

Summary
Neutralizing antibodies (NAbs) are traditionally thought to inhibit virus infection by preventing virion entry into target cells. Additionally, antibodies can engage Fc receptors (FcRs) on immune cells to activate antiviral responses. We describe a mechanism by which NAbs inhibit Chikungunya virus (CHIKV), the most common alphavirus infecting humans, by preventing virus budding from infected human cells and activating IgG-specific Fcγ receptors. NAbs bind to CHIKV glycoproteins on the infected cell surface and induce glycoprotein coalescence, preventing budding of nascent virions and leaving structurally heterogeneous nucleocapsids arrested in the cytosol. Furthermore, NAbs induce clustering of CHIKV replication spherules at sites of budding blockage. Functionally, these densely-packed glycoprotein-NAb complexes on infected cells activate Fcγ receptors, inducing a strong, antibody-dependent, cell-mediated cytotoxicity response from immune effector cells. Our findings describe a triply-functional antiviral pathway for NAbs that might be broadly applicable across virus-host systems, suggesting avenues for therapeutic innovation through antibody design.
Keywords: Chikungunya virus (CHIKV), neutralizing antibodies (NAbs), glycoproteins (GPs), nucleocapsid-like particles (NCLPs), stimulated emission depletion microscopy (STED), transmission electron microscopy (TEM), immunoelectron microscopy (IEM), cryo-electron tomography (cryoET), subtomogram averaging (STA)
eTOC
Jin et al. demonstrate that neutralizing antibodies (NAbs) inhibit chikungunya virus budding by inducing viral glycoprotein coalescence on infected cells. NAbs that crosslink glycoproteins engage Fc receptors on immune cells to activate antiviral responses. Considering the classical entry-inhibition function of NAbs, this work describes a triply-functional antiviral pathway for NAbs.
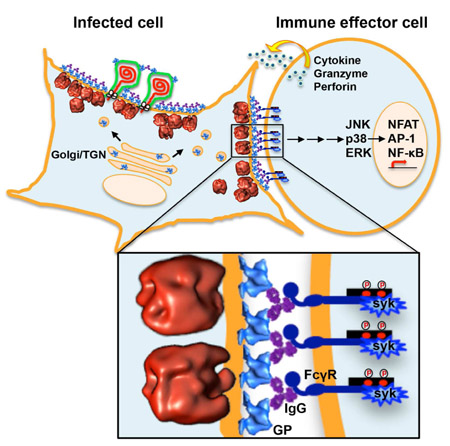
INTRODUCTION
Chikungunya virus (CHIKV) is the most common alphavirus infecting humans worldwide. A series of epidemics since 2004, of nearly pandemic proportions, have affected millions of people with severe, incapacitating, and often chronic arthralgia (Weaver et al., 2018). CHIKV, like other alphaviruses, is an enveloped, positive-stranded RNA virus. Its structural proteins [capsid (CP) and envelope (E1 and E2) glycoproteins (GPs)] assemble into mature virions that have T=4 quasi-icosahedral symmetry. Two icosahedral layers protect the positive-stranded RNA genome; namely, an outer GP shell of 240 E1-E2 heterodimers arranged into 80 trimeric GP spikes that are embedded in a lipid bilayer, and an internal nucleocapsid of 240 CPs encapsulating the genome (Sun et al., 2013). CHIKV virions enter target cells via endocytosis and the acidic environment in endosomes induces conformational changes in GPs allowing for fusion between the viral membrane and the host cell membrane (Solignat et al., 2009). Consequently, nucleocapsids (NCs) are released into the cytosol and quickly disassemble. Viral nonstructural proteins are then translated from the incoming viral genomic RNA in the cytosol and form replication complexes or “spherules” to synthesize viral RNA (Jose et al., 2009). Spherules are flask-shaped membrane compartments connecting to the cytosol through a narrow “neck”. Newly synthesized viral RNAs are exported through the neck into the cytosol. Viral structural proteins are synthesized from the subgenomic RNA in the cytosol where multiple copies of CP interact with genomic RNA to nucleate the assembly of NCs, which can occur independently of GPs in situ and in vitro (Mukhopadhyay et al., 2002). GPs assemble into trimeric spikes and are delivered to the plasma membrane through intracellular membrane trafficking. At the plasma membrane, horizontal interactions between GPs and vertical interactions between the cytosolic domain of E2 GPs with CPs are thought to drive the enveloping of assembled NCs and therefore the budding of nascent virions with outer icosahedral GP shells enclosing icosahedral NCs (Forsell et al., 1996; Forsell et al., 2000).
Compared with their well-studied entry process, the temporally and spatially orchestrated assembly/budding of alphaviruses is less well understood. Antiviral antibodies are traditionally screened for their ability to inhibit virus entry into target cells, from inhibition of receptor binding to prevention of conformational changes within the viral fusion proteins. Recently, we noticed an interesting antiviral effect for a panel of human and mouse anti-CHIKV neutralizing antibodies (NAbs) targeting at different epitopes; namely, NAbs inhibited the release of CHIKV particles from infected cells in addition to their expected canonical inhibition of virus entry into cells (Fox et al., 2015; Jin et al., 2015). However, the mechanism underlying this interesting phenotype and its functional consequences remained unresolved. In principle, NAbs could inhibit virus release by disrupting one or more of the following steps during virus production: 1) the intracellular assembly of NCs, 2) the recruitment of NCs to the plasma membrane, 3) the orchestrated assembly/budding of NCs into virions with NCs enveloped in the GP layer, 4) the pinching off of virions from the cell membrane, among others. Here, we visualized CHIKV-infected human cells using a combination of various microscopy techniques to determine the mechanism underlying the inhibition of CHIKV release by two ultra-potent human NAbs (C9 and IM-CKV063) targeting the CHIKV E2 glycoprotein.
Cryo-electron tomography (cryoET) and subtomogram averaging (STA) can visualize three-dimensional (3D) macromolecular structures at a resolution of a few nanometers in a near-native context (Galaz-Montoya and Ludtke, 2017). Indeed, cryoET is optimally suited to study conformationally and compositionally heterogeneous macromolecular complexes in vitro (Shahmoradian et al., 2013) and in situ (Asano et al., 2015; Baker et al., 2018; Dai et al., 2013; Pfeffer et al., 2015). Using cellular cryoET STA complemented with super-resolution stimulated emission depletion (STED) microscopy, transmission electron microscopy (TEM) and immuno-electron microscopy (IEM), we found that upon NAbs binding and crosslinking coalesced viral GPs failed to organize icosahedrally to drive virus budding, causing structurally heterogeneous nucleocapsid-like particles (NCLPs) to remain arrested in the cytosol.
Our study describes the mechanism by which NAbs inhibit virus budding from infected cell surface via binding to viral GPs and disrupting GPs driven envelopment of NCs. NAbs bound to coalesced GPs on the surface of infected cells activate Fcγ receptors via their Fc regions, inducing a strong, antibody-dependent, cell-mediated cytotoxicity (ADCC) response from immune effector cells. Therefore, our data suggest that these NAbs might exert a multi-fold, “triply functional” protective effect in vivo by blocking the assembly/budding of virions and eliciting the clearance of infected cells in addition to preventing the infection of new cells.
RESULTS
Neutralizing anti-CHIKV antibodies induced coalescence of viral GPs at the plasma membrane of mammalian cells.
We used sub-diffraction resolution STED microscopy to visualize U2OS cells that were infected with CHIKV vaccine strain 181/clone 25 (CHIKV 181) and treated with NAbs or with a non-neutralizing antibody (non-NAb) (Figure S1) (we observed n > 20 cells and analyzed three for each case). For all antibody treatment experiments, we administered the antibodies at 3 h post infection to allow for virus entry into the cells and initiation of viral genome replication, as described previously (Jin et al., 2015).
In cells treated with the non-NAb IM-CKV066, we observed that the antibody co-localized with viral structural proteins in distinct puncta that were distributed evenly over the plasma membrane, with some perinuclear accumulation assumed to correspond to endocytosed GP-antibody complexes (Figure S1A). Line profile analysis of the fluorescent puncta revealed sharp fluorescence intensity peaks of varying width (84-389 nm, n=418), with IM-CKV066 and CHIKV structural protein signals overlapping (Figure 1A). The diameter of virions is ~70 nm and the distance between antigen and fluorophore can range from 15 to 20 nm in a typical primary-secondary antibody complex binding to an antigen (Mennella et al., 2015). Therefore, we estimated that the size range of the puncta in the absence of NAbs (84-389 nm) corresponded to individual virions and small clusters of 2-5 virions. Importantly, the absence of a homogeneous fluorescent signal over the entire surface of the plasma membrane suggested that GPs were not distributed evenly on the plasma membrane; instead, they concentrated at distinct, relatively sparse sites of active virus budding.
Figure 1. NAbs crosslink viral glycoproteins into large patches on the surface of CHIKV-infected cells.

