

Abstract
Serine proteinase inhibitor, clade E, member 2 (SERPINE2), is a cell- and extracellular matrix–associated inhibitor of thrombin. Although SERPINE2 is a candidate susceptibility gene for chronic obstructive pulmonary disease, the physiologic role of this protease inhibitor in lung development and homeostasis is unknown. We observed spontaneous monocytic-cell infiltration in the lungs of Serpine2-deficient (SE2−/−) mice, beginning at or before the time of lung maturity, which resulted in lesions that resembled bronchus-associated lymphoid tissue (BALT). The initiation of lymphocyte accumulation in the lungs of SE2−/− mice involved the excessive expression of chemokines, cytokines, and adhesion molecules that are essential for BALT induction, organization, and maintenance. BALT-like lesion formation in the lungs of SE2−/− mice was also associated with a significant increase in the activation of thrombin, a recognized target of SE2, and excess stimulation of NF-κB, a major regulator of chemokine expression and inflammation. Finally, systemic delivery of thrombin rapidly stimulated lung chemokine expression in vivo. These data uncover a novel mechanism whereby loss of serine protease inhibition leads to lung lymphocyte accumulation.—Solleti, S. K., Srisuma, S., Bhattacharya, S., Rangel-Moreno, J., Bijli, K. M., Randall, T. D., Rahman, A., Mariani, T. J. Serpine2 deficiency results in lung lymphocyte accumulation and bronchus-associated lymphoid tissue formation.
Keywords: serine protease inhibitor, protease nexin-1, thrombin, chemokines, tertiary lymphoid structures
Chronic obstructive pulmonary disease (COPD) is a complex human disease that is likely influenced by environmental factors, multiple genes, and gene-by-smoking/environment interactions (1, 2). Prolonged exposure to tobacco smoke may add to the total burden of environmental factors that become sufficient to cause such chronic airway/parenchymal diseases as COPD, especially in individuals with high susceptibility, for example, genetic predisposition. COPD is principally an inflammatory lung disease, with pathologic features that include emphysema, chronic bronchitis, and the formation of bronchus-associated lymphoid tissue (BALT).
Deficiency in the activity of α1-antitrypsin/serine proteinase inhibitor, clade A, member 1 (SERPINA1), is a proven genetic risk factor for COPD but only contributes to 1% of COPD cases. Multiple genes, each with modest effect size, are also thought to contribute to COPD risk in the general population (3–5). We have previously identified serine proteinase inhibitor, clade E, member 2 (SERPINE2)/protease nexin-1 as a candidate genetic susceptibility factor for COPD on the basis of structural and expression variation (3), and these observations have subsequently been verified (6–10). However, the physiologic role of SERPINE2 in lung function and pathology is poorly understood.
Serine protease inhibitors (SERPINs) are key regulators in many intracellular and extracellular biologic processes (11). The SERPINE2 gene encodes a 45–50 kDa cell- and extracellular matrix–associated glycoprotein that acts as inhibitor of thrombin, trypsin, urokinase plasminogen activator (uPA), and plasmin, but not elastase (11, 12). It is expressed in many organs, such as the brain, olfactory system, male and female reproductive systems, kidneys, cartilage, spleen, muscle, heart, and lungs (12–14). SERPINE2 can regulate cellular differentiation (15–17) and can function as a regulator of angiogenesis (18), apoptosis (19, 20), and endothelial barrier function (20). SERPINE2 overexpression promotes development of an antiproteolytic vascular smooth muscle cell phenotype and might favor progressive aneurysmal dilation (21). Injury-related cytokines can increase SERPINE2 expression in different cell types (22, 23).
We and others have previously demonstrated that SERPINE2 in the lung is predominantly restricted to the airway (but not alveolar) parenchyma and vascular adventitia in humans and mice (3, 13). Similarities in expression suggest that the mouse may represent a reasonable model to understand the role of SERPINE2 in human lung disease. SERPINE2-deficient (SE2−/−) mice seem to develop normally; however, adults are susceptible to experimental epilepsy (24) and display male infertility (25).
Several reports have demonstrated that high levels of thrombin exist in the airway of patients with COPD (26, 27). Thrombin can be generated at sites of injury and plays a role in regulating inflammation (28), including induction of proinflammatory molecule release from resident lung cells (29, 30). We hypothesized that the absence of SERPINE2 activity may potentiate thrombin-promoted lung inflammatory responses that are associated with COPD. Here, we describe spontaneous chronic lung inflammation in SE2−/− mice that leads to the formation of BALT-like structures.
MATERIALS AND METHODS
Animals
SE2−/− mice were obtained from Dr. Denis Monard (Friedrich Miescher Institute, Basel, Switzerland) (24) and were maintained in a pure C57BL/6 background. As a result of male SE2−/− infertility, mice were bred by mating heterozygous males with knockout females. All animals were genotyped by PCR. Mice used for experiments were age and sex matched. All animal procedures were performed in adherence to the U.S. National Institutes of Health guidelines and approved by the University of Rochester Committee on Animal Resources.
Lung histology
Mice were euthanized by CO2 narcosis. Lungs were exposed, and the pulmonary vasculature was perfused with PBS. The right lung was sutured, resected, and flash frozen in liquid nitrogen for RNA and protein isolation, and the left lung was inflated via tracheal cannula to a fixed pressure of 25 cmH2O with 10% buffered formalin for 15 min. At the end of the fixation period, the trachea was ligated, and inflated lungs were removed en bloc and immersed in 30 ml 10% buffered formalin for 48 h at room temperature. After fixation, tissue was embedded in paraffin for histologic and morphologic analysis. Midsagittal sections (5 μm thick) were cut with a microtome. For quantitative assessment, one entire formalin-fixed, paraffin-embedded saggital section for each animal was stained with hematoxylin and eosin. Each stained section was analyzed in a blinded fashion, and the presence of mononuclear cell infiltrates was recorded, noting whether the lesion occurred adjacent to an airway or vessel.
Collection of bronchoalveolar lavage
Mice were euthanized by CO2 narcosis and lavaged with 1 volume (1 cc/25 g) of ice-cold PBS, without protease inhibitors, by tracheal catheter. Bronchoalveolar lavage (BAL) fluid collected from lungs of mice was centrifuged (3000 rpm for 3 min at 4°C) and the supernatant was retained for further analysis.
Immunohistochemical staining
Deparaffinized and rehydrated lung sections were heated in DAKO (Carpinteria, CA, USA) antigen retrieval solution for antigen retrieval. Nonspecific binding was blocked by incubating sections with 5–10% normal donkey serum and Fc block (2.4G2; 1 µg /ml) in buffered saline that included detergent (0.1–0.5% Triton X-100 and 0.1% Tween-20). Lung tissue sections were incubated with primary antibody overnight at 4°C in a humidified chamber. After being washed in buffered saline that included detergent, sections were incubated with fluorescently labeled secondary antibodies for 2 h at room temperature. Slides were mounted with SlowFade Gold Antifade (Thermo Fisher Scientific, Waltham, MA, USA) with DAPI, and images were taken with a Zeiss Axioplan 2 microscope and a Zeiss Axiocam digital camera (Zeiss, Jena, Germany).
