

Abstract
Trifluoromethylative difunctionalization and hydrofunctionalization of unactivated alkenes have been developed into powerful synthetic methodologies. On the other hand, methylative difunctionalization of olefins remains an unexplored research field. We report in this paper the Cu-catalyzed alkoxy methylation, azido methylation of alkenes using dicumyl peroxide (DCP), and di-tert-butyl peroxide (DTBP) as methyl sources. Using functionalized alkenes bearing a tethered nucleophile (alcohol, carboxylic acid, and sulfonamide), methylative cycloetherification, lactonization, and cycloamination processes are subsequently developed for the construction of important heterocycles such as 2,2-disubstituted tetrahydrofurans, tetrahydropyrans, γ-lactones, and pyrrolidines with concurrent generation of a quaternary carbon center. The results of control experiments suggest that the 1,2-alkoxy methylation of alkenes goes through a radical-cation crossover mechanism, whereas the 1,2-azido methylation proceeds via a radical addition and Cu-mediated azide transfer process.
While the trifluoromethylative difunctionalization of unactivated alkenes has been largely explored, methylative difunctionalization remains underinvestigated. Here, the authors report copper-catalyzed alkoxy- and azido-methylation reactions of alkenes leading to important synthetic building blocks and valuable O- and N-heterocycles.
Introduction
The so-called magic methyl effect has long been known in medicinal chemistry and has been frequently used to optimize the biological and pharmacological properties of a drug candidate1. In addition to traditional nucleophilic substitution reaction, transition metal-catalyzed cross-coupling reaction has recently been developed into a powerful tool for the methylation of (hetero)aromatics2,3. However, in comparison to the recent advances in the field of trifluoromethylation of organic compounds4,5, progress on the development of new methylation protocols has been much slower. While the importance of the CF3 group in pharmaceuticals and crop science is undeniable, the CH3 group deserved certainly equal attention. In fact, it was estimated that over 67% of 200 top-selling drugs bore at least one CH3 group, while <5% of the small molecule drugs in the same list contained a CF3 group6.
Most of the trifluoromethylative difunctionalization of alkenes involved the generation of electrophilic CF3 radical from the hypervalent iodine reagents7 followed by its addition to the electron-rich alkenes (Fig. 1a)3. Similarly, metal-catalyzed hydrofunctionalization of alkenes, pioneered by Mukaiyama in 1980s8, has been extensively investigated (Fig. 1b)9–16. By choosing an appropriate radical acceptor, Baran and co-workers developed a protocol for the hydromethylation of unactivated alkenes for the one-pot conversion of alkenes to alkanes (Fig. 1c)15. Interestingly, in spite of the known magic methyl effect in medicinal chemistry and its utility in natural product synthesis, the related methylative difunctionalization of unactivated olefins was, to the best of our knowledge, far less developed and a multistep sequence was generally needed to accomplish this endeavor. As illustrated in Fig. 1d, five steps were needed to convert alkene I to hydroxymethylated derivative II, an advanced intermediate on the way to vinigrol 17,18.
Fig. 1.

Functionalization of unactivated alkenes. a Trifluoromethylative difunctionalization of alkenes; b hydrofunctionalization of alkenes. c hydromethylation of alkenes; d Example of 1,2-hydroxy methylation of alkene in natural product synthesis. Five steps were required to accomplish this transformation; e methylative difunctionalization of electron-rich alkenes: radical-metal mediated ligand transfer and radical-cation crossover processes. Abbreviations: Fe(acac)3 iron (III) acetylacetonate; LAH lithium aluminum hydride; CDMT 2-chloro-4,6-dimethoxy-1,3,5-triazine; NMM N-methyl morpholine; AIBN azobisisobutyronitrile
Peroxides undergo homolytic cleavage of the O–O bond to generate acyloxy or alkoxy radicals, which can act as oxidants and radical initiators. These oxygen-centered radicals can also undergo further fragmentation to produce the alkyl radicals19. The groups of Kawazoe20 and Wong21 demonstrated in 1970s that the methyl radical generated from tert-butyl hydroperoxide and tert-butyl peracetate can methylate the protonated nucleobases via homolytic aromatic substitution (HAS) reaction. These pioneering studies, akin to Minisci reaction22, is in line with the nucleophilic nature of the methyl radical. Since then, conditions allowing the methylation of (hetero)arenes23–26, amides/carboxylic acids27–30, and isocyanides31–34 have been exploited. In addition, methylation of electron-deficient olefins such as N-arylacrylamides have also been developed35–38. In this latter case, the resulting electrophilic radical adduct underwent rapid intramolecular HAS with the tethered aromatic ring to afford 2,2-disubstituted oxindoles. On the other hand, methylative difunctionalization of unactivated double bonds using peroxide as methyl source has, to the best of our knowledge, not been reported39. This was probably due to the perception that methyl radical is nucleophilic, therefore, its addition to electron-rich alkenes would be polarity mismatched process40–43]. We report herein the realization of this endeavor by developing three-component 1,2-alkoxy methylation, 1,2-azido methylation, and methylative cycloetherification, lactonization, cycloamination of unactivated alkenes (Fig. 1e). The results of control experiments suggested that the 1,2-alkoxy methylation of alkenes went through a radical-cation crossover mechanism, whereas the azido methylation proceeded via a radical addition and Cu-mediated redox azide transfer process.
Results
Three-component 1,2-alkoxy methylation of alkenes
Examples of alkoxy alkylation of unactivated alkenes are rare. Wang and co-workers reported a rhenium-catalyzed 1,2-acetoxy methylation of styrene derivatives using phenyliodine diacetate (PIDA) as both the methyl and the acetoxy sources44, while Glorius45 and Bao46,47 reported the alkoxy alkylation of alkenes via decarboxylative generation of alkyl radicals.
