

Summary
We synthesized a mitochondria-targeted honokiol (Mito-HNK) that facilitates its mitochondrial accumulation; this dramatically increases its potency and efficacy against highly metastatic lung cancer lines in vitro, and in orthotopic lung tumor xenografts and brain metastases in vivo. Mito-HNK is >100-fold more potent than HNK in inhibiting cell proliferation, inhibiting mitochondrial complex ǀ, stimulating reactive oxygen species generation, oxidizing mitochondrial peroxiredoxin-3, and suppressing the phosphorylation of mitoSTAT3. Within lung cancer brain metastases in mice, Mito-HNK induced the mediators of cell death and decreased the pathways that support invasion and proliferation. In contrast, in the non-malignant stroma, Mito-HNK suppressed pathways that support metastatic lesions, including those involved in inflammation and angiogenesis. Mito-HNK showed no toxicity and targets the metabolic vulnerabilities of primary and metastatic lung cancers. Its pronounced anti-invasive and anti-metastatic effects in the brain are particularly intriguing given the paucity of treatment options for such patients either alone or in combination with standard chemotherapeutics.
Subject Areas: Natural Product Chemistry, Immunology, Medicinal and Aromatic Plants
Graphical Abstract
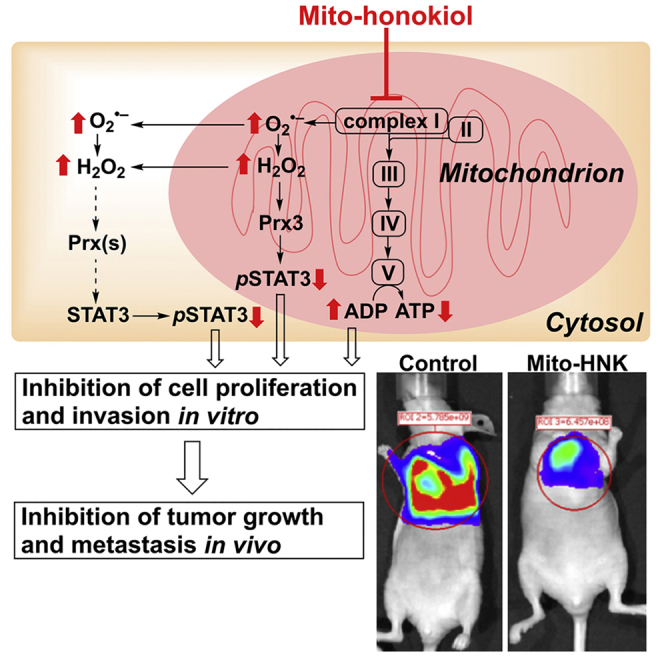
Highlights
-
•
Synthesis of mitochondria-targeted honokiol (Mito-HNK);
-
•
Mito-HNK inhibits mitochondrial complex I, and stimulates oxidizing peroxiredoxin-3
-
•
Mito-HNK suppresses the phosphorylation of mitoSTAT3
-
•
Mito-HNK has pronounced activity against lung cancer and its brain metastases
Natural Product Chemistry; Immunology; Medicinal and Aromatic Plants
Introduction
Lung cancer is the leading cause of cancer deaths worldwide. At the time of diagnosis, metastatic dissemination is seen in many patients with either non-small-cell lung cancer (NSCLC) or small cell lung cancer (SCLC) (Planchard and Giroux Leprieur, 2011). The overall 5-year survival rate for lung cancer is still only 10%–15% (Planchard and Giroux Leprieur, 2011). The brain is the most common site for lung cancer metastasis; 25%–30% of patients with NSCLC and 80% of patients with SCLC will develop brain metastasis during the course of their disease. Because the blood-brain barrier (BBB) is impermeable to most chemotherapeutic drugs used for lung cancer, the only available therapies for brain metastases include whole-brain irradiation or surgical resection in eligible patients. Although some small-molecule tyrosine kinase inhibitors have shown efficacy in a subset of patients, the results have not been consistent owing to the relatively low cerebrospinal fluid (CSF)-to-plasma ratios (Goldberg et al., 2015). Therefore, discovering novel small molecule agents that can inhibit both primary and metastatic lung cancer, including brain metastases, is a critical unmet need.
Although many tumor cells rely on aerobic glycolysis as the major source of adenosine triphosphate (ATP) to fuel cell proliferation (the Warburg effect), increasing evidence indicates that mitochondria are indeed functional in most tumor cells (Cheng et al., 2012, Cheng et al., 2013, Cheng et al., 2014, Cheng et al., 2015, Weinberg et al., 2010, Berridge et al., 2015). Recent profiling of cellular bioenergetics has provided new insights on the intermediacy of mitochondrial metabolism in tumor cells (Cheng et al., 2012, Cheng et al., 2013, Cheng et al., 2014, Cheng et al., 2015, Weinberg et al., 2010, Berridge et al., 2015). Cancer cells without mitochondrial DNA (mtDNA), called rho0 or ρ0 cells, show delayed tumor formation (Tan et al., 2015). However, mtDNA can be acquired by transfer of whole mitochondria from the host animal, which normalizes mitochondrial respiration and restores tumorigenic potential (Dong et al., 2017). Furthermore, several anti-cancer agents were recently identified that specifically target mitochondrial function, such as resveratrol (Widlund et al., 2017) or metformin (Wheaton et al., 2014). Thus, targeting mitochondrial bioenergetics is emerging as a promising and viable therapeutic approach to inhibit the proliferation of both poorly glycolytic and highly glycolytic cancer cells (Cheng et al., 2012, Cheng et al., 2013, Cheng et al., 2014, Cheng et al., 2015, Weinberg et al., 2010, Weinberg and Chandel, 2015). Honokiol (HNK), a key bioactive compound present in magnolia bark extracts, exhibits anti-cancer and anti-metastatic properties in a variety of in vitro and in vivo models (Pan et al., 2016, Arora et al., 2012). We previously demonstrated that high-dose HNK (37.5 μmol/kg) inhibits the development of squamous cell carcinoma (SCC) in a carcinogen-induced mouse model (Pan et al., 2014), in which HNK was shown to suppress mitochondrial respiration, decrease ATP levels, and increase the generation of reactive oxygen species (ROS) (Pan et al., 2014). Thus even though HNK is not specifically sequestered inside mitochondria, these findings revealed that HNK can directly target mitochondrial bioenergetics, leading to persistent inhibition of mitochondrial respiration in lung cancer cells (Pan et al., 2014). We therefore hypothesized that conjugating HNK to a compound that drives HNK into mitochondria should dramatically increase its antitumor efficacy and potency. There is ample evidence in the literature that supports the therapeutic efficacy of mitochondria-targeted triphenylphosphonium (TPP+)-linked agents in cellular and animal models and possibly in humans (Murphy and Smith, 2007, Gane et al., 2010, Tauskela, 2007, Snow et al., 2010, Murphy, 2016, Smith et al., 2012, Smith and Murphy, 2010). We therefore developed mitochondria-targeted honokiol (Mito-HNK) by attaching the bulky TPP+ group to HNK via a long alkyl chain, well separated from HNK's aromatic structure. Published reports also indicate that increasing the lipophilicity of TPP+-conjugated compounds enhances their cellular and mitochondrial uptake (Smith et al., 2003, Asin-Cayuela et al., 2004, Ross et al., 2008, Finichiu et al., 2015). Lipophilic cations, including TPP+-based agents, selectively accumulate in cancer cells in vitro and in vivo owing to an elevated mitochondrial membrane potential (Summerhayes et al., 1982, Lampidis et al., 1985, Rideout et al., 1989, Modica-Napolitano and Aprille, 2001, Cheng et al., 2013). Another rationale for using Mito-HNK in this lung cancer brain metastasis study is the observation that the TPP+-based compound, [11C]-triphenylmethylphosphonium (TPMP), selectively accumulates in brain tumors (Madar et al., 1999).
