

Summary
Adhesion G-protein-coupled receptors (aGPCRs) play critical roles in diverse cellular processes in neurobiology, development, immunity, and numerous diseases. The lack of molecular understanding of their activation mechanisms, especially with regard to the transmembrane domains, hampers further studies to facilitate aGPCR-targeted drug development. Latrophilin-1/ADGRL1 is a model aGPCR that regulates synapse formation and embryogenesis, and its mutations are associated with cancer and attention-deficit/hyperactivity disorder. Here, we established functional assays to monitor latrophilin-1 function and showed the activation of latrophilin-1 by its endogenous agonist peptide. Via a comprehensive mutagenesis screen, we identified transmembrane domain residues essential for latrophilin-1 basal activity and for agonist peptide response. Strikingly, a cancer-associated mutation exhibited increased basal activity and failed to rescue the embryonic developmental phenotype in transgenic worms. These results provide a mechanistic foundation for future aGPCR-targeted drug design.
Subject Areas: Molecular Biology, Membrane Architecture, Protein Structure Aspects
Graphical Abstract
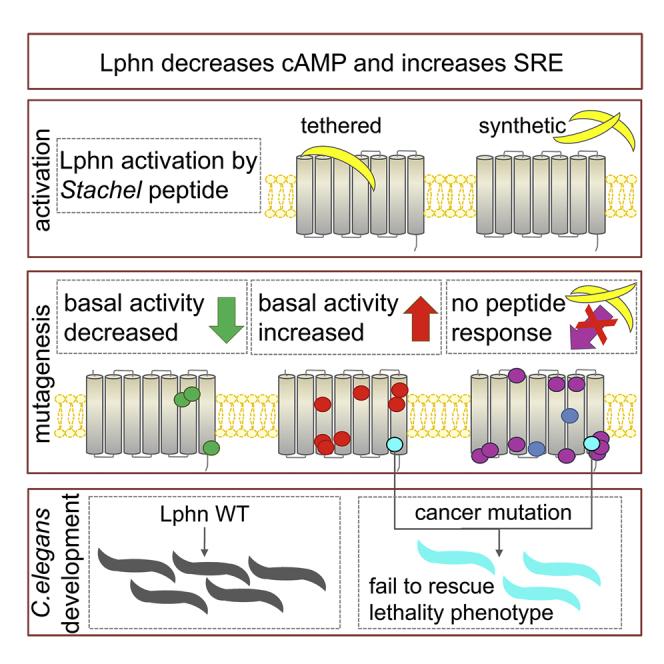
Highlights
-
•
Latrophilin-1/3 decreases cAMP levels and increases SRE levels in mammalian cells
-
•
Mutagenesis of the TM region reveals residues key for adhesion GPCR signaling
-
•
A cancer-associated mutation exhibits increased basal activity
-
•
The same cancer mutation abolishes latrophilin function in C. elegans development
Molecular Biology; Membrane Architecture; Protein Structure Aspects
Introduction
Adhesion G protein-coupled receptors (aGPCRs) are cell-surface molecules that mediate intercellular communication via cell-cell and cell-matrix interactions (Hamann et al., 2015, Prömel et al., 2013). With 33 members in humans, they make up the second largest GPCR family, but are the least studied and least understood (Fredriksson et al., 2003). Genetic studies suggest critical roles for aGPCRs in development and immunity and especially in neurobiology (such as brain development [Bae et al., 2014, Piao et al., 2004], synapse maturation and elimination [Bolliger et al., 2011], myelination of neurons [Monk et al., 2009], central nervous system [CNS] angiogenesis [Kuhnert et al., 2010], and neural tube development [Chae et al., 1999, Langenhan et al., 2009, Shima et al., 2004, Usui et al., 1999]), and link them to numerous diseases including neurodevelopmental disorders, deafness, male infertility, schizophrenia, and immune disorders (Langenhan et al., 2013, Prömel et al., 2013). In addition, many aGPCRs are found to be over- or underexpressed in various cancers (Kan et al., 2010, Shashidhar et al., 2005, Xu et al., 2006), and a recent study reports that aGPCRs are some of the frequently mutated genes in cancerous tumors (O'Hayre et al., 2013). Considering that many drugs target the transmembrane (TM) helices of GPCRs to regulate receptor activity, thereby eliciting the desirable therapeutic effects, aGPCRs may be promising targets for drugs to treat numerous diseases including cancer. Currently, there is no high-resolution structure for the TM domain of an aGPCR. GPCRs from the secretin family have the highest TM domain similarity to aGPCRs, suggesting that aGPCR TM domains might be activated via similar mechanisms (Fredriksson et al., 2003, Hollenstein et al., 2013, Rasmussen et al., 2011, Rosenbaum et al., 2007, Siu et al., 2013). In addition, the signaling pathways of aGPCRs are largely unknown, making any functional studies difficult to perform. Altogether, further studies of aGPCRs progress slowly owing to these obstacles, and the molecular mechanisms underlying aGPCR signal transduction remain unknown.
Unlike other GPCRs, aGPCRs have large extracellular regions (ECRs) that are autoproteolytically cleaved from their seven-pass TM regions within a conserved GPCR-autoproteolysis-inducing (GAIN) domain (Arac et al., 2012, Chang et al., 2003, Ichtchenko et al., 1999, Krasnoperov et al., 1997, Lin et al., 2004) (Figure 1A). Autoproteolysis occurs between the last two β stands of the GAIN domain, cleaving the receptor into two fragments (Arac et al., 2012): an N-terminal extracellular fragment comprising various adhesion domains and the GAIN domain (lacking its last β strand) and a membrane-anchored C-terminal fragment comprising the TM domain (tethered to the last β strand of the GAIN domain) and the cytoplasmic tail (Krasnoperov et al., 1997, Lelianova et al., 1997). Autoproteolysis occurs in the endoplasmic reticulum, and the cleaved fragments of an aGPCR stay associated as a heterodimer even after cleavage (Arac et al., 2012). One of the two models for the activation of aGPCRs is the tethered agonist model, which suggests that aGPCRs are activated by a short peptide that corresponds to the last β strand of the GAIN domain (called the Stachel peptide, Figure 1A) (Liebscher et al., 2014, Stoveken et al., 2015). Ligand binding to the ECR is believed to lead to shedding of the ECR, which results in exposure of the previously hidden Stachel peptide to the TM domain, leading to receptor activation.
Figure 1.

Lphn1 and Lphn3 Decrease cAMP Level and Increase SRE Level in Transfected Cells
(A) Schematic domain diagram of Lphns as a model aGPCR. All aGPCRs have a GAIN domain, a TM domain, and variable other domains. Stachel peptide is a tethered agonist. Yellow line indicates Stachel peptide and * indicates cleavage site.
(B) β2-adrenergic receptor assay to detect cAMP signaling of rat Lphn1 and human Lphn3 in transfected HEK293 cells. Lphn inhibits ISO-induced cAMP elevation. Signaling data are obtained from n = 3 independent experiments performed in triplicates and represented as means ± SEM. cAMP level was measured by GloSensor assay. Figure modified from Li et al. (2018). SEM, standard error of the mean.
(C) Forskolin assay to detect cAMP signaling of Lphn1 and Lphn3 in transfected HEK293 cells. Lphn1 inhibits forskolin-induced cAMP elevation.
(D) Basal activity of Lphn1 and Lphn3 as measured by the SRE-luciferase reporter assay.
NSp > 0.05; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001.