Flow cytometry analysis
Single-cell suspensions were prepared by digesting lungs with 6.25 mg/ml collagenase (C-7657; Sigma-Aldrich, St. Louis, MO, USA) and 0.295 mg/ml DNAse (D-5025-150KU; Sigma-Aldrich) for 30 min at 37°C. Digested tissue was passed through a metallic cell strainer. RBCs were lysed with ACK solution for 5 min at room temperature, neutralized with 10 ml fluorescence-activated cell sorting (FACS) medium (2% fetal bovine serum in PBS), and collected by centrifugation for 5 min at 1800 rpm. Single-cell suspensions were treated with Fc block for 10 min on ice, and antibodies were added to cells and incubated on ice for an additional 20 min. Cells were washed twice with FACS medium, centrifuged, and resuspended in FACS medium with 1 μg/ml propidium iodide. Cells were collected in an LSR II Flow Cytometer (BD Biosciences, San Jose, CA, USA), and data were analyzed with FlowJo (FlowJo, Ashland, OR, USA). Dead cells were excluded from the analysis, and lymphocytes were gated according to their classic forward scatter/side scatter profile. Cluster of differentiation (CD) 8 T cells were defined as live CD3+CD8+ cells, live CD3+CD4+ cells were considered CD4 T cells, and antibodies against CD19 were used to enumerate live B cells.
RNA isolation and real-time quantitative RT-PCR
Mouse lung tissue was homogenized in Trizol, and total RNA was extracted according to the manufacturer's protocol. Tissue RNA was repurified and rendered DNA free (Absolutely RNA; Agilent Technologies, La Jolla, CA, USA). RNA was reverse-transcribed by using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Quantitative real-time PCR (qPCR) analysis was performed by using a ViiA 7 Real-Time PCR System (Life Technologies, Grand Island, NY, USA) with SYBR green chemistry (Applied Biosystems, Danvers, MA, USA). Gene expression levels were calculated relative to Ppia (cyclophilin A) by using the ΔΔCt method as previously described (3, 31). Primer sequences were selected from primerbank (http://pga.mgh.harvard.edu/primerbank/), and sequences are given in Table 1.
TABLE 1.
Oligonucleotide primer sequences used for detection of murine gene expression in qPCR reactions
| Gene | Forward | Reverse |
|---|---|---|
| Ppia | GGTGGTGACTTTACACGCCA | TCTCCGTAGATGGACCTGCC |
| Tnfa | CCCTCACACTCAGATCATCTTCT | GCTACGACGTGGGCTACAG |
| Lta | CCACCTCTTGAGGGTGCTTG | CATGTCGGAGAAAGGCACGAT |
| Ltb | TGGCAGGAGCTACTTCCCT | TCCAGTCTTTTCTGAGCCTGT |
| Cxcl12 | TGCATCAGTGACGGTAAACCA | TTCTTCAGCCGTGCAACAATC |
| Cxcl13 | GGCCACGGTATTCTGGAAGC | GGGCGTAACTTGAATCCGATCTA |
| Cxcl9 | TCCTTTTGGGCATCATCTTCC | TTTGTAGTGGATCGTGCCTCG |
| Ccl19 | GGGGTGCTAATGATGCGGAA | CCTTAGTGTGGTGAACACAACA |
| Cxcl10 | CCAAGTGCTGCCGTCATTTTC | GGCTCGCAGGGATGATTTCAA |
| Cxcl11 | GGCTTCCTTATGTTCAAACAGGG | GCCGTTACTCGGGTAAATTACA |
| Ccl2 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| Ccl5 | GCTGCTTTGCCTACCTCTCC | TCGAGTGACAAACACGACTGC |
| Ccl8 | CTGGGCCAGATAAGGCTCC | CATGGGGCACTGGATATTGTT |
| Ccl9 | CCCTCTCCTTCCTCATTCTTACA | AGTCTTGAAAGCCCATGTGAAA |
| Ccl17 | GACGACAGAAGGGTACGGC | GCATCTGAAGTGACCTCATGGTA |
| Ccl20 | GCCTCTCGTACATACAGACGC | CCAGTTCTGCTTTGGATCAGC |
| Ccl21b | AGGCAGTGATGGAGGGGGA | GCTTAGAGTGCTTCCGGGGTA |
| Ccl22 | AGGTCCCTATGGTGCCAATGT | CGGCAGGATTTTGAGGTCCA |
| Cd34 | GGTAGCTCTCTGCCTGATGAG | TGGTAGGAACTGATGGGGATATT |
| Sele | ATGCCTCGCGCTTTCTCTC | GTAGTCCCGCTGACAGTATGC |
| Il2 | TGAGCAGGATGGAGAATTACAGG | GTCCAAGTTCATCTTCTAGGCAC |
| Il4 | GGTCTCAACCCCCAGCTAGT | GCCGATGATCTCTCTCAAGTGAT |
| Il6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| Il10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| Il21 | GGACCCTTGTCTGTCTGGTAG | TGTGGAGCTGATAGAAGTTCAGG |
| Il1b | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| Gmcsf | TCGTCTCTAACGAGTTCTCCTT | CGTAGACCCTGCTCGAATATCT |
| Tgfb1 | TTAGGAAGGACCTGGGTTGGA | ACTGTGTGTCCAGGCTCCAAAT |
| Itgb2 | CCTGGTCCAGTGAAGTTCAGC | CAGGAATGCACCAAGTACAAAGT |
| Icam1 | GGCATTGTTCTCTAATGTCTCCG | GCTCCAGGTATATCCGAGCTTC |
Microarray analysis
DNA-free RNA from multiple whole lungs was pooled (by genotype); targets were generated as described previously (32) and hybridized to the Mouse Expression Array 430 2.0 (Affymetrix, Santa Clara, CA, USA). A total of 6 arrays were interrogated, 3 for each genotype. Expression intensities were obtained from .CEL files by using Robust Multichip Average–generated normalized background-subtracted data and Microarray Suite 5.0 non-normalized background-subtracted data, implemented in Bioconductor (http://www.bioconductor.org/). Differential expression was assessed by using Student’s t test and significant analysis of microarray. Genes identified as differentially expressed by both significant analysis of microarray or Student’s t test were used for pathway analysis with Ingenuity Pathway Analysis software (Qiagen, Valencia, CA, USA). Canonical pathways were reported as significant if they achieved a value of P < 0.05 by using Fisher’s exact test.
Protein isolation and analysis
Mouse lung tissue was mechanically homogenized in either RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25 mM EDTA, 1% deoxycholic acid, 1% Triton X-100) that contained protease inhibitor cocktail (phosphatase inhibitor cocktail 2 and 3), PBS, or Tris-HCl buffer without any supplements. Tissue homogenates were kept on ice for 15–30 min to allow total cell lysis. Supernatants were collected by centrifugation at 10,000 g for 10 min. Protein concentrations were measured by bicinchoninic acid method and stored at −80°C until use. Levels of inflammatory molecules in tissue homogenates were measured by Luminex xMAP technology (Beadlyte; Millipore, Billerica, MA, USA) by using a mouse 12 plex magnetic bead panel according to the manufacturer instructions. For Western blot, isolated proteins (10–20 μg) were boiled for 10 min, resolved on SDS-PAGE, and transferred onto nitrocellulose membranes (Millipore). Membranes were blocked by incubation with 5% (w/v) nonfat dry milk in Tris-buffered saline with Tween-20 (10 mm Tris pH 8.0, 150 mm NaCl, and 0.05% Tween-20) for 1 h at room temperature. Membranes were then incubated with appropriate primary and secondary antibodies and developed by using an ECL kit (GE Healthcare, Pittsburgh, PA, USA). The intensity of each band in Western blot was quantified by using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Molecular activity assays
DNA-binding activity of p65 to a consensus NF-κB oligonucleotide was assessed in total lung protein by using ELISA-based TransAm kit (TransAm p65; Active Motif, Carlsbad, CA, USA). Absorbance was read at 450 nm with samples appropriately blanked. Thrombin activity in lung homogenates prepared in PBS and BAL fluid was determined by using the SensoLyte AFC Thrombin Activity Assay Kit (AnaSpec, Fremont, CA, USA). Plasminogen activator activity in lung homogenates was measured by using an indirect colorimetric uPA activity assay kit according to the manufacturer instructions (Chemicon; Millipore).
