

In a large, 10-year, population-based study of adolescents and adults with cystic fibrosis in the United States, we demonstrated that chronic Pseudomonas aeruginosa infection rates have decreased significantly, with no clear concomitant increase in other common airway pathogens.
Keywords: cystic fibrosis, epidemiology, Pseudomonas aeruginosa, population-based study, airway infections
Abstract
Background
Chronic Pseudomonas aeruginosa lung infection is associated with significant morbidity and mortality in cystic fibrosis (CF). It is not known whether recent advances in care have affected the rates of chronic infection. We aimed to determine if the rates of developing new chronic P. aeruginosa infection among adolescents and adults with CF significantly changed over time.
Methods
The cohort consisted of individuals with CF followed in the Cystic Fibrosis Foundation Patient Registry aged ≥13 years without chronic P. aeruginosa at baseline. Multivariable regression models accounting for within-patient correlation were used to assess the change in rate of developing chronic P. aeruginosa infection between 2003 and 2012.
Results
A total of 15504 individuals were followed for a median of 5 (interquartile range, 2–9) years. The annual rates of developing new chronic P. aeruginosa decreased from 14.3% in 2003 to 6.4% in 2012. After adjusting for potential confounders, relative risk (RR) of developing chronic P. aeruginosa infection decreased significantly over time compared to 2003 (P value test of trend < .001). Compared with 2003, the RR of developing chronic P. aeruginosa infection in 2012 was 0.33 (95% confidence interval, 0.30–0.37). No significant increases in risk of chronic infections with other major CF bacterial pathogens relative to 2003 were identified.
Conclusions
Among individuals with CF, a significant decrease in the risk and rates of developing chronic P. aeruginosa infection between 2003 and 2012 was observed. Whether this decline results in changes in clinical outcomes warrants further exploration.
Pseudomonas aeruginosa is considered the archetypal pathogen and results in chronic airway infections in up to 60%–70% of adults with cystic fibrosis (CF) [1–7]. Upon infection with P. aeruginosa, the clinical course typically consists of conversion to a mucoid phenotype (occurring at a median duration of 1–3 years) [8, 9], leading to greater frequency of exacerbations, increased treatment burden, and worsening clinical status [10–15]. Chronic P. aeruginosa infection in CF is associated with greater morbidity and mortality irrespective of lung function [13, 16]. Pseudomonas aeruginosa can be eradicated in early infection but, once established, can rarely be eliminated from the airway [17]. This feature of infection is likely due to the inherent abnormalities of airway clearance and the organism’s ability to form bacterial communities largely resistant to antibiotics [18, 19].
Advances in the diagnosis and management of CF are leading to significant improvements in survival, with increasing proportions of individuals living well into adulthood, including those with severe lung disease [5, 20]. Notably, 2015 US registry reports demonstrate that more than 50% of individuals with CF are aged >18 years [5]. In parallel, mortality decreased by 1.8% per year between 2000 and 2010. If this observed rate continues, individuals born with CF in 2010 are predicted to have life spans greater than 50 years [20]. This benchmark has already been surpassed in other countries such as Canada [1]. With these marked demographic shifts in CF, we have yet to understand the effects that these advances in care have had in the older CF population on the rate of developing chronic P. aeruginosa infections. A single-center study showed that the prevalence of chronic and mucoid P. aeruginosa decreased significantly between 2002 and 2012, but this effect was no longer significant after adjusting for confounders [21]. Thus, we aimed to determine if there has been a significant change in the rates of developing chronic P. aeruginosa infection among adolescents and adults in the United States over this same time period. We hypothesized that the rates of developing chronic P. aeruginosa infection have decreased in the CF populations over time.
METHODS
Study Population
The Cystic Fibrosis Foundation Patient Registry (CFFPR), first developed in the 1960s, contains prospectively collected demographic and clinical data on consenting individuals with CF who are receiving care at CF Foundation–accredited centers (>120) in the United States [5, 22]. In 2012, more than 27000 individuals provided data to the registry, representing approximately 81%–84% of the US CF population [5, 22]. We accessed data from the CFFPR for the period from January 2001 to December 2012 for purposes of this study. All individuals aged ≥13 years were considered for inclusion as this reflects the median acquisition age of mucoid P. aeruginosa and has been highly associated with chronic P. aeruginosa infection [11]. The population was dynamic in that individuals could enter the cohort throughout the study period if they reached age-inclusion criteria and/or had a new diagnosis of CF at ≥13 years. Alternatively, individuals could leave the population due to death, leaving the registry, or meeting exclusion criteria. Individuals were excluded from further observation after meeting the definition of chronic P. aeruginosa infection or following bilateral lung transplantation. Additionally, those with no clinical encounters in the study period were excluded. The study was approved by the University of Washington Institutional Review Board (45798).