Here we describe the synthesis and detailed in vitro and in vivo characterization of Mito-HNK. In human NSCLC lines derived from brain metastases and in an SCLC line with high metastatic potential, Mito-HNK proved to be more than 100-fold more potent than HNK in inhibiting cell proliferation, suppressing mitochondrial bioenergetics by inhibiting complex I, and stimulating ROS generation. To further support its mitochondrial effects, Mito-HNK oxidized mitochondrial peroxiredoxin-3 (Prx3) and suppressed the phosphorylated form of mitoSTAT3, which functions as a positive regulator of the mitochondrial electron transport chain through non-canonical STAT3 signaling in mitochondria (Yang and Rincon, 2016). Mito-HNK also effectively suppressed the invasion of NSCLC and SCLC cells. In vivo, low-dose Mito-HNK (3.75 μmol/kg) markedly suppressed the growth of NSCLC and SCLC lines in a lung orthotopic model and inhibited the growth and progression of lung cancer brain metastases. Within the brain metastases in mice, Mito-HNK induced mediators of cell death in the tumors, and it decreased pathways that support invasion and proliferation. In contrast, in the non-malignant stroma, Mito-HNK suppressed pathways that support metastatic lesions, including those involved in inflammation and angiogenesis. Even at doses 20-fold higher than those that were effective, Mito-HNK exhibited no toxicity in mice over 8 weeks of administration. Mito-HNK is therefore a safe agent that targets the metabolic vulnerabilities of primary and metastatic lung cancers. Its pronounced anti-invasive and anti-metastatic effects against brain metastases are particularly intriguing given the paucity of treatment options for such patients.
Results
Synthesis and Toxicity of Mitochondria-Targeted HNK
Two Mito-HNKs were synthesized by reacting HNK with the bromodecyl-TPP+ cation, generating HNK with one TPP+ (Mito-HNK) or two TPP+ (bis-Mito-HNK) moieties (Figure 1A). The products were isolated by gel chromatography and purified by preparative high-performance liquid chromatography (HPLC), and their structures were characterized by nuclear magnetic resonance (NMR) and mass spectrometry (MS) (Figures S1–S3; Figure S3 related to Figure 1). To assess the potential toxicity of Mito-HNK, an 8-week subchronic toxicity study was conducted in A/J mice. As neurons and muscle cells may have a relatively high plasma membrane potential that might enhance the uptake of Mito-HNK, we used a modified Irwin screen that was developed as a comprehensive observational battery to broadly screen for central nervous system (CNS) and neuromuscular effects of agents (Irwin, 1968, Crawley, 2000). Modifications of this test are extensively used in the pharmaceutical industry and in academic research to identify many possible changes in neurological function (Lindgren et al., 2008, Crawley, 2000). The screen we used employed 35 distinct measurements to assess sensorimotor, neurological, and autonomic nervous system function (Olsen et al., 2010). We included mice treated with vehicle control and with various doses of Mito-HNK (7.5, 37.5, and 75 μmol/kg, which represent 2×, 10×, and 20× of the effective dose, respectively) (Figure 1B), given via oral gavage 5 days per week for 8 weeks. After 8 weeks of treatment, we did not observe any significant differences between control mice and those treated with any dose of Mito-HNK, including the highest dose (75 μmol/kg) (Figures 1B and 1C; Table S1, related to Figure 1), for any of the 35 metrics tested. Furthermore, no histopathological changes were seen in either neural (frontal cortex and cerebellum) or muscle tissue (skeletal muscles including the soleus, plantaris, gastrocnemius, tibialis anterior, and quadriceps) (data not shown). Thus Mito-HNK did not show any indications of toxicity at a dose that is 20-fold higher than the effective dose.
Figure 1.

Design, Synthesis, and Subchronic Toxicity Screen of Mito-HNK
(A) Synthesis of Mito-HNK. Both mono-substituted (Mito-HNK) and double-substituted (Bis-Mito-HNK) mitochondria-targeted derivatives of honokiol were synthesized. The products were purified by HPLC and characterized by NMR and mass spectrometry. TPP+-linked compounds accumulate selectively in cancer cell mitochondria, according to the Nernst equation.
(B and C) Examples of subchronic toxicity screen of Mito-HNK. (B) Measured body weight (squares, left axis) and rectal temperature (circles, right axis). (C) Latency to fall in the inverted screen test measured after 8 weeks of treatment. The 2×, 10×, and 20× doses relate to the effective dose (1× = 3.5 μmol/kg) and correspond to 7.5, 37.5, and 75 μmol/kg, respectively. Error bars represent standard deviation (SD), n = 5 mice per group. Error bars indicate SE.
Mito-HNK Inhibits Lung Cancer Cell Proliferation and Tumor Growth
Before conducting lung cancer brain metastases studies, in vitro studies compared the effects of Mito-HNK and bis-Mito-HNK on the proliferation of lung cancer brain metastatic H2030-BrM3 and PC9-BrM3 cells. Mito-HNK inhibits H2030-BrM3 lung cancer cell proliferation at significantly lower levels (IC50 = 0.26 μM) than HNK (IC50 = 27 μM) (Figure 2A). The double-substituted HNK analog (bis-Mito-HNK) is also significantly more potent than HNK (IC50 = 0.8 μM, Figure 2A), but not as potent as Mito-HNK. Because Bis-Mito-HNK has both hydroxyl groups substituted, it is unlikely to be easily metabolized to a quinone methide, suggesting other mechanism(s) of action. In subsequent experiments, we used the more potent mono-substituted analog, Mito-HNK. We verified that the more than 100-fold enhancement in anti-proliferative properties of Mito-HNK versus HNK is due to its targeting to mitochondria, because of the attached TPP+ moiety. Liquid chromatography-mass spectrometry (LC-MS/MS) analysis of the fractions from H2030-BrM3 cells treated for 1 hr with 1 μM Mito-HNK showed mitochondrial accumulation of Mito-HNK (0.6 nmoles versus 4.3 nmoles in cytosolic versus mitochondrial fractions per 106 cells, respectively). Also, treatment of cells (0.1 μM, 24 hr) led to significantly higher mitochondrial accumulation of Mito-HNK than HNK (Figure 2B). Combinations of HNK or decyl-HNK with Me-TPP+ (see Figure 2C for chemical structures) failed to reproduce the anti-proliferative effects of Mito-HNK (Figures 2D and 2E). Thus, attaching TPP+ to HNK markedly increases mitochondrial accumulation and anti-proliferative activity in lung cancer cells.
Figure 2.

Mito-HNK Inhibits the Proliferation and Tumor Growth of Lung Cancer Cells
(A) Mito-HNK and bis-Mito-HNK inhibit the proliferation of H2030 cells at significantly lower concentrations than HNK. The IC50 values were determined at the point when untreated cells reached ∼95% confluence (∼4 days of incubation).
(B) Treatment of H2030-BrM3 cells (0.1 μM, 24 hr) leads to significantly higher mitochondrial accumulation of Mito-HNK than HNK (***P < 0.001 vs. HNK).
(C) The chemical structures of Mito-HNK and the control compounds (Dec-HNK and Me-TPP+) used for data in panels D and E.
(D and E) The combinations of HNK or decyl-HNK with Me-TPP+ fail to reproduce the anti-proliferative effects of Mito-HNK. The traces recorded during real-time monitoring of cell confluence are shown in (D), and the cell confluence after 5 days of incubation with the compounds (1 μM) is shown in (E).
(F) Representative bioluminescence live imaging of NSCLC orthotopic tumor growth in control or Mito-HNK-treated mice (3.75 μmol/kg each).
(G) Quantitative data for bioluminescence imaging of the orthotropic growth of NSCLC (H2030-Br3M) cells (n = 6 per group, observation duration = 21 days).
(H) Quantitative data for the bioluminescence imaging of the orthotropic growth of SCLC (DMS-273) cells (n = 6 per group, observation duration = 15 days).
(I) Left panel, representative immunohistochemistry staining of H2030-BrM3 orthotopic lung tumors for Ki-67 in control and Mito-HNK-treated groups; right panel, quantitative estimation of cell proliferation showing percentage of Ki-67+ cells (**p < 0.01 versus control).
(J) Representative H&E staining images of H2030-BrM3 orthotopic lung tumors taken from control and Mito-HNK groups. Error bars indicate SE.