Murine model of thrombin-induced lung inflammation
Lung inflammation was induced in adult mice by intraperitoneal thrombin injection (50 U/g body weight) (33). Lungs from mice were collected 1 h after thrombin administration, processed for RNA as described above, and analyzed for expression of inflammatory molecules by qPCR.
Statistical analysis
All experiments were performed with multiple replicates, and data are expressed as means ± se. Comparisons between experimental groups were made by either parametric Student’s t test or nonparametric Mann–Whitney U test. Differences in mean values were considered significant at P < 0.05.
RESULTS
Aged SE2−/− mice spontaneously develop lymphocytic aggregates
To understand the physiologic and pathophysiologic roles of Serpine2 in the lung, we studied SE2−/− mice. Whereas wild-type control (CTL) lung tissue displayed a normal histology that was devoid of overt inflammation (Fig. 1A, E), small areas of focal, peribronchial mononuclear-cell infiltration (<50 cells) were routinely observed in SE2−/− mice as early as 2 mo of age (Fig. 1B–D). By 8 mo, 85% of SE2−/− mice displayed large, consolidated mononuclear-cell aggregates (Fig.1F, G), whereas 20% of CTL mice displayed rare, small regions of infiltrates (P < 0.05; Fig. 1H). These infiltrates were within the lung tissue, subadjacent to large- or intermediate-sized airways and vessels.
Figure 1.

Spontaneous inflammatory cell accumulation in lungs of SE2−/− mice. A–G) Qualitative ontogeny of mononuclear cell accumulation in CTL and SE2−/− (SERPINE2 KO) mice. Hematoxylin and eosin staining of lung tissue from mice at 2 mo (A–D) or 8 mo (E–G) of age. At 2 mo of age, small numbers of mononuclear cell infiltrates (arrow) were noted adjacent to airways (AW; B–D) and vessels (v; B, C) exclusively in SE2−/− mice. At 8 mo of age, SE2−/− mice displayed large aggregates of mononuclear cells adjacent to airways (F) and vessels (G). H) Quantitative ontogeny of mononuclear cell accumulation. At 2 mo of age, 50% of SE2−/− mice displayed small peribronchial/perivascular cell infiltrates. Similar infiltrates were not observed in any CTL mice. At 8 mo of age, >80% of SE2−/− mice showed large aggregates of mononuclear cells, whereas <20% of CTL mice showed any infiltrates. X axis indicates the number of animals studied in each group. Shown are representative images from ≥3 animals of each genotype. Original magnification, ×10 (A, B, E–G), and ×40 (C, D).
Mononuclear cell aggregates in aged SE2−/− lungs resemble BALT-like structures
To better characterize the complexity of mononuclear cell aggregates in aged SE2−/− mice, we performed immunostaining of serial lung sections with antibodies against different cellular and stromal components of tertiary lymphoid organs (Fig. 2). Aggregates contained peanut agglutinin (PNA) d+ (peripheral node addressin), high endothelial venules, and Lyve-1+ (lymphatic vessel endothelial hyaluronan receptor 1) lymphatics (Fig. 2A). Aggregates contained numerous B lymphocytes (B220; Fig 2A, D, E), T lymphocytes (CD3; Fig. 2B), and follicular dendritic cell networks (CD21-35/follicular dendritic cell M1+; Fig. 2B, C, inset). CD3+ T cells were generally centrally located (Fig. 2B). Many of B220+ B cells expressed proliferating cell nuclear antigen (PCNA+) and bound PNA, and, thus, showed characteristics of proliferating germinal center B cells (Fig. 2D). Finally, large antibody-secreting cells (IgG) and lymphokine chemokine (C-X-C motif) ligand (Cxcl) 13 was detected within aggregates (Fig. 2E). These data suggest that SE2 deficiency leads to chronic lung lymphocyte accumulation, which results in the formation of BALT-like structures.
Figure 2.

Mononuclear cell aggregates in lungs of SE2−/− mice resemble BALT. Immunofluorescence staining of lung tissue sections identified the presence of multiple BALT-like features within the large mononuclear cell aggregates identified in SE2−/− mice. A, F) Aggregates were rich in B lymphocytes (B220) and contained elements of high endothelial venules (PNAd) and lymphatics (Lyve-1). B, C, G) Aggregates were also rich in T lymphocytes (CD3) and displayed follicular dendritic cell networks (CD21-CD35-follicular dendritic cell-M1). D, H) Many B lymphocytes (B220) within the lesions showed characteristics of proliferating germinal center B cells (PCNA+PNA+B220+). E, I) Aggregates also contained large, antibody-secreting cells (IgG) and stained for Cxcl13. Shown are representative images from ≥3 animals of each genotype. Original magnification, ×40.
We further enumerated the accumulation of T and B cells in the lungs of aged CTL and SE2−/− mice. Whole-lung tissue recovered from 1-y-old mice was digested with collagenase and assayed by flow cytometry (Fig. 3). We found a statistically significant increase in the number of CD3+ T lymphocytes (116.9 × 103 vs. 1065.7 × 103; P < 0.05) and CD19+ B lymphocytes (2.9 × 103 vs. 2147 × 103; P < 0.05) in SE2−/− mice. Within the CD3+ T cells, there were significant increases in both CD4+ (43.4 × 103 vs. 282 × 103; P < 0.05) and CD8+ (10.5 × 103 vs. 654.5 × 103; P < 0.05) populations.
Figure 3.

Quantitation of lymphocyte accumulation in lungs of SE2−/− mice. Lung digests from aged control wild-type (WT) and SE2−/− (KO) mice were stained with antibodies to detect lymphocyte-specific antigens and were analyzed by flow cytometry. A) Large increases in the number of total T (CD3+) and B (CD19+) lymphocytes were apparent in lungs of SE2−/− mice. B) Among T lymphocytes, increases in both CD4+ and CD8+ populations were observed. n = 4; data are presented as means ± sem. *P < 0.05.
Dysregulation of lymphocyte-associated chemokine and cytokine gene expression as a result of Serpine2 inhibition in vivo
To understand the pathophysiology of lungs that are affected by Serpine2 gene deficiency, we performed genome-wide transcriptional analysis of whole-lung tissue from the lungs of young (2 mo old) CTL and SE2−/− mice at a time when inflammatory aggregates were first detectable. The transcript for Serpine2 showed the greatest difference in expression (data not shown). Pathway analysis of differentially expressed genes suggested differences in biologic processes associated with lymphocyte development, activation, and maturation (Fig. 4). Some of these pathways include OX40 signaling, DC maturation, B-cell development and signaling, T-cell differentiation and signaling, and such pathways as ERK signaling and PI3K/Akt signaling.
Figure 4.