Enlarged cropped STED images of CHIKV-infected cells treated with (Ai) IM-CKV066 or (Bi) IM-CKV063. Cells were probed with Alexa647-conjugated (red) anti-human antibody and rabbit anti-CHIKV followed by Alexa594-conjugated (green) anti-rabbit antibody. Representative line profile analyses (along the blue arrows in Ai & Bi) of (Aii) fluorescent puncta or (Bii) patches. Size distributions of fluorescent (Aiii) puncta or (Biii) patches. The histogram insert in Biii shows the size distribution of patches larger than 1,000 nm. Scale bars: 1 μm.
In marked contrast, treatment with the NAb IM-CKV063 yielded some large patches of intense fluorescence (Figure S1B) of varying width (187 nm-12.7 μm, n=107) (Figure 1B) with overlapping CHIKV structural protein and IM-CKV063 signals. The NAb C9 induced a similar effect (not shown). The majority of patches were over 389 nm, the maximum width of the puncta seen in the non-NAb controls. Because the NAbs and non-NAb bind to CHIKV E2 GPs, our results suggested that NAbs induced the coalescence of GPs into large patches at the plasma membrane.
NAbs crosslinked GPs at the outer leaflet of the plasma membrane of CHIKV-infected cells, preventing the envelopment of NCs with plasma membrane.
To visualize virus budding in greater detail, we used thin-section electron microscopy to image CHIKV-infected RD cells that had been treated with anti-CHIKV monoclonal antibodies (Figures 2A, 2B & S2). In our controls [i.e., in the absence of antibodies (Figure S2A) or in the presence one of two non-NAbs targeting different epitopes (Fong et al., 2014), IM-CKV066 (Figures 2A & S2F) and IM-CKV062 (Figure S2G)], nascent budding particles and released virions were readily detected, yet relatively sparse, and sometimes found in small clusters of 2-5 particles, in agreement with the results obtained by STED microscopy (Figure 1A). In contrast, in the presence of human NAb C9 (Figures 2B & S2B) or IM-CKV063 (Figure S2C), NCLPs accumulated in the cytosol under the plasma membrane, which failed to curve around them; i.e., NAbs caused a phenotype that we henceforth will refer to as “NCLP arrest”. Two mouse NAbs CHK-9 (Figure S2D) and CHK-152 (Figure S2E) that we previously reported to inhibit CHIKV release caused the same “NCLP arrest” phenotype (Jin et al., 2015; Pal et al., 2013). In addition, our images captured flask-shaped viral spherules bulging from the cell surface of both control and NAb-treated cells (Figure S2). Viral spherules form early in the replication cycle, before GPs are translated, and their bulging out from the plasma membrane is thought to be GP-independent, driven by non-structural viral proteins and the synthesis of double-stranded RNA replication intermediates (Frolova et al., 2010). As expected, NAbs did not abrogate spherule formation in our experiments. Rather, NAbs thwarted virus budding by specifically inhibiting the envelopment of NCs, which normally requires for the plasma membrane incorporated with a GP lattice to curve around NCs (Garoff et al., 2004).
Figure 2. NAbs act on the outer leaflet of the plasma membrane to block CHIKV budding and leave a multitude of nucleocapsid-like particles arrested in the cytosol.

CHIKV-infected cells visualized by (A) TEM in the presence of non-NAb IM-CKV066 or (B) NAb C9, (C) IEM in the presence of non-NAb IM-CKV066 or (D) NAb C9 labeled with anti-human antibodies conjugated to 6-nm colloidal, and (E) cryoET (and corresponding annotation) in the absence of antibody treatment or (F) in the presence of NAb C9. Selected regions from representative cryoET tomograms are shown. A budding particle is indicated with a black arrow in C. Budding-arrested NCLPs are indicated with a red arrow in D. Virions in E and NCLPs in F are labeled in red. The cell membrane is labeled in yellow. Additional density on thickened plasma membrane patches is shown in grey blue in F, while viral spherules are labeled in green. Actin filaments and intermediate filaments are labeled in dark blue and magenta, respectively. Scale bars: 200 nm for A, B and D; 100 nm for C, E and F.
To better understand how NAbs induce NCLP arrest, we used IEM with anti-human antibodies conjugated to 6-nm colloidal gold to determine the localization of NAbs administered to infected cells. We observed gold particles lining the surface of infected cells and noticed a plethora of arrested NCLPs under the inner leaflet of the plasma membrane (Figures 2D, S3A & B). In contrast, in the presence of non-NAb, we readily detected released and budding virus particles labeled with gold-conjugated secondary antibodies (Figures 2C & S3C).
We previously reported that full IgG molecules but not Fab fragments of NAbs inhibit CHIKV release from cells (Jin et al., 2015). Therefore, considering such previous reports and our STED analyses above (Figure 1B), our IEM results here suggested that NAbs likely crosslinked viral spikes on the outer leaflet of the plasma membrane via bivalent binding of one IgG molecule to two E2 GPs in separate spikes. Consequently, GPs coalesced into large patches on the plasma membrane, which then failed to curve around NCs to enclose them, as required for virion assembly and budding.
In addition, our IEM images revealed the presence of GPs on the surface of viral replication spherules bound with monoclonal antibodies against E2 GP labeled with gold-conjugated anti-human antibodies (Figure S3B & C). The spherules bulging out from the cell surface often seemed to cluster at the sites of budding inhibition (Figure S3B), suggesting that NAbs might crosslink GPs on neighboring spherules. Released virions, budding particles, cytosolic NCLPs, and spherules, all of which are structural hallmarks of virus infection, were absent from control cells expressing CHIKV GPs only (Figure S3E).
To better understand the topology of CHIKV budding inhibition by NAbs, we collected TEM tiltseries of the same samples examined in Figure 2D and Figure S3A, B & D and reconstructed them into tomograms. The tomograms in the absence of NAbs showed normal virus budding and release (Movie S1 & Figure S5A). In contrast, the tomograms in the presence of NAbs (Movie S2) confirmed that NAbs bound to GPs at the outer surface of cells, leaving a host of NCLPs arrested in the cytosol (Figure S6). Furthermore, these tomograms also confirmed that spherules contained GPs on their surface (Figure S5Bi), which sometimes appeared to be crosslinked by NAbs (Figure S5Bii).
CryoET of CHIKV-infected cells suggested that treatment with NAbs might disrupt the normal organization of GPs at the plasma membrane.
Alphavirus GP spikes are proposed to be delivered to the plasma membrane as sheets of hexagonal arrays (Soonsawad et al., 2010). However, the native arrangement of GPs has never been directly observed on the plasma membrane of infected cells. To avoid artifacts from chemical fixation while visualizing CHIKV budding inhibition by NAbs in human cells, we collected 20 cryoET tiltseries from regions at the periphery of ice-embedded CHIKV-infected cells (i.e., the regions thin enough for cryoET imaging) without stains or fixatives, and reconstructed them into tomograms after motion and CTF correction (Figure 2E, 2F & S4). Consistent with our TEM and IEM results (Figures 2A-D, S2, 3, 5 & 6), our cryoET data revealed budding/released viral particles in the absence of NAbs (Figure 2E & Movie S3).
In our cryoET tomograms, the plasma membrane of CHIKV-infected cells in the absence of NAbs appeared smooth (Figures 2E & 3Ai, & Movie S3). STA of plasma membrane subtomograms extracted from CHIKV-infected cells in the absence of NAbs converged to an average that matched the expected thickness of mammalian cell membranes (Figure 3Aii). Low-density masses were present on the outer leaflet, which might be attributable to cell-surface proteins and/or sparse viral GPs, suggesting that GPs were not continuously distributed in high density on the cell surface, but instead concentrated at relatively dispersed and small active budding spots, readily enwrapping budding NCs (Figure 4A), in good agreement with results from our STED experiments (Figure 1A). The average of plasma membrane subtomograms in the absence of NAbs also showed strong, filamentous internal density beneath the plasma membrane that might correspond to cytoskeletal components (Figure 3Aii), which have been hypothesized to aid virus egress.
Figure 3. STA of coalesced NAb-GPs at the surface of CHIKV-infected cells.

Re-projections of representative subtomograms from the plasma membrane of CHIKV-infected cells in (Ai) the absence or (Bi) presence of NAb C9, and corresponding subtomogram averages (Aii, Bii).
Figure 4. STA of released virions.

(A) Tomographic slices from selected views of cryoET tomograms of CHIKV-infected cells showing small clusters of released virions. (B) Reference-free icosahedrally refined subtomogram average of released virions.