Both the secretin family and the aGPCR family receptors are activated by the N termini of short agonistic peptides (Hoare, 2005, Liebscher et al., 2014, Stoveken et al., 2015). However, Stachel peptides of aGPCRs are hydrophobic and prone to β strand formation (Arac et al., 2012), whereas the agonistic peptides of secretin-like receptors fold into α helices (Watkins et al., 2012), suggesting differences in the molecular details of the peptide-TM interaction. Although recent structures of secretin-like receptors in inactive and peptide-bound active conformations revealed the molecular mechanism of peptide recognition by the TM, little is known about the molecular details for the activation of aGPCRs by the Stachel peptide (Hollenstein et al., 2013, Liang et al., 2017, Rasmussen et al., 2011, Rosenbaum et al., 2007, Siu et al., 2013, Zhang et al., 2017). These secretin family structures are extremely valuable for aGPCR studies because more accurate models of aGPCR TMs can be constructed, facilitating studies like this one.
The knowledge of the signaling pathway of a receptor and an in vitro assay to monitor downstream signaling has been invaluable in characterizing and drugging the canonical GPCRs. The lack of a robust in vitro functional assay for aGPCRs has obstructed both structure/function studies, including the functional characterization of disease-causing mutations, and agonist/antagonist ligand discovery and characterization. In this study, we used latrophilin-1 (Lphn1)/ADGRL1, a key molecule in synapse formation and brain development, as a model aGPCR to study aGPCR function. We established two robust in vitro assays to monitor receptor signaling and showed that the endogenous agonist of Lphns, a 14-amino-acid peptide, binds to and activates the receptor. We studied a large set of bioinformatics-based point mutations and disease mutations on the TM region of Lphns and identified mutants that are constitutively active, constitutively inactive, or nonresponsive to the agonist peptide. Intriguingly, we identified a cancer-associated mutation that exhibited high basal activity and abolished the rescue of the embryonic development phenotype in transgenic worms. These results provide the basic groundwork for future drug design against aGPCRs.
Results
Establishment of In Vitro Functional Assays to Monitor Latrophilin Activity
Latrophilins (Lphn1–3) regulate synapse formation and brain development (O'Sullivan et al., 2012). Lphn1 was first identified as the calcium-independent receptor for α-latrotoxin, a black widow spider toxin component that triggers massive neurotransmitter release from nerve terminals (Deak et al., 2009, Krasnoperov et al., 1997, Lelianova et al., 1997, Sudhof, 2001, Sugita et al., 1999). Mutations of Lphns are associated with attention-deficit/hyperactivity disorder (ADHD) as well as numerous cancers in humans (Arcos-Burgos et al., 2010, Kan et al., 2010, O'Hayre et al., 2013). Lphns are one of the two aGPCRs that are conserved between vertebrates and invertebrates. In Caenorhabditis elegans, the Lphn homolog lat-1 is required for the alignment of cell division planes to the anterior-posterior axis during development (Langenhan et al., 2009, Muller et al., 2015). In Drosophila, Lphn/dCIRL sensitizes the chordotonal neurons to perceive mechanical signals (Scholz et al., 2015). In vertebrates, Lphns mediate excitatory synapse formation (Lu et al., 2015, O'Sullivan et al., 2012). The intracellular signaling of Lphns via G proteins was reported by several studies, including the activation of Gs by rat Lphn1 as well as LAT-1, one of the C. elegans homologs of Lphn (Muller et al., 2015); the decrease of cyclic adenosine monophosphate (cAMP) by Lphn/dCIRL in Drosophila (Scholz et al., 2017); and the increase of intracellular Ca2+ levels by rat Lphn1 (Silva et al., 2011); however, the assays used in these studies were not suitable for large-scale mutagenesis screens.
To measure Lphn activity in a signaling assay, we screened several assays that monitor downstream G protein signaling using the basal activity of full-length rat Lphn1 and full-length human Lphn3 (these constructs are used throughout this article unless stated otherwise). The first assay in which we robustly observed basal activity of Lphn was the GloSensor assay (Promega), which reports cAMP levels in mammalian expression systems. A comparison of the background cAMP level in control HEK293 cells (transfected with empty vector) with cells transfected with Lphn1 provided the first indication that Lphn1 may couple to Gi as Lphn1 overexpression decreased cAMP levels (Figure S1A).
We modified the GloSensor assay in two ways to reliably detect a decrease in cAMP levels: (1) we elevated cAMP level in cells by co-transfecting the cells with the Gs-coupled β2-adrenergic receptor (β2AR) and activating it with its agonist, isoproterenol (ISO) (Figures 1B and S1B) or (2) we elevated cAMP level in cells by adding forskolin to activate the endogenous adenylate cyclase (Figures 1C and S1C). Relative to empty vector, overexpression of Lphn1 or Lphn3 decreased the level of cAMP in cells, suggesting the possible activation of Gi. Lphn1 DNA concentration response curves for cell-surface expression of Lphn1 and for the decrease of cAMP levels in cells are as expected (Figures S1D and S1E). In addition, we performed direct G protein coupling experiments using human Lphn3 GAIN + TM domains embedded in insect cell membranes. Comparison of empty insect cell membranes with those embedded with Lphn3 showed an increase in binding to purified Gi proteins, suggesting that human Lphn3 couples to Gi (Figure S1F). When membranes were treated with urea to induce dissociation of the ECR and Stachel exposure, we observed increased coupling to Gi, suggesting that Lphns are sensitive to Stachel-mediated activation in this assay (Figure S1F).
The second assay in which we robustly observed basal activity of Lphn was a serum response element (SRE)-luciferase assay that reports, among others, activation of G12/13, which is upstream of SRE and RhoA (Luo et al., 2011, Salzman et al., 2016, Stoveken et al., 2015). Overexpression of Lphn1 or Lphn3 in HEK293T cells resulted in increased luminescence compared with cells transfected with the empty vector, showing that the basal activity of overexpressed Lphns can be detected in this assay (Figure 1D).
Latrophilins Are Activated by Their Stachel Peptide in the Established Assays
Several aGPCRs besides rat LPHN1 and its homologs in C. elegans and Drosophila, including GPR126, GPR133, GPR64, GPR114, GPR56, GPR110, and GPR116, have been reported to be activated by their Stachel peptide, which functions as a tethered agonist (Demberg et al., 2015, Demberg et al., 2017, Kishore et al., 2016, Liebscher et al., 2014, Muller et al., 2015, Scholz et al., 2017, Stoveken et al., 2015) (Figures 2A and 2B). Recent studies showed that addition of the synthesized Stachel peptide on full-length receptors in trans activated the receptors (Liebscher et al., 2014, Stoveken et al., 2015). Similarly, truncation constructs starting from the first residue after the autoproteolysis site, thus revealing the Stachel peptide free, also displayed dramatically increased signaling (Kishore et al., 2016, Paavola et al., 2011). To test whether these results can be generalized to other mammalian Lphns and to optimize our signaling assays for detecting Stachel peptide-dependent activation, we repeated similar experiments for Lphn1.
Figure 2.

Stachel Peptide is a Tethered Agonist for Lphn1
(A) Schematic diagram for Lphn1 constructs encoding full-length Lphn1, inactive-like TM domain of Lphn1 (TM), or active-like Stachel peptide/TM domain of Lphn1 (P + TM). The constructs are FLAG-tagged at the indicated positions. Stachel peptide is colored yellow. Asterisk represents autoproteolysis site within the GAIN domain.
(B) Sequence alignment of Stachel peptides from different Lphns. Identical residues are highlighted in gray.
(C) Signaling activity of Lphn1 constructs from (A) as measured by the β2-adrenergic receptor cAMP assay. Cells were pre-incubated with or without pertussis toxin and treated with 100 μM synthetic agonist peptide or solvent. Data are plotted as percentage of isoproterenol response of empty vector-transfected cells. Data are represented as mean ± SEM of three independent experiments, three repeats each (n = 9). SEM, standard error of the mean.