Study Design
A retrospective, cohort study was conducted between 2003 through 2012 to examine the annual rate of developing chronic P. aeruginosa infection. The primary predictor of interest was time designated and categorized by calendar year. CFFPR encounter data were summarized quarterly for each calendar year. In quarters with more than 1 clinical encounter, the quarter was considered positive for P. aeruginosa if any single encounter had a positive respiratory culture (sputum, oropharyngeal, or bronchoalveolar lavage) for the organism. Microbiologic testing was performed locally for patients in accordance with Cystic Fibrosis Foundation consensus guidelines [23].
The primary outcome of interest was the development of chronic P. aeruginosa infection. Each individual entering the cohort was assigned an initial status of P. aeruginosa infection on the basis of data using a 2-year roll-in screening period (ie, for 2003, data from 2001 to 2003 were used). Initial P. aeruginosa infection status was classified as never (no positive cultures), intermittent (at least 1 positive culture but not meeting chronic definition), or chronic infection (>50% positive cultures requiring a minimum of 3 cultures in 8 quarters) using a modified definition of chronic infection originally proposed by Lee et al [24]. As culture frequency is variable and low in the CFFPR, to decrease the risk of misclassification, a modified definition that was previously developed and since tested was used [21, 25]. Individuals who met the definition for chronic P. aeruginosa infection in the screening period were excluded from the analysis cohort. Those who met the never or intermittent status initially were followed with P. aeruginosa status assessment at each subsequent quarter during the observational period to identify the development of chronic infection using the same definitions.
As a secondary outcome, we assessed the development of mucoid P. aeruginosa infection over the study period as mucoidy has been previously demonstrated to be correlated with chronicity [26]. Mucoidy was defined as having at least 1 positive respiratory culture with a mucoid P. aeruginosa phenotype in a calendar year. We also assessed for the development of new chronic infection with other CF bacterial pathogens using the primary definition as for P. aeruginosa. The organisms examined included Burkholderia cepacia complex, Achromobacter xylosoxidans, Stenotrophomonas maltophilia, and methicillin-resistant Staphylococcus aureus (MRSA).
Statistical Analyses
Baseline demographics and clinical status were defined as the time of entry into the observed cohort. Descriptive statistics were used to summarize the cohort data. Continuous variables were reported as medians with interquartile ranges (IQR). Discrete variables were reported as proportions. The development of chronic P. aeruginosa infection was summarized by year; once an individual developed chronic infection (first event), he/she was excluded from further follow-up in a given analysis. Time-independent variables included age at diagnosis (years), age at entry, sex (male/female), pancreatic insufficiency (PI; yes/no), cystic fibrosis transmembrane conductance regulator (CFTR) functional class (I-III/IV-V/unclassified/unknown) [27], baseline lung function (forced expiratory volume in 1 second predicted [FEV1%]), and CF-related diabetes (CFRD). CFRD was defined as being present if an individual with CF was prescribed insulin; we used this conservative definition given the complexities in CFRD diagnosis. PI was defined as present by receipt of pancreatic enzyme replacement therapy. Multivariable generalized estimating equations (GEEs) using a Poisson distribution with log link, an independent working correlation structure, and robust variance estimation were constructed for each of the primary and secondary outcomes to account for repeated measures within a longitudinal dataset [18]. The GEE model was assumed to afford a valid inference for the outcomes as the population sample size to be used was large and annual event rates were expected to be low (approximately 10% or less) [28]. The multivariable models incorporated a priori confounders as above to determine the relative risk (RR) of developing chronic infection by calendar year relative to 2003. Additionally, for the primary outcome, we constructed 2 additional multivariable GEE models stratified by age (13–18/19–35/>35 years) and by CFTR functional class given their association with differential clinical status [29] (I-III/IV-V/unclassified/unknown), respectively, to determine if there were distinct changes in chronic P. aeruginosa infection risk over time.
As a sensitivity analysis to the primary outcome, a separate multivariable GEE model was developed to determine the RR of developing chronic P. aeruginosa infection in a cohort restricted to individuals who aged into the cohort at 13 years (less likely to have chronic infection at entry). All hypotheses and outcomes were predetermined, and a 2-tailed P value < .05 was considered statistically significant.
RESULTS
Patient Population
A total of 15504 individuals aged ≥13 years were analyzed for development of chronic P. aeruginosa infection over the study period (Figure 1). The median period of observation was 5 (IQR, 2–9) years. Baseline demographics for this cohort are summarized in Table 1. Compared to the 9192 (37%) individuals excluded from the observation group due to meeting the definition of chronic P. aeruginosa during the screening period, individuals free of chronic infection at the start of the observation period were younger, had better lung function, and a greater proportion had residual CFTR function (classes IV–VI). Changes within the cohort between 2003 and 2012 are summarized in Supplementary Table 1. Comparing the 2003 to 2012 analytic cohorts, mean lung function (FEV1%) improved from 73.2% to 81.5% and CFRD prevalence increased from 10.5% to 19.6%, respectively.