We began our studies by examining the 1,2-alkoxy methylation of α-methylstyrene (1a). After extensive survey of the reaction parameters varying the Cu sources, the ligands, the Cu/ligand ratio, the peroxides, the bases, the solvents, the concentration, and the reaction temperature, the optimum conditions found consisted of heating a MeOH solution of 1a (c 0.1 M) in a sealed tube in the presence of a catalytic amount of Cu(BF4)2•6H2O (0.2 equiv), 4,4-dimethoxy-2,2’-bipyridine (L1, 0.3 equiv) and Na2HPO4 (0.2 equiv) at 120 °C for 4 h. Under these conditions, 2a was isolated in 96% yield. We note that reaction using 2-hydroperoxy-2-methylbutane as ethyl donor under otherwise standard conditions provided a complex reaction mixture.
The scope of this 1,2-alkoxy methylation of alkenes is shown in Fig. 2. Electron-donating (Me and OMe) and electron-withdrawing (F and Cl) substituents at different positions of the phenyl ring of the α-methylstyrene derivatives were transformed into the corresponding methylated ethers (2b–2i) in excellent yields. Different alkyl groups (Et, iPr, and CH2CH2Ph) at the α-position of styrene were compatible (2j–2l) and the 1,1-diarylethylenes were similarly difunctionalized to afford the desired products (2m–2q) regardless of the electronic nature of the substituents on the aromatic ring. 2-Vinylnaphthalene and 1-methylene-1,2,3,4-tetrahydronaphthalene took part in the reaction to afford the three-component adducts without event (2r, 2s). However, styrene failed to give the desired 1,2-methoxy methylation product under standard conditions. Performing the reaction of 1a in ethanol and isopropanol under otherwise standard conditions afforded the ethyl ether (2t) and the isopropyl ether (2u), respectively. A gram scale experiment converted 1a to the three-component adduct 2a in 93% isolated yield.
Fig. 2.

1,2-Alkoxy methylation of unactivated alkenes. Unless specified, MeOH was used as solvent. (i) 140 °C; (ii) DTBP (4.0 equiv) was used instead of DCP; (iii) EtOH (2.0 mL, c 0.1 M); (iv) iPrOH (2.0 mL, c 0.1 M). Abbreviations: DTBP di-tert-butyl peroxide; DCP dicumyl peroxide
Three-component 1,2-azido methylation of alkenes
There were only few examples on the three-component carboazidation of alkenes with the concurrent formation of a C(sp3)-C(sp3) and a C(sp3)-N bond48. Renaud and co-workers developed a carboazidation of alkenes 1 employing electrophilic alkyl radicals generated from ethyl α-iodoacetate and phenylsulfonyl azide as the azide sources49,50. Three-component 1,2-azido alkylation of alkenes with nucleophilic alkyl radical is to the best of our knowledge unknown51–53. The α-methylstyrene (1a) was chosen as a test substrate for the optimization of the 1,2-azido methylation process. Using Cu(BF4)2•6H2O as catalyst (0.2 equiv), initial survey of the reaction conditions varying the methyl sources (DCP, DTBP, and tert-butyl peroxybenzoate), the azide sources (TMSN3, NaN3, KN3, and LiN3) and solvents (MeCN, tBuCN, DMF, DMSO, 1,4-dioxane, and tBuOH) prompted us to fix the following key parameters [LiN3, DTBP, tBuOH (c 0.1 M)] for further optimization. The 1,10-phenanthroline L2 turned out to be a superior ligand than L1 and CuSO4 stood out as the catalyst of choice among those copper salts screened [Cu(OAc)2, Cu(OTf)2, CuF2, and CuSO4]. Interestingly, reducing the loading of CuSO4 (0.01 equiv) gave a cleaner reaction mixture. Overall, under optimized conditions [CuSO4 (0.01 equiv), L2 (0.03 equiv), LiN3 (2.5 equiv), DTBP (4.0 equiv), tBuOH (c 0.1 M)], the desired compound 3a was isolated in 81% yield (Fig. 3). A similar yield of 3a (79%) was obtained when the 1,2-azido methylation of 1a was performed at 2.0 mmol scale. Once again, using 2-hydroperoxy-2-methylbutane as ethyl donor under otherwise standard conditions provided a complex reaction mixture.
Fig. 3.

1,2-Azido methylation of unactivated alkenes. The reaction scheme is shown above the table
The reaction was applicable to a variety of α-methylstyrene derivatives bearing electron-donating (Me and OMe) and -withdrawing groups (4-Cl, 4-Br, 4-F, 4-OCF3, 4-CN, and 4-NO2) on the phenyl ring. The presence of an o-methyl substituent in the substrate was also tolerated to afford 3k. α-Ethyl, α-isopropyl and α-phenethyl styrenes participated in the reaction without event (3m–3o). The 1,1-diarylethylenes bearing substituents with different electronic properties were similarly converted to the three-component adducts (3p–3u).
Methylative cycloetherification
Metal-catalyzed arylative cycloetherification and cycloamination has been well developed for the synthesis of functionalized oxa- and aza-heterocycles54,55. The alkylation-induced heterocyclization is, on the other hand, poorly documented. For instance, only few examples of alkylative cycloetherification have been reported in the literature 56.
We investigated the methylative cycloetherification of alkenes using 4-phenylpent-4-en-1-ol (4a, R=H, n = 1, Fig. 4a) as a test substrate. Gratefully, treatment of a tBuOH solution of 4a under conditions established for the 1,2-alkoxy methylation of alkenes afforded the tetrahydrofuran 5a in 58% yield. Replacing DCP with DTBP gave a similar yield of 5a (57%). Therefore, DTBP was used as a methyl source for further condition optimization as it provided a cleaner reaction mixture. Performing the reaction in tBuCN furnished 5a in only 30% yield. Other solvents such as tetrahydrofuran (THF), dimethoxyethane (DME) and N,N-dimethylformamide (DMF) led to the decomposition or polymerization of 4a. Finally, trifluoroethanol (TFE) turned out to be an optimum solvent and Cu(OTf)2 a slightly better catalyst than Cu(BF4)2•6H2O for this reaction. Overall, the reaction of 4a with DTBP in trifluoroethanol (c 0.1 M) in the presence of Cu(OTf)2 (0.2 equiv), 4,4’-dimethoxy-2,2’-dipyridine (L1, 0.3 equiv), Na3PO4 (0.2 equiv) at 120 °C afforded 2-ethyl-2-phenyltetrahydrofuran (5a) in 82% yield.