(D) Signaling activity of Lphn1 constructs as in (C) in the forskolin cAMP assay. Data are plotted as percentage of forskolin response of empty vector-transfected cells. Data are obtained from one typical experiment performed in triplicate and represented as mean ± SEM (n = 3).
NSp > 0.05; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. NS, Not Significant.
First, we added a synthesized 14-residue peptide (termed P14, TNFAVLMAHREIYQ) corresponding to the Stachel peptide of rat Lphn1 onto full-length rat Lphn1 and monitored receptor function in our cAMP assays. Addition of P14 onto Lphn1 decreased the cAMP level in both β2AR and forskolin assays (Figures 2C and 2D). Further addition of pertussis toxin (PTx), a specific inhibitor of Gi (Burns, 1988), abolished the effect of P14 and brought cAMP levels back to basal activity level, suggesting that Stachel peptide-dependent activation of Lphn1 is mediated by Gi (Figures 2C and 2D). However, PTx had no effect on the basal activity of Lphn1, suggesting that other signaling pathways mediate the decrease in cAMP levels upon Lphn1 overexpression. To confirm direct interaction between the Stachel peptide and the TM domain of Lphns rather than another domain of Lphn, we tested whether a construct encoding the TM domain of Lphn3 binds to a synthesized Stachel peptide. Purified and detergent-solubilized Lphn3 TM domain was mixed with synthesized Stachel peptide in the presence or absence of a cross-linking reagent, and peptide binding was detected by western blot analysis (Figures S2A and S2B). Taken together, these results suggest that Stachel peptide acts as an agonist for the TM domain of Lphns when added in trans.
Second, to test the role of the Stachel peptide as an agonist tethered to the Lphn TM domain, i.e., in cis, we designed an active-like construct encoding the TM domain with the tethered Stachel peptide of Lphn1 (termed P + TM) and an inactive-like construct encoding only the TM domain of Lphn1 (termed TM) (Figure 2A). To avoid expression problems and potential effects on signaling, both constructs were FLAG-tagged at the C terminus, rather than the N terminus. Overexpression of the P + TM construct displayed increased basal activity compared with full-length Lphn1, whereas the TM construct was less active, although not completely inactive. All mutants responded to P14 peptide addition in trans, and peptide response was sensitive to PTx exposure (Figures 2C and 2D). The cell-surface expression levels of TM and P + TM constructs were quantified by biotinylation experiments (Figures S2C and S2D). The cell-surface expression level of TM and P + TM were at similar levels, although both showed decreased expression when compared with the full-length Lphn1. These results suggest that the Stachel peptide acts as an agonist when presented to the receptor in a tethered conformation in cis.
Mutagenesis of the TM Domain Affects Basal Activity and Peptide Response
Ligand-induced conformational changes of GPCRs transduce extracellular signals to the intracellular part of the receptor and facilitate G protein interaction. Conserved motifs in the rhodopsin family of GPCRs (such as the CWxP motif in TM6, NPxxY motif in TM7, and DRY motif in TM3) are critical for receptor activation upon ligand binding (Audet and Bouvier, 2012). To identify essential residues for basal activity and for agonist peptide response in adhesion GPCRs, we performed a mutagenesis screen of the TM domain within the full-length Lphn1 construct using our β2AR cAMP-based signaling assay (Figure S1C). A homology model of Lphn1 TM domain was generated based on the crystal structure of the corticotropin-releasing factor receptor (CRFR), a member of the secretin family with 29% sequence identity to Lphn1 TM domain (Ebejer et al., 2013, Hollenstein et al., 2013). Over 60 point mutations were designed based on various criteria, including homology to functionally important conserved motifs in the rhodopsin family (Audet and Bouvier, 2012), homology to the secretin family in general (Krishnan et al., 2012, Schioth et al., 2011), homology to CRFR residues reported to interact with its agonist peptide (Coin et al., 2013), conservation within aGPCRs (including the following motifs: GWGxP [helix IV], K/RKLH [intracellular loop I], and discontinuous RHE motif [helix II]) (Peeters et al., 2016), and visual analysis of extracellular loops. In addition, previously reported mutations associated with cancer and ADHD were generated (Kan et al., 2010, O'Hayre et al., 2013) (Figure S3 and Table S1).
The effect of mutations on the function of the full-length Lphn1 was monitored by performing the β2AR cAMP-based signaling assay (Figures 3A, 3D, and 3G). Cell-surface expression level for all mutants was quantified using flow cytometry by detecting N-terminally FLAG-tagged Lphn1 on non-permeabilized cells (Figures 3C, 3F, and 3I). Approximately half of the mutations had an effect on Lphn1 signaling in the cAMP assay. These mutants were further tested in the SRE-luciferase assay (Figures 3B, 3E, and 3H). Our screen revealed mutants that either increased basal activity (colored red throughout Figures 4 and 6 and Table S1), decreased basal activity (colored green), or affected response of the receptor to the agonist peptide (colored magenta for confirmed mutants and orange for likely mutants). Some mutants had an effect on both the basal activity and response to the peptide (colored blue). All cAMP signaling, SRE signaling, and cell-surface expression data for each mutant as well as the information about the location and conservation of the mutated residues can be found in Figures 3 and 5, Table S1, and Figures S5 and S6. Please note that color coding of residues in figures and tables oversimplifies the results, and we recommend referring to the raw data presented in Figures 3 and 5 and Table S1.
Figure 3.

TM Mutations That Affect Basal Activity of Lphn1
(A–I) TM mutations that have an effect on the basal activity of full-length Lphn1 are shown for the cAMP signaling assay (A, D, and G), SRE signaling assay (B, E, and H), and cell-surface expression quantification (C, F, and I). The mutations are categorized as: (A–C) mutations that map to the indicated conserved motifs from rhodopsin, secretin, and adhesion families; (D–F) mutations that lead to constitutive activity; and (G–I) previously reported cancer-associated mutations that affect Lphn1 signaling. Mutations that abolish receptor response to the agonist peptide are shown in Figure 5. One mutation may belong to more than one category. See Table S1 for raw data and for other mutants that had no effect. See Figure 4 for structural visualization of basal activity mutants. See Figures S4 and S5 for raw cell-surface expression data. Mutations that are introduced into transgenic worm are indicated by a cyan star. Basal activity in cAMP assay was detected as percent of wild-type (WT) Lphn1 after activation with 50 nM isoproterenol. The effect of the agonist peptide was detected by pre-incubation with 100 μM synthetic agonist peptide for 5 min before isoproterenol activation. Signaling data are obtained from three independent β2AR co-expression experiments performed in triplicates and represented as means ± SE Basal activity in SRE assay was normalized to empty vector-transfected cells. Signaling data are obtained from three independent SRE experiments performed in triplicates and presented as means ± SE Cell-surface expression for each mutant was obtained from three independent flow cytometry experiments using the same cells as those used for the cAMP assay and presented as mean ± SE Additional DNA titration experiments for some of the mutants with very low or high expression levels are presented on the right side of the panels and were aimed to measure receptor signaling at expression levels comparable to WT. SE, standard error.
NSp > 0.05; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. NS, Not Significant.
Figure 4.

Basal Activity Mutations Mapped on the Modeled TM Structure of Lphn1
(A) Snake plot for visualization of basal activity mutations on the transmembrane helices.
(B) All basal activity mutations mapped on the Lphn1 TM domain, which is modeled based on the inactive CRFR structure (PDB ID: 4K5Y). Mutations that increase basal activity are colored red, mutations that decrease basal activity are colored green, and mutations that increase basal activity and also decrease response to the agonist peptide are colored blue.
(C) A cytoplasmic view of the modeled Lphn1 structure showing the basal activity mutants and the mutants that map to conserved region homologous to the NPxxY motif.