Figure 1.

CONSORT flow diagram. Abbreviations: CF, cystic fibrosis; CFFPR, Cystic Fibrosis Foundation Patient Registry.
Table 1.
Comparison of Baseline Characteristics Between Individuals With and Without Chronic Pseudomonas aeruginosa Infection
| Demographics | Free of Chronic Infection | Chronic Infection at Baseline | ||
|---|---|---|---|---|
| N = 15504 | N = 9192 | |||
| Median age, years (IQR) | 15.6 | (13.6–24.5) | 20.0 | (13.0–29.0) |
| Median age of diagnosis, years (IQR) | 0.86 | (0.21–7.17) | 0.59 | (0.17–3.39) |
| Gender, female N (%) | 7096 | (45.8) | 4588 | (49.9) |
| Race, white N (%) | 14513 | (93.6) | 8784 | (95.6) |
| Ethnicity, Hispanic N (%) | 843 | (5.4) | 487 | (5.3) |
| f508dela status | ||||
| Homozygous, N (%) | 6166 | (39.8) | 4207 | (50.8) |
| Heterozygous, N (%) | 5876 | (37.9) | 3133 | (37.8) |
| Other/Unknown, N (%) | 3462 | (22.3) | 1635 | (10.3) |
| CFTR Functional classificationb | ||||
| Minimal, N (%) | 9301 | (60.0) | 6207 | (67.5) |
| Residual, N (%) | 1964 | (12.7) | 418 | (4.6) |
| Other/Unknown, N (%) | 4239 | (27.3) | 2567 | (27.9) |
| Forced expiratory volume in 1 second, % predicted, mean (standard deviation) | 81.9 | (24.1) | 67.8 | (25.7) |
| Comorbidities | ||||
| Cystic fibrosis–related diabetes,c N (%) | 1165 | (10.2) | 1741 | (19.3) |
| Pancreatic insufficiency,d N (%) | 10018 | (92.8) | 8227 | (89.5) |
| Microbiology | ||||
| Pseudomonas aeruginosa, N (%) | 4392 | (41.9) | 7229 | (91.3) |
| Mucoid Pseudomonas phenotype, N (%) | 2474 | (56.3) | 5797 | (80.2) |
| Staphylococcus aureus, N (%) | 7455 | (71.2) | 4207 | (53.0) |
| Methicillin-resistant Staphylococcus aureus, N (%) | 6264 | (84.0) | 3199 | (76.0) |
| Methicillin-resistant Staphylococcus aureus, N (%) | 1947 | (26.1) | 1422 | (33.8) |
| Haemophilus influenzae, N (%) | 1656 | (15.8) | 592 | (7.5) |
| Burkholderia sp.,e N (%) | 422 | (4.0) | 237 | (3.0) |
| Alcaligenes xylosoxidans,e N (%) | 747 | (7.1) | 562 | (7.1) |
| Stenotrophomonas,e N (%) | 1628 | (15.5) | 859 | (10.8) |
Abbreviation: CFTR, cystic fibrosis transmembrane conductance regulator; IQR, interquartile range.
aHomozygous: both alleles containing the delta F508 mutation; Heterozygous: one allele containing the delta F508 mutation; other/unknown: alleles containing mutations that are different or unknown.
bMinimal: both alleles containing mutations resulting in minimal CFTR function (class 1, 2, or 3); residual: at least 1 allele containing mutation resulting in partial CFTR function (class 4 or 5); unclassified: at least 1 allele with unknown CFTR function and if other allele function known, mutation resulting in minimal CFTR function.
cCystic fibrosis–related diabetes: use of insulin.
dPancreatic insufficiency: use of pancreatic enzymes.
eBurkholderia: Burkholderia species, Achromobacter: Alcaligenes xylosoxidans, Stenotrophomonas: Stenotrophomonas Maltophilia.