Next, we evaluated the efficacy of Mito-HNK in lung tumor growth using an orthotopic model of lung cancer in NOD/SCID mice. At 8 weeks of age, H2030-BrM3 (NSCLC) or DMS-273 (SCLC) cells were injected into the left lung of mice. After 1 week, mice were treated by oral gavage 5 days per week with Mito-HNK (3.75 μmol/kg) or vehicle (corn oil). During the treatment, the progression of lung tumors was monitored by bioluminescence imaging. As shown in Figures 2F–2H, Mito-HNK (3.75 μmol/kg) reduced tumor size by ∼70% (H2030-BrM3 cells) or ∼40% (DMS-273 cells) when compared with control mice, whereas the same dose of HNK failed to decrease lung tumor growth (data not shown). However, in our previous study, a 10-fold higher dose of HNK (37.5 μmol/kg) was effective in decreasing lung tumor growth (Pan et al., 2017). Immunohistochemistry of serial sections showed a significant reduction in the proliferation marker Ki-67 in Mito-HNK-treated tumors (Figure 2I). Hematoxylin and eosin (H&E) staining did not reveal other morphological changes upon Mito-HNK treatment (Figure 2J).
Mito-HNK Inhibits Invasion and Brain Metastasis of Lung Cancer Cells
The Boyden chamber invasion assay, which was used to assess anti-metastatic potential, showed that Mito-HNK (48 hr) suppressed the in vitro invasion of both brain metastatic NSCLC lines in a dose-dependent manner (Figures 3A and 3C ). Similarly, Mito-HNK inhibited the invasion of the SCLC line in a dose dependent manner (DMS-273) (Figures 3B and 3D) and was over 100-fold more potent than HNK (Pan et al., 2017). To rule out the possibility of pro-apoptotic or cytotoxic effects of Mito-HNK, the invasion assay was also conducted in lung cancer cells pretreated with Mito-HNK; as shown in Figure S4A; wells pretreated with Mito-HNK had significantly less cell invasion compared to control wells, while at these doses Mito-HNK did not cause significant cell death or pro-apoptotic activities, but rather blocked cell cycling machinery (Figures S4B and S4C, related to Figure 3), indicating that Mito-HNK directly inhibits cancer cell invasion.
Figure 3.

Mito-HNK Inhibits Invasion and Brain Metastasis of Lung Cancer Cells
(A and C) The anti-invasive effects of Mito-HNK on NSCLC lines H2030-BrM3 and PC9-BrM3 were assessed via the Boyden chamber invasion assay after 48-hr treatment. Representative images are shown in (A) and the quantitative data are shown in (C, *P < 0.05, **P < 0.01, ***P < 0.001 vs.control).
(B and D) Representative images (B) and quantitative data (D) of the invasion assay for SCLC DMS-273 cells indicate that Mito-HNK is at least 100-fold more potent than HNK (*P < 0.05, **P < 0.01 vs.control).
(E) High-resolution echocardiography to visualize the needle position within the left cardiac ventricle, for injecting lung cancer cells in the in vivo experiments to establish brain metastases.
(F) Representative bioluminescence, GFP expression, and H&E staining images of brains taken from control, HNK, and Mito-HNK (3.75 μmol/kg each) mice.
(G and H) Quantitative data for the bioluminescence imaging of brain metastases over time. Mito-HNK treatment was started 1 day after injection of H2030-BrM3 cells (G, n = 6 for control, and n = 5 for Mito-HNK, observation duration = 27 days) or DMS-273 cells (H, n = 7 each, observation duration = 14 days). (G, inset) shows the LC-MS trace from a brain extract indicating the presence of Mito-HNK (65 pmol/g of whole brain tissue). Error bars indicate SE.
To investigate the effect of Mito-HNK on in vivo metastasis, we used a previously characterized experimental lung cancer brain metastasis model to generate brain metastases in mice (Nguyen et al., 2009b, Pan et al., 2017). We generated green fluorescent protein (GFP)-luciferase-expressing variants of each lung cancer cell line (both NSCLC and SCLC) and confirmed that they efficiently formed brain metastases. We used an ultrasound-guided procedure to secure the precise injection of brain-seeking lung cancer cells into the left cardiac ventricle of NOD/SCID mice as described previously (Pan et al., 2017). These injected lung cancer cells rapidly colonized the cerebrum and cerebellum, forming many nodules in animals treated with vehicle control (Figure 3F). Mito-HNK (3.75 μmol/kg) was administered by oral gavage starting 1 day after engrafting lung cancer cells in the arterial circulation; brain metastases were monitored by bioluminescence upon injection of luciferin. Mito-HNK inhibited the growth of lung cancer brain metastases of both NSCLC (H2030-BrM3) (Figure 3G) and SCLC (DMS-273) (Figure 3H) cells. Metastases were confirmed by ex vivo GFP imaging, H&E, and GFP immunostaining (Figure 3F). LC-MS analyses of brain extracts (Figure 3G) confirm that Mito-HNK crosses the BBB. These results show that Mito-HNK is a potent inhibitor of lung cancer brain metastases for both the NSCLC and SCLC models used.
Mito-HNK Inhibits Complex I, Promotes ROS Generation, and Oxidizes Mitochondrial Peroxiredoxin-3
We previously found that HNK blocks squamous cell lung cancer progression by inhibiting cellular respiration (or oxygen consumption rate [OCR]) and mitochondrial function (Pan et al., 2014). Here, we determined the effect of Mito-HNK on mitochondrial respiration. To test the activity on mitochondrial complexes, the cells were pretreated for 24 hr with Mito-HNK or HNK, followed by measuring the activities of mitochondrial respiratory complexes using the Seahorse XF96 extracellular flux analyzer. This approach provides high throughput, does not require cell fractionation, and can monitor the effect on complexes I and II in a single run. While establishing the model systems, we observed that rotenone (complex I inhibitor) blocks OCR, which can be restored by adding succinate (complex II substrate), whereas in the presence of malonate (complex II inhibitor), the addition of succinate does not stimulate OCR (Figure S5, related to Figure 4). Antimycin A (complex III inhibitor) decreases both pyruvate- and succinate-driven OCR. These studies established the validity of these mitochondrial complex assays, in agreement with the published protocol (Salabei et al., 2014). Next, we compared the ability of HNK and Mito-HNK to inhibit mitochondrial complex I in H2030-BrM3 and DMS-273 cell lines. Mito-HNK was greater than 100-fold more potent than HNK in inhibiting complex I both in H2030-BrM3 (Figure 4A) and DMS-273 (Figure 4B) cells.
Figure 4.

Effects of Mito-HNK on Mitochondrial Complex I Activity, ROS, and the Redox State of Peroxiredoxins
To measure complex I activity, cells were pretreated for 24 hr with Mito-HNK and HNK, the cell membrane was permeabilized, and OCR was measured upon the addition of mitochondrial substrates/inhibitors.
(A and B) Both HNK (IC50 = 30 μM for both cell lines) and Mito-HNK (IC50 = 0.1 μM for both cell lines) inhibit complex I in H2030-BrM3 NSCLC cells (A) and DMS-273 SCLC cells (B).
(C and D) The effect of Mito-HNK (1 μM, 24 hr treatment) on cellular ROS production, as measured by HPLC-based profiling of the oxidation products of the HE probe in H2030-BrM3 (C) and DMS-273 (D) cells. The compound 2-hydroxyethidium (2-OH-E+) is a specific product for superoxide and diethidium (E+-E+) is a marker product for one-electron oxidants. HPLC traces are shown in the left panels and the quantitative results in the right panels (**p < 0.01, ***p < 0.001).
(E and F) The 24-hr treatment of H2030-BrM3 cells with 0.2 μM Mito-HNK (E) or DMS-273 cells with 0.3 μM Mito-HNK (F) leads to significant oxidation of mitochondrial Prx3, whereas the oxidation state of cytosolic Prx1 is not significantly affected (**P < 0.01, ***P < 0.001 vs.control). Error bars indicate SD.