(D) A view of the modeled Lphn1 structure showing the basal activity mutants that map to conserved regions homologous to the DRY motif and the ionic lock. I1045N mutation (colored magenta) affects peptide response.
(E) Cancer-associated mutations on Lphn1 TM domain that affect signaling. V1094I (V1095I in human Lphn1 in upper aerodigestive tract carcinoma), R885L (R886L in human Lphn1 in endometrioid carcinoma), C1098Y (C1098Y in human Lphn1 in endometrioid carcinoma), Y1001F (Y1019F in human Lphn3 in lung adenocarcinoma), and I1045N (I1146N in human Lphn1 in hepatocellular carcinoma) were reported previously (Kan et al., 2010, O'Hayre et al., 2013) (colored yellow).
Mutations that are introduced into transgenic worm are labeled with a cyan star.
Figure 6.

Peptide Response Mutations Mapped on the Modeled TM Structure of Lphn1
(A) Snake plot for visualization of peptide response mutations on the transmembrane helices.
(B) All mutations that affect response of the receptor to the agonist peptide mapped on the Lphn1 TM domain, which is modeled based on the peptide-bound active GLP receptor structure (PDB ID: 5VAI). Mutations that affect peptide response are colored magenta, mutations that affect both peptide response and basal activity are colored blue, and mutations that likely affect peptide response are colored orange.
(C) Superimposition of the model of Lphn1 TM domain (gray) with the GLP1 peptide (red)-bound GLP1 receptor/G protein (green/tan) complex structure (PDB ID: 5VAI). Lphn1 residues are colored as in B.
(D) Close-up extracellular view of (C) showing the modeled Lphn1 TM domain and the GLP1 peptide. Peptide response mutants that map to the extracellular side of the receptor as well as GLP peptide residues important for its interaction with the GLP receptor are shown by sticks.
(E) Close-up cytoplasmic view of (C) showing the modeled Lphn1 TM domain and the G protein. Peptide response mutants that map to the cytoplasmic side of the receptor are shown by sticks.
Mutations that are introduced into transgenic worms are labeled with cyan asterisk.
Figure 5.

TM Mutations That Affect Response of Lphn1 to Agonist Peptide
(A–C) TM mutations that have an effect on response of full-length Lphn1 to the agonist peptide are shown in the cAMP signaling assay (A), SRE signaling assay (B), and cell-surface expression quantification (C). One mutation may belong to more than one category and might be listed in Figure 3 as well. Mutations that are introduced into transgenic worm are indicated by a cyan star. See Table S1 for raw data and for other mutations that had no effect. See Figure 6 for structural visualization of peptide response mutations. See Figures S4 and S5 for cell-surface expression data. See Figure 3 legend for details about the cAMP assay, SRE assay, and cell-surface expression quantification.
NSp > 0.05; ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. NS, Not Significant.
Homologous Yet Different Molecular Features within the TM Mediate Basal Activity
Although aGPCRs do not have the motifs conserved in the rhodopsin family, mutagenesis of homologous residues resulted in altered basal activity in Lphn1, suggesting that aGPCRs are activated by molecular mechanisms that are mediated by homologous motifs (Figures 3, 4, and S4). For instance, the NPxxY motif that is conserved in the rhodopsin family is replaced by “IFVFH” in Lphn1 (Figures 3A–3C). Mutations in this region affected basal activity severely (Figures 3A and 4C). V1094I and nearby V1090A increased basal activity, whereas H1096A decreased basal activity (Figures 3 and 4C). Furthermore, I1092A, V1094I, and nearby C1097Y abolished response to the peptide. Among all mutants, the most active mutant, V1094I, is one that was associated with upper aerodigestive tract cancer, previously (O'Hayre et al., 2013) (Figures 3G–3I and 4E). C1097Y is also associated with endometrium cancer. These results suggest that a motif homologous to the NPxxY motif is critical for aGPCR activation.
Another example of a unique aGPCR motif that might act similarly to a homologous one in the rhodopsin family is the DRY motif, which is replaced by a more hydrophobic “HLY” motif in Lphn1 (Figures 3A–3C and 4D). In conventional GPCRs, the DRY motif on TM3 and a glutamate residue from TM6 (replaced by D1043 in Lphn1) contribute to an ionic lock that traps the receptor in an inactive conformation (Ballesteros et al., 2001, Greasley et al., 2002). In Lphn1, both L947A and Y948A mutations within the “HLY” motif lead to increased basal activity (Figures 3A–3C). Furthermore, mutation of a hydrophobic residue on TM6, I1045N, a liver carcinoma mutation, abolished response of the receptor to the agonist peptide (Figures 3G–3I and 4E). The altered activity upon mutagenesis of these hydrophobic residues in Lphn1 suggests a possible hydrophobic lock in aGPCRs instead of the classical ionic lock.
Overall analysis of the dataset showed that the majority of basal activity-altering mutations resulted in increased basal activity, suggesting that mutations disturb the inactive conformation and promote an active conformation (Figures 3 and 4B). For instance, only W1063A, G1066A, and H1096A decreased basal activity, whereas V1010A, W939A, V1090A, M1077A, Y970A, and Y948A are among the many mutations that increased basal activity. Mapping these basal activity-altering mutations on the Lphn1 model revealed that the mutations were distributed throughout the TM domain, including distant positions, suggesting long-distance allosteric pathways within the TM fold (Figure 4B). Our results largely agree with the results of a recent study that mutagenized the active-like truncated construct (P + TM) for another aGPCR, GPR112, with some differences that are likely due to the use of full-length versus truncated constructs in these studies (Peeters et al., 2016).
Numerous Residues within the TM Are Involved in the Response to Agonist Peptide
To identify the residues that are involved in the response of the receptor to the agonist peptide, we looked for mutations that have similar basal activity in the presence or absence of the exogenously added peptide on the full-length receptor (Figure 5). These residues were mapped onto the modeled structure of Lphn1 that was generated based on the agonist peptide-bound active structure of the Glucagon-like peptide (GLP1) receptor (Zhang et al., 2017) (Figure 6). Visual analysis revealed that residues that affect peptide response map onto the extracellular face, the middle layers, or the cytoplasmic face of the receptor (Figures 6A and 6B). We expect that lack of receptor response to the agonist peptide can be caused by the following different mechanisms. Residues that are on the extracellular face (such as Y1001F, F1069A, A1078G, and A926G) are likely involved in direct peptide binding, which their mutagenesis may abolish (Figures 6B–6D). Noticeably, Y1001 is a lung cancer-associated mutation (Kan et al., 2010). Residues that map to the core of the receptor (such as G1060A, G1008A, G1089A, G969A, H946A, and V1094I) likely mediate transmission of the signal from the extracellular face to the intracellular face (Figure 6B). These glycine residues likely enable the flexibility of the receptor, which allows the signal to be transduced down the protein. Finally, residues that map to the cytoplasmic face of the receptor (such as R891A, H1096A, H895A, R885A, H946A, Y964A, Y965A, I1092A, and C1097Y) likely mediate receptor activation and G protein binding (Figures 6C and 6E). Several mutants including G1060A and V1094I were constitutively active and did not respond to exogenous peptide (Figure 5). These results suggest that a large set of residues are involved in the response of the receptor to the agonist peptide.