Primary Outcome—Development of Chronic P. aeruginosa Infection
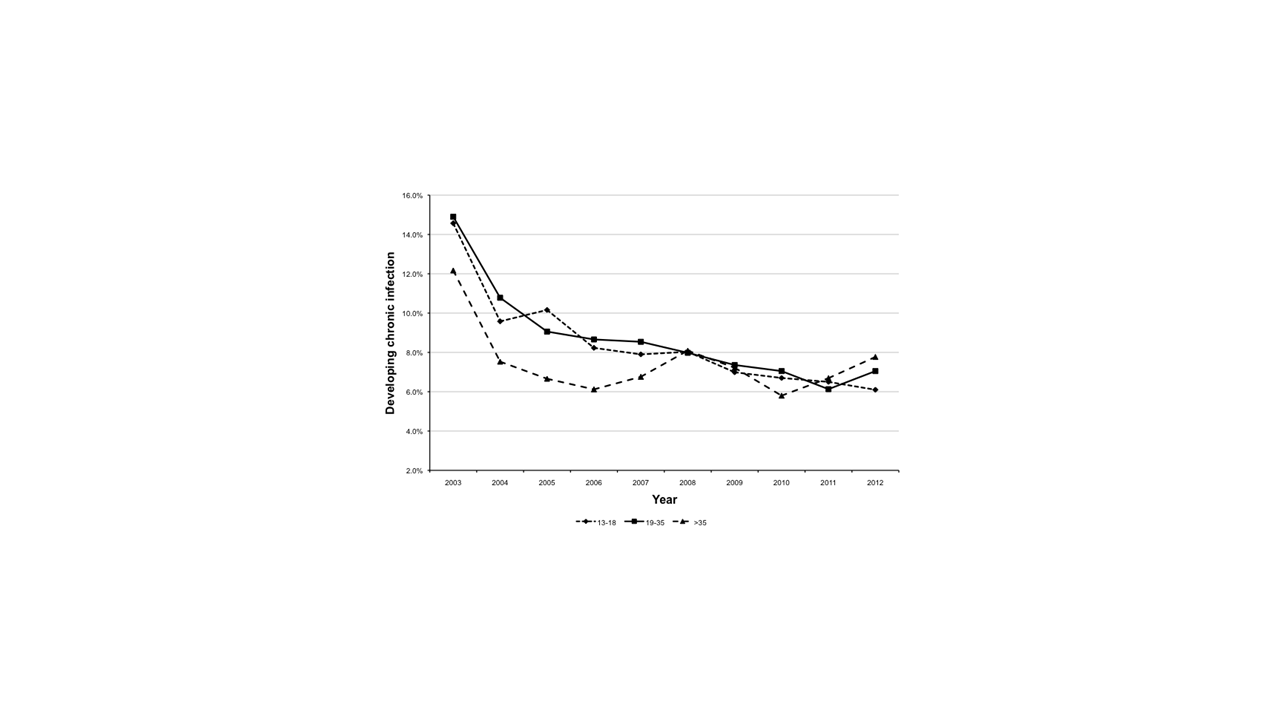
The annual rate of developing chronic P. aeruginosa infection among the cohort was 14.3% (N = 1347) in 2003 and decreased to 6.4% (N = 522) in 2012. There was a significant decrease in the development of chronic P. aeruginosa infection throughout the observation period (P value test of trend < .001). Compared to 2003, the relative risk of developing chronic P. aeruginosa infection in 2012 was 0.45 (95% confidence interval [CI], 0.41–0.49), with the intervening years also having a significant decrease in incidence (Table 2). After adjusting for age, age at diagnosis, sex, CFTR functional class, baseline FEV1%, CFRD, and PI, the decrease in RR of developing chronic P. aeruginosa infection remained significant (P value test of trend < .001; Table 2). The decrease in development of new chronic P. aeruginosa infection was comparable among age groups (13–18, 19–35, >35) and CFTR functional groups (I-III, IV-V, other) (Supplementary Figures 1 and 2).
Table 2.
Unadjusted and Adjusted Relative Risk of Developing Chronic Pseudomonas aeruginosa Infection Compared to 2003
| Year | Unadjusted | Adjusteda | ||
|---|---|---|---|---|
| RRb | (95% CI) | RRc | (95% CI) | |
| 2003 | - | - | - | - |
| 2004 | 0.68 | (0.63–0.74) | 0.70 | (0.65–0.76) |
| 2005 | 0.65 | (0.60–0.70) | 0.62 | (0.58–0.68) |
| 2006 | 0.56 | (0.52–0.62) | 0.51 | (0.46–0.56) |
| 2007 | 0.56 | (0.51–0.61) | 0.50 | (0.45–0.55) |
| 2008 | 0.56 | (0.51–0.61) | 0.49 | (0.45–0.55) |
| 2009 | 0.50 | (0.45–0.55) | 0.41 | (0.37–0.46) |
| 2010 | 0.47 | (0.43–0.51) | 0.41 | (0.37–0.45) |
| 2011 | 0.45 | (0.41–0.50) | 0.38 | (0.34–0.43) |
| 2012 | 0.45 | (0.41–0.49) | 0.35 | (0.31–0.39) |
Abbreviations: CI, confidence interval; RR, relative risk.
aAdjusted for age at entry, age at diagnosis, gender, cystic fibrosis transmembrane conductance regulator functional class, baseline forced expiratory volume in 1 second, cystic fibrosis–related diabetes at entry, and pancreatic insufficiency at entry.
bTrend test P < .001.
c Trend test P < .001.