We hypothesized that inhibition of mitochondrial complex I by Mito-HNK would lead to increased ROS levels in lung cancer cells. Indeed, 2-OH-E+, the O2⋅–-specific product of hydroethidine (HE) oxidation, was significantly increased in H2030-BrM3 and DMS-723 cells treated with Mito-HNK (Figures 4C, 4D, and S6; Figure S6 related to Figure 4). A strong induction of one-electron oxidation of HE with the formation of diethidium (E+-E+) was also observed, indicating that Mito-HNK also induces the generation of another stronger oxidant in both H2030-BrM3 and DMS-273 cells (Figures 4C and 4D). Mito-HNK induced oxidant production at a 100-fold lower concentration than is required for HNK (Figure S6, related to Figure 4). To assess if mitochondrial oxidative stress occurs as a result of Mito-HNK-induced ROS, we tested the oxidation status of cytosolic (Prx1) and mitochondrial (Prx3) peroxiredoxins in lung cancer cells treated for 24 hr with Mito-HNK, using doses that inhibit complex I by 60%–80% (as shown in Figures 4A and 4B). In all three lung cancer lines, these treatments caused significant oxidation of mitochondrial Prx3 but relatively little effect on the redox status of cytosolic Prx1 (examples shown in Figures 4E and 4F, with the data for PC9-BrM3 cells shown in Figure S7; Figure S7 related to Figure 4). This implies that Mito-HNK-induced ROS formation occurs primarily in the mitochondria, consistent with its site of accumulation.
Mitochondrial DNA Depletion Abrogates Mito-HNK's Effect on Cancer Cell Proliferation
Mitochondria are vital organelles and the powerhouse of eukaryotic cells. mtDNA, encodes for 13 proteins of the ∼1,500 total that comprise the mitochondrial proteome. All 13 mtDNA-encoded proteins are subunits of mitochondrial respiratory complexes and are therefore critical for the bioenergetic machinery. Mutations and copy number changes in mtDNA are often linked with cancer development. A recent study showed that tumor cells without mtDNA (ρ0 cells) have delayed tumor growth (Tan et al., 2015, Dong et al., 2017), and the transfer of intact mitochondria with their mtDNA from host cells to the tumor cells can restore the tumor cells' respiration and tumor formation efficacy. To delineate the involvement of mitochondria in Mito-HNK's anti-cancer effects, we tested Mito-HNK in these same ρ0 cells. ρ0 Cells showed no mitochondrial complex I and complex II activities compared with parental cells (Figures 5A and 5B, left panels), which is consistent with the absence of mitochondria in ρ0 cells. PicoGreen DNA staining revealed a complete loss of mtDNA in both ρ0 cell lines (Figures 5A and 5B, right panel, bottom row, arrows). In contrast, the parental lines that had mitochondria showed numerous cytoplasmic green punctae located within the mitochondrial fibrillar network (Figures 5A and 5B, right panel, top row, arrows) representing mtDNA nucleoids that co-localized with MitoTracker Deep Red staining. Mito-HNK treatment inhibited cell proliferation in both parental cell lines by >50% (Figures 5C and 5D), but showed only minimal effects on both ρ0 cell lines, indicating that loss of mitochondrial function abrogates the anti-proliferative effects of Mito-HNK. We further validated these findings using another approach, i.e., depleting host mtDNA with a truncated form of a viral UL12.5 gene (Figure S10, related to Figure 5). These results strongly suggest that the anti-cancer effects of Mito-HNK are dependent on mitochondrial function.
Figure 5.

Depletion of mtDNA Abrogates the Anti-Proliferative Effects of Mito-HNK in Lung Cancer Cells
(A and B) Validation of the loss of activities of the mitochondrial complexes in ρ0 cells. (A and B, left panels) Permeabilized cells were assayed in medium containing 10 mM pyruvate and 1.5 mM malate (substrates for complex I) in mannitol and sucrose (MAS) buffer. The complex I-related oxygen consumption rate (OCR) was assayed immediately and verified by injecting rotenone (complex I inhibitor) as indicated. Then, complex II-related OCR was measured by supplying cells with succinate (substrate for complex II, 10 mM). Both malonate (complex II inhibitor, 10 mM) and antimycin A (complex III inhibitor, 20 μM) were injected where indicated. (A and B, right panels) Validation of the absence of mitochondria in ρ0 cells with PicoGreen staining for mtDNA. Arrows indicate mitochondria fibrillar network.
(C and D) mtDNA depletion abrogates the anti-proliferative effects of Mito-HNK (0.4 μM, 48 hr) in both B16 and 143B ρ0 cells. (***P < 0.001 vs. B16 or 143b parental cells). Error bars indicate SD.
Role of mitoSTAT3 in Mediating the Effects of Mito-HNK on Lung Cancer Cells
Potential additional components of the mechanism by which Mito-HNK inhibits lung cancer brain metastasis were examined via the PathScan RTK Signaling Antibody Array (Figure S8A, related to Figure 6), which has been used extensively to study mechanisms of candidate cancer drugs (Pan et al., 2017, Vazquez-Martin et al., 2013). This array identified mitoSTAT3 as a potential target of Mito-HNK (Figure 6A). We validated the effects of Mito-HNK on mitoSTAT3 phosphorylation in lung cancer cells via western blot (Figures 6B and S8B; Figure S8B related to Figure 6). Since AMPK and mitoSTAT3 can play important roles in regulating mitochondrial activity, apoptosis, proliferation, and migration (Sobotta et al., 2015, Feng et al., 2014, Liu et al., 2003, Zhang et al., 2013, Wen et al., 1995), we examined the effects of Mito-HNK on AMPKThr172 and mitoSTAT3Ser727 phosphorylation by western blot (Figure 6B). Compared with HNK, Mito-HNK induced much stronger AMPK phosphorylation and potently inhibited mitoSTAT3Ser727 phosphorylation in both PC9-BrM3 and H2030-BrM3 cells. We then used the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas9) system to knock out STAT3 (Figure S9, related to Figure 6). Cell lines were screened by polymerase chain reaction (PCR) and sequencing, and knockouts were confirmed by western blotting (Figure 6C). CRISPR significantly decreased STAT3 protein by >80% (Figure 6C), and the anti-invasive and anti-proliferation effects of Mito-HNK (24 hr) were abrogated in the STAT3-deficient cells (Figures 6D and 6E). However, no direct binding of Mito-HNK to STAT3 was detected (not shown) using an anti-TPP+ antibody (obtained from Dr. Michael P. Murphy, Mitochondrial Biology Unit, Cambridge, UK).
Figure 6.

Role of STAT3 in the Anti-proliferative and Anti-invasive Effects of HNK and Mito-HNK in Lung Cancer Cells
(A) Results of the receptor tyrosine kinase proteomic array of H2030-BrM3 cells treated with HNK (20 μM) or Mito-HNK (0.2 μM) for 24 hr.
(B) Western blot analysis of the effect of HNK (20 μM) and Mito-HNK (0.2 μM) on AMPK and STAT3 phosphorylation status in PC9-BrM3 and H2030-BrM3 cells.
(C) Western blot to verify knockout and knockdown of STAT3 in H2030-BrM3 cells.
(D and E) STAT3 knockout abrogates the anti-invasive and anti-proliferation effects of Mito-HNK (0.2 μM) in H2030-BrM3 cells. (D) Representative images and (E) the quantitative results of the cell invasion assay, ***p < 0.001. Error bars indicate SE.