Fig. 4.

Methylative heterocyclization of alkenes. a Methylative cycloetherification: 4 (0.2 mmol), Cu(OTf)2 (0.2 equiv), L1 (0.3 equiv), Na3PO4 (0.2 equiv), DTBP (4.0 equiv), CF3CH2OH (c 0.1 M), 120 °C. Yields refer to isolated products. b Methylative lactonization: 6 (0.2 mmol), CuSO4 (0.2 equiv), L2 (0.3 equiv), Na3PO4 (0.3 equiv), DTBP (4.0 equiv), tBuOH (c 0.1 M), 120 °C. c Methylative cycloamination: 8, Cu(OAc)2 (0.2 equiv), L2 (0.3 equiv), Na3PO4 (0.2 equiv), DTBP (4.0 equiv), tBuOH (c 0.1 M), 120 °C
Under the above-optimized conditions, a diverse set of 4-aryl substituted pent-4-en-1-ols 4 underwent methylative cycloetherification to afford the 2,2-disubstituted tetrahydrofurans (5b–5m) in good yields (Fig. 4a). The reaction tolerated the presence of both electron-donating (Me, OMe, Ph, and iPr) and electron-withdrawing groups (F, Cl, Br, and CN) at different positions of the aryl ring. The 5-phenylhex-5-en-ol underwent the similar methylative cycloetherification to afford 2-ethyl-2-phenyltetrahydro-2H-pyran (5n) in 76% yield.
Methylative lactonization
γ-Butyrolactones are found in many bioactive compounds57 and are also useful building blocks in organic synthesis58. Consequently, many different synthetic routes have been developed for the synthesis of this important heterocycle59–64. Encouraged by the aforementioned results, the methylative lactonization of alkenes was next investigated. The optimum reaction conditions we found consisted of heating a solution of 6 in tBuOH (c 0.1 M) in the presence of CuSO4 (0.2 equiv), 1,10-Phen (L2, 0.3 equiv), DTBP (4.0 equiv) and Na3PO4 (0.2 equiv) at 120 °C. As it is shown in Fig. 4b, electron-donating (Me, OMe, Ph, and iPr) and electron-withdrawing groups (F, Br, Cl, and CN) on the phenyl ring of the α-methyl styrene derivatives were well tolerated leading to γ-lactones (7a–7i) in good yields. The (E)-4-phenylbut-3-enoic acid (6j), a 1,2-disubstituted alkene, underwent regioselective methylative lactonization to afford the 4,5-trans-disubstituted γ-lactone 7j as a single isolable diastereomer in 41% yield together with the methyl ester of 6j (24%).
Methylative cycloamination
While trifluoromethylative cycloamination of alkenes have been reported recently65,66, the methylative counterpart is to the best of our knowledge unknown. We therefore set out to examine this reaction using sulfonamide as internal nucleophile. Optimum conditions found for the methylative cycloamination of 8a with DTBP (4.0 equiv) consisted of heating a solution of 8a in tBuOH (c 0.1 M) in the presence of Cu(OAc)2 (0.2 equiv), 1,10-Phen (L2) and Na3PO4 (0.2 equiv) at 120 °C. Under these conditions, the pyrrolidine 9a was isolated in 71% yield. As it is shown in Fig. 4c, electron-donating (Me, OMe, Ph, and iPr) and electron-withdrawing groups (F, Br, Cl, and CN) on the phenyl ring of the α-methyl styrene derivatives were well tolerated leading to 2,2-disubstituted pyrrolidines (9a–9k) in good yields.
Mechanistic studies
Possible reaction pathways for the 1,2-alkoxy methylation and 1,2-azido methylation of alkenes are depicted in Fig. 5a. Reduction of peroxide (DCP or DTBP) by the in situ generated Cu(I)X salt A would produce the tert-alkoxy radical B and Cu(II) salt C. Alternatively, thermal decomposition of peroxide would generate two molecules of alkoxy radical B. β-Scission of B would generate ketone D and methyl radical E. Addition of E to the alkene would produce the benzyl radical F which would be oxidized by Cu(II) salt C to the carbenium G with the concurrent regeneration of the Cu(I)X salt. Trapping of the carbenium G by nucleophile would then afford the observed products (route a). Alternatively, radical F could be directly converted to the adduct via a Cu-centered redox transfer process (route b) or via radical rebound of F with C followed by reductive elimination of the resulting Cu(III) species H (route c) 64.
Fig. 5.

Mechanistic proposal and control experiments. a Possible reaction pathways. b Radical trapping experiment. c Radical clock experiment. d Super sensitive radical probe experiment. Conditions a: 1a (0.2 mmol), Cu(BF4)2•6H2O (0.2 equiv), L1 (0.3 equiv), DCP (4.0 equiv), Na2HPO4 (0.2 equiv), MeOH (2.0 mL, c 0.1 M), 120 °C, 4 h; Conditions b: 1a (0.2 mmol), DTBP (0.8 mmol), LiN3 (0.5 mmol), CuSO4 (0.002 mmol, 0.1 equiv), L2 (0.06 mmol), tBuOH (2.0 mL, c 0.1 M), 120 °C
Several experimental observations and the results of control experiments were in line with the proposed reaction pathway. First, 1,2-methoxy methylation of 1a was completely inhibited in the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, 10). 1-(Methoxy)-2,2,6,6-tetramethylpiperidine (11) was instead isolated in 29% yield (Fig. 5b). Second, submitting (1-cyclopropylvinyl)benzene (12) to the standard 1,2-methoxy methylation conditions afforded dihydronaphthalene 13 in 43% yield (Fig. 5c). These two experiments clearly indicated the existence of both the methyl radical (Me•) and the adduct radical 14 in this three-component process. To gain further insight on the reaction mechanism, the 2-tert-butoxy-3-(1-phenylvinyl)cyclopropyl)benzene (15), developed by Newcomb as a supersensitive radical probe, was synthesized67,68. It has been demonstrated that the cyclopropane will be opened at the phenyl-bearing carbon in a radical mechanism and at the oxygen-bearing carbon in a cationic mechanism. Eventually, treatment of 15 under our methoxy methylation conditions afforded a quite complex reaction mixture from which 1,4-disubstituted naphthalene 16 was isolated in 17% yield. On the other hand, compound 15 was converted, under 1,2-azido methylation conditions, cleanly to 16 in 56% yield (Fig. 5d). Formation of benzyl radical 17 followed by regioselective ring opening to 18 and intramolecular HAS reaction would provide dihydronaphthalene 20 which, upon elimination of tBuOH, would afford naphthalene 16. The observed regioselective ring opening of cyclopropane supported the involvement of the benzylic radical 17 as a possible reactive intermediate.