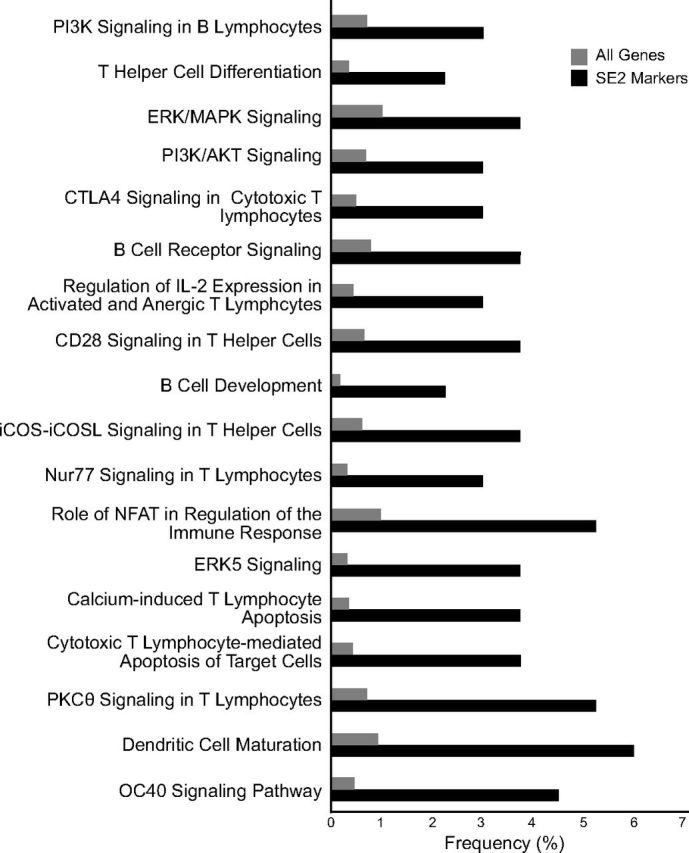
Canonical pathways dysregulated in lungs of SE2−/− mice. Genome-wide transcriptomic analysis was performed on 2-mo-old SE2−/− mice by using Affymetrix microarrays. To estimate biologic mechanisms that are altered in lungs of SE2−/− mice, we performed pathways analysis for genes identified as differentially expressed. Shown here are significantly (P < 0.05) affected canonical pathways, in which the x axis represents the frequency (percentage) of genes within each canonical pathway for the entire genome (gray) or those differentially expressed in lungs of SE2−/− mice (black). CTLA, cytotoxic T-lymphocyte–associated protein; iCOS, inducible T-cell costimulator; iCOSL, inducible T-cell costimulor ligand; NFAT, nuclear factor of activated T cells.
We further explored expression of genes that control lymphocyte accumulation and activation in SE2−/− mice by using qPCR (Fig. 5). Consistent with microarray analysis, substantive changes in the expression of chemokines, cytokines, and adhesion molecules were observed at 2 mo of age. Genes involved in cellular adhesion were significantly up-regulated, including Cd34 (1.4-fold; P < 0.05), E-Selectin (Sele; 4.1-fold; P < 0.05), and lymphocyte function-associated antigen (Lfa1/Itgb2; 24-fold; P < 0.09; Fig. 5A). Genes that are essential for tertiary lymphoid organ formation, including TNF family members Tnfa (5.2-fold; P < 0.05) and Lymphotaxin (Lt-a; 3.7-fold; P < 0.05), as well as homeostatic chemokines, Cxcl13 (2.1-fold; P < 0.05), chemokine (C- C motif) ligand (Ccl) 19 (4.2-fold; P < 0.01), and Ccl21b (1.8-fold; P < 0.05) were also elevated in lungs of SE2−/− mice (Fig. 5B). While a modest nonsignificant increase in Cxcl12 was detected (1.5-fold), expression of Ltb (1 vs. 0.6-fold; P < 0.005) was significantly decreased. In addition, chemokines for recruiting innate and adaptive immune cells, including Ccl2 (3.7-fold; P < 0.05), Ccl9 (2.6-fold; P < 0.01), Ccl20 (4.4-fold; P < 0.05), Ccl5 (2.1-fold; P < 0.01), Ccl8 (8.4-fold; P < 0.01), Ccl17 (4.7-fold; P < 0.01), Ccl22 (3.3-fold; P < 0.05), Cxcl9 (12.7-fold; P < 0.01), Cxcl10 (4.6-fold; P < 0.05), and Cxcl11 (3.2-fold; P < 0.01) were significantly increased in lungs of SE2−/− mice (Fig. 5C). No significant changes in expression of Ccl4, Cxcl1, and Cxcl2 at the mRNA level were detected (data not shown).
Figure 5.

Increased mRNA levels of numerous proinflammatory molecules in lungs of young SE2−/− mice. We studied the expression of inflammatory chemokines, cytokines, and adhesion molecules in 2-mo-old SE2−/− (KO) and CTL mouse lung tissue at the RNA (A–D) level. A) Expression of adhesion molecules. B) Expression of genes involved in the development of ectopic lymphoid structures. C) Expression of chemokines responsible for recruitment of inflammatory cells. D) Expression of cytokines produced by immune cells at inflammatory sites (n ≥ 5). Data are presented as means ± sem. #P < 0.05; ##P < 0.09 by Mann–Whitney U test.
Additional cytokine gene expression was also altered in lungs of SE2−/− mice (Fig. 5D). Whereas expression of Th1 cytokines, Ifng (3.9-fold; P < 0.05) and Il2 (2.1-fold; P < 0.05), were elevated, levels of Th2 cytokine, il4 (0.51-fold; P < 0.001), were modestly reduced. Of interest, anti-inflammatory cytokines, Tgfb (5.5-fold; P < 0.09) and Il10 (5.3-fold; P < 0.05), were also elevated. Il17-related molecules, including Il1b (2.6-fold; P < 0.05) and Il21 (5.3-fold; P < 0.09), were elevated, whereas Gmcsf (0.56-fold; P < 0.001) expression levels were modestly decreased. No significant changes in expression of Il6 and Il17 were detected (data not shown).
Finally, we attempted to confirm differences in expression for distinct but overlapping selected cytokines and chemokines at the protein level (Fig. 6). We found significant differences in the expression of Tnfα (7.7-fold; P < 0.05), Il6 (3.1-fold; P < 0.05), Il17 (34.2-fold; P < 0.05), Cxcl2 (1.4-fold; P < 0.05), Cxcl9 (2-fold; P < 0.05), Ccl4 (2.1-fold; P < 0.05), and Ccl5 (1.3-fold; P < 0.05) in lungs of SE2−/− mice.
Figure 6.

Increased protein levels of proinflammatory molecules in lungs of young SE2−/− mice. We studied protein concentrations (Conc.) of inflammatory chemokines and cytokines in 2-mo-old SE2−/− (KO) and CTL mouse lung tissue. A) Total lung protein concentrations of cytokines. B) Total lung protein concentrations of chemokines (n ≥ 5). Data are presented as means ± sem. *P < 0.05 by Student’s t test.
Increased thrombin activity in SE2−/− mice
Thrombin, a main target of Serpine2 inhibitory activity, plays an important role in regulating various biologic processes, including acting as a stimulator of lung inflammation (33, 34). Whereas additional Serpine2 targets exist (e.g., trypsin and prostasin), because thrombin is clearly elevated and the most likely to promote a proinflammatory response, we pursued thrombin as a candidate target protease for mediating the pathology observed in lungs of SE2−/− mice. We investigated whether Serpine2 deficiency was associated with excess thrombin activity in lungs of SE2−/− mice (Fig. 7). Homogenates from lungs of SE2−/− mice at 2 mo of age displayed an 8-fold increase (P < 0.01) in thrombin-specific proteolytic activity (Fig. 7A). BAL fluid also showed slightly elevated thrombin activity, which was greater in aged mice (data not shown). Conversely, we observed a much more modest and nonsignificant increase in plasmin activity (∼2-fold; P > 0.05) in lungs of SE2−/− mice at 2 mo of age (Fig. 7B), and reduced expression (0.5-fold; P < 0.05) of both lung urokinase plasminogen activator (Plau/uPA) and tissue plasminogen activator (Plat/tissue plasminogen activator) genes (Fig. 7C). These observations are consistent with Serpine2 being the major tissue-localized thrombin inhibitor, whereas numerous tissue-localized plasmin inhibitors are appreciated.
Figure 7.