In marked contrast, the membrane of CHIKV-infected cells treated with NAbs was coated with extra density on the outer leaflet at sites of NCLP arrest (Figures 2F & 3Bi, & Movie S4). Given our IEM observation that NAbs bound to GPs on the outer leaflet of the plasma membrane (Figures 2D & S3A), we attributed the extra density to coalesced NAb-GP complexes. The organization of GP spikes into an icosahedron during virion assembly at the plasma membrane has been hypothesized to drive virus budding (Forsell et al., 1996; Forsell et al., 2000). Therefore, we investigated whether GPs would exhibit any symmetric organization in the presence of NAbs at the sites of NCLP arrest where there was clear extra density coating the plasma membrane. We extracted subtomograms containing thickened membrane patches and subjected them to STA. Our results showed extra density beyond the cell membrane, on both the inner and the outer leaflets (Figure 3Bii). The extra density in the cytosol might be attributable to a combination of intracellular GP tails, components of the cellular milieu, and arrested NCLPs. Of note, these intracellular densities lacked the cytoskeleton-like order seen in the absence of NAbs (Figure 3Aii). On the outer leaflet of the plasma membrane, the ~8–9 nm thick layer of extra density matched the protruding height of GPs above the lipid bilayer in mature virions (Sun et al., 2013), and was thus attributable to coalesced viral spikes whereas the lighter layer extending further out might be attributable to floppy NAbs on top (Figure 3Bii). Of note, coalesced GPs did not appear to be arranged symmetrically, suggesting that they were conformationally and/or compositionally heterogeneous. However, if GPs were indeed natively hexagonally ordered in flat sheets on the plasma membrane as previously hypothesized (Soonsawad et al., 2010), our results here suggested that NAb binding disrupted such organization, thereby inhibiting CHIKV budding.
Arrested NCLPs inside CHIKV-infected cells in the presence of NAb were pleomorphic and heterogeneous.
The tomograms of fixed, CHIKV-infected cells treated with NAbs allowed to measure the size of the NCLPs in 3D, which can be challenging or impractical in two dimensions because of overlapping densities that might come from different Z planes in the specimen. In our IEM tomograms, the NCLPs (n=98) were structurally heterogeneous, with spherically averaged radii (or “radius of gyration”) ranging from 5.3 to 16.0 nm (μ=12.8 nm, σ=1.78 nm) (Figure S6B), in good agreement with manual measurements from the mid-Z slice of representative NCLPs taken directly from the tomograms. The known radius of the CHIKV nucleocapsid core inside mature virions is ~19.3 nm, while the radius of a subpopulation of icosahedrally symmetric particles among purified Venezuelan equine encephalitis virus (VEEV, another alphavirus) NCs was ~21 nm in two independent studies (Lamb et al., 2010; Paredes et al., 2003). In addition to the icosahedrally symmetric map derived from a small set of 90 projection images, Lamb et al., 2010 also reported a wide size range for purified NCs (15.5-27 nm in radius, μ=21.5 nm, σ=1.75 nm). This suggested that budding-arrested NCLPs in our IEM tomograms were either incomplete (either not fully assembled or starting to disassemble) and/or shrunk as an artifact due to chemical fixation during specimen preparation.
Our cryoET tomograms of CHIKV-infected cells in the absence or presence of NAbs allowed us to visualize normal virus budding and budding-arrested NCLPs without artifacts from chemical fixation. We first used self-symmetry alignment (SSA) (Dai et al., 2013; Galaz-Montoya et al., 2015) to create a reference-free initial model for released virions (n=47) in the absence of NAbs. Iterative-refinement STA of virions using this initial model yielded an icosahedrally symmetric map (Figure 4B) resolved to ~3.4 nm resolution (Figure S7A), in good agreement with the known structure of CHIKV (Sun et al., 2013). Next, from our cryoET tomograms of CHIKV-infected cells treated with NAbs, we extracted 1,163 budding-arrested NCLPs (Figure 5), which appeared structurally heterogeneous and pleomorphic upon visual inspection (Figure 5A). We could not rule out the possibility that a subset of NCLPs might be icosahedrally symmetric; however, after extensive analyses (see STAR Methods), including the use of a symmetric initial model, we could not find any evidence for strict icosahedral symmetry among this set of in situ NCLPs. Since refinement (iterative alignment and averaging) of structurally heterogeneous particles together can prevent them from converging to a robust average, we classified the NCLPs by size and carried out STA among particles with the same radius (the classes were 7.9-27.9 nm in radius, μ=18.3 nm, σ=3.0 nm) (Figure 5B). The size distribution for NCLPs was shifted towards larger radii compared to that for particles from our IEM tomograms and was much closer to the size range reported for purified NCs (15.5-27 nm in radius) (Lamb et al., 2010). In our cryoET tomograms, most NCLPs had sizes near the mean, within a small (~5%) difference from the radius of NCs inside virions. The size classes at either end of the spectrum (representing particles with radii ~8-15 nm and ~23-28 nm) were sparsely populated and failed to yield good subtomogram averages. On the other hand, five classes within the 15.8-22.2 nm radius range (Figure 5C, 49 < n < 107 per class) converged to structures at ~3.6-4 nm resolutions (Figure S7B), comparable to the resolution obtained for the subtomogram average of released virions (~3.4 nm) (Figure S7A), considering that NCLPs were not icosahedrally averaged.
Figure 5. Arrested NCLPs in the cytosol of CHIKV-infected cells are structurally heterogeneous.

(A) ~1.5 nm-thick slice through a cryoET tomogram of a CHIKV-infected cell in the presence of NAb C9 (scale bar: 130 nm). (B) Histogram of spherically averaged radii of NCLPs. (C) Isosurfaces of subtomogram averages for five classes of NCLPs classified by size (n=49, 86, 107, 75, 61).
NAbs induced clustering of viral spherules at the cell surface.
Consistent with the results from TEM and IEM experiments (Figures S3B & S6), cryoET revealed clusters of viral spherules in the presence of NAbs, often close to the sites of NCLP arrest (Figures 2F & 6A, & Movie S4). The spherules (n=99) spanned a wide range of sizes (~400,000 nm3-5,000,000 nm3 in volume)(Figure 6B). Furthermore, their shapes often deviated from spherical, being ellipsoidal or pleomorphic. Spherules that were not clustered by NAbs appeared smaller and more uniform, closer to a spheroid (with minor and major axes ranging between 47-58 nm and 56-68 nm, respectively), in good agreement with the reported diameter of 58 nm for Semliki Forest virus (SFV) spherules (Kallio et al., 2013). In contrast, spherules clustered by NAbs were larger and more strongly ellipsoidal on average (with minor and major axes ranging between 45-103 nm and 69-132 nm, respectively) (Figures 6B). In our cryoET tomograms, the spherules seemed to contain wrapped fibrillar densities (Figure 6A) resembling those reported for dsRNA inside spherules of Flock House Nodavirus (Ertel et al., 2017). On the other hand, in our TEM and IEM experiments, chemical fixation condensed the dsRNA into a small clump in the middle of the spherules with a few fibrils connected to the center (Figures S2, S3 & S5).
Figure 6. CryoET of viral spherules.

(A) Slices of representative spherules from cryoET tomograms of CHIKV-infected cells treated with NAb C9 as shown in Figure 3A. Red arrows indicate connections between spherules. Yellow arrows indicate elongated spherules. White arrows indicate wrapped fibrillar densities inside spherules. (B) Size distribution of CHIKV spherules by volume and by major and minor axis length. Scale bars: 50 nm.
NAbs that crosslinked CHIKV GPs at the cell surface induced ADCC from immune effector cells against CHIKV-infected human cells
Results from our microscopy experiments above suggested that NAbs crosslinked GPs at the surface of CHIKV-infected cells, inducing the formation of large, dense patches of NAb-GP complexes. We therefore hypothesized that this dense layer of coalesced NAb-GPs should be able to bind and crosslink Fc receptors on the surface of immune effector cells, leading to the induction of ADCC. To test this hypothesis, we incubated reporter cells expressing FcγRIIIa with antibody-treated, CHIKV-infected cells. We quantified the induction of ADCC using the luciferase reporter that was produced as the result of activating the nuclear factor of activated T-cells (NFAT) pathway. We found that NAbs C9 and IM-CKV063 were indeed able to specifically induce strong ADCC from effector cells bearing FcγRIIIa in a concentration-dependent manner, while their N297Q mutants failed to activate ADCC response due to a lack of the glycosylation required for interaction with FcγRIIIa (Figure 7A). Two non-NAbs IM-CKV066 and IM-CKV062 (which did not crosslink GPs into large, dense patches) only induced a weak ADCC response at very high concentrations. A control NAb against dengue virus failed to induce ADCC against CHIKV-infected cells (Figure 7A). These functional results support our hypothesis that IgGs crosslink GPs at the surface of CHIKV-infected cells via the Fab regions to block virus budding, while the Fc regions crosslink Fcγ receptors at the surface of effector cells to induce cytotoxicity against infected cells (Figure 7B).
Figure 7. NAbs induce ADCC against CHIKV-infected cells.