The significant difference in the yield of 16 from radical clock probe 15 under the methoxy methylation and azido methylation conditions was intriguing. We tentatively attributed to the different oxidation power of the copper salts. CuSO4 is known to be a weaker oxidant than Cu(BF4)2•6H2O, the benzylic radical generated under the azido methylation conditions (CuSO4-catalyzed) would, therefore, have a longer half-life than that generated under methoxy methylation conditions [Cu(BF4)2•6H2O-catalyzed], hence the clean formation of product 16. This led us to hypothesize that the C–O bond formation in the present alkoxy methylation went through cationic intermediate (route a, Fig. 5a), whereas the C–N bond formation in the azido methylation proceeded via the Cu-mediated azide transfer process (route b or c, Fig. 5a).
In accordance with the aforementioned reaction manifolds, 1,2-methoxy methylation of 1-methyl-1-(4-nitrophenyl)ethylene (21) under standard conditions afforded a significant amount of dimer 22 and only a trace amount of the desired methoxy methylation product (Fig. 6a). The presence of the strong electron-withdrawing nitro group on the phenyl ring might significantly reduce the rate of the oxidation of benzyl radical 23 to carbenium, blocking therefore the methoxylation process. It underwent instead the dimerization to afford 22. On the other hand, treatment of 21 under standard azido methylation conditions afforded the three-component adduct 3f in 78% yield together with a small amount of dimer 22 (Fig. 6b). The result supported the notion that oxidation of radical to cation is not involved in the azidation step and the azido group was transferred directly to the radical 23 via presumably a Cu-mediated redox transfer process. The azide transfer reaction was apparently faster than the dimerization process under our optimized azido methylation conditions. It is also worth noting that dimer was rarely observed under the optimized methoxy methylation of alkenes due presumably to the rapid oxidation of benzyl radical to benzyl cation (except for 21), while it was very often observed as a side product in the azido methylation process due to the relatively long-lived benzyl radical species. Finally, performing the azidomethylation of α-methylstyrene (1a) in MeOH and tBuOH/MeOH (v/v = 4:1) under otherwise standard conditions afforded the desired product 3a in yields of 46 and 62%, respectively. The potential competitive reaction leading to the 1,2-methoxy methylated product 2a was not observed. This result reinforced the hypothesis that benzyl cation might not be involved in the azidomethylation of alkenes.
Fig. 6.
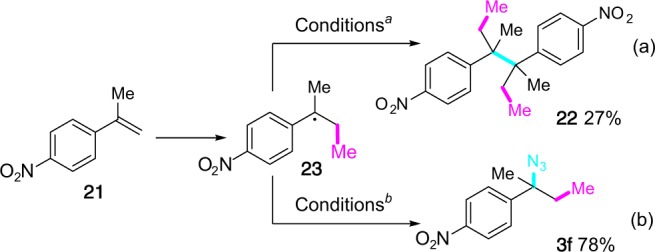
Mechanistic divergence between methoxy methylation and azido methylation. a: 21 (0.2 mmol), Cu(BF4)2•6H2O (0.2 equiv), L1 (0.3 equiv), DCP (4.0 equiv), Na2HPO4 (0.2 equiv), MeOH (2.0 mL, c 0.1 M), 120 °C, 4 h; b: 21 (0.2 mmol), DTBP (0.8 mmol), LiN3 (0.5 mmol), CuSO4 (0.002 mmol, 0.1 equiv), L2 (0.06 mmol), tBuOH (2.0 mL, c 0.1 M), 120 °C
At the outset of this research, we were concerned about the hydrogen abstraction of MeOH by tert-alkoxy radical B to generate the hydroxymethyl radical I (route d, Fig. 5a). This process has indeed been exploited in the difunctionalization of activated alkenes69,70. Two pathways, namely, thermal decomposition and reduction by Cu(I) salt, may contribute to the generation of the radical B from the peroxide. The formal process generates two molecules of alkoxy radical B, while the latter produces one molecule of B and one molecule of copper tert-butoxide C. Therefore, it was difficult to quantify the ratio of β-scission of B (generating Me•) vs H-abstraction of MeOH by B (leading to •CH2OH) based on the ratio of acetophenone (D) vs 2-phenylpropan-2-ol (J). Nevertheless, the high J/D ratio (3/1) we obtained for the methylative methoxylation of α-methylstyrene (1a) indicated that route d, a thermodynamically favorable process (BDE of H-CH2OH: 96.06 ± 0.15 kcal/mol; tBuO-H: 106.3 ± 0.7 kcal/mol), was indeed occurring in parallel. However, the so-generated hydroxymethyl radical I did not interfere with the methylation process probably due to the pronounced nucleophilic nature of this radical or its rapid oxidation to formaldehyde.
In summary, we reported the Cu-catalyzed carboalkoxylation, carboazidation, carbocycloetherification, carbolactonization, and carbocycloamination of alkenes using dicumyl peroxide (DCP) or di-tert-butyl peroxide (DTBP) as methyl sources. A diverse set of styrene derivatives were converted to the methylated ethers, azides, tetrahydrofurans, tetrahydropyrans, γ-lactones, and pyrrolidines with concurrent generation of a quaternary carbon in good to excellent yields. The results of control experiments suggested that the 1,2-alkoxy methylation of alkenes went through a radical-cation crossover mechanism, whereas the azido methylation proceeded via a radical addition and Cu-mediated redox azide transfer process. This mechanistic insight would serve as a guideline in our searching for new alkene difunctionalization protocols.