Serpine2 deficiency results in increased lung thrombin activity. A, B) Total lung protein and BAL fluid collected from CTL or SE2−/− (KO) mice were tested for serine protease activity. A) Total lung protein from 2-mo-old KO mice showed an 8-fold increase in thrombin-specific proteolytic activity, whereas BAL fluid from similar aged mice had less profound increases. B) Total lung protein from KO mice showed a modest nonsignificant increase in uPA activity. C) Expression of plasmin/plasminogen pathway genes was assessed in whole-lung RNA from CTL or KO mice at 2 mo of age by qPCR. SE2−/− mice showed decreased expression of both uPA and tissue plasminogen activator mRNA (n ≥ 5). Data are presented as means ± sem. #P < 0.05 by Mann–Whitney U test; *P < 0.05; **P < 0.09 by Student’s t test.
Increased NF-κB pathway activity in lungs of SE2−/− mice in vivo
Expression of proinflammatory molecules is regulated by various transcription factors. Of these, NF-κB has been identified as critical (28, 35). Thrombin is known to induce NF-κB activation in resident lung cells, which leads to cellular responses that promote lung inflammation (28, 33, 36); therefore, we assessed the activation status of NF-κB in lungs of SE2−/− mice (Fig. 8A–C). Serpine2 deficiency resulted in enhanced NF-κB–associated DNA binding activity (1.4-fold; P < 0.003) in freshly isolated lung protein homogenates from 2 mo old mice (Fig 8A). Western blot analysis also suggested significantly increased p65 phosphorylation (Ser536) and IKKα/β phosphorylation (Ser176) in lung homogenates from SE2−/− mice (Fig. 8B, C). Phosphorylation of IKK was associated with concomitant degradation of IκBα (Fig. 8B, C).
Figure 8.

Proinflammatory signaling pathways are altered in lungs of SE2−/− mice. Total lung protein was isolated from 2-mo-old CTL or SE2−/− (KO) mice and tested for NF-κB, Akt, and ERK1/2 pathway activity. A) Lungs of SE2−/− mice demonstrate a significant increase in p65 DNA binding activity. B, C) Lungs of SE2−/− mice showed evidence of increased IKKα/β (Ser176) phosphorylation and increased degradation of IκBα. Whereas total p65 levels were not changed, we noticed increased p65 (Ser536) phosphorylation. D, E) Increased Akt and ERK phosphorylation was observed in lungs of SE2−/− mice (n = 4–5). Data are presented as means ± sem. #P < 0.05 by Mann–Whitney U test.
Numerous reports suggest that Akt and ERK pathways are also involved in regulation of expression of inflammatory mediators. Furthermore, our transcriptomics analyses suggested regulation of these pathways in SE2−/− mice. We used Western blot analysis of lung homogenates to investigate activation status of these pathways (Fig. 8D, E). Akt, which is an upstream regulator of NF-κB, demonstrated significantly increased phosphorylation (Ser473) in lungs of SE2−/− mice. We also observed a significant increase in phosphorylation of ERK1/2 (Thr202/Tyr204, Thr185, and Tyr187) in lungs of SE2−/− mice.
Thrombin challenge increases lung chemokine expression in vivo
Acute, systemic thrombin challenge results in lung inflammation in mice (33), which involves activation of NF-κB. We tested whether systemic thrombin challenge induces expression of cytokines and chemokines associated with lymphocyte accumulation observed in SE2−/− mice (Fig. 9). In adult CTL mice, intraperitoneal thrombin injection resulted in increased expression of Cxcl10 (3.4-fold; P < 0.05), Cxcl13 (1.6-fold; P < 0.08), Ccl9 (2.3-fold; P < 0.05), Tnfa (5498-fold; P < 0.05), Il1b (3.2-fold; P < 0.005), Il6 (4.7-fold; P < 0.05), and Icam1 (4.7-fold; P < 0.05).
Figure 9.

Exogenous thrombin induces lung chemokine expression in vivo. CTL mice were i.p. injected with either PBS (CTL-Sal) or 50 U/g body weight of α-thrombin (CTL-αT), and lungs were harvested for gene expression analysis after 1 h. A) Expression of CXC class chemokines Cxcl9, Cxcl10, Cxcl12 and Cxcl13. B) Expression of CC class chemokines Ccl5, Ccl9, Ccl19, Ccl20 and Ccl21b. C) Expression of Tnfa. D) Expression of Il1b and Il6. E) Expression of Icam1 (n ≥ 4). Data are presented as means ± sem. #P < 0. 05 by Mann–Whitney U test; **P < 0.09 by Student’s t test.
DISCUSSION
In this study, we report the spontaneous and persistent accumulation of lymphocytes in the lungs of SE2−/− mice, which leads to the formation of BALT-like structures. The initiation of lymphocyte accumulation is associated with excess thrombin activity, increased NF-κB activation, and overproduction of lung chemokines and cytokines in vivo. We also demonstrate that exogenous, systemic thrombin delivery to CTL mice induces the expression of chemokines and cytokines dysregulated in the lungs of SE2−/− mice. Spontaneous accumulation of activated lymphocytes in aging mice has been previously reported (37–39). Whereas inflammation associated with injury has been reported in SE2−/− mice (40), spontaneous inflammation has not been reported and was not evident in a survey of other organs. Our data suggest a previously unappreciated role for serine proteases and their inhibitors in regulating lymphocyte trafficking in the lung.
On the basis of genetic structural variation in humans and gene expression variation in humans and mice, we previously implicated SERPINE2 as a candidate COPD susceptibility gene (3). These results have subsequently been independently verified (6–10); however, we still lack an understanding of the function of SERPINE2 in homeostasis and disease. Here, we describe the unanticipated observation that Serpine2 deficiency is associated with the spontaneous accumulation of lymphocytes in the peribronchiolar and perivascular space, which eventually leads to the formation of BALT-like structures, in unchallenged mice. COPD involves the inflammatory destruction of alveoli or remodeling of conducting airways (41) and has more recently been associated with increased BALT formation (42–46). The increase in the presence of these tertiary lymphoid structures in human COPD and in SE2−/− mice is currently of unknown functional consequence.
We believe that increases in SERPINE2 expression in COPD represent a compensatory mechanism, in response to the overwhelmed protease burden and associated antiprotease functional insufficiency. Similar to our findings with Serpine2, recently Baraldo et al. (47) demonstrated enhanced T- and B-lymphocyte accumulation and exaggerated lymphoid follicle formation in humans with SERPINA1/α1-antitrypsin deficiency. It should be noted that many, though not all, disease-related effects of SERPINA1 are mediated via its ability to regulate elastase activity, whereas SERPINE2 does not inhibit elastase. Together, our results suggest the involvement of SERPINs in complex adaptive immune responses associated with COPD pathology, beyond the regulation of tissue destruction and emphysema. The identification of where these 2 serine protease inhibition pathways converge to regulate COPD-related lymphocyte trafficking is a focus of future investigation with potential for therapeutic implications. Interestingly, we also find a modest, but significant, congenital airspace enlargement in SE2−/− mice (unpublished results). This difference in respiratory structure is nonprogressive and is not associated with evidence of neutrophil or macrophage inflammation.
