

Abstract
Inflammatory activation precedes and correlates with accumulating τ lesions in Alzheimer’s disease and tauopathies. However, the relationship between neuroinflammation and etiology of pathologic τ remains elusive. To evaluate whether inflammatory signaling may promote or accelerate neurofibrillary tangle pathology, we explored the effect of recombinant adeno-associated virus (rAAV)-mediated overexpression of a master inflammatory cytokine, IFN-γ, on τ phosphorylation. In initial studies in primary neuroglial cultures, rAAV-mediated expression of IFN-γ did not alter endogenous τ production or paired helical filament τ phosphorylation. Next, we tested the effect of rAAV-mediated expression of IFN-γ in the brains of 2 mouse models of tauopathy: JNPL3 and rTg4510. In both models, IFN-γ increased 1) signal transducer and activator of transcription 1 levels and gliosis, and 2) hyperphosphorylation and conformational alterations of soluble τ compared with control cohorts. However, sarkosyl-insoluble phosphorylated τ levels and ubiquitin staining were unaltered in the IFN-γ cohorts. Notably, IFN-γ–induced τ hyperphosphorylation was associated with release of the inhibitory effect of glycogen synthase kinase 3β function by decreasing Ser9 phosphorylation. Our data suggest that type II IFN signaling can promote τ phosphorylation by modulating cellular kinase activity, though this is insufficient in accelerating neuritic tangle pathology.—Li, A., Ceballos-Diaz, C., DiNunno, N., Levites, Y., Cruz, P. E., Lewis, J., Golde, T. E., Chakrabarty, P. IFN-γ promotes τ phosphorylation without affecting mature tangles.
Keywords: interferon-γ, neuroinflammation, τ tangle, GSK3β
Aggregates of the hyperphosphorylated forms of the microtubule-associated protein τ (MAPT), referred to as neurofibrillary tangles (NFTs), are implicated in the loss of axonal integrity and synaptic connectivity leading to cognitive deficit and neurodegeneration in tauopathies including Alzheimer’s disease (AD) (1). Although rare mutations directly underlie primary neurodegenerative tauopathy in fronto-temporal dementia, the factors that induce τ pathology in sporadic and secondary tauopathies remain enigmatic.
Several recent studies suggest that inflammatory signaling promotes tauopathy (2). Imaging and histochemical studies show that chronic glial activation precedes and is correlated with NFT burden in AD and preclinical mouse models of tauopathy (3, 4). Interestingly, cortical astrocytes and oligodendroglia themselves display typical NFT pathology in human tauopathies (5). Neuronal overexpression of τ in rodents leads to τ inclusions and neurodegeneration (6), which is preceded by inflammatory changes, suggesting the involvement of both non–cell autonomous and cell-autonomous mechanisms in the neuroimmune regulation of tauopathy. Although the mechanisms remain obscure, it is thought that hyperphosphorylated τ can aggregate inside the cells and can also be released into the extracellular space (7). Such increased intracellular proteostasis as well as extracellular τ could potentially be recognized as non-self and may activate different arms of the immune system, such as complements, TLRs, and glial surface receptors in the CNS (8).
In rodents, inflammatory cytokines, such as IL-1β, induce τ hyperphosphorylation, whereas immunosuppressants FK506 or minocycline attenuate tauopathy in τ mice (9–11). Bacterial LPS or viral infection mediated inflammation exacerbates τ hyperphosphorylation in MAPT transgenic mice and wild-type (WT) mice (12–14). Few studies have directly addressed the question of whether inflammatory signaling actually accelerates the disease process and contributes to sarkosyl-insoluble τ species that is biochemically comparable to the NFTs characteristic of tauopathies. Initially, using a multiplex array technology (NanoString, Technologies, Seattle, WA, USA), we established that major changes in innate immune gene expression occur in the brains and spinal cords of symptomatic and asymptomatic mutant P301L τ transgenic JNPL3 mice, when compared with age-matched nontransgenic mice. Following this, we tested whether inflammatory signaling triggers tauopathy in the brains of 2 mouse models of tauopathies. Toward this end, we expressed a master inflammatory cytokine, IFN-γ, using intracerebroventricular injection of recombinant adeno-associated virus (rAAV) [rAAV 2 plasmid packaged in serotype 1 (rAAV2/1) –IFN-γ] in neonatal MAPT transgenic mice. IFN-γ is a type II IFN that has antiviral and immunoregulatory properties (15). IFN-γ signaling proceeds via the Janus-activated kinase 2/signal transducer and activator of transcription 1 (STAT1) pathway and leads to activation of immune cells via production of various immunomodulatory factors (such as IL-12, MIP1α/β, and MIG), complement factors, and apoptotic factors (such as TNF-α receptor, Fas/FasL, and caspase-1/IL-1β–converting enzyme). Many of these IFN-γ–responsive genes are up-regulated in AD (16, 17) and in preclinical models of amyloid β (Aβ) deposition (18, 19). We have demonstrated previously that IFN-γ expression attenuates Aβ deposition in APP (amyloid precursor protein) mice via synergistic activation of glia and innate immune system components (20). Given that immune signaling pathways can potentially affect AD pathologies, such as τ NFTs and Aβ plaques, in disparate ways (21), and this can have important implications in immune-based therapies, we attempted to delineate how IFN-γ would affect τ pathology in this study. Here, we show that IFN-γ expression increases hyperphosphorylation of soluble τ in AD-relevant phospho-epitopes [paired helical filament (PHF1) and CP13] but does not affect sarkosyl-insoluble τ or silver-impregnated NFTs in the 2 MAPT transgenic mouse models tested. Our results suggest that IFN-γ–induced inflammatory signaling increases the levels of soluble phosphorylated τ (phospho τ) via activation of glycogen synthase kinase 3β (GSK3β) without accelerating inclusion pathology.