Cells were infected with CHIKV before being cultured in the presence of NAb C9 or IM-CKV063, or their aglycosylated mutants C9 (N297Q) and IM-CKV063 (N297Q), or non-NAb IM-CKV066, or a control NAb against DENV. FcγRIIIa-bearing reporter effector cells were added to target CHIKV-infected cells at an E:T ratio of 6. (A) Fold of induction of ADCC quantified by luciferase expression driven by NFAT. (B) A model diagram of the NAb-induced budding inhibition and ADCC against CHIKV-infected cells.
DISCUSSION
CHIKV is transmitted to humans by Aedes mosquitoes and causes an acute febrile illness that is typically accompanied by severe arthralgia, with relapses that last weeks to years (Weaver et al., 2018), resulting in great economic losses due to loss of productivity. In the past decade, CHIKV has spread from endemic areas of Africa and Asia to Europe and the Americas, with millions of individuals infected during the 2000s, resulting in thousands of deaths (Johansson, 2015). Although we, and others, have isolated CHIKV NAbs and demonstrated their ability to protect against infection (Fong et al., 2014; Jin et al., 2015; Pal et al., 2013; Smith et al., 2015), there is currently no approved vaccine or treatment against CHIKV infection. Previous studies have evaluated antibodies for their ability to block virus entry in vitro, following the traditional paradigm in immunology. Although we previously reported a dual antiviral effect in which CHIKV NAbs block virus entry and virus release (Jin et al., 2015), the mechanism for blocking virus release was not resolved, and its functional implications were not explored. Here, we report the structural basis underlying the mechanism by which NAbs inhibit CHIKV release. Importantly, the model we described here for NAb function targeting infected cells kills two birds with one stone: inhibition of nascent virus budding at the surface of infected cells and activation of immune effector cells to clear infected cells. Considering that these NAbs also block virus entry into cells, their protection seems to be triply-functional, providing a mechanistic explanation to their known antiviral super-potency.
Inhibition of viral release by antibodies has been reported for viruses from a number of different families (Corboba et al., 2000; Kajihara et al., 2012; Masrinoul et al., 2014; Vanderplasschen et al., 1997); however, the underlying mechanisms remain largely unknown. Previous thin-section TEM studies with two non-neutralizing antibodies against Marburg virus (Kajihara et al., 2012) and a broadly reactive antibody against influenza A virus (Yamayoshi et al., 2017) have suggested a tethering model for the inhibition of virus release. It was proposed that those antibodies might allow for normal virus budding while tethering released virions to viral GPs on the cell surface, but the underlying mechanisms for this proposed tethering model are yet to be elucidated. On the other hand, here we showed NAbs crosslinking viral GPs to inhibit virus budding, leaving a myriad of NCs arrested inside cells. Therefore, we propose that a lower concentration of NAbs might be required to block virion budding than to tether every budded virion to the cell surface, or to completely neutralize virus entry. Furthermore, the inhibition of virus budding through the mechanism we describe here elicited a strong ADCC response from immune effector cells, a response not yet demonstrated for antibodies that tether virions to the surface of cells. Of note, the anti-budding mechanism we describe here might apply to other viruses whose budding is driven by GPs at the cell surface. Furthermore, the activation of ADCC suggests that these antibodies might promote the clearance of infected cells in vivo, warranting further studies to test such hypothesis.
Although CPs can independently assemble into NCs both in cells and in vitro, nucleocapsid preassembly in the cytosol is not a prerequisite for alphavirus budding. Indeed, the budding of assembly-deficient NCs can be rescued by horizontal spike-spike interactions and vertical spike-capsid interactions (Forsell et al., 2000), suggesting that those interactions drive virus budding. In situ electron tomography of chemically fixed SFV-infected cells in a previous study suggested that trimeric GP spikes might already be assembled and organized into hexagonal lattices in cellular membrane compartments prior to being delivered to the plasma membrane, where they are required for virus budding (Soonsawad et al., 2010). Mutations in the D-loop of the SFV E2 protein that disrupt the contact between the viral E1 and E2 proteins inhibit SFV budding, further suggesting that perturbations to the native organization of the GP lattice can result in budding arrest (Byrd and Kielian, 2017). Here, we demonstrated that NAbs can crosslink viral GPs on the extracellular side of the plasma membrane to induce their coalescence. STA of dense NAb-GP patches from cryoET tomograms yielded averages with no hexagonal organization or any regular pattern, suggesting that NAb crosslinking disrupts the normal organization of GPs, which are hypothesized to drive alphavirus budding (Forsell et al., 1996; Forsell et al., 2000).
Alphavirus nucleocapsid cores purified from cells vary in size and density and are less structurally stable than mature virions (Lamb et al., 2010). In two previous studies, a subpopulation of VEEV NCs isolated from virions stripped from their envelope (Paredes et al., 2003) or purified from infected cells (Lamb et al., 2010) was icosahedrally symmetric when visualized in vitro by cryoEM single particle analysis (SPA). Yet, in both studies the organization of CPs in the isolated NCs differed from that inside mature virions, suggesting that NCs are highly plastic and undergo GP-driven and/or lipid-driven structural changes upon envelopment. Importantly, the previous studies did not address structural heterogeneity among NCs. Indeed, Lamb et al. (2010) discarded many particles due to poor correlation against the average refined with icosahedral symmetry imposed. The discarded particles included those that differed in size or were otherwise distorted/damaged, as is commonly done in SPA aiming to computationally purify specific conformations of macromolecular complexes in order to determine their structure at the highest possible resolution. However, a more recent study reported that even NCs inside mature Ross River virus (another alphavirus) virions presented structural heterogeneity deviating from icosahedral symmetry (Wang et al., 2015). For in vitro studies, there is always the concern that harsh chemical manipulation to purify macromolecular complexes can result in structural damages, giving rise to artificial, non-native heterogeneity. On the other hand, the heterogeneity that we observed here across budding-arrested NCLPs in situ in the presence of NAbs might be attributable to a combination of assembly/disassembly intermediates, natural structural variations among fully assembled NCs, and both of these types of NCLPs interacting with host factors inside the cytosol, where molecular crowding can have dramatic effects on the composition, conformation, and function of macromolecular complexes (Grunewald et al., 2003).
Resolving CHIKV assembly/disassembly intermediates in 4D (a time course study combined with 3D cryoET) inside cells in the absence of NAbs might reveal mechanistic aspects of virus production in great detail, such as the interactions of NCs with cellular host factors, and how the organization of GPs on the plasma membrane drives the envelopment of NCs. However, CHIKV budding appears to be a relatively sparse and quick process that is challenging to capture by high-magnification cryoET restricted to the thin cell periphery. Indeed, intracellular NCs were incredibly scant in our TEM and cryoET data in the absence of NAbs. Therefore, the budding arrest induced in our system by NAb treatment, which leads to the accumulation of arrested NCLPs inside the cytosol, provides a unique opportunity to study alphavirus nucleocapsid assembly/disassembly intermediates in situ, warranting future infection time-course studies (Murata et al., 2017), with much larger datasets, and enhancing the contrast and resolution, possibly with the aid of phase plates (Asano et al., 2015; Dai et al., 2013). The advent of structural biology in situ (Galaz-Montoya and Ludtke, 2017) brought about by the recent emergence of revolutionary technology in cryo electron microscopy and tomography such as direct electron detection cameras (McMullan et al., 2014), phase plates (Danev and Baumeister, 2017), correlative light and electron microscopy, and focused ion beam milling (Wang et al., 2012), will increasingly yield insights into viral infection and other biological processes in a native cellular context.
The synthesis of viral dsRNA is required for the formation of spherules at the plasma membrane very early during alphavirus infection, before the synthesis of viral structural proteins (Frolova et al., 2010). Unlike SFV and Sindbis virus spherules, which are internalized at later time points during infection, the majority of CHIKV spherules remain on the cell surface throughout the course of infection (Thaa et al., 2015). In our TEM images, spherules seemed to be distributed dispersedly on the plasma membrane in the absence of NAbs. Unexpectedly, spherules in tomograms of NAb-treated cells agglomerated in clusters. Since our 2D and 3D TEM/IEM images and tomograms revealed the presence of GPs bound with NAbs on the surface of spherules, spherule clustering might result from NAbs crosslinking GPs on neighboring spherules. Interestingly, the spherules in intertwined clusters were enlarged compared with SFV spherules for which the size of the viral genome is thought to be the main determinant of spherule size (Kallio et al., 2013). Since the CHIKV and SFV genomes are similar in length (11.8 kb for CHIKV vs 11.4 kb for SFV), the size discrepancy between SFV spherules and the enlarged, clustered CHIKV spherules we observed might arise from differences in experimental factors (chemical fixation vs near-native preservation by plunge freezing) and data analysis (reporting only one parameter for spherule size vs measuring the major and minor axes of the central cross section of each ellipsoidal spherule). Alternatively, the clustering of spherules through NAb crosslinking might cause them to swell by perturbing their normal function and dynamics.