Methods
Three-component 1,2-alkoxy methylation of alkenes
A screw cap tube was charged with Cu(BF4)2•6H2O (13.8 mg, 0.0400 mmol), 4,4’-dimethoxy-2,2’-bipyridine L1 (13.0 mg, 0.0601 mmol), Na2HPO4 (5.7 mg, 0.0402 mmol) and R3OH (2.0 mL). The mixture was stirred at room temperature for 30 min, then substrate 1 (0.2 mmol, 1.0 equiv) and DCP (216.2 mg, 0.800 mmol) or DTBP (0.15 mL, 4.0 equiv) were added to the above mixture. After being stirred for 4 h at 120 °C under N2 atmosphere, the reaction mixture was quenched with water and the aqueous phase was extracted with EtOAc. The organic extracts were washed with brine, dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by flash chromatography to give 2.
Three-component 1,2-azido methylation of alkenes
A screw cap tube was charged with CuSO4 (0.32 mg, 0.002 mmol, 0.01 equiv), 1,10-phenanthroline L2 (1.08 mg, 0.003 mmol, 0.03 equiv) and tBuOH (2.0 mL). The mixture was stirred at 40 °C for 30 min, then cooled to room temperature. Substrate 1 (0.2 mmol, 1.0 equiv), LiN3 (20% w/w, 0.12 mL, 2.5 equiv) and DTBP (0.15 mL, 4.0 equiv) were added to the above mixture, and the reaction mixture was stirred at 120 °C for 8 h under N2 atmosphere. The reaction was quenched with water and the aqueous phase was extracted with EtOAc. The organic extracts were washed with brine, dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by flash chromatography to give 3.
Methylative cycloetherification
A screw cap tube was charged with Cu(OTf)2 (14.5 mg, 0.04 mmol, 0.2 equiv), L1 (13.0 mg, 0.06 mmol, 0.03 equiv) and CF3CH2OH (2.0 mL). The mixture was stirred at room temperature for 30 min. Substrate 4 (0.2 mmol, 1.0 equiv), Na3PO4 (6.5 mg, 0.04 mmol, 0.2 equiv) and DTBP (0.15 mL, 4.0 equiv) were added to the above mixture, and the reaction mixture was stirred at 120 °C for 6 h under N2 atmosphere. The reaction was quenched with water, and the aqueous phase was extracted with EtOAc. The organic extracts were washed with brine, dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by flash chromatography to give 5.
Methylative lactonization
A screw cap tube was charged with CuSO4 (6.4 mg, 0.04 mmol, 0.2 equiv), 1,10-phenanthroline L2 (10.8 mg, 0.06 mmol, 0.03 equiv) and CF3CH2OH (2.0 mL). The mixture was stirred at room temperature for 30 min. Substrate 6 (0.2 mmol, 1.0 equiv), Na3PO4 (9.8 mg, 0.06 mmol, 0.3 equiv) and DTBP (0.15 mL, 4.0 equiv) were added to the above mixture, and the reaction mixture was stirred at 120 °C for 6 h under N2 atmosphere. The reaction was quenched with water and extracted with EtOAc. The organic extracts were washed with brine, dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by flash chromatography to give 7.
Methylative cycloamination
A screw cap tube was charged with Cu(OAc)2 (7.3 mg, 0.04 mmol, 0.2 equiv), 1,10-phenanthroline L2 (10.8 mg, 0.06 mmol, 0.03 equiv), and tBuOH (2.0 mL). The mixture was stirred at room temperature for 30 min. Substrate 8 (0.2 mmol, 1.0 equiv), Na3PO4 (6.5 mg, 0.04 mmol, 0.2 equiv) and DTBP (0.15 mL, 4.0 equiv) were added to the above mixture, and the reaction mixture was stirred at 120 °C for 3 h under N2 atmosphere. The reaction was quenched with water and the mixture was extracted with EtOAc. The organic extracts were washed with brine, dried over Na2SO4. The solvent was removed under reduced pressure. The residue was purified by flash chromatography to give 9.
Electronic supplementary material
Acknowledgements
We thank EPFL (Switzerland), the Swiss National Science Foundation (SNSF 20020_155973; SNSF 20021_178846) and the Swiss National Centers of Competence in Research (NCCR- Chemical Biology) for financial support.
Author contributions
X.B., T.Y., T.M.H., Q.W., and J.Z. conceived and designed the experiments. X.B., T.Y., and T.M.H. carried out the experiments. X.B., T.Y., T.M.H., Q.W., and J.Z. interpreted the results and X.B., Q.W., and J.Z. co-wrote the manuscript.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and Supplementary Information, as well as from the authors upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41467-018-06246-6.
References
- 1.Schönherr H, Cernak T. Profound methyl effects in drug discovery and a call for new C-H methylation reactions. Angew. Chem., Int. Ed. 2013;52:12256–12267. doi: 10.1002/anie.201303207. [DOI] [PubMed] [Google Scholar]
- 2.Yan G, Borah AJ, Wang L, Yang M. Recent advances in transition metal-catalyzed methylation reactions. Adv. Synth. Catal. 2015;357:1333–1350. doi: 10.1002/adsc.201400984. [DOI] [Google Scholar]
- 3.Hu L, Liu YA, Liao X. Recent progress in methylation of (hetero)arenes by cross-coupling or C–H activation. Synlett. 2018;29:375–382. doi: 10.1055/s-0037-1609093. [DOI] [Google Scholar]
- 4.Studer AA. “Renaissance” in radical trifluoromethylation. Angew. Chem., Int. Ed. 2012;51:8950–8958. doi: 10.1002/anie.201202624. [DOI] [PubMed] [Google Scholar]
- 5.Merino E, Nevado C. Addition of CF3 across unsaturated moieties: a powerful functionalization tool. Chem. Soc. Rev. 2014;43:6598–6608. doi: 10.1039/C4CS00025K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGrath NA, Brichacek M, Njardarson JT. A graphical journey of innovative organic architectures that have improved our lives. J. Chem. Educ. 2010;87:1348–1349. doi: 10.1021/ed1003806. [DOI] [Google Scholar]
- 7.Charpentier J, Früh N, Togni A. Electrophilic trifluoromethylation by use of hypervalent iodine reagents. Chem. Rev. 2015;115:650–682. doi: 10.1021/cr500223h. [DOI] [PubMed] [Google Scholar]
- 8.Mukaiyama, T., et al. Oxidation-reduction hydration of olefins with molecular oxygen and 2-propanol catalyzed by bis(acetylacetonato)cobalt(II). Chem. Lett.18, 449–452 (1989).