Secondary Outcomes
Using the mucoidy definition for chronic P. aeruginosa, the annual rate was 10.3% in 2003 (N = 786) and decreased to 6.5% (N = 505), in keeping with the primary outcome. This was a significant trend even after adjusting for confounders as with the primary outcome (P value test of trend < .001). When other CF bacterial pathogens of B. cepacia complex, A. xylosoxidans, S. maltophilia, and MRSA were examined for RR of new chronic infection compared to 2003, an increase in parallel to the decrease observed with P. aeruginosa was not observed. Conversely, all organisms with the exception of MRSA had an overall decreased RR of developing new chronic infections over time (Figure 2).
Figure 2.

Development of chronic infections with other cystic fibrosis pathogens compared to 2003 in study period. Abbreviations: MRSA, methicillin-resistant Staphylococcus aureus.
Sensitivity Analysis
As a sensitivity analysis, the change in rates of developing chronic P. aeruginosa during the observation period was compared only among individuals who aged into the cohort at 13 years (n = 6789). Over the 10-year observation period, there was a significant decrease in the rate of developing chronic P. aeruginosa infection (P value test of trend < .001) (Figure 3).
Figure 3.

Risk of chronic Pseudomonas aeruginosa among individuals entering the cohort at age 13 years by year relative to 2003 adjusting for age, age at diagnosis, gender, cystic fibrosis transmembrane conductance regulator functional class, baseline forced expiratory volume in 1 second, predicted, cystic fibrosis–related diabetes, and pancreatic insufficiency. Abbreviations: CI, confidence interval; RR, relative risk.
DISCUSSION
Among a large cohort of adolescents and adults with CF, we demonstrated a significant decrease in the risk of developing chronic P. aeruginosa infection between 2003 through 2012, even with adjustment for potential confounders. When we used an alternate definition of mucoidy for P. aeruginosa chronic infection, we demonstrated a similar significant decrease over time. There appeared to be no concurrent increase in other CF bacterial pathogens over the same period, but this warrants further study.
Recent observational studies have suggested that the prevalence of P. aeruginosa infection is decreasing in both adolescents and adults [7, 30, 31]. Both chronic infection and the mucoid phenotype of P. aeruginosa are associated with increased morbidity and mortality when compared to those without P. aeruginosa infection or those with nonchronic or nonmucoid infections [10]. Our study is one of the first contemporary evaluations of new chronic infections (as a more sensitive measure compared to prevalent infections) of P. aeruginosa in a large CF population while accounting for within-person correlations and confounders. In a prior study in which a small cohort of adults from a single center was used, we identified a trend toward decreasing prevalence of chronic P. aeruginosa infection, although it did not reach our predetermined significance level [21]. Other regional and national studies have also demonstrated a decline in the frequency of chronic P. aeruginosa infection over time, though these were both in earlier cohorts and applied alternative definitions of chronic infection [32, 33].
The observed decreased incidence in P. aeruginosa infections may be attributable to several changes. As noted, the study period occurred when inhaled antibiotics were frequently used. In the setting of newly acquired P. aeruginosa infection, inhaled anti-pseudomonal antibiotics can eradicate it from the subsequent respiratory culture about 75% of the time [24, 25, 28, 34–36], and those with successful eradication have delayed reacquisition [37, 38]. In the study by Mayer-Hamblett et al, those who remained free of P. aeruginosa for more than 12 months after eradication therapy had a significantly reduced risk of developing chronic P. aeruginosa infection (hazard ratio, 0.26; 95% CI, 0.17–0.40) [37]. Although the majority of P. aeruginosa eradication studies targeted the pediatric CF population, a recent study demonstrated that eradication was successful in adults [39]. Concordantly, we noted that a significant decrease in the incidence of developing chronic P. aeruginosa occurred during the observation period for both the 18–35 and >35 age groups.
During this study period, an increased proportion of individuals with residual function CFTR mutations were diagnosed and included in the registry data. This likely occurred because of advances in CF diagnostics resulting in more individuals being diagnosed as adults and may have contributed to lower incidence of chronic P. aeruginosa infection [40]. Further, the introduction of CFTR modulator agents, such as ivacaftor (not used during the study period), which improves CFTR function, may have a similar result. Using recent data from the G551D observational cohort, Heltshe et al demonstrated significantly reduced odds of isolating P. aeruginosa from the airways and a 23% reduction in mucoid P. aeruginosa following ivacaftor therapy compared to culture data prior to initiation (odds ratio, 0.65; P < .001) [41]. However, when we assessed new chronic P. aeruginosa infection among the CFTR functional groups in our study cohort, similar decreases were noted across groups, arguing against the theory that changes in the CF genetic composition over time significantly affected our results.