There exists considerable complexity in the inflammatory milieu in lungs of SE2−/− mice. We first tested adhesion molecules that were expressed by inflamed endothelium, which are key in initial steps of leukocyte migration, and found a significant increase in the expression of CD34 and Sele. TNF family members like Tnfa and Lta, which are crucial in the induction of homeostatic chemokines, also demonstrated increased expression. There was a parallel increase in expression of homeostatic chemokines Cxcl13, Ccl19, and Ccl21b, which are critical for B- and T-cell recruitment and organization and function of BALT (48). In addition, we found an induction of multiple chemokines that are key in the recruitment of cells from the innate (Ccl2, Ccl9, and Ccl20) and adaptive immune system (Ccl5, Ccl8, Ccl17, and Ccl22). In summary, the lungs of SE2−/− mice show gene expression signs of tertiary lymphoid organ formation (Tnfa, Lta, Cxcl13, Ccl19, and Ccl21b), active recruitment of cells from innate (Ccl2, Ccl9, and Ccl20) and adaptive immune systems (Ccl4, Ccl5, Ccl8, Ccl17, and Ccl22), local production of cytokines and chemokines that are involved in type 1 responses (Tnfa, Ifng, Il2, Cxcl9, Cxcl10, and Cxcl11), evidence of a supportive IL17 milieu (Il1b, Il6, and Il17), and modulation of the inflammatory response by classic anti-inflammatory cytokines such as Il10 and Tgfb.
Specific mechanisms that lead to accumulation of lymphocytes in the lungs of SE2−/− mice are not completely clear. Thrombin plays a critical role in thrombosis and hemostasis but also promotes a wide range of cellular responses, including inflammation and tissue repair (34, 49). We chose to pursue thrombin as a candidate target protease for mediation of the pathology observed in lungs of SE2−/− mice because 1) Serpine2 is the predominant tissue-localized inhibitor of thrombin, although numerous tissue-localized plasmin inhibitors exist; 2) we found large increases in thrombin activity in the lung tissue of younger SE2−/− mice; and 3) unlike other protease targets, such as trypsin or prostasin, thrombin can signal via resident lung cell NF-κB (29, 33) to initiate an inflammatory response. Thrombin activity is increased in lung tissue of younger SE2−/− mice, where it is available to act upon multiple cell types to promote an inflammatory phenotype. Thrombin levels are elevated in patients with COPD (26, 27), lung fibrosis (50, 51), and asthma (52), where they correlate with inflammatory cytokine production (53). Acute and chronic lung diseases are accompanied by evidence of inflammation and vascular injury. As a result of the slowly progressive nature of lesions in SE2−/− mice, it is likely that thrombin is released in a homeostatic mechanism, where in the absence of SE2 it is persistently available to act upon multiple cell types to potentiate inflammation.
Our data do not support the conclusion that thrombin-mediated activation of NF-κB is exclusively responsible for the regulation of all relevant chemokines and cytokines from resident lung cells. In fact, thrombin stimulates multiple intracellular signaling pathways, including PI3K/Akt (30, 54), ERK1/2 (55), C/EBPβ (56), and p38 (57). Induction of some of these additional signaling pathways in lungs of SE2−/− mice is consistent with this interpretation. Furthermore, it is possible that additional extracellular processes are likely to contribute to the observed phenotype. For instance, enhanced activity of the protease prostasin, an alternate target for SE2 inhibition, can increase ENaC activity (58), which can result in lung lymphocyte accumulation (59). In fact, preliminary evidence suggests altered barrier function in the lungs of SE2−/− mice (unpublished results). Dissection of serine protease–dependent cellular and molecular mechanisms that are causally involved in control of lung lymphocyte accumulation is the focus of current and future studies. Our results suggest that multiple pathways likely contribute to the complex BALT-like lung pathology associated with excess thrombin and elevated NF-κB activity in SE2−/− mice.
Acknowledgments
This work was supported by Flight Attendant Medical Research Institute, U.S. National Institutes of Health (NIH), National Institute of Environmental Health Sciences (Grant ES014372), and NIH National Heart, Lung, and Blood Institutes (Grants HL094431 and HL116632). The authors thank Dr. Mohammad Minhajuddin, Dr. Fabeha Fazal, Dr. Maria da la Luz Garcia-Hernandez, Valerie Lunger, Ashley Lopez, Swati Shimpi, Jared Mereness, and Muranda Maurer (University of Rochester Medical Center, Rochester, NY, USA) and Temana Andalcio, Jungha Hong, and Roberto Landazury (Brigham and Women's Hospital, Harvard Medical School, Boston, MA, USA) for assistance.
Glossary
- BAL
broncho-alveolar lavage
- BALT
bronchus-associated lymphoid tissue
- CCL
chemokine (C- C motif) ligand
- CD
cluster of differentiation
- COPD
chronic obstructive pulmonary disease
- CTL
wild-type control
- CXCL
chemokine (C-X-C motif) ligand
- FACS
fluorescence-activated cell sorting
- qPCR
quantitative real-time PCR
- PNA
peanut agglutinin
- SERPIN
serine protease inhibitor
- SERPINA1
serine proteinase inhibitor, clade A, member 1
- SERPINE2/SE2
serine proteinase inhibitor, clade E, member 2
- uPA
urokinase plasminogen activator
REFERENCES
- 1.Lomas D. A., Silverman E. K. (2001) The genetics of chronic obstructive pulmonary disease. Respir. Res. , 20–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleeberger S. R., Peden D. (2005) Gene-environment interactions in asthma and other respiratory diseases. Annu. Rev. Med. , 383–400 [DOI] [PubMed] [Google Scholar]
- 3.Demeo D. L., Mariani T. J., Lange C., Srisuma S., Litonjua A. A., Celedon J. C., Lake S. L., Reilly J. J., Chapman H. A., Mecham B. H., Haley K. J., Sylvia J. S., Sparrow D., Spira A. E., Beane J., Pinto-Plata V., Speizer F. E., Shapiro S. D., Weiss S. T., Silverman E. K. (2006) The SERPINE2 gene is associated with chronic obstructive pulmonary disease. Am. J. Hum. Genet. , 253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunninghake G. M., Cho M. H., Tesfaigzi Y., Soto-Quiros M. E., Avila L., Lasky-Su J., Stidley C., Melén E., Söderhäll C., Hallberg J., Kull I., Kere J., Svartengren M., Pershagen G., Wickman M., Lange C., Demeo D. L., Hersh C. P., Klanderman B. J., Raby B. A., Sparrow D., Shapiro S. D., Silverman E. K., Litonjua A. A., Weiss S. T., Celedón J. C. (2009) MMP12, lung function, and COPD in high-risk populations. N. Engl. J. Med. , 2599–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sørheim I. C., DeMeo D. L., Washko G., Litonjua A., Sparrow D., Bowler R., Bakke P., Pillai S. G., Coxson H. O., Lomas D. A., Silverman E. K., Hersh C. P.; International COPD Genetics Network Investigators (2010) Polymorphisms in the superoxide dismutase-3 gene are associated with emphysema in COPD. COPD , 262–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castaldi P. J., Hersh C. P., Reilly J. J., Silverman E. K. (2009) Genetic associations with hypoxemia and pulmonary arterial pressure in COPD. Chest , 737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu G., Warren L., Aponte J., Gulsvik A., Bakke P., Anderson W. H., Lomas D. A., Silverman E. K., Pillai S. G.; International COPD Genetics Network (ICGN) Investigators (2007) The SERPINE2 gene is associated with chronic obstructive pulmonary disease in two large populations. Am. J. Respir. Crit. Care Med. , 167–173 [DOI] [PubMed] [Google Scholar]