- 9.Waser J, Gaspar B, Nambu H, Carreira EM. Hydrazines and azides via the metal-catalyzed hydrohydrazination and hydroazidation of olefins. J. Am. Chem. Soc. 2006;128:11693–11712. doi: 10.1021/ja062355+. [DOI] [PubMed] [Google Scholar]
- 10.Barker TJ, Boger DL. Fe(III)/NaBH4-mediated free radical hydrofluorination of unactivated alkenes. J. Am. Chem. Soc. 2012;134:13588–13591. doi: 10.1021/ja3063716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shigehisa, H., et al. Catalytic synthesis of saturated oxygen heterocycles by hydrofunctionalization of unactivated olefins: unprotected and protected strategies. J. Am. Chem. Soc.138, 10597–10604 (2016). [DOI] [PubMed]
- 12.Girijavallabhan V, Alvarez C, Njoroge FG. Regioselective cobalt-catalyzed addition of sulfides to unactivated alkenes. J. Org. Chem. 2011;76:6442–6446. doi: 10.1021/jo201016z. [DOI] [PubMed] [Google Scholar]
- 13.Green SA, Matos JLM, Yagi A, Shenvi RA. Branch-selective hydroarylation: iodoarene–olefin cross-coupling. J. Am. Chem. Soc. 2016;138:12779–12782. doi: 10.1021/jacs.6b08507. [DOI] [PubMed] [Google Scholar]
- 14.King SM, Ma X, Herzon SB. A method for the selective hydrogenation of alkenyl halides to alkyl halides. J. Am. Chem. Soc. 2014;136:6884–6887. doi: 10.1021/ja502885c. [DOI] [PubMed] [Google Scholar]
- 15.Dao HT, Li C, Michaudel Q, Maxwell BD, Baran PS. Hydromethylation of unactivated olefins. J. Am. Chem. Soc. 2015;137:8046–8049. doi: 10.1021/jacs.5b05144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crossley SWM, Obradors C, Martinez RM, Shenvi RA. Mn-, Fe-, and Co-catalyzed radical hydrofunctionalizations of olefins. Chem. Rev. 2016;116:8912–9000. doi: 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maimone TJ, Shi J, Ashida S, Baran PS. Total synthesis of vinigrol. J. Am. Chem. Soc. 2009;131:17066–17067. doi: 10.1021/ja908194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poulin J, Grisé-Bard CM, Barriault L. A formal synthesis of vinigrol. Angew. Chem., Int. Ed. 2012;51:2111–2114. doi: 10.1002/anie.201108779. [DOI] [PubMed] [Google Scholar]
- 19.Gray P, Williams A. The thermochemistry and reactivity of alkoxyl radicals. Chem. Rev. 1959;59:239–328. doi: 10.1021/cr50026a002. [DOI] [Google Scholar]
- 20.Maeda M, Nushi K, Kawazoe Y. Studies on chemical alterations of nucleic acids and their components-VII: C-alkylation of purine bases through free radical process catalyzed by ferrous ion. Tetrahedron. 1974;30:2677–2682. doi: 10.1016/S0040-4020(01)97428-9. [DOI] [Google Scholar]
- 21.Zady MF, Wong JL. Kinetics and mechanism of carbon-8 methylation of purine bases and nucleosides by methyl radical. J. Am. Chem. Soc. 1977;99:5096–5101. doi: 10.1021/ja00457a033. [DOI] [PubMed] [Google Scholar]
- 22.Minisci F, Vismara E, Fontana F. Recent developments of free-radical substitutions of heteroaromatic bases. Heterocycles. 1989;28:489–519. doi: 10.3987/REV-88-SR1. [DOI] [Google Scholar]
- 23.Zhang Y, Feng J, Li CJ. Palladium-catalyzed methylation of aryl C−H bond by using peroxides. J. Am. Chem. Soc. 2008;130:2900–2901. doi: 10.1021/ja0775063. [DOI] [PubMed] [Google Scholar]
- 24.Kubo T, Chatani N. Dicumyl peroxide as a methylating reagent in the Ni-catalyzed methylation of ortho C–H bonds in aromatic amides. Org. Lett. 2016;18:1698–1701. doi: 10.1021/acs.orglett.6b00658. [DOI] [PubMed] [Google Scholar]
- 25.Zhang, P.-Z., et al. Metal-free radical C–H methylation of pyrimidinones and pyridinones with dicumyl peroxide. Green. Chem.19, 919–923 (2017).