Although we conducted a large population-based study of incident chronic P. aeruginosa infection in CF, we have several limitations to consider. A number of definitions for chronic P. aeruginosa infection have been evaluated in the literature [24, 42, 43], most using the original or modified form of the Leeds criteria. As the average number of months with available culture data vastly differed in our CFFPR-based cohort (Supplementary Table 2) compared to the initial validation study for the Leeds criteria, strict application of this score to our study may have resulted in frequent misclassification. Thus, we applied a modified version of the Leeds criteria to our cohort [24] and have successfully used it for P. aeruginosa classification in CF in the past [21]; it has since been demonstrated to perform similarly to the original score, including in relation to clinical outcomes [25]. As the frequency of quarterly cultures in our cohort was low, even if misclassification occurred, it would have biased results toward the null. In regard to the other bacterial pathogens assessed, we used a conservative definition of chronic infection as applied to P. aeruginosa, and it is possible that the incidence of other bacterial pathogens was underestimated by this approach. Next, as we observed a marked drop in incidence of chronic P. aeruginosa infections between 2003 and 2004, this may have been due to left censoring of the cohort with individuals either aging into the cohort at 13 years or entering at a later age following a new diagnosis. In our restricted cohort sensitivity analysis that was based on the probability of increasing chronic infection risk with age, the initial drop in incident chronic P. aeruginosa was no longer observed; however, the significant decrease over the study period persisted. Third, as the definition of chronic infection is dependent on number of cultures over time, there was potential for individuals with worsened lung function to undergo more respiratory cultures. We summarized culture data quarterly to minimize this effect. Further, the number of total cultures increased over time, which may have resulted in increasing incidence of P. aeruginosa infection captured and thus would have again biased the results toward the null. Last, with use of the registry data, we captured approximately 84% of the CF population in the United States; such a subset, albeit large, could be different from the entire CF population in the United States. However, patients are more likely to be seen in specialty CF centers captured in the CFFPR as they get sicker and require more specialized care. We also acknowledge that given the use of registry data, missingness and loss to follow-up may impact the results in an unpredictable manner. Regardless, this was a large CF population–based study to robustly examine incident chronic P. aeruginosa infections over a 10-year period and improves our understanding of the epidemiologic trends of this organism.
In conclusion, our large registry-based study in the United States demonstrated a significant decline in incident chronic P. aeruginosa infections in adolescents and adults with CF between 2003 through 2012. We did not observe a parallel increase in other characteristic bacterial airway pathogens over the same period. Additional studies are needed to determine whether these changes in chronic P. aeruginosa infection continue and how they correlate with future clinical outcomes in CF.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
Presented in part: North American Cystic Fibrosis Conference. Phoenix, AZ, October 2015. Pediatr Pulmonol; 50(S41):385.
Notes
Author contributions. E. C. and M. C. were primarily responsible for data management and analysis. M. C., R. S., and K. R. were responsible for the creation of the manuscript. C. H. G., A. R., N. M. H., and M. C. were responsible for the project’s inception and supervision. All authors contributed to development of the final manuscript. C. H. G. serves as guarantor of the work.
Acknowledgments. We thank the Cystic Fibrosis Foundation (CFF) for use of The Cystic Fibrosis Foundation Patient Registry (CFFPR) data to conduct this study. Additionally, we thank the patients, care providers, and clinic coordinators at CF centers throughout the United States for their contributions to the CFFPR.
Financial support. C. H. G. receives funding from the CFF, the National Institutes of Health (NIH; grantsR01HL103965, R01HL113382, R01AI101307, UM1HL119073, P30DK089507), and the US Food and Drug Administration (grant R01FD003704). N. M. H. reports grants from the NIH and CFF. R. S. is supported by fellowship funding from Cystic Fibrosis Canada, the Canadian Institutes of Health Research, and Alberta Innovates-Health Solutions. K. J. R. is supported by fellowship funding from the CFF.