The peptide hormone-bound structures of GLP1 receptor and calcitonin receptor revealed that the peptide agonist engages the receptor by binding to a hydrophobic pocket that is generated by the large outward movement of the extracellular ends of TM helices 6 and 7 (Liang et al., 2017). This conformation is accompanied by a 60° kink in helix 6. A large outward movement of the intracellular end of this helix opens the TM to accommodate interactions with the α5 helix of Gαs (Liang et al., 2017, Zhang et al., 2017). In Lphn1, A926 in TM3, Y1001 in TM5, F1069 in TM6, and A1078 in TM7 are residues on the extracellular face of the receptor (Figure 6D). Superimposition of the modeled Lphn1 structure with the peptide-bound GLP1 receptor structure suggests that these residues might be involved in direct peptide binding (Figures 6C and 6D). F1069 and A1078 are located at the tips of TM6 and TM7, respectively, where the helices bend out in the calcitonin receptor (Liang et al., 2017). G1060, on the other hand, sits in the middle of the kink in TM6, which enables opening of the TM bundle (Liang et al., 2017). Although Stachel peptides of aGPCRs are more hydrophobic and more prone to β strand formation than agonist peptides of secretin receptors, our results suggest that they may share similar molecular signatures for binding to their agonist peptides.
A Cancer-Associated Mutation with Increased Basal Activity Abolishes Lphn Function in C. elegans Development
To get insights in the physiological impact of point mutations that alter Lphn1 signaling on an entire organism, we selected the mutations that exhibited the most effect in our in vitro assay and mutated them in the Lphn homolog lat-1 of the nematode C. elegans: (1) V1094I (L790A in C. elegans), which severely increased basal activity and abolished the response of the receptor to the agonist peptide; importantly, this is a mutation associated with upper aerodigestive tract cancer and maps to TM7 at the NPxxY homology region (O'Hayre et al., 2013). (2) H1096A (H792A in C. elegans), which is one of the few mutations that decreased basal activity, although not completely; this mutation abolished agonist peptide response and maps to TM7 at the NPxxY homology region. (3) F1069A (F763A in C. elegans), which maps to the extracellular end of TM6 and results in abolished agonist peptide response (these three residues are colored cyan or indicated by a cyan asterisk throughout Figures 3, 4, 5, and 6). All three mutations yield receptors with expression levels and localization similar to wild-type lat-1 in C. elegans (Figure 7A).
Figure 7.

An Overactive Cancer-Associated Mutant Fails to Rescue the Developmental Phenotype of LAT-1 Knockdown in C. elegans
(A) Expression and protein localization of all three point mutation-containing variants is indistinguishable from wild-type lat-1::gfp. Fluorescence images show presence of LAT-1 and the variants at the plasma membrane of pharyngeal muscle cells, neurons in the nerve ring, and the pharyngeal nervous system. Scale bars represent 10 μm.
(B and C) The point mutations within the TM of LAT-1 lead to different abilities to rescue fertility (brood size, B) and lethality (individuals reaching adulthood, C) of lat-1 mutants. All three variants ameliorate the fertility defects observed in lat-1 mutants similar to a wild-type lat-1 transgene or a construct comprising the extracellular region tethered to the membrane via the first TM (lat-1 [aa1–581]) (B). Only LAT-1(L790A) does not rescue lethality, while LAT-1(F763A) and LAT-1(H792A) display the same functionality as a wild-type lat-1 transgene. Data are shown as percentage of the original brood sizes (C). The wild-type lat-1 transgene, which rescues fertility and lethality, and a lat-1(aa1–581) construct, which only rescues fertility, served as controls. Data are shown as means ± SEM, n ≥ 20, n.s., not significant; **p < 0.01; ***p < 0.001. SEM, standard error of the mean.
LAT-1 plays essential roles in fertility as well as in oriented cell division in embryogenesis (Langenhan et al., 2009, Prömel et al., 2012). During embryonic development, LAT-1 transduces a signal cell autonomously via the TM and the cytoplasmic tail mediating a G protein-dependent signal (Muller et al., 2015, Prömel et al., 2012), whereas its role in fertility requires only the ECR (Prömel et al., 2012). Consistently, a lat-1 null mutant, lat-1(ok1565) (hereafter referred to as lat-1 mutant), shows severe defects in fertility, characterized by a reduced brood size, and in cell polarity during embryogenesis, reflected in a high lethality rate (Langenhan et al., 2009, Prömel et al., 2012) (Figures 7B and 7C). Expression of transgenic lat-1 constructs ameliorates these phenotypes and leads to a rescue. This rescue can be read out through the total brood size and the level of developmental lethality in the progeny of transformants. We used these assays to assess the functionality of the three generated LAT-1 variants F763A, L790A, and H792A in vivo. All three mutations rescue the fertility defects of lat-1 mutants and thus are able to fulfill LAT-1 function in this context (Figure 7B). Variants carrying F763A and H792A are fully functional receptors as they also rescue the lethality phenotype (Figure 7C). Strikingly, the variant carrying L790A failed to rescue the lethality phenotype. Thus, the point mutation L790A leads to a receptor that is not capable of transducing signals into the cell, suggesting that L790A mutation, which corresponds to the V1094I cancer-associated mutation in rat and exhibits high basal activity and no response to the agonist, is critical for proper LAT-1 function during C. elegans development.
Discussion
GPCRs are the largest family of cell-surface receptors and are the targets for approximately 30% of currently marketed drugs that treat a very broad spectrum of diseases including neuropsychiatric, cardiovascular, pulmonary, and metabolic disorders; cancer; and acquired immune deficiency syndrome (AIDS). Adhesion GPCRs recently emerged as a novel subfamily of GPCRs with important roles in diverse cellular processes, and they carry the potential to be the targets for the next generation of drugs. However, a molecular understanding of their activation mechanisms is a first step for moving forward. In this study, we focused on a model aGPCR subfamily, Lphns, and completed a thorough study that links in vitro molecular understanding of the receptor to in vivo function in whole animals.
Our data show an activation of a Gi signaling cascade by Lphn1, which can be stimulated by a 14-amino-acid endogenous agonist peptide. This peptide compares well with a similar 13-amino-acid-long peptide, which has already been used successfully in previous studies on Lphn1 (Muller et al., 2015). The work by Muller et al. has shown that the receptor activates a Gs cascade leading to the formation of cAMP. These results might be explained by the different assays used in these studies. Owing to the different cell systems, expression levels can vary, and thus the possibility of coupling to other G protein families was not fully excluded in the study. We successfully monitored Lphn downstream signaling by intracellular cAMP and SRE-luciferase assays (Figure 1). A carefully designed mutagenesis screen performed on both assays revealed that approximately half of the mutated residues have an effect on the activity of the receptor (Figures 3 and 5). Importantly, the aGPCR residues that are homologous to the conserved motifs in the rhodopsin family that have diverged significantly, such as the DRY and the NPxxY motifs, are indeed critical for aGPCR function (Figures 4C and 4D). For instance, the ionic lock that the DRY motif seems to be replaced by is a possible hydrophobic lock that is still critical for receptor function. Analysis of the entire dataset suggests that although the TM fold of the aGPCRs carries a similar skeleton and works in a similar fashion to other GPCR families, there are major differences in the fine details within the fold.
To study the mechanism of aGPCR activation by the agonist peptide, we first identified the agonist peptide and showed that the peptide can bind and activate Lphn1 both in trans (i.e., as added synthetic peptide) and in cis (i.e., as tethered to the receptor) (Figure 2). The residues that are involved in the response of the receptor localize to the entire receptor, as they are likely involved in direct peptide binding, signal transduction, or interaction with the G protein (Figure 6). This large dataset is consistent with the numerous allosteric pathways within the TM fold (DeVree et al., 2016). Comparison of the GLP receptor bound to GLP with a model of Lphn1 suggests that the mutations on the extracellular face (A926, Y1001, F1069, and A1078) are likely involved in direct peptide binding (Figure 6D). However, the structure that the agonist peptide adopts when bound to an aGPCR might be different from that of secretin receptor peptides.