Pathway Analysis of the Downstream Mediators of Mito-HNK Therapy
As shown in Figure 3, Mito-HNK treatment for 4 weeks dramatically reduced the brain metastatic burden of H2030-BrM3 cells. We used SSRS (species-specific RNAseq analysis) to profile the gene expression changes in the H2030-BrM3 brain metastatic lesions remaining after 4 weeks of Mito-HNK treatment compared with the brain metastatic lesions in vehicle-treated control animals. The H2030-BrM3 cells remaining in the brain metastatic lesions of Mito-HNK-treated animals had 4,660 differentially expressed (DE) genes (false discovery rate [FDR] <0.05) compared with vehicle-treated animals (Table S2; related to Figure 7). The nonmalignant stroma of the remaining brain metastatic lesions in Mito-HNK-treated animals had 6,544 DE genes (FDR <0.05) compared with the vehicle-treated mice (Table S3, related to Figure 7), of which only 22% (1,444 DE genes) overlapped with DE genes in the human malignant cells. We used the Ingenuity IPA tool (Qiagen, Redwood City, CA) to assess the biological and molecular pathways that were differentially regulated between the malignant tumor cells and the nonmalignant stroma (Figure 7). Mito-HNK treatment strongly induced mediators of cell death (Z score: +2.5; p < 10−57) in the human malignant H2030-BrM3 cells but not in the nonmalignant stroma (Figure 7A), indicating that the pro-apoptotic effects of Mito-HNK are largely specific to the malignant tumor. In addition, Mito-HNK treatment also significantly downregulated the pathways involved in tumor invasion and proliferation and altered multiple molecular pathways that have been implicated in all three cellular processes (Figure 7B). In comparison, the nonmalignant stroma of the H2030-BrM3 brain lesions in Mito-HNK-treated mice had significant downregulation of inflammatory and angiogenic pathways, as well as altered expression of multiple key pathways that contribute to these processes (Figure 7C). Collectively, these data suggest that, in addition to Mito-HNK's pro-apoptotic and anti-proliferative effects on the tumor cells, Mito-HNK also inhibits stromal cell pathways that typically support the formation and progression of metastatic lesions in the brain.
Figure 7.

Analysis of Pathways Regulated by Mito-HNK Treatment in the Malignant Tumor Cells and Nonmalignant Stroma of H2030-BrM3 Brain Metastatic Lesions
(A) RNAseq analysis to compare differentially expressed (DE) genes that regulate cell death in the malignant tumor and nonmalignant stroma of brain metastatic lesions of mice treated with the vehicle control (C) or Mito-HNK (MH).
(B and C) Analysis of molecular pathways and cellular functions that are positively and negatively enriched (indicated by Z score) following Mito-HNK treatment in the malignant tumor cells (B) and nonmalignant stroma (C). The raw Z scores were normalized within tumor and stromal groups. Note that the significant enrichment of DE genes that induce cell death are highly upregulated only in the Mito-HNK-treated malignant tumor cells (Z score: +2.5; p < 10−57). p Values are indicated on the graphs. Black bars indicate molecular pathways, and red bars indicate cellular function pathways.
Discussion
Although higher dose of HNK can induce mitochondria-dependent apoptosis in cancer cells (Lin et al., 2012, Pan et al., 2014, Martin et al., 2013), it is not specifically sequestered into cell mitochondria. By conjugating HNK with TPP+ to generate Mito-HNK, we markedly enhanced mitochondrial accumulation and the ability to block proliferation and invasion of brain metastatic human lung cancer cells. Low micromolar levels of Mito-HNK inhibit complex I, stimulate ROS, oxidize mitochondrial Prx3, and suppress mitoSTAT3Ser727 phosphorylation. These findings provide the first evidence that, by directly targeting mitochondrial complex I, Mito-HNK can have exceptional preventive and therapeutic efficacy to inhibit lung cancer growth and brain metastasis.
Mito-HNK (at 3.75 μmol/kg) significantly inhibited lung cancer metastases from growing in the brain, whereas this same dose of HNK does not inhibit brain metastasis. A 10-fold higher dose of HNK (37.5 μmol/kg, which equals to 10 mg/kg) is needed to inhibit brain metastasis (Pan et al., 2014, Pan et al., 2017). Mito-HNK (3.75 μmol/kg) also inhibited lung tumor growth in orthotopic models of both NSCLC and SCLC. These results are consistent with the markedly greater potency and efficacy of Mito-HNK in inhibiting proliferation, migration, and invasion of lung cancer cells. These results further suggest that Mito-HNK can be effective regardless of the driver mutation status (mutant EGFR in PC9-BrM3 cells; mutant KRAS in H2030-BrM3 cells). Therefore this study constitutes a critical step toward the clinical translation of Mito-HNK for treating lung cancer and its metastases.
The constitutive activation (phosphorylation) of STAT3 in many tumors is important for tumor growth, survival, progression (Poli and Camporeale, 2015), and metastasis (Kamran et al., 2013). Our data imply that mitoSTAT3 is another important molecular target of Mito-HNK. Mito-HNK (0.2 μM) suppressed the phosphorylation of STAT3 at Ser727 (Figure 6), and STAT3 knockout abrogated its anti-invasive effects (Figures 6D and 6E). Phosphorylation of STAT3 on Tyr705 promotes its nuclear translocation and its canonical activity as a transcription factor (Sobotta et al., 2015, Feng et al., 2014, Liu et al., 2003, Zhang et al., 2013, Wen et al., 1995). Phosphorylation at Ser727 also has important mitochondrial implications that are independent of its transcriptional role (Poli and Camporeale, 2015, Yang and Rincon, 2016). MitoSTAT3 can inhibit the mitochondrial permeability transition pore and can promote optimal electron transport (Poli and Camporeale, 2015, Wegrzyn et al., 2009, Szczepanek et al., 2011). The effects of p-STAT3Ser727 on mitochondrial electron transport may not be direct, however, given that complexes I and II are about 105 more abundant than mitoSTAT3 (Phillips et al., 2010). Of particular note, p-STAT3Ser727 can promote complex I activity and the transformation and growth of multiple cancers (Yu et al., 2015, Gough et al., 2009, Zhang et al., 2013, Capron et al., 2014). These effects are not dependent on p-STAT3Tyr705 (Yu et al., 2015, Gough et al., 2009, Zhang et al., 2013, Capron et al., 2014). The suppression of p-STAT3Ser727 could therefore be important for Mito-HNK's ability to suppress lung tumor growth, invasion, and metastasis. Cancer metastasis is a complex, multistep process including cell proliferation, invasion, self-renewal, and tumor cell immune evasion. Compelling evidence published in recent years has demonstrated that STAT3 activation plays a critical role in every step of metastasis (Devarajan and Huang, 2009). Activated STAT3 could directly bind to the promoter of MMP (matrix metalloproteinases) genes, increase the expression of multiple MMPs, and lead to the extracellular matrix formation, ultimately promote cellular invasion (Dechow et al., 2004, Xie et al., 2004, Itoh et al., 2006, Li et al., 2011). Furthermore, STAT3 also modulates Rac1 or Rho activity to maintain directional persistence during migration (Debidda et al., 2005, Teng et al., 2009). STAT3 activation also upregulates the secretion of various immunosuppressive factors such as interleukin-6 and tumor necrosis factor-α (Nguyen et al., 2009a) and reduces the activity of NK cells (Wang et al., 2004), and thereby protecting tumor cells during circulation.
Although there are multiple events by which Mito-HNK might function to decrease p-STAT3Ser727, it is unlikely that Mito-HNK directly affects STAT3, as no direct binding of Mito-HNK to STAT3 was detected. Rather, the data suggest that complex I inhibition, which occurs within minutes of Mito-HNK treatment, is upstream of the effects on mitoSTAT3. Specifically, Mito-HNK still inhibited complex I in cells in which STAT3 was knocked down (not shown), indicating that STAT3 is not required for complex I inhibition. Based on the data, we postulate a mechanism (Figure 8) in which complex I inhibition is a key initiating event that promotes the other observed effects. It is well recognized that complex I inhibition can stimulate mitochondrial ROS generation, consistent with the ROS data (Figures 4C and 4D) and the selective oxidation of mitochondrial Prx3 (Figures 4E and 4F). Since Prx3 represents ∼90% of total mitochondrial peroxidase activity (Cox et al., 2009), these data indicate that Mito-HNK-induced ROS generation overwhelms the mitochondrial capacity to degrade peroxides. STAT3 is susceptible to oxidative inactivation (Li et al., 2010, Shaw, 2010), and the levels of active p-STAT3Ser727 can be enhanced by antioxidants (e.g., glutathione, N-acetylcysteine) (Capron et al., 2014). Although peroxides might conceivably oxidize STAT3 directly (Li et al., 2010), it is unlikely that STAT3 thiols would initially compete with Prxs for cellular H2O2 given the highly favorable kinetics of Prxs (Cox et al., 2009). Oxidized Prxs can oxidize various proteins (Bindoli et al., 2008, Forman et al., 2010, Forman et al., 2014), including STAT3 (Sobotta et al., 2015), so pronounced Prx3 oxidation in Mito-HNK-treated cells is a logical link between enhanced mitochondrial ROS generation and inhibiting mitoSTAT3 function.