Potential conflicts of interest. C. H. G. has consulted for Vertex Pharmaceuticals and has participated on the boards of Transave Inc, KalBios Pharmaceuticals, and Boehringer Ingelheim Pharm GmbH & Co outside of the submitted work. In her role as co-director of the Therapeutics Development Network Coordinating Center, N. M. H.’s institution receives other funding from AbbVie Inc. Achaogen AlgiPharma AS Aridis Pharmaceuticals LLC Bayer HealthCare AG Catabasis Celtaxsys Corbus Pharmaceuticals Covance, Inc. CURx Pharmaceuticals, Inc. Flatley Discovery Lab LLV Gilead Sciences, Inc. Horizon Pharma KaloBios La Jolla Pharmaceutical Laurent Pharmaceuticals Life Science Strategies Mylene Gencey Nivalis Therapeutics, Inc. Novartis Pharmaceuticals Corp. ProQR Therapeutics B.V. Protalix Ltd. Proteostasis Therapeutics, Inc. Pulmatrix Quintiles, Inc. Sanofi Savara Pharmaceuticals Synedgen, Inc. Teva Branded Pharmaceutical Products R&D Vertex Pharmaceuticals Incorporated, outside the submitted work. All remaining authors: no reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. 2013 Annual Report - The Canadian Cystic Fibrosis Registry. Cystic Fibrosis Canada. Available at: http://www.cysticfibrosis.ca/cf-care/cf-registry/. Accessed on 15 August 2015. [Google Scholar]
- 2. Razvi S, Quittell L, Sewall A, Quinton H, Marshall B, Saiman L. Respiratory microbiology of patients with cystic fibrosis in the United States, 1995 to 2005. Chest 2009; 136:1554–60. [DOI] [PubMed] [Google Scholar]
- 3. Lambiase A, Raia V, Del Pezzo M, Sepe A, Carnovale V, Rossano F. Microbiology of airway disease in a cohort of patients with cystic fibrosis. BMC Infect Dis 2006; 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Emerson J, McNamara S, Buccat AM, Worrell K, Burns JL. Changes in cystic fibrosis sputum microbiology in the United States between 1995 and 2008. Pediatr Pulmonol 2010; 45:363–70. [DOI] [PubMed] [Google Scholar]
- 5. Cystic Fibrosis Foundation Patient Registry. 2015 annual data report. Bethesda, MD: Cystic Fibrosis Foundation, 2015. [Google Scholar]
- 6. 2013 Annual Report - The ECFS Patient Registry. European Cystic Fibrosis Society. Available at: https://www.ecfs.eu/sites/default/files/images/ECFSPR_Report2013_02.2016.pdf. Accessed on 15 February 2016. [Google Scholar]
- 7. Salsgiver EL, Fink AK, Knapp EA et al. . Changing epidemiology of the respiratory bacteriology of patients with cystic fibrosis. Chest 2016; 149:390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jelsbak L, Johansen HK, Frost AL et al. . Molecular epidemiology and dynamics of Pseudomonas aeruginosa populations in lungs of cystic fibrosis patients. Infect Immun 2007; 75:2214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Govan JR, Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia.Microbiol Rev 1996; 60:539–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Henry RL, Mellis CM, Petrovic L. Mucoid Pseudomonas aeruginosa is a marker of poor survival in cystic fibrosis. Pediatr Pulmonol 1992; 12:158–61. [DOI] [PubMed] [Google Scholar]
- 11. Li Z, Kosorok MR, Farrel PM et al. . Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. J Pediatr 2005; 138:699–704. [DOI] [PubMed] [Google Scholar]
- 12. Kosorok MR, Zeng L, West SE et al. . Acceleration of lung disease in children with cystic fibrosis after Pseudomonas aeruginosa acquisition. Pediatr Pulmonol 2001; 32:277–87. [DOI] [PubMed] [Google Scholar]
- 13. Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 2002; 34:91–100. [DOI] [PubMed] [Google Scholar]
- 14. Konstan MW, Morgan WJ, Butler SM et al. ; Scientific Advisory Group and the Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr 2007; 151: 134–9, 139.e1. [DOI] [PubMed] [Google Scholar]
- 15. Nixon GM, Armstrong DS, Carzino R et al. . Clinical outcome after early Pseudomonas aeruginosa infection in cystic fibrosis. J Pediatr 2001; 138:699–704. [DOI] [PubMed] [Google Scholar]
- 16. Corey M FV. Determinants of mortality from cystic fibrosis in Canada, 1970–1989. Am J Epidemiol 1996; 143:1007–17. [DOI] [PubMed] [Google Scholar]
- 17. Döring G, Hoiby N; Consensus Study Group Early intervention and prevention of lung disease in cystic fibrosis: a European consensus. J Cyst Fibros 2004; 3:67–91. [DOI] [PubMed] [Google Scholar]
- 18. Treggiari MM, Rosenfeld M, Retsch-Bogart G, Gibson R, Ramsey B. Approach to eradication of initial Pseudomonas aeruginosa infection in children with cystic fibrosis. Pediatr Pulmonol 2007; 42:751–6. [DOI] [PubMed] [Google Scholar]
- 19. Staudinger BJ, Muller JF, Halldórsson S et al. . Conditions associated with the cystic fibrosis defect promote chronic Pseudomonas aeruginosa infection. Am J Respir Crit Care Med 2014; 189:812–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. MacKenzie T, Gifford AH, Sabadosa KA et al. . Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the Cystic Fibrosis Foundation patient registry. Ann Intern Med 2014; 161:233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Crull MR, Ramos KJ, Caldwell E, Mayer-Hamblett N, Aitken ML, Goss CH. Change in Pseudomonas aeruginosa prevalence in cystic fibrosis adults over time. BMC Pulm Med 2016; 16:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knapp EA, Fink AK, Goss CH et al. . The Cystic Fibrosis Foundation patient registry. Design and methods of a national observational disease registry. Ann Am Thorac Soc 2016; 13:1173–9. [DOI] [PubMed] [Google Scholar]