Aberrant expression and altered signaling profile of mutated GPCRs contribute to cancer progression and metastasis (O'Hayre et al., 2013). Mutant GPCRs are often overexpressed and constitutively active in various cancers (Dorsam and Gutkind, 2007, Kan et al., 2010, O'Hayre et al., 2013). Disease-associated mutations were previously reported to change other adhesion GPCR signaling (Kishore and Hall, 2017, Purcell et al., 2017). We tested cancer-associated mutations of Lphns reported in different types of carcinomas (Kan et al., 2010, O'Hayre et al., 2013) and found that the cancer-associated mutations affected basal activity and/or peptide response (Figure 4E). Remarkably, V1094I (O'Hayre et al., 2013) mutant was constitutively active in both the cAMP and SRE assays, suggesting a possible role for high activity of Lphns in cancer progression.
Introduction of this highly active cancer-associated mutation to transgenic worms (corresponding to L790A mutation in C. elegans Lat-1) led to a LAT-1 variant devoid of its function in cell polarity during embryonic development but not in fertility (Figures 7B and 7C). As it has been shown that the role of LAT-1 in fertility is solely dependent on its ECR but independent of TM or cytoplasmic tail (Prömel et al., 2012), it is conceivable that any mutation within the TM does not impair LAT-1 function in this context. However, cell polarity during embryogenesis is controlled by LAT-1 via a signal requiring TM and cytoplasmic tail (Muller et al., 2015, Prömel et al., 2012). L790/V1094 is located in TM7 at a position homologous to the NPxxY motif essential for transducing the signal into the cell. An amino acid change at this position likely alters LAT-1 signaling capabilities in a way such that it cannot regulate cell polarity.
Introduction of the V1094I mutation into rat LPHN1 leads to an increase in basal activity, whereas the equivalent mutation (L790A) in the homolog LAT-1 in C. elegans leads to a loss of receptor function in vivo. Several reasons can account for this fact. First, in a living organism, over-activation of the receptor might not have scorable effects. It also cannot be excluded that a highly active variant is detrimental to the organism and that the observed developmental lethality is a consequence of this rather than to a non-active LAT-1. Furthermore, it is conceivable that the motif identified in mammalian Lphns and its impact on receptor integrity and importance for signaling is not comparable to its function in C. elegans LAT-1. Both homologs could have evolved differently during evolution, and thus the variants might not exhibit the same properties.
Altogether, these results provide the groundwork for a thorough characterization of the model aGPCR, Lphn1. The data shed light on signaling pathways and Stachel peptide-mediated activation mechanism of aGPCRs, and correlate disease-associated mutations to in vivo function.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dean Staus for the GloSensor plasmid as a kind gift and for discussions during the identification of the coupled G proteins. We thank Antony Boucard for providing his expertise and discussions on mammalian expression systems. We thank Chuan He for use of his luminescence plate reader. Supported by Brain Research Foundation (D.A.), Big Ideas Generator (D.A.), and NIH grants R01-GM120322 (D.A.), F30-GM116455 (G.S.), and T32GM007183.
Author Contributions
O.N. identified the signaling pathway and performed the signaling assays. A.K. performed bioinformatics analysis, designed mutations, and performed mutagenesis (with assistance from G.S. and P.N.). J.W. and S.P. designed and performed C. elegans experiments. H.S. and G.T. designed and performed direct G protein coupling experiments with assistance from K.L. and Y.L. K.L., K.M., and Y.L. performed the peptide-binding experiments. D.A. designed all experiments, performed data analysis, and wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: May 25, 2018
Footnotes
Supplemental Information includes Transparent Methods, six figures, and one table and can be found with this article online at https://doi.org/10.1016/j.isci.2018.04.019.
Supporting Citations
The following references appear in the Supplemental Information: Brenner, 1974, Dolphin and Hope, 2006, Lee et al., 2001, Mello and Fire, 1995, Mello et al., 1991, Tursun et al., 2009, Warming et al., 2005.
Supplemental Information
Table encodes full raw data. Annotated for each mutation is the location of the mutation, the justification for performing the mutation, the cell-surface expression, signaling (raw and normalized), whether there is an observed peptide response, and possible references. Basal activity means cAMP level without addition of ligand peptide, and it was calculated by taking the luminescence value of wild-type (WT) over the luminescence value of the mutant. A value <1 means mutant is more active than WT, whereas a value >1 means mutant is less active than WT. The slashes between values separate individual experiments. Each individual experiment is repeated at least thrice. Colors match to the residue colors in main figures.
References
- Arac D., Boucard A.A., Bolliger M.F., Nguyen J., Soltis S.M., Sudhof T.C., Brunger A.T. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 2012;31:1364–1378. doi: 10.1038/emboj.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcos-Burgos M., Jain M., Acosta M.T., Shively S., Stanescu H., Wallis D., Domene S., Velez J.I., Karkera J.D., Balog J. A common variant of the latrophilin 3 gene, LPHN3, confers susceptibility to ADHD and predicts effectiveness of stimulant medication. Mol. Psychiatry. 2010;15:1053–1066. doi: 10.1038/mp.2010.6. [DOI] [PubMed] [Google Scholar]
- Audet M., Bouvier M. Restructuring G-protein- coupled receptor activation. Cell. 2012;151:14–23. doi: 10.1016/j.cell.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Bae B.I., Tietjen I., Atabay K.D., Evrony G.D., Johnson M.B., Asare E., Wang P.P., Murayama A.Y., Im K., Lisgo S.N. Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science. 2014;343:764–768. doi: 10.1126/science.1244392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J.A., Jensen A.D., Liapakis G., Rasmussen S.G., Shi L., Gether U., Javitch J.A. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- Bolliger M.F., Martinelli D.C., Sudhof T.C. The cell-adhesion G protein-coupled receptor Bai3 is a high-affinity receptor for C1q-like proteins. Proc. Natl. Acad. Sci. USA. 2011;108:2534–2539. doi: 10.1073/pnas.1019577108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns D.L. Subunit structure and enzymic activity of pertussis toxin. Microbiol. Sci. 1988;5:285–287. [PubMed] [Google Scholar]
- Chae J., Kim M.J., Goo J.H., Collier S., Gubb D., Charlton J., Adler P.N., Park W.J. The Drosophila tissue polarity gene starry night encodes a member of the protocadherin family. Development. 1999;126:5421–5429. doi: 10.1242/dev.126.23.5421. [DOI] [PubMed] [Google Scholar]
- Chang G.W., Stacey M., Kwakkenbos M.J., Hamann J., Gordon S., Lin H.H. Proteolytic cleavage of the EMR2 receptor requires both the extracellular stalk and the GPS motif. FEBS Lett. 2003;547:145–150. doi: 10.1016/s0014-5793(03)00695-1. [DOI] [PubMed] [Google Scholar]
- Coin I., Katritch V., Sun T., Xiang Z., Siu F.Y., Beyermann M., Stevens R.C., Wang L. Genetically encoded chemical probes in cells reveal the binding path of urocortin-I to CRF class B GPCR. Cell. 2013;155:1258–1269. doi: 10.1016/j.cell.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak F., Liu X., Khvotchev M., Li G., Kavalali E.T., Sugita S., Sudhof T.C. Alpha-latrotoxin stimulates a novel pathway of Ca2+-dependent synaptic exocytosis independent of the classical synaptic fusion machinery. J. Neurosci. 2009;29:8639–8648. doi: 10.1523/JNEUROSCI.0898-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demberg L.M., Rothemund S., Schoneberg T., Liebscher I. Identification of the tethered peptide agonist of the adhesion G protein-coupled receptor GPR64/ADGRG2. Biochem. Biophys. Res. Commun. 2015;464:743–747. doi: 10.1016/j.bbrc.2015.07.020. [DOI] [PubMed] [Google Scholar]