Figure 8.

Proposed Mechanisms of the Antiproliferative and Anti-invasive Effects of HNK and Mito-HNK
NADH dehydrogenase (complex I) plays a key role in regulating and maintaining mitochondrial function and energy production (ATP). Suppressing complex I activity can increase ROS production and signaling through ROS-associated pathways. We propose that one potential mechanism of action of Mito-HNK is that it inhibits complex I in lung cancer cells; stimulates ROS generation, which promotes oxidation of mitochondrial Prx3; activates AMPK; and inhibits STAT3ser727 phosphorylation and cell proliferation. The results indicate that Mito-HNK mediates these events more robustly and at much lower concentrations than does HNK.
We have shown that the increased in vitro potency of Mito-HNK over HNK is clear, and the antitumor activity of Mito-HNK in the in vivo tumor models (Figures 2F–2H and 3F–3H) is impressive. Our observation that the compound has limited effects on normal cells or tissues (Figure 1) is most likely due to the unusually greater uptake and retention of mito-targeted compounds by cancer cells as a consequence of highly elevated mitochondrial and plasma membrane potentials. Thus the preferential accumulation of Mito-HNK in the mitochondria of cancer cells leads to highly potent anti-cancer effects with no observed toxicity in mice given 20-fold more than the effective dose for 8 weeks. Similarly, other mitochondria-targeted agents are less toxic to normal cells than tumorigenic cells (Modica-Napolitano and Aprille, 2001, Cheng et al., 2013) and show no adverse effects with long-term administration in mice and humans (Gane et al., 2010, McManus et al., 2011). Combining Mito-HNK with standard chemotherapeutics, such as cisplatin, could significantly increase the killing of cancer cells and, meanwhile, lower the effective doses of these chemotherapeutics (Figure S11, related to Figure 3). The low toxicity of Mito-HNK, coupled with its ability to cross the BBB and the blood-CSF barrier (Wang et al., 2011, Lin et al., 2012), make it a highly attractive therapeutic agent, alone or in combination with standard therapy, for lung cancer and its metastases to the brain.
The SSRS analysis of brain metastatic H2030-BrM3 cells in treated mice further demonstrate the on-target antitumor effects of Mito-HNK to activate cell death and anti-proliferative signaling pathways in the metastatic tumor cells (Figures 7A and 7B), whereas similar pathways were not induced in the nonmalignant stromal cells (Figures 7A and 7C). In the nonmalignant stroma, Mito-HNK significantly attenuated inflammatory and pro-angiogenic pathways (Figure 7C), which resembles similar stromal-dependent antitumor mechanisms reported for HNK (Kim and Cho, 2008, Bai et al., 2003). Thus Mito-HNK is substantially more potent than HNK in targeting tumor cell mitochondria while maintaining its ability to suppress key components of the tumor stroma that can promote malignancy.
In summary, we demonstrate here the pronounced anti-cancer and anti-metastatic potential of Mito-HNK using both in vitro and in vivo lung cancer models. Mito-HNK potently inhibits orthotopic lung tumor growth as well as lung cancer metastasis to the brain. Mechanistically, we discovered, for the first time, that the ability of Mito-HNK to concentrate in the mitochondria renders it highly effective at inhibiting complex I, which corresponds with its ability to enhance mitochondrial ROS generation, promote Prx3 oxidation, suppress the phosphorylation of mitoSTAT3, and suppress the proliferation and invasion of cancer cells. The low toxicity of Mito-HNK, coupled with its ability to cross the BBB, makes it a highly attractive preventive and therapeutic agent for lung cancer and its brain metastases. Targeting of cancer cell mitochondrial bioenergetics represents a new strategy for treating lung cancer.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Patent
PCT/US2016/036827 “Mito-Honokiol Compounds and Methods of Synthesis and Use Thereof”.
Acknowledgments
We are grateful to Dr. Joan Massage (Memorial Sloan Kettering Cancer Center) for providing the H2030-BrM3 and PC9-BrM3 cells, to O.V. (Roskilde University) for providing the 143B and 143B ρ0 cells, and to M.B. (Czech Academy of Sciences) for providing the B16 and B16 ρ0 cells. This work was funded by the NIH grant R01CA208648.
Author Contributions
M.Y., B.K., J.P., and J.Z. were responsible for the overall experimental design with input from Y.L., G.C., D.X., M.F., C.M.O., and C.R.M. The project was supervised by M.Y., B.K., J.P., and J.Z. M.H. synthesized and characterized the mito-honokiols. C.M.O., J.P., and Q.Z. assessed the animal toxicity of Mito-HNK. G.C., J.P., and Y.L. did the cell proliferation and transwell invasion studies. J.P. and Y.L. did the tyrosine kinase assays, western blots, and STAT3 knockout and knockdown experiments. J.P., Y.L., Q.Z., and Y.W. conducted the orthotopic lung cancer and brain metastasis animal studies. M.F., D.X., and S.-W.T. did the RNA-seq analysis. J.P. and G.C. conducted the experiments with ρ0 cells. G.C. and J.Z. analyzed respiratory enzyme activity and intracellular oxidants. C.R.M. did the peroxiredoxin experiments. M.B., O.V., and J.N. provided rho0 or ρ0 cells. M.Y. and B.K. acquired the funding. The following were largely responsible for writing, reviewing, and editing the manuscript: J.P., G.C., J.Z., M.H., M.F., C.R.M., B.K., and M.Y.
Declaration of Interests
The authors declare that there are no financial or other conflicts of interest.
Published: May 25, 2018
Footnotes
Supplemental Information includes Transparent Methods, 11 figures, and 3 tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.04.013.
Data and Software Availability
The authors declare that all data supporting the findings of this study are available within the article and its Supplemental Information, and from the corresponding author upon reasonable request. The RNA-seq raw sequence reads data from the H2030-BrM3 brain metastatic lesions of nude mice treated with Mito-HNK or vehicle control for four weeks (n = 3 per group, total N = 6) as described in the Supplementary file have been deposited in the Sequence Read Archive under accession number of SRP144372. Mito-HNK will be freely distributed upon request to qualified academic investigators for non-commercial research. Primary or luciferase-tagged cancer cells will be distributed following completion of a material transfer agreement agreed upon by the Medical College of Wisconsin and the recipient organization. Costs to ship Mito-HNK or cancer cell lines generated by this project will be incurred by the requestor and will be shipped following all Federal guidelines and precautions to minimize loss.
