (d, J = 5.8 Hz, 2 H), 3.90 (s, 2H), 2.93 (s, 6H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 171.7, 168.15, 164.0, 151.2, 140.8, 139.2, 131.7, 128.6, 128.3, 127.3, 127.15, 127.0, 126.82, 126.86, 121.9, 110.95, 42.1, 39.7 ppm. Anal. Calcd. for C26H29N4O4: 461.2183 [M+H]+, found: 461.2182.
Inhibitory activities.
The in vitro inhibitory activities (IC50 values) of compounds 1, 2, and 4 (Bavarostat) against HDAC isozymes have been previously reported.30,31 The in vitro inhibitory activities of compound 3 against HDAC6 and HDAC1 were measured using a previously described protocol.35 OptiPlate-96 black microplates (Perkin Elmer) were used with an assay volume of 50 μL. A total of 5 μL 3 or control, diluted in assay buffer [50 mM Tris-HCl (pH 8.0), 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.1 mg/mL BSA], were incubated with 35 μL of the fluorogenic substrate ZMAL (ZLys(Ac)-AMC)36 (21.43 μM in assay buffer) and 10 μL of human recombinant HDAC1 (BPS Bioscience, Catalog #50051) or HDAC6 (BPS Bioscience, Catalog #50006) at 37 °C. After an incubation time of 90 min, 50 μL of 0.4 mg/mL trypsin in trypsin buffer [50 mM Tris-HCl (pH 8.0), 100 mM NaCl] were added, followed by further incubation at 37 °C for 30 min. Fluorescence was measured with an excitation wavelength of 355 nm and an emission wavelength of 460 nm using a Fluoroskan Ascent microplate reader (Thermo Scientific). Compound 3 was evaluated in duplicate in two independent experiments.
Crystallization.
Catalytic domain 2 of HDAC6 from Danio rerio (henceforth simply “HDAC6”) was recombinantly expressed in the E. coli BL21 (DE3) strain using the His6-MBPTEV-HDAC6-pET28a(+) vector and purified as previously described.22,24 All HDAC6-inhibitor complexes were crystallized in sitting drops by the vapor diffusion method at 4°C.
For cocrystallization of the HDAC6–1 complex, a 5 μL drop of protein solution [5 mg/mL HDAC6, 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1 mM tris(2-carboxyethyl)phosphine (TCEP), saturated 1 (approximately 0.5 mM), and 5% DMSO (v/v)] was added to 5 μL of precipitant solution [400 mM NaF and 15% polyethylene glycol (PEG) 3,350 (w/v)] and equilibrated against 500 μL of precipitant solution. Crystals appeared within 2 days.
For cocrystallization of the HDAC6–2 complex, a 350 nL drop of protein solution [10 mg/mL HDAC6, 50 mM HEPES (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1 mM TCEP, saturated 2 (approximately 1.0 mM), and 5% DMSO (v/v)] was added to 350 nL of precipitant solution [2% tacsimate (pH 6.0; w/v), 0.1 M BisTris (pH 6.5), and 20% PEG 3,350 (w/v)] and equilibrated against 80 μL of precipitant solution. Crystals appeared within 2 days.
For cocrystallization of the HDAC6–3 complex, a 350 nL drop of protein solution [10 mg/mL HDAC6, 50 mM HEPES (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1 mM TCEP, saturated 3 (approximately 0.5 mM), and 5% DMSO (v/v)] was added to 350 nL of precipitant solution [0.2 M ammonium tartrate dibasic pH 7.0 and 20% PEG 3,350 (w/v)] and equilibrated against 80 μL of precipitant solution. Crystals appeared within 2 days.
For cocrystallization of the HDAC6–4 (Bavarostat) complex, a 350 nL drop of protein solution [10 mg/mL HDAC6, 50 mM HEPES (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1 mM TCEP, saturated Bavarostat (approximately 0.5 mM), and 5% DMSO (v/v)] was added to 350 nL of precipitant solution [200 mM L-proline, 100 mM HEPES (pH 7.5), and 24% PEG 1,500 (w/v)] and equilibrated against 80 μL of precipitant solution. Crystals appeared within 2 days.
All crystals were soaked in a cryoprotectant solution containing mother liquor supplemented with 20% ethylene glycol prior to flash-cooling in liquid nitrogen.
Data collection and Structure Determination.