- Demberg L.M., Winkler J., Wilde C., Simon K.U., Schon J., Rothemund S., Schoneberg T., Promel S., Liebscher I. Activation of adhesion G Protein-coupled receptors: AGONIST SPECIFICITY OF STACHEL SEQUENCE-DERIVED PEPTIDES. J. Biol. Chem. 2017;292:4383–4394. doi: 10.1074/jbc.M116.763656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVree B.T., Mahoney J.P., Velez-Ruiz G.A., Rasmussen S.G., Kuszak A.J., Edwald E., Fung J.J., Manglik A., Masureel M., Du Y. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature. 2016;535:182–186. doi: 10.1038/nature18324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin C.T., Hope I.A. Caenorhabditis elegans reporter fusion genes generated by seamless modification of large genomic DNA clones. Nucleic Acids Res. 2006;34:e72. doi: 10.1093/nar/gkl352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsam R.T., Gutkind J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- Ebejer J.P., Hill J.R., Kelm S., Shi J., Deane C.M. Memoir: template-based structure prediction for membrane proteins. Nucleic Acids Res. 2013;41:W379–W383. doi: 10.1093/nar/gkt331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R., Lagerstrom M.C., Lundin L.G., Schioth H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Greasley P.J., Fanelli F., Rossier O., Abuin L., Cotecchia S. Mutagenesis and modelling of the alpha(1b)-adrenergic receptor highlight the role of the helix 3/helix 6 interface in receptor activation. Mol. Pharmacol. 2002;61:1025–1032. doi: 10.1124/mol.61.5.1025. [DOI] [PubMed] [Google Scholar]
- Hamann J., Aust G., Arac D., Engel F.B., Formstone C., Fredriksson R., Hall R.A., Harty B.L., Kirchhoff C., Knapp B. International union of basic and clinical pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol. Rev. 2015;67:338–367. doi: 10.1124/pr.114.009647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoare S.R. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov. Today. 2005;10:417–427. doi: 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- Hollenstein K., Kean J., Bortolato A., Cheng R.K., Dore A.S., Jazayeri A., Cooke R.M., Weir M., Marshall F.H. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K., Bittner M.A., Krasnoperov V., Little A.R., Chepurny O., Holz R.W., Petrenko A.G. A novel ubiquitously expressed alpha-latrotoxin receptor is a member of the CIRL family of G-protein-coupled receptors. J. Biol. Chem. 1999;274:5491–5498. doi: 10.1074/jbc.274.9.5491. [DOI] [PubMed] [Google Scholar]
- Kan Z., Jaiswal B.S., Stinson J., Janakiraman V., Bhatt D., Stern H.M., Yue P., Haverty P.M., Bourgon R., Zheng J. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- Kishore A., Hall R.A. Disease-associated extracellular loop mutations in the adhesion G protein-coupled receptor G1 (ADGRG1; GPR56) differentially regulate downstream signaling. J. Biol. Chem. 2017;292:9711–9720. doi: 10.1074/jbc.M117.780551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore A., Purcell R.H., Nassiri-Toosi Z., Hall R.A. Stalk-dependent and stalk-independent signaling by the adhesion G protein-coupled receptors GPR56 (ADGRG1) and Bai1 (ADGRB1) J. Biol. Chem. 2016;291:3385–3394. doi: 10.1074/jbc.M115.689349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnoperov V.G., Bittner M.A., Beavis R., Kuang Y., Salnikow K.V., Chepurny O.G., Little A.R., Plotnikov A.N., Wu D., Holz R.W. alpha-Latrotoxin stimulates exocytosis by the interaction with a neuronal G-protein-coupled receptor. Neuron. 1997;18:925–937. doi: 10.1016/s0896-6273(00)80332-3. [DOI] [PubMed] [Google Scholar]
- Krishnan A., Almen M.S., Fredriksson R., Schioth H.B. The origin of GPCRs: identification of mammalian like rhodopsin, adhesion, glutamate and frizzled GPCRs in fungi. PLoS One. 2012;7:e29817. doi: 10.1371/journal.pone.0029817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnert F., Mancuso M.R., Shamloo A., Wang H.T., Choksi V., Florek M., Su H., Fruttiger M., Young W.L., Heilshorn S.C. Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science. 2010;330:985–989. doi: 10.1126/science.1196554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenhan T., Promel S., Mestek L., Esmaeili B., Waller-Evans H., Hennig C., Kohara Y., Avery L., Vakonakis I., Schnabel R. Latrophilin signaling links anterior-posterior tissue polarity and oriented cell divisions in the C. elegans embryo. Dev. Cell. 2009;17:494–504. doi: 10.1016/j.devcel.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenhan T., Aust G., Hamann J. Sticky signaling–adhesion class G protein-coupled receptors take the stage. Sci. Signal. 2013;6:re3. doi: 10.1126/scisignal.2003825. [DOI] [PubMed] [Google Scholar]
- Lee E.C., Yu D., Martinez de Velasco J., Tessarollo L., Swing D.A., Court D.L., Jenkins N.A., Copeland N.G. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73:56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- Lelianova V.G., Davletov B.A., Sterling A., Rahman M.A., Grishin E.V., Totty N.F., Ushkaryov Y.A. Alpha-latrotoxin receptor, latrophilin, is a novel member of the secretin family of G protein-coupled receptors. J. Biol. Chem. 1997;272:21504–21508. doi: 10.1074/jbc.272.34.21504. [DOI] [PubMed] [Google Scholar]
- Li J., Shalev-Benami M., Sando R., Jiang X., Kibrom A., Wang J., Leon K., Katanski C., Nazarko O., Lu Y.C. Structural basis for teneurin function in circuit-wiring: a toxin motif at the synapse. Cell. 2018;173:735–748.e15. doi: 10.1016/j.cell.2018.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.L., Khoshouei M., Radjainia M., Zhang Y., Glukhova A., Tarrasch J., Thal D.M., Furness S.G.B., Christopoulos G., Coudrat T. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature. 2017;546:118–123. doi: 10.1038/nature22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebscher I., Schon J., Petersen S.C., Fischer L., Auerbach N., Demberg L.M., Mogha A., Coster M., Simon K.U., Rothemund S. A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep. 2014;9:2018–2026. doi: 10.1016/j.celrep.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.H., Chang G.W., Davies J.Q., Stacey M., Harris J., Gordon S. Autocatalytic cleavage of the EMR2 receptor occurs at a conserved G protein-coupled receptor proteolytic site motif. J. Biol. Chem. 2004;279:31823–31832. doi: 10.1074/jbc.M402974200. [DOI] [PubMed] [Google Scholar]