MATERIALS AND METHODS
Mice
All animal husbandry procedures performed were approved by the Institutional Animal Care and Use Committee. Pregnant mice (for rTg4510 litters) were maintained by J.L. as described previously (22). In a blinded procedure, one scientist (J.L.) evaluated potential gender bias in the rTg4510 mice contributing to hippocampal neurodegeneration by rating hematoxylin and eosin-stained brain sections. Hemizygous JNPL3 mice were maintained by breeding transgenic females with male Swiss Webster mice (Harlan Laboratories, Indianapolis, IN, USA) (23). Only female JNPL3 mice were used in this study. Homozygous JNPL3 mice (on Swiss Webster background) were maintained by breeding nonsibling males and females.
rAAV2/1 preparation and injection
rAAV2/1 viruses expressing IFN-γ (Image clone 8733812) or enhanced green fluorescent protein (EGFP) under the control of the cytomegalovirus enhancer/chicken β-actin promoter were generated as described previously (20). Neonates were injected on neonatal day P2 and aged as described before (20).
RNA isolation and NanoString analysis
Eight-month-old female Swiss Webster mice and homozygous JNPL3 mice were analyzed. Mice displaying frank hindlimb paralysis were considered as symptomatic, whereas cage mates with no obvious paralysis were termed asymptomatic. Mice were euthanized by physical methods, and the whole spinal cord and forebrain were collected and flash frozen for RNA analysis. Total RNA from mouse brain was purified using TRIzol and the RNAqueous kit (Ambion, Thermo Fisher Scientific, Waltham, MA, USA) and reverse transcribed using Superscript III (Invitrogen, Life Technologies, Carlsbad, CA, USA). Multiplex analysis of RNA was done using the nCounter GX mouse inflammation kit (NanoString Technologies, Seattle, WA, USA) using 100 ng total RNA from mouse brains using the manufacturer’s recommendation (24). A list of the actual target genes is available at the manufacturer's website (http://www.nanostring.com).
WT primary neuroglia cultures
Briefly, cortices were isolated from B6/C3H newborn mice (Harlan Laboratories). Tissue was dissociated by digestion with papain solution (Worthington Biochemical, Lakewood, NJ, USA) and 50 μg/ml DNase I (Sigma-Aldrich, St. Louis, MO, USA) in a 37°C water bath for 20 min. Digested tissue was washed 3 times with HBSS (Life Technologies, Thermo Fisher Scientific) to remove papain and resuspended in growth medium consisting of Neurobasal Medium (Life Technologies) supplemented with 0.02% NeuroCult SM1 (Stemcell Technologies, Vancouver, BC, Canada), 0.5 mM GlutaMAX (Gibco, Life Technologies, Grand Island, NY, USA), 5% fetal bovine serum (HyClone Laboratories, Logan, UT, USA), and 0.01% Pen Strep (Gibco, Life Technologies). The tissue was triturated in the same medium, and dissociated cells were plated in 8-well chamber slides (Nunc Lab-Tek CCII Chamber slides; Thermo Fisher Scientific) at a density of 20,000 cells per well for imaging and in poly-d-lysine (Sigma-Aldrich)-coated 6-well plates (Costar; Thermo Fisher Scientific) for biochemical analysis. Cells were maintained at 37°C in 5% CO2 in a humidified incubator.