Functionally, our study demonstrated that NAbs that crosslink viral GPs on the plasma membrane of CHIKV-infected cells were able to engage Fc receptors on effector cells to induce ADCC against CHIKV-infected cells. The density of IgG molecules coating the infected cells is a critical factor for the crosslinking and activation of Fcγ receptors as well as for the downstream induction of ADCC (Bournazos and Ravetch, 2015). Our results here, taken together with our previous studies (Jin et al., 2015; Fox et al., 2015), lead to a model in which bivalent binding of IgGs to viral GPs via Fab regions induces coalescence of viral spikes on the surface of CHIKV-infected human cells. In turn, a dense display of IgGs on coalesced GP patches activates Fcγ receptors on the surface of effector cells via their Fc regions. Fcγ receptor-mediated cellular activation of effector leukocytes, such as monocytes, macrophages, neutrophils, and NK cells, should result in ADCC, phagocytosis, and cytokine and chemokine release, facilitating the cytotoxic lysis and clearance of infected cells. Our results here provide an answer to one of the central questions in a recent review of super-potent antibodies regarding why in vitro neutralization does not always correlate well with in vivo efficiency (Walker and Burton, 2018). As we showed here, NAbs can inhibit pathogens at different stages of their life cycle. These observations warrant further studies in vivo to determine whether the same or similar clearance mechanisms can better protect organisms against infection and the associated pathogenesis compared to only preventing the entry of virus particles into cells.
In summary, the results of our study suggest a model that explains the mechanism by which NAbs inhibit CHIKV release from infected cells. We found that the specific step of the CHIKV production process that is inhibited by NAbs is the envelopment/budding of NCs. IgG molecules crosslink viral GP spikes on the plasma membrane of CHIKV-infected cells, likely disrupting the normal organization of GPs that drives the plasma membrane to curve around NCs. As a result, structurally heterogeneous NCLPs remain arrested under the plasma membrane, and Fc receptors are activated to induce ADCC from immune effector cells to clear virus-infected cells. Our findings provide a better understanding of super, multi-functional antibodies and shed light on alphavirus assembly in and budding from cells, highlighting the importance of evaluating antibody functions throughout the complete virus replication cycle. This paradigm shift towards a comprehensive model of virus inhibition, deviating from the traditional view that focuses heavily on strength of entry neutralization, opens avenues for improved antiviral therapy development.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Graham Simmons (gsimmons@bloodsystems.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human muscle-cell line RD (Cat. #CCL-136), bone-cell line U2OS cells (Cat. #HTB-96) and adenovirus 5 DNA transformed human kidney epithelial cell line 293 c18 (Cat. #CRL-10852) are all female cell lines purchased from American Type Culture Collection (ATCC). Hamster fibroblast cell line BHK21 cells (Cat. #CCL-10) were purchased from ATCC. Cells were maintained at 37 °C with 5% humidified CO2 in DMEM (Invitrogen) supplemented with penicillin and streptomycin, 10 mM HEPES, non-essential amino acids, and 10% FBS (Hyclone).
Virus strains
CHIKV strain 37997 and vaccine strain 181/clone 25 (CHIKV 181) were amplified in BHK21 cells.
METHOD DETAILS
Transfection
RD or 293 c18 cells were transfected using TransIT 2020 transfection reagent (Mirus) at a ratio of 3 μl of TransIT 2020 to 1 μg DNA diluted in Opeti-MEM (Thermo Fisher Scientific). Cells were at ~80-90% confluency.
Antibody production
Sequences for all antibody light chain (LC) and heavy chain (HC) were sub-cloned in pTT5 expression vector. Paired LC and HC expressing vectors were transfected into 293 c18 cells and antibodies were purified from the supernatant of the transfected cells. N297Q mutation was introduced to HC of C9 and IM-CKV063 by PCR-based site-direct mutagenesis to generate aglycosylated C9 and IM-CKV063.
Antibody labeling
Alexa 488-conjugated C9 was generated with the Alexa Fluor™ 488 protein labeling kit, per the manufacturer’s instructions (Invitrogen).
Fluorescence microscopy of CHIKV-infected cells
Antibody concentration and treatment time and multiplicity of infection (MOI) were optimized to reach the most efficient inhibition of virus release by NAbs with a majority of cells infected to facilitate microscopy observation. U2OS cells were infected with CHIKV 181 at a multiplicity of infection (MOI) of 10 for 3 h. After extensive washing, the cells were cultured for 7 h in the presence of 10 μg/ml NAb C9 or IM-CKV063, or the non-NAb IM-CKV066 as a control. At 10 h post infection, the cells were fixed, permeabilized, and then probed with rabbit anti-CHIKV 181 polyclonal antibody (IBT Bioservices, Gaithersburg, MD, USA) followed by staining with Alexa 594-conjugated anti-rabbit and Alexa 647-conjugated anti-human antibodies (Invitrogen). Confocal and STED super-resolution microscopy images were taken with a Leica TCS SP8 STED 3X Nanoscope.
TEM of CHIKV-infected cells
RD cells were infected with CHIKV 37997 at an MOI of 1 for 3 h. After extensive washing, the cells were cultured in the presence or absence of 2 μg/ml human NAb C9, IM-CKV063, or mouse NAb CHK9, CHK152, or the human non-NAb IM-CKV066, IM-CKV062. Then, 20 mM NH4Cl were added to prevent further rounds of infection. At 18 h post infection, the cells were fixed with 2.5% glutaraldehyde. After dehydration, the cells were embedded in an epoxy resin, sectioned ultra-thin (60-80 nm), and examined using a transmission electron microscope.
IEM
RD cells were infected with CHIKV 37997 at an MOI of 1 for 3 h. After extensive washing, the cells were cultured in the presence or absence of 2 μg/ml NAb C9, IM-CKV063, or the non-NAb IM-CKV066. Then, 20 mM NH4Cl were added to prevent further rounds of infection. At 18 h post infection, the cells were fixed with 2% paraformaldehyde and 0.025% glutaraldehyde. RD cells were transfected with pCAGGS CHIKV E3/E2/E1 plasmid (Salvador et al., 2009). At 6 h post transfection, the cells were cultured in the presence or absence of 2 μg/ml NAb C9. At 18 h post transfection, the cells were fixed with 2% paraformaldehyde and 0.025% glutaraldehyde. Cells were probed with goat anti-human antibodies conjugated to 6-nm colloidal gold before post-fixation with osmium tetroxide. After dehydration, the cells were embedded in an epoxy resin, sectioned ultra-thin (60-80 nm), and examined using a transmission electron microscope.
IEM tomography
Electron tomography was performed using the IEM samples. In those experiments, semi-thick sections (~150 nm thick) were cut, and dual-axis tiltseries were collected using a Tecnai TF30 microscope operated at 300 kV. Tomographic reconstruction was carried out using the IMOD software package as previously described (Kremer et al., 1996).
CryoET of CHIKV-infected cells
U2OS cells grown on fibronectin-coated gold R2/2 Quantifoil finder EM grids were infected with CHIKV 181 at an MOI of 10 for 3 h. After extensive washing, the cells were cultured for 7 h in the presence or absence of 10 μg/ml Alexa 488-conjugated C9. At 10 h post infection, the grids were imaged with a fluorescence microscope to assess antibody binding to infected cells. Upon confirming binding of antibodies, the grids were one-side blotted with filter paper and plunged into liquid ethane for rapid vitrification. After optimizing vitrification having screened multiple grids in different days, we collected 20 cryoET tiltseries at the regions of interest in the presence or absence of NAbs from different grids during multiple imaging sessions on different days using a JEM2200FS (JEOL Ltd., 3-1-2 Musashino, Akishima, Tokyo 196-8558, JAPAN) electron microscope operated at 200 kV equipped with an in-column energy filter (omega type), using a condenser lens aperture of 100 mm, no objective aperture, spot size 3. The energy filter slit was set to a width of 20 eV. Micrographs were recorded using a DE-20 direct electron detector (Direct Electron, LP, San Diego, CA, USA) at a recording rate of 25 raw frames per second and a total exposure time of 0.6 s, yielding 15 frames per stack at each tilt angle. Tiltseries were acquired semi-automatically using SerialEM software (Mastronarde, 2005) in low-dose mode with a target defocus at the tilt axis of −6 μm. For each tiltseries, the images were acquired from a target range of −60° to +60° in increments of 2°. Frames were motion corrected using DE software (Direct Electron LP, San Diego, CA, USA). The magnification was set to 10,000x for six tiltseries to acquire a larger field of view, yielding images with a sampling size of 5.74 Å/pixel, and to 15,000x for the other 14 tiltseries, yielding images with a sampling size of 3.58 Å/pixel (total cumulative dose for the latter ~ 60 e/Å2).