- 8.Cha S. I., Kang H. G., Choi J. E., Kim M. J., Park J., Lee W. K., Kim C. H., Jung T. H., Park J. Y. (2009) SERPINE2 polymorphisms and chronic obstructive pulmonary disease. J. Korean Med. Sci. , 1119–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kukkonen M. K., Tiili E., Hämäläinen S., Vehmas T., Oksa P., Piirilä P., Hirvonen A. (2011) SERPINE2 haplotype as a risk factor for panlobular type of emphysema. BMC Med. Genet. , 157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.An L., Yang T., Zhang Y., Lin Y., Zhang H., Jiao X., Hua L., Dai H., Wang C. (2012) Association of SERPINE2 gene with the risk of chronic obstructive pulmonary disease and spirometric phenotypes in northern Han Chinese population. Mol. Biol. Rep. , 1427–1433 [DOI] [PubMed] [Google Scholar]
- 11.Bouton M. C., Boulaftali Y., Richard B., Arocas V., Michel J. B., Jandrot-Perrus M. (2012) Emerging role of serpinE2/protease nexin-1 in hemostasis and vascular biology. Blood , 2452–2457 [DOI] [PubMed] [Google Scholar]
- 12.Baker J. B., Low D. A., Simmer R. L., Cunningham D. D. (1980) Protease-nexin: a cellular component that links thrombin and plasminogen activator and mediates their binding to cells. Cell , 37–45 [DOI] [PubMed] [Google Scholar]
- 13.Mansuy I. M., van der Putten H., Schmid P., Meins M., Botteri F. M., Monard D. (1993) Variable and multiple expression of Protease Nexin-1 during mouse organogenesis and nervous system development. Development , 1119–1134 [DOI] [PubMed] [Google Scholar]
- 14.Bouton M. C., Richard B., Rossignol P., Philippe M., Guillin M. C., Michel J. B., Jandrot-Perrus M. (2003) The serpin protease-nexin 1 is present in rat aortic smooth muscle cells and is upregulated in L-NAME hypertensive rats. Arterioscler. Thromb. Vasc. Biol. , 142–147 [DOI] [PubMed] [Google Scholar]
- 15.Bédard J., Brûlé S., Price C. A., Silversides D. W., Lussier J. G. (2003) Serine protease inhibitor-E2 (SERPINE2) is differentially expressed in granulosa cells of dominant follicle in cattle. Mol. Reprod. Dev. , 152–165 [DOI] [PubMed] [Google Scholar]
- 16.Feutz A. C., Barrandon Y., Monard D. (2008) Control of thrombin signaling through PI3K is a mechanism underlying plasticity between hair follicle dermal sheath and papilla cells. J. Cell Sci. , 1435–1443 [DOI] [PubMed] [Google Scholar]
- 17.Drapkin P. T., Monard D., Silverman A. J. (2002) The role of serine proteases and serine protease inhibitors in the migration of gonadotropin-releasing hormone neurons. BMC Dev. Biol. , 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKee C. M., Xu D., Muschel R. J. (2013) Protease nexin 1: a novel regulator of prostate cancer cell growth and neo-angiogenesis. Oncotarget , 1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKee C. M, Ding Y., Zhou J., Li C., Huang L., Xin X., He J., Allen J. E., El-Deiry W. S., Cao Y., Muschel R. J., and Xu D. (2015) Protease nexin 1 induces apoptosis of prostate tumor cells through inhibition of X-chromosome-linked inhibitor of apoptosis protein. Oncotarget , 3784–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulaftali Y., François D., Venisse L., Jandrot-Perrus M., Arocas V., Bouton M. C. (2013) Endothelial protease nexin-1 is a novel regulator of A disintegrin and metalloproteinase 17 maturation and endothelial protein C receptor shedding via furin inhibition. Arterioscler. Thromb. Vasc. Biol. , 1647–1654 [DOI] [PubMed] [Google Scholar]
- 21.Gomez D., Kessler K., Borges L. F., Richard B., Touat Z., Ollivier V., Mansilla S., Bouton M. C., Alkoder S., Nataf P., Jandrot-Perrus M., Jondeau G., Vranckx R., Michel J. B. (2013) Smad2-dependent protease nexin-1 overexpression differentiates chronic aneurysms from acute dissections of human ascending aorta. Arterioscler. Thromb. Vasc. Biol. , 2222–2232 [DOI] [PubMed] [Google Scholar]
- 22.Guttridge D. C., Lau A. L., Cunningham D. D. (1993) Protease nexin-1, a thrombin inhibitor, is regulated by interleukin-1 and dexamethasone in normal human fibroblasts. J. Biol. Chem. , 18966–18974 [PubMed] [Google Scholar]
- 23.Vaughan P. J., Cunningham D. D. (1993) Regulation of protease nexin-1 synthesis and secretion in cultured brain cells by injury-related factors. J. Biol. Chem. , 3720–3727 [PubMed] [Google Scholar]
- 24.Lüthi A., Van der Putten H., Botteri F. M., Mansuy I. M., Meins M., Frey U., Sansig G., Portet C., Schmutz M., Schröder M., Nitsch C., Laurent J. P., Monard D. (1997) Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term potentiation. J. Neurosci. , 4688–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murer V., Spetz J. F., Hengst U., Altrogge L. M., de Agostini A., Monard D. (2001) Male fertility defects in mice lacking the serine protease inhibitor protease nexin-1. Proc. Natl. Acad. Sci. USA , 3029–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashitani J., Mukae H., Arimura Y., Matsukura S. (2002) Elevated plasma procoagulant and fibrinolytic markers in patients with chronic obstructive pulmonary disease. Intern. Med. , 181–185 [DOI] [PubMed] [Google Scholar]
- 27.Jankowski M., Undas A., Kaczmarek P., Butenas S. (2011) Activated factor XI and tissue factor in chronic obstructive pulmonary disease: links with inflammation and thrombin generation. Thromb. Res. , 242–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahman A., Fazal F. (2011) Blocking NF-κB: an inflammatory issue. Proc. Am. Thorac. Soc. , 497–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rahman A., True A. L., Anwar K. N., Ye R. D., Voyno-Yasenetskaya T. A., Malik A. B. (2002) Galpha(q) and Gbetagamma regulate PAR-1 signaling of thrombin-induced NF-kappaB activation and ICAM-1 transcription in endothelial cells. Circ. Res. , 398–405 [DOI] [PubMed] [Google Scholar]
- 30.Lin C. H., Cheng H. W., Ma H. P., Wu C. H., Hong C. Y., Chen B. C. (2011) Thrombin induces NF-kappaB activation and IL-8/CXCL8 expression in lung epithelial cells by a Rac1-dependent PI3K/Akt pathway. J. Biol. Chem. , 10483–10494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solleti S. K., Simon D. M., Srisuma S., Arikan M. C., Bhattacharya S., Rangasamy T., Bijli K. M., Rahman A., Crossno J. T. Jr., Shapiro S. D., Mariani T. J. (2015) Airway epithelial cell PPARγ modulates cigarette smoke-induced chemokine expression and emphysema susceptibility in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. , L293–L304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srisuma S., Bhattacharya S., Simon D. M., Solleti S. K., Tyagi S., Starcher B., Mariani T. J. (2010) Fibroblast growth factor receptors control epithelial-mesenchymal interactions necessary for alveolar elastogenesis. Am. J. Respir. Crit. Care. Med. , 838–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fazal F., Bijli K. M., Murrill M., Leonard A., Minhajuddin M., Anwar K. N., Finkelstein J. N., Watterson D. M., Rahman A. (2013) Critical role of non-muscle myosin light chain kinase in thrombin-induced endothelial cell inflammation and lung PMN infiltration. PLoS One , e59965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levi M., van der Poll T., Büller H. R. (2004) Bidirectional relation between inflammation and coagulation. Circulation , 2698–2704 [DOI] [PubMed] [Google Scholar]