- Lu Y.C., Nazarko O.V., Sando R., 3rd, Salzman G.S., Sudhof T.C., Arac D. Structural basis of latrophilin-FLRT-UNC5 interaction in cell adhesion. Structure. 2015;23:1678–1691. doi: 10.1016/j.str.2015.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R., Jeong S.J., Jin Z., Strokes N., Li S., Piao X. G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination. Proc. Natl. Acad. Sci. USA. 2011;108:12925–12930. doi: 10.1073/pnas.1104821108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello C., Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- Mello C.C., Kramer J.M., Stinchcomb D., Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk K.R., Naylor S.G., Glenn T.D., Mercurio S., Perlin J.R., Dominguez C., Moens C.B., Talbot W.S. A G protein-coupled receptor is essential for Schwann cells to initiate myelination. Science. 2009;325:1402–1405. doi: 10.1126/science.1173474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller A., Winkler J., Fiedler F., Sastradihardja T., Binder C., Schnabel R., Kungel J., Rothemund S., Hennig C., Schoneberg T. Oriented cell division in the C. elegans embryo is coordinated by G-protein signaling dependent on the adhesion GPCR LAT-1. PLoS Genet. 2015;11:e1005624. doi: 10.1371/journal.pgen.1005624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hayre M., Vazquez-Prado J., Kufareva I., Stawiski E.W., Handel T.M., Seshagiri S., Gutkind J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer. 2013;13:412–424. doi: 10.1038/nrc3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan M.L., de Wit J., Savas J.N., Comoletti D., Otto-Hitt S., Yates J.R., 3rd, Ghosh A. FLRT proteins are endogenous latrophilin ligands and regulate excitatory synapse development. Neuron. 2012;73:903–910. doi: 10.1016/j.neuron.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paavola K.J., Stephenson J.R., Ritter S.L., Alter S.P., Hall R.A. The GPR56 N-terminus controls receptor signaling activity. J. Biol. Chem. 2011;286:28914–289121. doi: 10.1074/jbc.M111.247973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters M.C., Mos I., Lenselink E.B., Lucchesi M., IJzerman A.P., Schwartz T.W. Getting from A to B-exploring the activation motifs of the class B adhesion G protein-coupled receptor subfamily G member 4/GPR112. FASEB J. 2016;30:1836–1848. doi: 10.1096/fj.201500110. [DOI] [PubMed] [Google Scholar]
- Piao X., Hill R.S., Bodell A., Chang B.S., Basel-Vanagaite L., Straussberg R., Dobyns W.B., Qasrawi B., Winter R.M., Innes A.M. G protein-coupled receptor-dependent development of human frontal cortex. Science. 2004;303:2033–2036. doi: 10.1126/science.1092780. [DOI] [PubMed] [Google Scholar]
- Prömel S., Frickenhaus M., Hughes S., Mestek L., Staunton D., Woollard A., Vakonakis I., Schöneberg T., Schnabel R., Russ A.P. The GPS motif is a molecular switch for bimodal activities of adhesion class G protein-coupled receptors. Cell Rep. 2012;2:321–331. doi: 10.1016/j.celrep.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prömel S., Langenhan T., Arac D. Matching structure with function: the GAIN domain of adhesion-GPCR and PKD1-like proteins. Trends Pharmacol. Sci. 2013;34:470–478. doi: 10.1016/j.tips.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Purcell R.H., Toro C., Gahl W.A., Hall R.A. A disease-associated mutation in the adhesion GPCR Bai2 (ADGRB2) increases receptor signaling activity. Hum. Mutat. 2017;38:1751–1760. doi: 10.1002/humu.23336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S.G., DeVree B.T., Zou Y., Kruse A.C., Chung K.Y., Kobilka T.S., Thian F.S., Chae P.S., Pardon E., Calinski D. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D.M., Cherezov V., Hanson M.A., Rasmussen S.G., Thian F.S., Kobilka T.S., Choi H.J., Yao X.J., Weis W.I., Stevens R.C. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Salzman G.S., Ackerman S.D., Ding C., Koide A., Leon K., Luo R., Stoveken H.M., Fernandez C.G., Tall G.G., Piao X. Structural basis for regulation of GPR56/ADGRG1 by its alternatively spliced extracellular domains. Neuron. 2016;91:1292–1304. doi: 10.1016/j.neuron.2016.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schioth H.B., Nordstrom K.J., Fredriksson R. The adhesion GPCRs; gene repertoire, phylogeny and evolution. Adv. Exp. Med. Biol. 2011;706:1–13. doi: 10.1007/978-1-4419-7913-1_1. [DOI] [PubMed] [Google Scholar]
- Scholz N., Gehring J., Guan C., Ljaschenko D., Fischer R., Lakshmanan V., Kittel R.J., Langenhan T. The adhesion GPCR latrophilin/CIRL shapes mechanosensation. Cell Rep. 2015;11:866–874. doi: 10.1016/j.celrep.2015.04.008. [DOI] [PubMed] [Google Scholar]
- Scholz N., Guan C., Nieberler M., Grotemeyer A., Maiellaro I., Gao S., Beck S., Pawlak M., Sauer M., Asan E. Mechano-dependent signaling by Latrophilin/CIRL quenches cAMP in proprioceptive neurons. Elife. 2017;6 doi: 10.7554/eLife.28360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashidhar S., Lorente G., Nagavarapu U., Nelson A., Kuo J., Cummins J., Nikolich K., Urfer R., Foehr E.D. GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene. 2005;24:1673–1682. doi: 10.1038/sj.onc.1208395. [DOI] [PubMed] [Google Scholar]
- Shima Y., Kengaku M., Hirano T., Takeichi M., Uemura T. Regulation of dendritic maintenance and growth by a mammalian 7-pass transmembrane cadherin. Dev. Cell. 2004;7:205–216. doi: 10.1016/j.devcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Silva J.P., Lelianova V.G., Ermolyuk Y.S., Vysokov N., Hitchen P.G., Berninghausen O., Rahman M.A., Zangrandi A., Fidalgo S., Tonevitsky A.G. Latrophilin 1 and its endogenous ligand Lasso/teneurin-2 form a high-affinity transsynaptic receptor pair with signaling capabilities. Proc. Natl. Acad. Sci. USA. 2011;108:12113–12118. doi: 10.1073/pnas.1019434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu F.Y., He M., de Graaf C., Han G.W., Yang D., Zhang Z., Zhou C., Xu Q., Wacker D., Joseph J.S. Structure of the human glucagon class B G-protein-coupled receptor. Nature. 2013;499:444–449. doi: 10.1038/nature12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoveken H.M., Hajduczok A.G., Xu L., Tall G.G. Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist. Proc. Natl. Acad. Sci. USA. 2015;112:6194–6199. doi: 10.1073/pnas.1421785112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof T.C. alpha-Latrotoxin and its receptors: neurexins and CIRL/latrophilins. Annu. Rev. Neurosci. 2001;24:933–962. doi: 10.1146/annurev.neuro.24.1.933. [DOI] [PubMed] [Google Scholar]
- Sugita S., Khvochtev M., Sudhof T.C. Neurexins are functional alpha-latrotoxin receptors. Neuron. 1999;22:489–496. doi: 10.1016/s0896-6273(00)80704-7. [DOI] [PubMed] [Google Scholar]
- Tursun B., Cochella L., Carrera I., Hobert O. A toolkit and robust pipeline for the generation of fosmid-based reporter genes in C. elegans. PLoS One. 2009;4:e4625. doi: 10.1371/journal.pone.0004625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T., Shima Y., Shimada Y., Hirano S., Burgess R.W., Schwarz T.L., Takeichi M., Uemura T. Flamingo, a seven-pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell. 1999;98:585–595. doi: 10.1016/s0092-8674(00)80046-x. [DOI] [PubMed] [Google Scholar]
- Warming S., Costantino N., Court D.L., Jenkins N.A., Copeland N.G. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins H.A., Au M., Hay D.L. The structure of secretin family GPCR peptide ligands: implications for receptor pharmacology and drug development. Drug Discov. Today. 2012;17:1006–1014. doi: 10.1016/j.drudis.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Xu L., Begum S., Hearn J.D., Hynes R.O. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc. Natl. Acad. Sci. USA. 2006;103:9023–9028. doi: 10.1073/pnas.0602681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Sun B., Feng D., Hu H., Chu M., Qu Q., Tarrasch J.T., Li S., Sun Kobilka T., Kobilka B.K. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature. 2017;546:248–253. doi: 10.1038/nature22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table encodes full raw data. Annotated for each mutation is the location of the mutation, the justification for performing the mutation, the cell-surface expression, signaling (raw and normalized), whether there is an observed peptide response, and possible references. Basal activity means cAMP level without addition of ligand peptide, and it was calculated by taking the luminescence value of wild-type (WT) over the luminescence value of the mutant. A value <1 means mutant is more active than WT, whereas a value >1 means mutant is less active than WT. The slashes between values separate individual experiments. Each individual experiment is repeated at least thrice. Colors match to the residue colors in main figures.