X-ray diffraction data for HDAC6 complexes with 1 and 2 were collected on NE-CAT beamline 24-ID-E at the Advanced Photon Source, Argonne National Lab. Diffraction data were collected from crystals of the HDAC6–3 and HDAC6–4 complexes on beamline 9–2 at the Stanford Synchrotron Radiation Laboratory (SSRL), Stanford University. Data were indexed and integrated using iMosflm37 and data scaling was carried out using Aimless in the CCP4 program suite.38
The atomic coordinates of unliganded HDAC6 (PDB 5EEM)22 were used as a search model to phase each crystal structure by molecular replacement using the program Phaser.39 Atomic models were built and manipulated using the graphics program Coot40 and crystallographic refinement was executed using Phenix.41 Inhibitor molecules were built into well-defined electron density in later rounds of refinement. The quality of each model was assessed using MolProbity42 and PROCHECK.43 Data collection and refinement statistics are recorded in Table S1, Supporting Information.
Supplementary Material
ACKNOWLEDGMENT
Experimental support by Andrea Schöler is gratefully acknowledged. Additionally, we thank Dr. R. Rajashankar and synchrotron beamline staff at the Northeastern Collaborative Access Team beamlines (supported by Grants P1 GM103403 and S10 RR02905) at the Advanced Photon Source (APS), a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory (ANL) under Contract DE-AC02–06CH11357. We also thank Dr. S. Russi at the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, a facility supported by the DOE Office of Science, Office of Basic Energy Sciences, under contract DE-ACO2–76SF00515. The Structural Molecular Biology Program at SSRL is supported by the DOE Office of Biological and Environmental Research, and by the National Institute of General Medical Sciences (Grant P41GM103393).
Funding Sources
This research was supported by US National Institutes of Health (NIH) grant GM49758 to D.W.C. and Deutsche Forschungsgemeinschaft (DFG) grant HA 7783/1–1 to F.K.H. N.J.P. received financial support from the NIH through Chemistry-Biology Interface Training Grant T32 GM071339.
Abbreviations
- DMSO
dimethylsulfoxide
- HDAC
histone deacetylase
- HDAC6 CD2, or HDAC6
histone deacetylase 6, catalytic domain 2
- HEPES
4-(2-hydroxyethyl)-1-piperazineethane-sulfonic acid
- PEG
polyethylene glycol
- TCEP
tris(2-carboxyethyl)phosphine
- HPOB
N-hydroxy-4-(2-[(2-hydroxyethyl)(phenyl)amino]-2-oxoethyl)benzamide
- HPB
N-hydroxy-4-[(N(2-hydroxyethyl)-2-phenylacetamido)methyl)-benzamide)]
ASSOCIATED CONTENT
SUPPORTING INFORMATION
Table S1, crystallographic data collection and refinement statistics.
Accession codes
The atomic coordinates and crystallographic structure factors of HDAC6 complexes with inhibitors 1, 2, 3, and 4 have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 6DVL, 6DVM, 6DVN, and 6DVO, respectively. Authors will release the atomic coordinates upon article publication.
N.J.P., J.D.O., D.D., T.K., F.K.H., and D.W.C. declare no competing financial interests. J.M.H. is a co-founder of Eikonizo Therapeutics, Inc., which has licensed intellectual property related to Bavarostat.
REFERENCES
- 1.Kouzarides T Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000, 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 2010, 327:1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M The growing landscape of lysine acetylation links metabolism and cell signaling. Nat. Rev. Mol. Cell Biol 2014, 15, 536–550. [DOI] [PubMed] [Google Scholar]
- 5.Gregoretti IV, Lee YM, Goodson HV Molecular evolution of the histone deacetylase family: functional implication of phylogenetic analysis. J. Mol. Biol 2004, 338, 17–31. [DOI] [PubMed] [Google Scholar]
- 6.Lombardi PM, Cole KE, Dowling DP, and Christianson DW Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol 2011, 21, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernick M, Fierke CA Zinc hydrolases: the mechanisms of zinc-dependent deacetylases. Arch. Biochem. Biophys 2005, 433, 71–84. [DOI] [PubMed] [Google Scholar]
- 8.López JE, Sullivan ED, Fierke CA Metal-dependent deacetylases: cancer and epigenetic regulators. ACS Chem. Biol 2016, 11, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan H, Marmorstein R Structural basis for sirtuin activity and inhibition. J. Biol. Chem 2012, 287, 42428–42435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dokmanovic M, Clarke C, Marks PA Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res 2007, 5, 981–989. [DOI] [PubMed] [Google Scholar]
- 11.Gupta P, Reid RC, Iyer A, Sweet MJ, Fairlie DP Towards isozyme-selective HDAC inhibitors for interrogating disease. Curr. Top. Med. Chem 2012, 12, 1479–1499. [DOI] [PubMed] [Google Scholar]
- 12.Falkenberg KJ, Johnstone RW Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Disc 2014, 13, 673–691. [DOI] [PubMed] [Google Scholar]