- 23. Farrell PM, Rosenstein BJ, White TB et al. ; Cystic Fibrosis Foundation Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr 2008; 153:S4–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee TW, Brownlee KG, Conway SP, Denton M, Littlewood JM. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J Cyst Fibros 2003; 2:29–34. [DOI] [PubMed] [Google Scholar]
- 25. Beckett V, Heltshe S, Skalland M, Khan U, Rosenfeld M. New pseudomonas chronicity score: evaluating association with clinical outcomes and comparing to Leeds criteria [abstract]. Pediatr Pulmonol 2017; 52:338. [Google Scholar]
- 26. Pressler T, Frederiksen B, Skov M, Garred P, Koch C, Høiby N. Early rise of anti-pseudomonas antibodies and a mucoid phenotype of Pseudomonas aeruginosa are risk factors for development of chronic lung infection–a case control study. J Cyst Fibros 2006; 5:9–15. [DOI] [PubMed] [Google Scholar]
- 27. McKone EF, Emerson SS, Edwards KL, Aitken ML. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet 2003; 361:1671–6. [DOI] [PubMed] [Google Scholar]
- 28. Thomsen JL, Parner ET. Methods for analysing recurrent events in health care data. Examples from admissions in Ebeltoft Health Promotion Project. Fam Pract 2006; 23:407–13. [DOI] [PubMed] [Google Scholar]
- 29. Bonadia LC, de Lima Marson FA, Ribeiro JD et al. . CFTR genotype and clinical outcomes of adult patients carried as cystic fibrosis disease. Gene 2014; 540:183–90. [DOI] [PubMed] [Google Scholar]
- 30. Salsgiver EL, Fink AK, Knapp EA et al. . Changing epidemiology of the respiratory bacteriology of patients with cystic fibrosis. Chest 2016; 149(2):390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramsay KA, Sandhu H, Geake JB et al. . The changing prevalence of pulmonary infection in adults with cystic fibrosis: a longitudinal analysis. J Cyst Fibros 2017; 16:70–7. [DOI] [PubMed] [Google Scholar]
- 32. Lee TW, Brownlee KG, Denton M, Littlewood JM, Conway SP. Reduction in prevalence of chronic Pseudomonas aeruginosa infection at a regional pediatric cystic fibrosis center. Pediatr Pulmonol 2004; 37:104–10. [DOI] [PubMed] [Google Scholar]
- 33. Frederiksen B, Koch C, Høiby N. Changing epidemiology of Pseudomonas aeruginosa infection in Danish cystic fibrosis patients (1974–1995). Pediatr Pulmonol 1999; 28:159–66. [DOI] [PubMed] [Google Scholar]
- 34. Gibson RL, Emerson J, McNamara S et al. ; Cystic Fibrosis Therapeutics Development Network Study Group Significant microbiological effect of inhaled tobramycin in young children with cystic fibrosis. Am J Respir Crit Care Med 2003; 167:841–9. [DOI] [PubMed] [Google Scholar]
- 35. Ratjen F, Munck A, Kho P, Angyalosi G; ELITE Study Group Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis: the ELITE trial. Thorax 2010; 65:286–91. [DOI] [PubMed] [Google Scholar]
- 36. Treggiari MM, Retsch-Bogart G, Mayer-Hamblett N et al. ; Early Pseudomonas Infection Control Investigators Comparative efficacy and safety of 4 randomized regimens to treat early Pseudomonas aeruginosa infection in children with cystic fibrosis. Arch Pediatr Adolesc Med 2011; 165:847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mayer-Hamblett N, Kloster M, Rosenfeld M et al. . Impact of sustained eradication of new Pseudomonas aeruginosa infection on long-term outcomes in cystic fibrosis. Clin Infect Dis 2015; 61:707–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gibson RL, Emerson J, Mayer-Hamblett N et al. . Duration of treatment effect after tobramycin solution for inhalation in young children with cystic fibrosis. Pediatr Pulmonol 2007; 42:610–23. [DOI] [PubMed] [Google Scholar]
- 39. Kenny SL, Shaw TD, Downey DG, Moore JE, Rendall JC, Elborn JS. Eradication of Pseudomonas aeruginosa in adults with cystic fibrosis. BMJ Open Respir Res 2014; 1:e000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Keating CL, Liu X, Dimango EA. Classic respiratory disease but atypical diagnostic testing distinguishes adult presentation of cystic fibrosis. Chest 2010; 137:1157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heltshe SL, Mayer-Hamblett N, Burns JL et al. ; the G551D Observation-AL Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis 2015; 60:703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mayer-Hamblett N, Rosenfeld M, Gibson RL et al. . Pseudomonas aeruginosa in vitro phenotypes distinguish cystic fibrosis infection stages and outcomes. Am J Respir Crit Care Med 2014; 190:289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoiby N, Flensborg EW, Beck B, Friis B, Jacobsen SV, Jacobsen L. Pseudomonas aeruginosa infection in cystic fibrosis. Diagnostic and prognostic significance of Pseudomonas aeruginosa precipitins determined by means of crossed immunoelectrophoresis. Scand J Respir Dis 1977; 58:65–79. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.