ADCC assay
An ADCC reporter bioassay core kit (Promega) was used to measure the induction of ADCC by NAbs that crosslinked viral spikes at the surface of CHIKV-infected cells. U2OS cells grown on 96-well plates (1.25×104 cells/well) were infected with CHIKV 181 at an MOI of 5 for 3 h. After extensive washing, the cells were cultured in the presence or absence of serial dilutions of anti-CHIKV or control anti-DENV antibodies. At 7 h post infection, 7.5×104 effector cells that stably expressed FcγRIIIa and an NFAT response element driving the expression of firefly luciferase were added to each well. Luciferase activity was measured 10 h later according to the manufacturer’s instructions (Promega). The fold of induction was calculated as relative luciferase unit (RLU) (induced-background)/RLU (no antibody-background).
QUANTIFICATION AND STATISTICAL ANALYSIS
We analyzed following features in our datasets: 418 fluorescent puncta in CHIKV-infected cells treated with non-NAb and 107 fluorescent patches in CHIKV-infected cells treated with NAb in STED dataset; 98 NCLPs in CHIKV-infected cells treated with NAb in IEM tomogram dataset; 47 egressed virions and 82 membrane patches in CHIKV-infected cells, and 1,163 NCLPs, 243 membrane patches and 99 spherules in CHIKV-infected cells treated with NAb in cryoET dataset. Detailed classification and parameters used for STA are described below.
NCLP size measurement from IEM tomograms of fixed CHIKV-infected RD cells
Nucleocapsid-like particle (NCLP) subtomograms (n=98) were manually picked and extracted from the IEM tomograms using e2spt_boxer.py, publicly available through EMAN2. The NCLPs appeared pleomorphic, varying widely in size. To quantify the variation in size, we computed radial density profile plots from spherically averaged subtomograms and measured the hydrodynamic radius (i.e., the radius of gyration, or spherically averaged radius) of individual particles by identifying the radius at which the steepest slope occurred after the maximum peak in their corresponding radial density profiles. Lacking adequate models for heterogeneous NCLPs, we fine-tuned the centering of the manually picked particles by aligning them translationally to a spherical blob with a soft Gaussian falloff. The particles were low-pass filtered to 40 Å and spherically masked with a soft Gaussian mask to optimize their translational alignment prior to computing their radial density profiles.
Reconstruction of cryoET tiltseries into tomograms
CryoET tiltseries were subjected to fiducial-based alignment in IMOD and CTF corrected using EMAN2 (Galaz-Montoya et al., 2016) with defocus values derived through CTF fitting for the images and tiltseries for which the power spectrum showed clear CTF oscillations. Otherwise, the average defocus value between flanking images in the tiltseries was used based on the assumption of defocus trends. For tiltseries for which the CTF could be fitted only in a minority of images, or could not be fit in any of them, the target defocus at the tilt axis was assumed to be approximately correct. The tiltseries were reconstructed into tomograms using a compressed sensing algorithm implemented in the ICON-GPU package (Chen et al., 2017b). The tiltseries were pre-filtered with a band-pass filter (a low pass at 100 Å, and high-pass zeroing out the first four pixels in Fourier space) to improve gold fiducial visibility and tracking during tiltseries alignment; however, the alignment parameters were applied to the unfiltered tiltseries, and gold fiducials were erased with IMOD prior to tomographic reconstruction with ICON-GPU. The tiltseries were shrunk by a factor of two using Fourier cropping to increase the reconstruction speed with ICON-GPU. Tomographic annotation was performed semi-automatically using the neural network algorithm in EMAN2 (Chen et al., 2017a). We used Chimera UCSF (Pettersen et al., 2004) for all isosurface visualizations.
STA
All particle picking was done manually using e2spt_boxer.py available in EMAN2 on 4x down-sampled and heavily filtered tomograms (low-pass to 100 Å, high-pass erasing the first four Fourier pixels). Subtomogram alignment and averaging, and all related analyses and controls, were performed using the tools for single-particle tomography in EMAN2 (Galaz-Montoya et al., 2015) as follows.
Released virions were extracted from control tomograms in the absence of NAbs (n=47). Reference-free initial models were built using self-symmetry alignment (Dai et al., 2013) and used to seed iterative, gold-standard refinement (Galaz-Montoya et al., 2015) until convergence was achieved.
For “membrane particles” (patches of membrane) from the control tomograms in the absence of NAbs (n=82) and from tomograms in the presence of NAbs (n=243), an initial model was built using hierarchical ascendant classification for each case, after which the particles in each set (with or without NAbs) were iteratively refined against each respective initial model and averaged.
We also extracted arrested nucleocapsid-like particle (NCLP) subtomograms (n=1,163) from tomograms of CHIKV-infected cells in the presence of NAbs. Given previous reports of icosahedral subpopulations existing among isolated nucleocapsids (NCs) in vitro (Lamb et al., 2010; Paredes et al., 2003), we hypothesized that assembled NCs inside cells might also acquire an icosahedrally symmetric conformation. To test this hypothesis, we carried out extensive STA analyses of the NCLPs from tomograms at the highest magnification and with the best contrast for STA (n=763). We first subjected the arrested NCLPs to self-symmetry alignment but the algorithm failed to produce any consistent icosahedrally symmetric initial models using the same method that worked on released virions, even when the algorithm was run for many more iterations (n=110) than those needed to align typical icosahedral viruses (n=10-50). Given our successful alignment of released virions using the same published methodology (Dai et al., 2013) and software (Galaz-Montoya et al., 2015; Galaz-Montoya et al., 2016) as well as tomograms reconstructed using the same pipeline from tiltseries of similar quality, the results for the arrested NCLPs suggested that the vast majority were indeed not icosahedrally symmetric but rather pleomorphic and structurally heterogeneous. The absence of readily detectable symmetry in NCLPs persisted even for trials in which we computationally deleted the central region of NCLPs (at a radius of ~20 pixels, corresponding to ~14 nm) with a soft mask to abrogate any influence from the genome on alignment, since the genome has been hypothesized to lack an icosahedral arrangement, even in mature virions. Given that the averages after self-symmetry alignment did not resemble the known structure in mature virions, we performed visual examination of randomly selected individual particles (n=~100). We did not find any NCLPs resembling the known structure of mature NCs in budded virions. While some individual subtomograms exhibited angular features and some of the symmetry imposed structures might correspond to legitimate conformations for a few individual NCLPs (not shown) even if they differ from the known structure inside mature virions, further analyses or studies with larger datasets at higher resolution are needed to validate them. We then iteratively refined the entire set of NCLPs without enforcing symmetry. The average of the NCLPs converged to a low-resolution globular structure, ~20 nm in radius, that did not resemble the known structure inside mature virions. Quintile classification based on correlation after alignment against the global average, followed by iterative refinement and averaging of the particles within each quintile, yielded five structures that were all globular yet structurally pleomorphic in different ways. This strongly suggested that presence of extensive structural heterogeneity among the NCLPs in our specimens. To determine whether a “good” initial model could help to computationally isolate a consistent, icosahedrally symmetric subpopulation, we aligned all of the NCLP subtomograms to the known icosahedral structure of the nucleocapsid in mature CHIKV. The initial average resembled the known structure due to model bias. However, when we iteratively refined the data to prevent model bias, the average structure across iterations drifted quickly from the initial biased average, reverting to a low-resolution, globular density. This strongly suggested that the bulk of NCLPs were not in a conformation like that of NCs in mature virions. We then took the 10% of particles that had the best correlation with the known structure after a single round of alignment and aligned them de novo to each other, in an all-versus-all fashion. The average structure did not resemble that of the nucleocapsid inside mature virions. These analyses suggested that there was no significant subpopulation among our in situ budding-arrested NCLPs dataset with the same icosahedral arrangements as those seen in NCs isolated in vitro and in NCs inside mature virions. Indeed, in our cryoET tomograms, the NCLPs appeared pleomorphic upon visual inspection, in agreement with the observations from our 2D and 3D TEM/IEM experiments with fixed cells. Next, we again used radial density profiles, as described above for the analysis of NCLPs in IEM tomograms, to measure the size distribution of the NCLPs and classify them by size. Of the 17 size classes by radius, those at the tails at either end of the distribution (representing particles with radii between ~8-15 nm, and ~23-28 nm) were sparsely populated (n<20 per class) and failed to yield converging subtomogram averages. On the other hand, all 10 classes ranging from ~15.8 nm to ~22.2 nm in radius (23 < n < 107 per class) yielded dense, globular averages upon symmetry-free, reference-free, gold-standard iterative refinement. Of these ten globular classes, five yielded gold-standard FSC curves indicating very low resolutions (< 50 Å), suggesting the presence of extensive conformational heterogeneity still lingering within those size classes. The remaining five classes converged to averages yielding gold-standard FSCs indicating resolutions between ~36 Å and ~40 Å, which is comparable to the resolution obtained for the released virion average (~34 Å), taking into account that the NCLP averages had no symmetry imposed (and therefore it is natural that the resolution would be at least slightly worse).