Supplemental Information
References
- Arora S., Singh S., Piazza G.A., Contreras C.M., Panyam J., Singh A.P. Honokiol: a novel natural agent for cancer prevention and therapy. Curr. Mol. Med. 2012;12:1244–1252. doi: 10.2174/156652412803833508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asin-Cayuela J., Manas A.R.B., James A.M., Smith R.A.J., Murphy M.P. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004;571:9–16. doi: 10.1016/j.febslet.2004.06.045. [DOI] [PubMed] [Google Scholar]
- Bai X., Cerimele F., Ushio-Fukai M., Waqas M., Campbell P.M., Govindarajan B., Der C.J., Battle T., Frank D.A., Ye K. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J. Biol. Chem. 2003;278:35501–35507. doi: 10.1074/jbc.M302967200. [DOI] [PubMed] [Google Scholar]
- Berridge M.V., Dong L., Neuzil J. Mitochondrial DNA in tumor initiation, progression, and metastasis: role of horizontal mtDNA transfer. Cancer Res. 2015;75:3203–3208. doi: 10.1158/0008-5472.CAN-15-0859. [DOI] [PubMed] [Google Scholar]
- Bindoli A., Fukuto J.M., Forman H.J. Thiol chemistry in peroxidase catalysis and redox signaling. Antioxid. Redox Signal. 2008;10:1549–1564. doi: 10.1089/ars.2008.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capron C., Jondeau K., Casetti L., Jalbert V., Costa C., Verhoeyen E., Masse J.M., Coppo P., Bene M.C., Bourdoncle P. Viability and stress protection of chronic lymphoid leukemia cells involves overactivation of mitochondrial phosphoSTAT3Ser727. Cell Death Dis. 2014;5:e1451. doi: 10.1038/cddis.2014.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G., Zielonka J., Dranka B.P., Mcallister D., Mackinnon A.C., Jr., Joseph J., Kalyanaraman B. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012;72:2634–2644. doi: 10.1158/0008-5472.CAN-11-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G., Zielonka J., Mcallister D., Hardy M., Ouari O., Joseph J., Dwinell M.B., Kalyanaraman B. Antiproliferative effects of mitochondria-targeted cationic antioxidants and analogs: role of mitochondrial bioenergetics and energy-sensing mechanism. Cancer Lett. 2015;365:96–106. doi: 10.1016/j.canlet.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G., Zielonka J., Mcallister D., Tsai S., Dwinell M.B., Kalyanaraman B. Profiling and targeting of cellular bioenergetics: inhibition of pancreatic cancer cell proliferation. Br. J. Cancer. 2014;111:85–93. doi: 10.1038/bjc.2014.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G., Zielonka J., Mcallister D.M., Mackinnon A.C., Jr., Joseph J., Dwinell M.B., Kalyanaraman B. Mitochondria-targeted vitamin E analogs inhibit breast cancer cell energy metabolism and promote cell death. BMC Cancer. 2013;13:285. doi: 10.1186/1471-2407-13-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox A.G., Winterbourn C.C., Hampton M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2009;425:313–325. doi: 10.1042/BJ20091541. [DOI] [PubMed] [Google Scholar]
- Crawley J.N. Wiley-Liss; 2000. What's Wrong with My Mouse? Behavioral Phenotyping of Transgenic and Knockout Mice. [Google Scholar]
- Debidda M., Wang L., Zang H., Poli V., Zheng Y. A role of STAT3 in Rho GTPase-regulated cell migration and proliferation. J. Biol. Chem. 2005;280:17275–17285. doi: 10.1074/jbc.M413187200. [DOI] [PubMed] [Google Scholar]
- Dechow T.N., Pedranzini L., Leitch A., Leslie K., Gerald W.L., Linkov I., Bromberg J.F. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc. Natl. Acad. Sci. USA. 2004;101:10602–10607. doi: 10.1073/pnas.0404100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarajan E., Huang S. STAT3 as a central regulator of tumor metastases. Curr. Mol. Med. 2009;9:626–633. doi: 10.2174/156652409788488720. [DOI] [PubMed] [Google Scholar]
- Dong L.F., Kovarova J., Bajzikova M., Bezawork-Geleta A., Svec D., Endaya B., Sachaphibulkij K., Coelho A.R., Sebkova N., Ruzickova A. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife. 2017;6:e22187. doi: 10.7554/eLife.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Ke C., Tang Q., Dong H., Zheng X., Lin W., Ke J., Huang J., Yeung S.C., Zhang H. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis. 2014;5:e1088. doi: 10.1038/cddis.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finichiu P.G., Larsen D.S., Evans C., Larsen L., Bright T.P., Robb E.L., Trnka J., Prime T.A., James A.M., Smith R.A.J. A mitochondria-targeted derivative of ascorbate: MitoC. Free Radic. Biol. Med. 2015;89:668–678. doi: 10.1016/j.freeradbiomed.2015.07.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman H.J., Maiorino M., Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835–842. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman H.J., Ursini F., Maiorino M. An overview of mechanisms of redox signaling. J. Mol. Cell Cardiol. 2014;73:2–9. doi: 10.1016/j.yjmcc.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gane E.J., Weilert F., Orr D.W., Keogh G.F., Gibson M., Lockhart M.M., Frampton C.M., Taylor K.M., Smith R.A., Murphy M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- Goldberg S.B., Contessa J.N., Omay S.B., Chiang V. Lung cancer brain metastases. Cancer J. 2015;21:398–403. doi: 10.1097/PPO.0000000000000146. [DOI] [PubMed] [Google Scholar]
- Gough D.J., Corlett A., Schlessinger K., Wegrzyn J., Larner A.C., Levy D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin S. Comprehensive observational assessment: Ia. a systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacologia. 1968;13:222–257. doi: 10.1007/BF00401402. [DOI] [PubMed] [Google Scholar]
- Itoh M., Murata T., Suzuki T., Shindoh M., Nakajima K., Imai K., Yoshida K. Requirement of STAT3 activation for maximal collagenase-1 (MMP-1) induction by epidermal growth factor and malignant characteristics in T24 bladder cancer cells. Oncogene. 2006;25:1195–1204. doi: 10.1038/sj.onc.1209149. [DOI] [PubMed] [Google Scholar]
- Kamran M.Z., Patil P., Gude R.P. Role of STAT3 in cancer metastasis and translational advances. Biomed. Res. Int. 2013;2013:421821. doi: 10.1155/2013/421821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B.H., Cho J.Y. Anti-inflammatory effect of honokiol is mediated by PI3K/Akt pathway suppression. Acta Pharmacol. Sin. 2008;29:113–122. doi: 10.1111/j.1745-7254.2008.00725.x. [DOI] [PubMed] [Google Scholar]
- Lampidis T.J., Hasin Y., Weiss M.J., Chen L.B. Selective killing of carcinoma cells "in vitro" by lipophilic-cationic compounds: a cellular basis. Biomed. Pharmacother. 1985;39:220–226. [PubMed] [Google Scholar]
- Li H., Huang C., Huang K., Wu W., Jiang T., Cao J., Feng Z., Qiu Z. STAT3 knockdown reduces pancreatic cancer cell invasiveness and matrix metalloproteinase-7 expression in nude mice. PLoS One. 2011;6:e25941. doi: 10.1371/journal.pone.0025941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Cheung S.H., Evans E.L., Shaw P.E. Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Res. 2010;70:8222–8232. doi: 10.1158/0008-5472.CAN-10-0894. [DOI] [PubMed] [Google Scholar]
- Lin J.W., Chen J.T., Hong C.Y., Lin Y.L., Wang K.T., Yao C.J., Lai G.M., Chen R.M. Honokiol traverses the blood-brain barrier and induces apoptosis of neuroblastoma cells via an intrinsic bax-mitochondrion-cytochrome c-caspase protease pathway. Neuro Oncol. 2012;14:302–314. doi: 10.1093/neuonc/nor217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren S., Bass A.S., Briscoe R., Bruse K., Friedrichs G.S., Kallman M.J., Markgraf C., Patmore L., Pugsley M.K. Benchmarking safety pharmacology regulatory packages and best practice. J. Pharmacol. Toxicol. Methods. 2008;58:99–109. doi: 10.1016/j.vascn.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Liu H., Ma Y., Cole S.M., Zander C., Chen K.H., Karras J., Pope R.M. Serine phosphorylation of STAT3 is essential for Mcl-1 expression and macrophage survival. Blood. 2003;102:344–352. doi: 10.1182/blood-2002-11-3396. [DOI] [PubMed] [Google Scholar]
- Madar I., Anderson J.H., Szabo Z., Scheffel U., Kao P.F., Ravert H.T., Dannals R.F. Enhanced uptake of [11C]TPMP in canine brain tumor: a PET study. J. Nucl. Med. 1999;40:1180–1185. [PubMed] [Google Scholar]
- Martin S., Lamb H.K., Brady C., Lefkove B., Bonner M.Y., Thompson P., Lovat P.E., Arbiser J.L., Hawkins A.R., Redfern C.P. Inducing apoptosis of cancer cells using small-molecule plant compounds that bind to GRP78. Br. J. Cancer. 2013;109:433–443. doi: 10.1038/bjc.2013.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus M.J., Murphy M.P., Franklin J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease. J. Neurosci. 2011;31:15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modica-Napolitano J.S., Aprille J.R. Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv. Drug Deliv. Rev. 2001;49:63–70. doi: 10.1016/s0169-409x(01)00125-9. [DOI] [PubMed] [Google Scholar]