- 26.Li, Q., et al. Cobalt-catalyzed C(sp2)-H methylation by using Dicumyl peroxide as both the methylating reagent and hydrogen acceptor. Chem. Eur. J.22, 12286–12289 (2016). [DOI] [PubMed]
- 27.Xia Q, Liu X, Zhang Y, Chen C, Chen W. Copper-catalyzed N-methylation of amides and O-methylation of carboxylic acids by using peroxides as the methylating reagents. Org. Lett. 2013;15:3326–3329. doi: 10.1021/ol401362k. [DOI] [PubMed] [Google Scholar]
- 28.Zhu, Y., et al. Copper-catalyzed methyl esterification reactions via C–C bond cleavage. J. Org. Chem.78, 9898–9905 (2013). [DOI] [PubMed]
- 29.Teng F, Cheng J, Yu JT. Copper-catalyzed N-methylation/ethylation of sulfoximines. Org. Biomol. Chem. 2015;13:9934–9937. doi: 10.1039/C5OB01558H. [DOI] [PubMed] [Google Scholar]
- 30.Bao, Y., et al. Copper-catalyzed radical methylation/C–H amination/oxidation cascade for the synthesis of quinazolinones. J. Org. Chem.80, 4736–4742 (2015). [DOI] [PubMed]
- 31.Xu Z, Yan C, Liu ZQ. A free-radical cascade methylation/cyclization of N-arylacrylamides and isocyanides with dicumyl peroxide. Org. Lett. 2014;16:5670–5673. doi: 10.1021/ol502738a. [DOI] [PubMed] [Google Scholar]
- 32.Dai, Q., et al. Di-tert butyl peroxide-promoted sequential methylation and intramolecular aromatization of isonitriles. Adv. Synth. Catal.356, 3341–3346 (2014).
- 33.Xu Z, Hang Z, Liu ZQ. Free-radical triggered ordered domino reaction: an approach to C-C bond formation via selective functionalization of α-hydroxyl-(sp3)C-H in fluorinated alcohols. Org. Lett. 2016;18:4470–4473. doi: 10.1021/acs.orglett.6b01946. [DOI] [PubMed] [Google Scholar]
- 34.Zhang, X., et al. Selective oxidative coupling reaction of isocyanides using peroxide as switchable alkylating and alkoxylating reagent. Adv. Synth. Catal.360, 272–277 (2018).
- 35.Fan JH, Zhou MB, Liu Y, Wei WT, Ouyang XH, Song RJ, Li JH. Iron-catalyzed oxidative arylmethylation of activated alkenes using a peroxide as the methyl source. Synlett. 2014;25:657–660. doi: 10.1055/s-0033-1340671. [DOI] [Google Scholar]
- 36.Dai, Q., et al. The carbomethylation of arylacrylamides leading to 3-ethyl-3-substituted indolin-2-one by cascade radical addition/cyclization. Chem. Commun.50, 3865–3867 (2014). [DOI] [PubMed]
- 37.Tan FL, Song RJ, Hu M, Li JH. Metal-free oxidative 1, 2-arylmethylation cascades of N-(arylsulfonyl)acrylamides using peroxides as the methyl resource. Org. Lett. 2016;18:3198–3201. doi: 10.1021/acs.orglett.6b01419. [DOI] [PubMed] [Google Scholar]
- 38.Tan FL, Hu M, Song RJ, Li JH. Metal-free annulation cascades of 1,7-enynes using di-tert-butyl peroxide as the methyl source towards the synthesis of polyheterocyclic scaffolds. Adv. Synth. Catal. 2017;359:3602–3610. doi: 10.1002/adsc.201700699. [DOI] [Google Scholar]
- 39.Dai Q, Jiang Y, Yu JT, Cheng J. Peroxide: A novel methylating reagent. Synthesis. 2016;48:329–339. doi: 10.1055/s-0035-1562795. [DOI] [Google Scholar]
- 40.Herk L, Stefani A, Szwarc M. Methyl affinities of some compounds related to acrylates and acrylonitriles. reactivities of conjugated systems involving atoms other than carbon. J. Am. Chem. Soc. 1961;83:3008–3011. doi: 10.1021/ja01475a007. [DOI] [Google Scholar]
- 41.Minisci F, Mondelli R, Gardini GP, Porta O. Nucleophilic character of alkyl radicals—VII: substituent effects on the homolytic alkylation of protonated heteroaromatic bases with methyl, primary, secondary and tertiary alkyl radicals. Tetrahedron. 1972;28:2403–2413. doi: 10.1016/0040-4020(72)80077-2. [DOI] [Google Scholar]
- 42.Zytowski T, Fischer H. Absolute rate constants for the addition of methyl radicals to alkenes in solution: new evidence for polar interactions. J. Am. Chem. Soc. 1996;118:437–439. doi: 10.1021/ja953085q. [DOI] [Google Scholar]
- 43.Zhu N, Zhao J, Bao H. Iron catalyzed methylation and ethylation of vinyl arenes. Chem. Sci. 2017;8:2081–2085. doi: 10.1039/C6SC04274K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang, Y., et al. Alkene oxyalkylation enabled by merging rhenium catalysis with hypervalent iodine(III) reagents via decarboxylation. J. Am. Chem. Soc.135, 18048–18051 (2013). [DOI] [PubMed]
- 45.Tlahuext-Aca A, Garza-Sanchez RA, Glorius F. Multicomponent oxyalkylation of styrenes enabled by hydrogen-bond-assisted photoinduced electron transfer. Angew. Chem., Int. Ed. 2017;56:3708–3711. doi: 10.1002/anie.201700049. [DOI] [PubMed] [Google Scholar]
- 46.Jian W, Ge L, Jiao Y, Qian B, Bao H. Iron-catalyzed decarboxylative alkyl etherification of vinylarenes with aliphatic acids as the alkyl source. Angew. Chem., Int. Ed. 2017;56:3650–3654. doi: 10.1002/anie.201612365. [DOI] [PubMed] [Google Scholar]
- 47.Li, Y., et al. Copper-catalyzed regioselective 1,2-alkylesterification of dienes to allylic esters. Org. Lett.18, 392–395 (2016). [DOI] [PubMed]