DATA AND SOFTWARE AVAILABILITY
All software packages used in this study are publicly available. The algorithms and methods for STA and NCLP size analyses are available in EMAN2. All the subtomogram averages presented in this study have been made accessible through the public repository for electron microscopy density maps, the Electron Microscopy Data Bank (EMDB). The accession numbers are as follows: EMD-7513 for released CHIKV virions; EMD-7515 for NCLPs.
Supplementary Material
The movie shows consecutive slices in Z through the tomogram. Gold particles were used as markers for tiltseries alignment during three-dimensional reconstruction. Scale bar: 100 nm.
The movie shows consecutive slices in Z through the tomogram. Gold particles were used as markers for tiltseries alignment during three-dimensional reconstruction. Note the accumulation of gold-conjugated anti-human IgG at the outer surface of the infected cell and at the membrane of viral spherules. Scale bar: 200 nm.
The movie shows consecutive slices in Z through the tomogram. Scale bar: 100 nm.
The movie shows consecutive slices in Z through the tomogram. Scale bar: 100 nm.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-CHIKV 181 polyclonal antibody | IBT Bioservices | Cat. #04-0008 |
| Alexa Fluor 594-conjugated anti-rabbit IgG | Thermo Fisher Scientific | Cat. #A-11012 |
| Alexa Fluor 647-conjugated anti-human IgG | Thermo Fisher Scientific | Cat. #A-21445 |
| C9 | Selvarajah, 2013 | N/A |
| IM-CKV063, IM-CKV066, IM-CKV062 | Fong, 2014 | N/A |
| CHK-9, CHK-152 | Pal, 2013 | N/A |
| Human monoclonal anti-DENV | This manuscript | N/A |
| 6 nm gold conjugated anti-human IgG | Electron Microscopy Sciences | Cat. #25203 |
| Bacterial and Virus Strains | ||
| CHIKV vaccine strain 181/clone 25 | BEI Resources | Cat. #NR-13222 |
| CHIKV strain 37997 | Dr. Stephen Higgs, Kansas State University |
N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 16% Paraformaldehyde | Electron Microscopy Sciences | Cat. #15710 |
| 8% Glutaraldehyde | Electron Microscopy Sciences | Cat. #16019 |
| ProLong Gold Antifade Mountant | Thermo Fisher Scientific | Cat. #P36934 |
| Fibronectin solution | Sigma-Aldrich | Cat. #F1141 |
| TransIT-2020 transfection reagent | Mirus | Cat. #MIR 5404 |
| Critical Commercial Assays | ||
| Alexa Fluor 488 protein labeling kit | Thermo Fisher Scientific | Cat. #A10235 |
| ADCC reporter bioassay core kit | Promega | Cat. #G7010 |
| Deposited Data | ||
| Egressed CHIKV virion subtomogram average |
This paper | EMD-7513 |
| NCLP subtomogram average | This paper | EMD-7515 |
| Experimental Models: Cell Lines | ||
| Human RD cell line | ATCC | Cat. #CCL-136 |
| Human U2OS cell line | ATCC | Cat. #HTB-96 |
| Human 293 c18 cell line | ATCC | Cat. #CRL-10852 |
| BHK21 cell line | ATCC | Cat. #CCL-10 |
| Oligonucleotides | ||
| N297Q_for: AGGAGCAGTACCAGAGCACGTACCGTGTGGTC AGC |
Integrated DNA Technologies | N/A |
| N297Q_rev: CGGTACGTGCTCTGGTACTGCTCCTCCCGCGGC |
Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pTT5 | Nova lifetech | Cat. #PVT 1032 |
| pCAGGS CHIKV E3/E2/E1 | Salvador, 2009 | N/A |
| Software and Algorithms | ||
| Leica LAS X lite | Leica microsystems | https://leica.box.com/v/Leica-LasX-Small |
| SerialEM | Mastronarde, 2003, 2005 | http://bio3d.colorado.edu/SerialEM/ |
| DE Software (motion correction script) | Direct Electron LP | http://www.directelectron.com/scripts/de_process frames |
| IMOD | Kremer, 1996; Mastronarde, 1997 | http://bio3d.colorado.edu/imod/ |
| ICON | Deng, 2016; Chen, 2017 | http://feilab.ibp.ac.cn/LBEMSB/ICON.html |
| EMAN2 | Tang, 2007; Galaz-Montoya, 2015. |
http://blake.bcm.tmc.edu/EMAN2/ |
| Chimera | Pettersen, 2005. | https://www.cgl.ucsf.edu/chimera/ |
| Segger | Pintilie, 2010. |
http://ncmi.bcm.edu/ncmi/software/segger/docs |
Highlights.
NAbs inhibit CHIKV budding by inducing viral glycoprotein coalescence on infected cells
Budding arrested nucleocapsid-like particles are structurally heterogeneous
NAbs induce clustering of CHIKV replication spherules at sites of budding blockage
Coalesced glycoprotein-NAb complexes activate Fc receptors on effector cells
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (NIH) grants R01 AI119056 (to G.S.), P41GM103832 (to W.C.), R24AI120942 (to S.C.W.), and contract HHSN272200900055C (via Integral Molecular Inc.). The authors thank Dr. David Agard (UCSF) for helpful discussion, Dr. Michael Diamond (Washington University) for generous gifts of mouse NAbs CHK-9 and CHK-152, and Drs. David Castaneda-Castellanos and Manu Mabila for technical support for STED microscopy. Plastic section tomography was performed in the Boulder Electron Microscopy Services facility at the University of Colorado. We thank Dr. Michael Schmid (SLAC National Accelerator Laboratory) for his critical reading of the manuscript.
The findings and conclusions are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention (CDC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
G.S. is listed as a co-inventor on a patent that includes in part claims relating to C9 and IM-CKV063.
REFERENCES
- Asano S, Fukuda Y, Beck F, Aufderheide A, Forster F, Danev R, and Baumeister W (2015). Proteasomes. A molecular census of 26S proteasomes in intact neurons. Science 347, 439–442. [DOI] [PubMed] [Google Scholar]
- Baker LA, Sinnige T, Schellenberger P, de Keyzer J, Siebert CA, Driessen AJM, Baldus M, and Grunewald K (2018). Combined (1)H-Detected Solid-State NMR Spectroscopy and Electron Cryotomography to Study Membrane Proteins across Resolutions in Native Environments. Structure 26, 161–170 e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournazos S, and Ravetch JV (2015). Fcgamma receptor pathways during active and passive immunization. Immunol Rev 268, 88–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd EA, and Kielian M (2017). An Alphavirus E2 Membrane-Proximal Domain Promotes Envelope Protein Lateral Interactions and Virus Budding. mBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Dai W, Sun SY, Jonasch D, He CY, Schmid MF, Chiu W, and Ludtke SJ (2017a). Convolutional neural networks for automated annotation of cellular cryo-electron tomograms. Nature methods 14, 983–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang Z, Zhang J, Li L, Wan X, Sun F, and Zhang F (2017b). Accelerating electron tomography reconstruction algorithm ICON with GPU. Biophys Rep 3, 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corboba P, Grutadauria S, Cuffini C, and Zapata M (2000). Neutralizing monoclonal antibody to the E1 glycoprotein epitope of rubella virus mediates virus arrest in VERO cells. Viral immunology 13, 83–92. [DOI] [PubMed] [Google Scholar]
- Dai W, Fu C, Raytcheva D, Flanagan J, Khant HA, Liu X, Rochat RH, Haase-Pettingell C, Piret J, Ludtke SJ, et al. (2013). Visualizing virus assembly intermediates inside marine cyanobacteria. Nature 502, 707–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danev R, and Baumeister W (2017). Expanding the boundaries of cryo-EM with phase plates. Current opinion in structural biology 46, 87–94. [DOI] [PubMed] [Google Scholar]
- Ertel KJ, Benefield D, Castano-Diez D, Pennington JG, Horswill M, den Boon JA, Otegui MS, and Ahlquist P (2017). Cryo-electron tomography reveals novel features of a viral RNA replication compartment. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong RH, Banik SS, Mattia K, Barnes T, Tucker D, Liss N, Lu K, Selvarajah S, Srinivasan S, Mabila M, et al. (2014). Exposure of epitope residues on the outer face of the chikungunya virus envelope trimer determines antibody neutralizing efficacy. Journal of virology 88, 14364–14379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsell K, Griffiths G, and Garoff H (1996). Preformed cytoplasmic nucleocapsids are not necessary for alphavirus budding. The EMBO journal 15, 6495–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsell K, Xing L, Kozlovska T, Cheng RH, and Garoff H (2000). Membrane proteins organize a symmetrical virus. The EMBO journal 19, 5081–5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JM, Long F, Edeling MA, Lin H, van Duijl-Richter MK, Fong RH, Kahle KM, Smit JM, Jin J, Simmons G, et al. (2015). Broadly Neutralizing Alphavirus Antibodies Bind an Epitope on E2 and Inhibit Entry and Egress. Cell 163, 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova EI, Gorchakov R, Pereboeva L, Atasheva S, and Frolov I (2010). Functional Sindbis virus replicative complexes are formed at the plasma membrane. Journal of virology 84, 11679–11695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaz-Montoya JG, Flanagan J, Schmid MF, and Ludtke SJ (2015). Single particle tomography in EMAN2. Journal of structural biology 190, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaz-Montoya JG, Hecksel CW, Baldwin PR, Wang E, Weaver SC, Schmid MF, Ludtke SJ, and Chiu W (2016). Alignment algorithms and per-particle CTF correction for single particle cryo-electron tomography. Journal of structural biology 194, 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaz-Montoya JG, and Ludtke SJ (2017). The advent of structural biology in situ by single particle cryo-electron tomography. Biophys Rep 3, 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garoff H, Sjoberg M, and Cheng RH (2004). Budding of alphaviruses. Virus research 106, 103–116. [DOI] [PubMed] [Google Scholar]