- Murphy M.P. Understanding and preventing mitochondrial oxidative damage. Biochem. Soc. Trans. 2016;44:1219–1226. doi: 10.1042/BST20160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M.P., Smith R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- Nguyen D.X., Bos P.D., Massague J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- Nguyen D.X., Chiang A.C., Zhang X.H., Kim J.Y., Kris M.G., Ladanyi M., Gerald W.L., Massague J. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009;138:51–62. doi: 10.1016/j.cell.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen C.M., Childs D.S., Stanwood G.D., Winder D.G. Operant sensation seeking requires metabotropic glutamate receptor 5 (mGluR5) PLoS One. 2010;5:e15085. doi: 10.1371/journal.pone.0015085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J., Lee Y., Wang Y., You M. Honokiol targets mitochondria to halt cancer progression and metastasis. Mol. Nutr. Food Res. 2016;60:1383–1395. doi: 10.1002/mnfr.201501007. [DOI] [PubMed] [Google Scholar]
- Pan J., Lee Y., Zhang Q., Xiong D., Wan T.C., Wang Y., You M. Honokiol decreases lung cancer metastasis through inhibition of the STAT3 signaling pathway. Cancer Prev. Res. (Phila.) 2017;10:133–141. doi: 10.1158/1940-6207.CAPR-16-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J., Zhang Q., Liu Q., Komas S.M., Kalyanaraman B., Lubet R.A., Wang Y., You M. Honokiol inhibits lung tumorigenesis through inhibition of mitochondrial function. Cancer Prev. Res. (Phila.) 2014;7:1149–1159. doi: 10.1158/1940-6207.CAPR-14-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D., Reilley M.J., Aponte A.M., Wang G., Boja E., Gucek M., Balaban R.S. Stoichiometry of STAT3 and mitochondrial proteins: implications for the regulation of oxidative phosphorylation by protein-protein interactions. J. Biol. Chem. 2010;285:23532–23536. doi: 10.1074/jbc.C110.152652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchard D., Giroux Leprieur E. [Biomarkers and targeted therapies in non-small cell lung cancer: present and future treatments] Rev. Pneumol Clin. 2011;67(Suppl 1):S36–S40. doi: 10.1016/S0761-8417(11)70009-8. [DOI] [PubMed] [Google Scholar]
- Poli V., Camporeale A. STAT3-Mediated metabolic reprograming in cellular transformation and implications for drug resistance. Front. Oncol. 2015;5:121. doi: 10.3389/fonc.2015.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout D.C., Calogeropoulou T., Jaworski J.S., Dagnino R., Jr., Mccarthy M.R. Phosphonium salts exhibiting selective anti-carcinoma activity in vitro. Anticancer Drug Des. 1989;4:265–280. [PubMed] [Google Scholar]
- Ross M.F., Prime T.A., Abakumova I., James A.M., Porteous C.M., Smith R.A.J., Murphy M.P. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem. J. 2008;411:633–645. doi: 10.1042/BJ20080063. [DOI] [PubMed] [Google Scholar]
- Salabei J.K., Gibb A.A., Hill B.G. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014;9:421–438. doi: 10.1038/nprot.2014.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P.E. Could STAT3 provide a link between respiration and cell cycle progression? Cell Cycle. 2010;9:4294–4296. doi: 10.4161/cc.9.21.13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R.A., Porteous C.M., Gane A.M., Murphy M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R.A.J., Hartley R.C., Cochemé H.M., Murphy M.P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012;33:341–352. doi: 10.1016/j.tips.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Smith R.A.J., Murphy M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N.Y. Acad. Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- Snow B.J., Rolfe F.L., Lockhart M.M., Frampton C.M., O'sullivan J.D., Fung V., Smith R.A.J., Murphy M.P., Taylor K.M. A double-blind, placebo-controlled study to assess the mitochondria- targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov. Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- Sobotta M.C., Liou W., Stocker S., Talwar D., Oehler M., Ruppert T., Scharf A.N., Dick T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015;11:64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- Summerhayes I.C., Lampidis T.J., Bernal S.D., Nadakavukaren J.J., Nadakavukaren K.K., Shepherd E.L., Chen L.B. Unusual retention of rhodamine 123 by mitochondria in muscle and carcinoma cells. Proc. Natl. Acad. Sci. USA. 1982;79:5292–5296. doi: 10.1073/pnas.79.17.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanek K., Chen Q., Derecka M., Salloum F.N., Zhang Q., Szelag M., Cichy J., Kukreja R.C., Dulak J., Lesnefsky E.J. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J. Biol. Chem. 2011;286:29610–29620. doi: 10.1074/jbc.M111.226209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A.S., Baty J.W., Dong L.F., Bezawork-Geleta A., Endaya B., Goodwin J., Bajzikova M., Kovarova J., Peterka M., Yan B. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21:81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Tauskela J.S. MitoQ–a mitochondria-targeted antioxidant. IDrugs. 2007;10:399–412. [PubMed] [Google Scholar]
- Teng T.S., Lin B., Manser E., Ng D.C., Cao X. Stat3 promotes directional cell migration by regulating Rac1 activity via its activator betaPIX. J. Cell Sci. 2009;122:4150–4159. doi: 10.1242/jcs.057109. [DOI] [PubMed] [Google Scholar]
- Vazquez-Martin A., Cufi S., Oliveras-Ferraros C., Torres-Garcia V.Z., Corominas-Faja B., Cuyas E., Bonavia R., Visa J., Martin-Castillo B., Barrajon-Catalan E. IGF-1R/epithelial-to-mesenchymal transition (EMT) crosstalk suppresses the erlotinib-sensitizing effect of EGFR exon 19 deletion mutations. Sci. Rep. 2013;3:2560. doi: 10.1038/srep02560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Niu G., Kortylewski M., Burdelya L., Shain K., Zhang S., Bhattacharya R., Gabrilovich D., Heller R., Coppola D. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- Wang X., Duan X., Yang G., Zhang X., Deng L., Zheng H., Deng C., Wen J., Wang N., Peng C. Honokiol crosses BBB and BCSFB, and inhibits brain tumor growth in rat 9L intracerebral gliosarcoma model and human U251 xenograft glioma model. PLoS One. 2011;6:e18490. doi: 10.1371/journal.pone.0018490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn J., Potla R., Chwae Y.J., Sepuri N.B., Zhang Q., Koeck T., Derecka M., Szczepanek K., Szelag M., Gornicka A. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F., Hamanaka R., Wheaton W.W., Weinberg S., Joseph J., Lopez M., Kalyanaraman B., Mutlu G.M., Budinger G.R., Chandel N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg S.E., Chandel N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z., Zhong Z., Darnell J.E., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- Wheaton W.W., Weinberg S.E., Hamanaka R.B., Soberanes S., Sullivan L.B., Anso E., Glasauer A., Dufour E., Mutlu G.M., Budigner G.S. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlund A.L., Baral K., Dalgaard L.T., Vang O. Functional mitochondria are important for the effect of resveratrol. Molecules. 2017;22:847. doi: 10.3390/molecules22050847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T.X., Wei D., Liu M., Gao A.C., Ali-Osman F., Sawaya R., Huang S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004;23:3550–3560. doi: 10.1038/sj.onc.1207383. [DOI] [PubMed] [Google Scholar]
- Yang R., Rincon M. Mitochondrial Stat3, the need for design thinking. Int. J. Biol. Sci. 2016;12:532–544. doi: 10.7150/ijbs.15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C., Huo X., Agoston A.T., Zhang X., Theiss A.L., Cheng E., Zhang Q., Zaika A., Pham T.H., Wang D.H. Mitochondrial STAT3 contributes to transformation of Barrett's epithelial cells that express oncogenic Ras in a p53-independent fashion. Am. J. Physiol. Gastrointest. Liver Physiol. 2015;309:G146–G161. doi: 10.1152/ajpgi.00462.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Raje V., Yakovlev V.A., Yacoub A., Szczepanek K., Meier J., Derecka M., Chen Q., Hu Y., Sisler J. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013;288:31280–31288. doi: 10.1074/jbc.M113.505057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.