- 13.Ganai SA, Ramadoss M, Mahadevan V Histone deacetylase (HDAC) inhibitors – emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr. Neuropharmacol 2016, 14, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grozinger CM, Hassig CA, Schreiber SL Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verdel A, Khochbin S Identification of a new family of higher eukaryotic histone deacetylases: coordinate expression of differentiation-dependent chromatin modifiers. J. Biol. Chem 1999, 274, 2440–2445. [DOI] [PubMed] [Google Scholar]
- 16.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP HDAC6 is a microtubule-associated deacetylase. Nature 2002. 417,455–458. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J 2003, 22, 1168–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aldana-Masangkay GI, Sakamoto KM The role of HDAC6 in cancer. J. Biomed. Biotechnol 2011, 875824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seidel C, Schnekenburger M, Dicato M, Diederich M Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [DOI] [PubMed] [Google Scholar]
- 20.Yang PH, Zhang L, Zhang YJ, Zhang J, Xu WF HDAC6: physiological function and its selective inhibitors for cancer treatment. Drug Disc. Ther 2013, 7, 233–242. [DOI] [PubMed] [Google Scholar]
- 21.Trüe O, Matthias P Interplay between histone deacetylases and autophagy – from cancer therapy to neurodegeneration. Immunol. Cell Biol 2012, 90, 78–84. [DOI] [PubMed] [Google Scholar]
- 22.Hai Y, Christianson DW Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol 2016, 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyake Y, Keusch JJ, Wang L, Saito M, Hess D, Wang X, Melancon BJ, Helquist P, Gut H, Matthias P Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol 2016, 12, 748–754. [DOI] [PubMed] [Google Scholar]
- 24.Porter NJ, Mahendran A, Breslow R, Christianson DW Unusual zinc binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. USA, 2017, 114, 13459–13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagner FF, Olson DE, Gale JP, Kaya T, Weïwer M, Aidoud N, Thomas M, Davoine EL, Lemercier BC, Zhang Y-L, Holson EB Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J. Med. Chem 2013, 56, 1772–1776. [DOI] [PubMed] [Google Scholar]
- 26.Porter NJ, Wagner FF, Christianson DW Entropy as a driver of selectivity for inhibitor binding to histone deacetylase 6. Biochemistry 2018, 57, DOI: 10.1021/acs.biochem.8b00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JH, Mahendran A, Yao Y, Ngo L, Venta-Perez G, Choy ML, Kim N, Ham WS, Breslow R, Marks PA Development of a histone deacetylase 6 inhibitor and its biological effects. Proc. Natl. Acad. Sci. USA 2013, 110, 15704–15709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JH, Yao Y, Mahendran A, Ngo L, Venta-Perez G, Choy ML, Breslow R, Marks PA Creation of a histone deacetylase 6 inhibitor and its biological effects. Proc. Natl. Acad. Sci. USA 2015, 112, 12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diedrich D, Hamacher A, Gertzen CG, Alves Avelar LA, Reiss GJ, Kurz T, Gohlke H, Kassack MU, Hansen FK Rational design and diversity-oriented synthesis of peptoid-based selective HDAC6 inhibitors. Chem. Commun 2016, 52, 3219–3222. [DOI] [PubMed] [Google Scholar]
- 31.Strebl MG, Campbell AJ, Zhao W-N, Schroeder FA, Riley MM, Chindavong PS, Morin TM, Haggarty SJ, Wagner FF, Ritter T, Hooker JM HDAC6 brain mapping with [18F]Bavarostat enabled by a Ru-mediated deoxyfluorination. ACS Cent. Sci 2017, 3, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watson PJ, Millard CJ, Riley AM, Robertson NS, Wright LC, Godage HY, Cowley SM, Jamieson AG, Potter BV, Schwabe JWR Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat. Commun 2016, 7, 11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, De Jong R, O’Connell S, Grimshaw CE, Naver M, Gangloff AR Exploration of the HDAC2 foot pocket: synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett 2010, 20, 3142–3145. [DOI] [PubMed] [Google Scholar]
- 34.Watson PJ, Fairall L, Santos GM, Schwabe JWR Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012, 481, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mackwitz MKW, Hamacher A, Osko JD, Held J, Schöler A, Christianson DW, Kassack MU, Hansen FK Multicomponent synthesis and binding mode of imidazo[1,2-a]pyridine-capped selective HDAC6 inhibitors. Org. Lett 2018, 20, 3255–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heltweg B, Dequiedt F, Verdin E, Jung M Nonisotopic substrate for assaying both human zinc and NAD+-dependent histone deacetylases. Anal. Biochem 2003,319, 42–48. [DOI] [PubMed] [Google Scholar]
- 37.Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ Phaser crystallographic software. J. Appl. Cryst 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P, Lohkamp B, Scott WG, Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwillinger TC, Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen VB, Arendal WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystal-logr 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laskowski RA, MacArthur MW, Moss DS, Thornton JM PROCHECK: A program to check the stereochemical quality of protein structures. J Appl. Cryst 1993, 26, 283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.