- Grunewald K, Medalia O, Gross A, Steven AC, and Baumeister W (2003). Prospects of electron cryotomography to visualize macromolecular complexes inside cellular compartments: implications of crowding. Biophys Chem 100, 577–591. [DOI] [PubMed] [Google Scholar]
- Jin J, Liss NM, Chen DH, Liao M, Fox JM, Shimak RM, Fong RH, Chafets D, Bakkour S, Keating S , et al. (2015). Neutralizing Monoclonal Antibodies Block Chikungunya Virus Entry and Release by Targeting an Epitope Critical to Viral Pathogenesis. Cell reports 13, 2553–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson MA (2015). Chikungunya on the move. Trends Parasitol 31, 43–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose J, Snyder JE, and Kuhn RJ (2009). A structural and functional perspective of alphavirus replication and assembly. Future Microbiol 4, 837–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajihara M, Marzi A, Nakayama E, Noda T, Kuroda M, Manzoor R, Matsuno K, Feldmann H, Yoshida R, Kawaoka Y , et al. (2012). Inhibition of Marburg virus budding by nonneutralizing antibodies to the envelope glycoprotein. Journal of virology 86, 13467–13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio K, Hellstrom K, Balistreri G, Spuul P, Jokitalo E, and Ahola T (2013). Template RNA length determines the size of replication complex spherules for Semliki Forest virus. Journal of virology 87, 9125–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, and McIntosh JR (1996). Computer visualization of three-dimensional image data using IMOD. Journal of structural biology 116, 71–76. [DOI] [PubMed] [Google Scholar]
- Lamb K, Lokesh GL, Sherman M, and Watowich S (2010). Structure of a Venezuelan equine encephalitis virus assembly intermediate isolated from infected cells. Virology 406, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masrinoul P, Puiprom O, Tanaka A, Kuwahara M, Chaichana P, Ikuta K, Ramasoota P, and Okabayashi T (2014). Monoclonal antibody targeting chikungunya virus envelope 1 protein inhibits virus release. Virology 464-465, 111–117. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. Journal of structural biology 152, 36–51. [DOI] [PubMed] [Google Scholar]
- McMullan G, Faruqi AR, Clare D, and Henderson R (2014). Comparison of optimal performance at 300keV of three direct electron detectors for use in low dose electron microscopy. Ultramicroscopy 147, 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennella V, Hanna R, and Kim M (2015). Subdiffraction resolution microscopy methods for analyzing centrosomes organization. Methods in cell biology 129, 129–152. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Chipman PR, Hong EM, Kuhn RJ, and Rossmann MG (2002). In vitro-assembled alphavirus core-like particles maintain a structure similar to that of nucleocapsid cores in mature virus. Journal of virology 76, 11128–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata K, Zhang Q, Gerardo Galaz-Montoya J, Fu C, Coleman ML, Osburne MS, Schmid MF, Sullivan MB, Chisholm SW, and Chiu W (2017). Visualizing Adsorption of Cyanophage P-SSP7 onto Marine Prochlorococcus. Sci Rep 7, 44176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal P, Dowd KA, Brien JD, Edeling MA, Gorlatov S, Johnson S, Lee I, Akahata W, Nabel GJ, Richter MK, et al. (2013). Development of a Highly Protective Combination Monoclonal Antibody Therapy against Chikungunya Virus. PLoS pathogens 9, e1003312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes A, Alwell-Warda K, Weaver SC, Chiu W, and Watowich SJ (2003). Structure of isolated nucleocapsids from venezuelan equine encephalitis virus and implications for assembly and disassembly of enveloped virus. Journal of virology 77, 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pfeffer S, Woellhaf MW, Herrmann JM, and Forster F (2015). Organization of the mitochondrial translation machinery studied in situ by cryoelectron tomography. Nature communications 6, 6019. [DOI] [PubMed] [Google Scholar]
- Salvador B, Zhou Y, Michault A, Muench MO, and Simmons G (2009). Characterization of Chikungunya pseudotyped viruses: Identification of refractory cell lines and demonstration of cellular tropism differences mediated by mutations in E1 glycoprotein. Virology 393, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahmoradian SH, Galaz-Montoya JG, Schmid MF, Cong Y, Ma B, Spiess C, Frydman J, Ludtke SJ, and Chiu W (2013). TRiC's tricks inhibit huntingtin aggregation. eLife 2, e00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SA, Silva LA, Fox JM, Flyak AI, Kose N, Sapparapu G, Khomadiak S, Ashbrook AW, Kahle KM, Fong RH, et al. (2015). Isolation and Characterization of Broad and Ultrapotent Human Monoclonal Antibodies with Therapeutic Activity against Chikungunya Virus. Cell host & microbe 18, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solignat M, Gay B, Higgs S, Briant L, and Devaux C (2009). Replication cycle of chikungunya: a re-emerging arbovirus. Virology 393, 183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soonsawad P, Xing L, Milla E, Espinoza JM, Kawano M, Marko M, Hsieh C, Furukawa H, Kawasaki M, Weerachatyanukul W, et al. (2010). Structural evidence of glycoprotein assembly in cellular membrane compartments prior to Alphavirus budding. Journal of virology 84, 11145–11151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Xiang Y, Akahata W, Holdaway H, Pal P, Zhang X, Diamond MS, Nabel GJ, and Rossmann MG (2013). Structural analyses at pseudo atomic resolution of Chikungunya virus and antibodies show mechanisms of neutralization. eLife 2, e00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaa B, Biasiotto R, Eng K, Neuvonen M, Gotte B, Rheinemann L, Mutso M, Utt A, Varghese F, Balistreri G, et al. (2015). Differential Phosphatidylinositol-3-Kinase-Akt-mTOR Activation by Semliki Forest and Chikungunya Viruses Is Dependent on nsP3 and Connected to Replication Complex Internalization. Journal of virology 89, 11420–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderplasschen A, Hollinshead M, and Smith GL (1997). Antibodies against vaccinia virus do not neutralize extracellular enveloped virus but prevent virus release from infected cells and comet formation. The Journal of general virology 78 (Pt 8), 2041–2048. [DOI] [PubMed] [Google Scholar]
- Walker LM, and Burton DR (2018). Passive immunotherapy of viral infections: 'super-antibodies' enter the fray. Nat Rev Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Chen C, Rayaprolu V, Mukhopadhyay S, and Zlotnick A (2015). Self-Assembly of an Alphavirus Core-like Particle Is Distinguished by Strong Intersubunit Association Energy and Structural Defects. ACS Nano 9, 8898–8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Strunk K, Zhao G, Gray JL, and Zhang P (2012). 3D structure determination of native mammalian cells using cryo-FIB and cryo-electron tomography. Journal of structural biology 180, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Charlier C, Vasilakis N, and Lecuit M (2018). Zika, Chikungunya, and Other Emerging Vector-Borne Viral Diseases. Annu Rev Med 69, 395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamayoshi S, Uraki R, Ito M, Kiso M, Nakatsu S, Yasuhara A, Oishi K, Sasaki T, Ikuta K, and Kawaoka Y (2017). A Broadly Reactive Human Anti-hemagglutinin Stem Monoclonal Antibody That Inhibits Influenza A Virus Particle Release. EBioMedicine 17, 182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The movie shows consecutive slices in Z through the tomogram. Gold particles were used as markers for tiltseries alignment during three-dimensional reconstruction. Scale bar: 100 nm.
The movie shows consecutive slices in Z through the tomogram. Gold particles were used as markers for tiltseries alignment during three-dimensional reconstruction. Note the accumulation of gold-conjugated anti-human IgG at the outer surface of the infected cell and at the membrane of viral spherules. Scale bar: 200 nm.
The movie shows consecutive slices in Z through the tomogram. Scale bar: 100 nm.
The movie shows consecutive slices in Z through the tomogram. Scale bar: 100 nm.