- 48.Wu K, Liang Y, Jiao N. Azidation in the difunctionalization of olefins. Molecules. 2016;21:352. doi: 10.3390/molecules21030352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Renaud P, Ollivier C, Panchaud P. Radical carboazidation of alkenes: an efficient tool for the preparation of pyrrolidinone derivatives. Angew. Chem., Int. Ed. 2002;41:3460–3462. doi: 10.1002/1521-3773(20020916)41:18<3460::AID-ANIE3460>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 50.Weidner K, Giroult A, Panchaud P, Renaud P. Efficient carboazidation of alkenes using a radical desulfonylative azide transfer process. J. Am. Chem. Soc. 2010;132:17511–17515. doi: 10.1021/ja1068036. [DOI] [PubMed] [Google Scholar]
- 51.Bunescu A, Ha TM, Wang Q, Zhu J. Copper-catalyzed three-component carboazidation of alkenes with acetonitrile and sodium azide. Angew. Chem., Int. Ed. 2017;56:10555–10558. doi: 10.1002/anie.201705353. [DOI] [PubMed] [Google Scholar]
- 52.Qian B, Chen S, Wang T, Zhang X, Bao H. Iron-catalyzed carboamination of olefins: synthesis of amines and disubstituted β-amino acids. J. Am. Chem. Soc. 2017;139:13076–13082. doi: 10.1021/jacs.7b06590. [DOI] [PubMed] [Google Scholar]
- 53.Liu YY, Yang XH, Song RJ, Luo S, Li JH. Oxidative 1,2-carboamination of alkenes with alkyl nitriles and amines toward γ-amino alkyl nitriles. Nat. Commun. 2017;8:14720–14725. doi: 10.1038/ncomms14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolfe, J. P. Palladium-catalyzed carboetherification and carboamination reactions of γ-hydroxy- and γ-aminoalkenes for the synthesis of tetrahydrofurans and pyrrolidines. Eur. J. Org. Chem. 571-582 (2007). [PMC free article] [PubMed]
- 55.Chemler SR, Fuller PH. Heterocycle synthesis by copper facilitated addition of heteroatoms to alkenes, alkynes and arenes. Chem. Soc. Rev. 2007;36:1153–1160. doi: 10.1039/b607819m. [DOI] [PubMed] [Google Scholar]
- 56.Ha TM, Wang Q, Zhu J. Copper-catalysed cyanoalkylative cycloetherification of alkenes to 1,3-dihydroisobenzofurans: development and application to the synthesis of citalopram. Chem. Commun. 2016;52:11100–11103. doi: 10.1039/C6CC06356J. [DOI] [PubMed] [Google Scholar]
- 57.Connolly, J. D. & Hill, R. A. Dictionary of terpenoids. Vol. 1, 476–545 (Chapman and Hall, London, 1991).
- 58.Collins I. Saturated and unsaturated lactones. J. Chem. Soc., Perkin Trans. 1999;1:1377–1395. doi: 10.1039/a808137i. [DOI] [Google Scholar]
- 59.Corey EJ, Kang MC. A new and general synthesis of polycyclic γ-lactones by double annulation. J. Am. Chem. Soc. 1984;106:5384–5385. doi: 10.1021/ja00330a076. [DOI] [Google Scholar]
- 60.Huang L, Jiang H, Qi C, Liu X. Copper-catalyzed intermolecular oxidative [3+2] cycloaddition between alkenes and anhydrides: a new synthetic approach to γ-lactones. J. Am. Chem. Soc. 2010;132:17652–17654. doi: 10.1021/ja108073k. [DOI] [PubMed] [Google Scholar]
- 61.Wei, X.-J., et al. A novel intermolecular synthesis of γ-lactones via visible-light photoredox catalysis. Org. Lett.15, 6054–6057 (2013). [DOI] [PubMed]
- 62.Ha TM, Chatalova-Sazepin C, Wang Q, Zhu J. Copper-catalyzed formal [2+2+1] heteroannulation of alkenes, alkylnitriles, and water: method development and application to a total synthesis of ( ± )-Sacidumlignan D. Angew. Chem., Int. Ed. 2016;55:9249–9252. doi: 10.1002/anie.201604528. [DOI] [PubMed] [Google Scholar]
- 63.Sha W, Ni S, Han J, Pan Y. Access to alkyl-substituted lactone via photoredox-catalyzed alkylation/lactonization of unsaturated carboxylic acids. Org. Lett. 2017;19:5900–5903. doi: 10.1021/acs.orglett.7b02899. [DOI] [PubMed] [Google Scholar]
- 64.Zhu R, Buchwald SL. Versatile enantioselective synthesis of functionalized lactones via copper-catalyzed radical oxyfunctionalization of alkenes. J. Am. Chem. Soc. 2015;137:8069–8077. doi: 10.1021/jacs.5b04821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Egami H, Kawamura S, Miyazaki A, Sodeoka M. Trifluoromethylation reactions for the synthesis of β-trifluoromethylamines. Angew. Chem., Int. Ed. 2013;52:7841–7844. doi: 10.1002/anie.201303350. [DOI] [PubMed] [Google Scholar]
- 66.Lin, J.-S., et al. A dual-catalytic strategy to direct asymmetric radical aminotrifluoromethylation of alkenes. J. Am. Chem. Soc.138, 9357–9360 (2016). [DOI] [PubMed]
- 67.Newcomb M, Chestney DL. A hypersensitive mechanistic probe for distinguishing between radical and carbocation intermediates. J. Am. Chem. Soc. 1994;116:9753–9754. doi: 10.1021/ja00100a052. [DOI] [Google Scholar]
- 68.Um C, Chemler SR. Synthesis of 2-aryl- and 2-vinylpyrrolidines via copper-catalyzed coupling of styrenes and dienes with potassium β-aminoethyl trifluoroborates. Org. Lett. 2016;18:2515–2518. doi: 10.1021/acs.orglett.6b01259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meng Y, Guo LN, Wang H, Duan XH. Metal-free oxidative hydroxyalkylarylation of activated alkenes by direct sp3 C-H functionalization of alcohols. Chem. Commun. 2013;49:7540–7542. doi: 10.1039/c3cc44055a. [DOI] [PubMed] [Google Scholar]
- 70.Cheng JK, Loh TP. Copper- and cobalt-catalyzed direct coupling of sp3 α-carbon of alcohols with alkenes and hydroperoxides. J. Am. Chem. Soc. 2015;137:42–45. doi: 10.1021/ja510635k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and Supplementary Information, as well as from the authors upon request.