- 35.Oeckinghaus A., Hayden M. S., Ghosh S. (2011) Crosstalk in NF-κB signaling pathways. Nat. Immunol. , 695–708 [DOI] [PubMed] [Google Scholar]
- 36.Bijli K. M., Fazal F., Minhajuddin M., Rahman A. (2008) Activation of Syk by protein kinase C-delta regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via tyrosine phosphorylation of RelA/p65. J. Biol. Chem. , 14674–14684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun H., Lu B., Li R. Q., Flavell R. A., Taneja R. (2001) Defective T cell activation and autoimmune disorder in Stra13-deficient mice. Nat. Immunol. , 1040–1047 [DOI] [PubMed] [Google Scholar]
- 38.Davalos-Misslitz A. C., Rieckenberg J., Willenzon S., Worbs T., Kremmer E., Bernhardt G., Förster R. (2007) Generalized multi-organ autoimmunity in CCR7-deficient mice. Eur. J. Immunol. , 613–622 [DOI] [PubMed] [Google Scholar]
- 39.Kelly F. M., Reddy R. N., Roberts B. R., Gangappa S., Williams I. R., Gooch J. L. (2009) TGF-beta upregulation drives tertiary lymphoid organ formation and kidney dysfunction in calcineurin A-alpha heterozygous mice. Am. J. Physiol. Renal Physiol. , F512–F520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lino M. M., Atanasoski S., Kvajo M., Fayard B., Moreno E., Brenner H. R., Suter U., Monard D. (2007) Mice lacking protease nexin-1 show delayed structural and functional recovery after sciatic nerve crush. J. Neurosci. , 3677–3685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barnes P. J. (2008) Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. , 183–192 [DOI] [PubMed] [Google Scholar]
- 42.Hogg J. C., Chu F., Utokaparch S., Woods R., Elliott W. M., Buzatu L., Cherniack R. M., Rogers R. M., Sciurba F. C., Coxson H. O., Paré P. D. (2004) The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. , 2645–2653 [DOI] [PubMed] [Google Scholar]
- 43.Demoor T., Bracke K. R., Maes T., Vandooren B., Elewaut D., Pilette C., Joos G. F., Brusselle G. G. (2009) Role of lymphotoxin-alpha in cigarette smoke-induced inflammation and lymphoid neogenesis. Eur. Respir. J. , 405–416 [DOI] [PubMed] [Google Scholar]
- 44.Brusselle G. G., Demoor T., Bracke K. R., Brandsma C. A., Timens W. (2009) Lymphoid follicles in (very) severe COPD: beneficial or harmful? Eur. Respir. J. , 219–230 [DOI] [PubMed] [Google Scholar]
- 45.Polverino F., Cosio B. G., Pons J., Laucho-Contreras M., Tejera P., Iglesias A., Rios A., Jahn A., Sauleda J., Divo M., Pinto-Plata V., Sholl L., Rosas I. O., Agusti A., Celli B. R., Owen C. A. (2015) B cell-activating factor. an orchestrator of lymphoid follicles in severe chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care. Med. , 695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bracke K. R., Verhamme F. M., Seys L. J., Bantsimba-Malanda C., Cunoosamy D. M., Herbst R., Hammad H., Lambrecht B. N., Joos G. F., Brusselle G. G. (2013) Role of CXCL13 in cigarette smoke-induced lymphoid follicle formation and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. , 343–355 [DOI] [PubMed] [Google Scholar]
- 47.Baraldo S., Turato G., Lunardi F., Bazzan E., Schiavon M., Ferrarotti I., Molena B., Cazzuffi R., Damin M., Balestro E., Luisetti M., Rea F., Calabrese F., Cosio M. G., Saetta M. (2015) Immune activation in α1-antitrypsin-deficiency emphysema. Beyond the protease-antiprotease paradigm. Am. J. Respir. Crit. Care Med. , 402–409 [DOI] [PubMed] [Google Scholar]
- 48.Foo S. Y., Phipps S. (2010) Regulation of inducible BALT formation and contribution to immunity and pathology. Mucosal Immunol. , 537–544 [DOI] [PubMed] [Google Scholar]
- 49.Chen D., Dorling A. (2009) Critical roles for thrombin in acute and chronic inflammation. J. Thromb. Haemost. (Suppl 1), 122–126 [DOI] [PubMed] [Google Scholar]
- 50.Hernández-Rodríguez N. A., Cambrey A. D., Harrison N. K., Chambers R. C., Gray A. J., Southcott A. M., duBois R. M., Black C. M., Scully M. F., McAnulty R. J., et al. (1995) Role of thrombin in pulmonary fibrosis. Lancet , 1071–1073 [DOI] [PubMed] [Google Scholar]
- 51.François D., Venisse L., Marchal-Somme J., Jandrot-Perrus M., Crestani B., Arocas V., Bouton M. C. (2014) Increased expression of protease nexin-1 in fibroblasts during idiopathic pulmonary fibrosis regulates thrombin activity and fibronectin expression. Lab. Invest. , 1237–1246 [DOI] [PubMed] [Google Scholar]
- 52.Gabazza E. C., Taguchi O., Tamaki S., Takeya H., Kobayashi H., Yasui H., Kobayashi T., Hataji O., Urano H., Zhou H., Suzuki K., Adachi Y. (1999) Thrombin in the airways of asthmatic patients. Lung , 253–262 [DOI] [PubMed] [Google Scholar]
- 53.Terada M., Kelly E. A., Jarjour N. N. (2004) Increased thrombin activity after allergen challenge: a potential link to airway remodeling? Am. J. Respir. Crit. Care Med. , 373–377 [DOI] [PubMed] [Google Scholar]
- 54.Minhajuddin M., Bijli K. M., Fazal F., Sassano A., Nakayama K. I., Hay N., Platanias L. C., Rahman A. (2009) Protein kinase C-delta and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-kappaB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells. J. Biol. Chem. , 4052–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin C. H., Yu M. C., Chiang C. C., Bien M. Y., Chien M. H., Chen B. C. (2013) Thrombin-induced NF-κB activation and IL-8/CXCL8 release is mediated by c-Src-dependent Shc, Raf-1, and ERK pathways in lung epithelial cells. Cell. Signal. , 1166–1175 [DOI] [PubMed] [Google Scholar]
- 56.Lin C. H., Nai P. L., Bien M. Y., Yu C. C., Chen B. C. (2014) Thrombin-induced CCAAT/enhancer-binding protein β activation and IL-8/CXCL8 expression via MEKK1, ERK, and p90 ribosomal S6 kinase 1 in lung epithelial cells. J. Immunol. , 338–348 [DOI] [PubMed] [Google Scholar]
- 57.Marin V., Farnarier C., Grès S., Kaplanski S., Su M. S., Dinarello C. A., Kaplanski G. (2001) The p38 mitogen-activated protein kinase pathway plays a critical role in thrombin-induced endothelial chemokine production and leukocyte recruitment. Blood , 667–673 [DOI] [PubMed] [Google Scholar]
- 58.Wakida N., Kitamura K., Tuyen D. G., Maekawa A., Miyoshi T., Adachi M., Shiraishi N., Ko T., Ha V., Nonoguchi H., Tomita K. (2006) Inhibition of prostasin-induced ENaC activities by PN-1 and regulation of PN-1 expression by TGF-beta1 and aldosterone. Kidney Int. , 1432–1438 [DOI] [PubMed] [Google Scholar]
- 59.Livraghi A., Grubb B. R., Hudson E. J., Wilkinson K. J., Sheehan J. K., Mall M. A., O’Neal W. K., Boucher R. C., Randell S. H. (2009) Airway and lung pathology due to mucosal surface dehydration in beta-epithelial Na+ channel-overexpressing mice: role of TNF-alpha and IL-4Ralpha signaling, influence of neonatal development, and limited efficacy of glucocorticoid treatment. J. Immunol. , 4357–4367 [DOI] [PMC free article] [PubMed] [Google Scholar]