Preparation of tissue homogenate for immunoblotting and ELISA
Brains were sagittally dissected, and the left forebrains were snap frozen. These brains were subsequently sequentially extracted as described previously (22). Briefly, forebrains were homogenized in Tris-buffered saline (TBS) (pH 7.5) containing a protease/phosphatase cocktail (Roche, Indianapolis, IN, USA) and separated at 22,000 rpm (∼27,000 g) for 20 min at 4°C. The pellet (P1) was homogenized in Tris-sucrose buffer and similarly separated. The supernatant (S2) was incubated in 1% sarkosyl for 60 min at 37°C and was centrifuged at 60,000 rpm (150,000 g) for 30 min at 4°C. The pellet (P3) was resuspended in Tris-EDTA buffer. Protein samples were separated on Bis-Tris 4–12% XT gels (Bio-Rad, Hercules, CA, USA) and probed with the phospho τ antibodies [PHF1, pSer 396/404, CP13, and pSer202, provided by P. Davies (Feinstein Institute for Medical Research, Manhasset, NY, USA)], Tau5 (clone C17, 1:1000; Santa Cruz Biotechnology, Dallas, TX, USA), glial fibrillary acidic protein (GFAP, 1:500; Chemicon, Billerica, MA, USA), STAT1 (1:500; Cell Signaling Technology, Beverly, MA, USA), pSer9GSK3β (1:500; Cell Signaling Technology), GSK3β (1:1000; Cell Signaling Technology), microtubule-associated protein kinase p38 (p38MAPK, 1:500; Cell Signaling Technology), p35/p25 (1:500; Cell Signaling Technology), synaptophysin (1:500; Abcam, San Francisco, CA, USA), postsynaptic density protein 95 (PSD95, 1:500; Abcam), CD68 (1:500; AbD Serotec, Raleigh, NC, USA), glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 1:1000; Abcam), and β-actin (1:1000; Sigma-Aldrich). Protein bands were detected using the multiplex Li-Cor Odyssey Infrared Imaging system (Li-Cor Biosciences, Lincoln, NE, USA) or SuperSignal West Femto reagent (Pierce, Rockford, IL, USA). Relative band intensity was quantified using ImageJ software (NIH, Bethesda, MD).
Primary neuroglial cells were collected 4 d following rAAV transduction, washed in warm saline, and lysed in TBS (pH 7.5) containing a protease/phosphatase cocktail. Supernatants were used as the soluble fraction. Pellets were subsequently extracted in 2% sodium dodecyl sulfate (SDS) solution, and the supernatant was used in experiments as the insoluble fraction.
Cytokine ELISA was performed with the soluble TBS (pH 7.5) lysates or cell culture medium using OptiEIA reagents (Becton Dickinson, Franklin Lakes, NJ, USA). ELISA results were analyzed using SoftMax Pro software (Molecular Devices, Sunnyvale, CA, USA).
Immunohistochemical imaging and image processing
Brains were sagittally dissected, and the right brain was fixed in 4% paraformaldehyde. Whole spinal columns were immersion fixed in 4% paraformaldehyde. Immunohistochemical staining was done on paraffin-embedded brain or spinal sections using phospho τ antibodies (PHF1, CP13, Alz50, and amino acids 2–10 and 312–342, 1:500; provided by P. Davies), ionized calcium binding adapter protein 1 (Iba-1, 1:1000; Abcam), GFAP (1:500), and ubiquitin (1:2000; Dako, Carpenteria, CA, USA). Color development was done using Vector ImmPress reagents (Vector Laboratories, Burlingame, CA, USA). Silver staining was performed using a modified form of the Campbell-Switzer method. Briefly, sections were incubated in a silver/pyridine/carbonate solution containing 1% silver nitrate and then color was developed using the procedure of Gallyas (25).
For primary neuroglial cultures grown in chamber slides, formaldehyde-fixed cells were stained with anti-τ antibodies (1:500; see above) and microtubule-associated protein 2 (MAP2, 1:2000; Sigma-Aldrich) with DAPI used for nuclear staining. Secondary Alexa Fluor-labeled antibodies were obtained from Invitrogen (Thermo Fisher Scientific).
Immunohistochemically and fluorescent-stained sections were captured using the ScanScope XT or FL image scanner (Aperio, Leica Microsystems, Buffalo Grove, IL, USA) and analyzed using the ImageScope program (Aperio). Brightness and contrast alterations were applied identically on captured images using Photoshop CS3 (Adobe Systems, San Jose, CA, USA).
Statistical analysis
Unpaired 2-tailed Student’s t test or 1-way ANOVA was used for statistical comparison. Graphic analyses were done using Prism 4 (GraphPad Software, La Jolla, CA, USA), and final images were created using Photoshop CS2 (Adobe Systems).
RESULTS
Activation of innate immune pathways in asymptomatic and end-stage JNPL3 mice
To demonstrate the extent of astroglial activation in the brain and spinal cords of the homozygous P301L mouse model of tauopathy (JNPL3), we performed Iba-1 (microglia-specific) and GFAP (astrocyte-specific) immunohistochemistry. Compared with nontransgenic mice, both asymptomatic and symptomatic (hindlimb-paralyzed) female JNPL3 mice showed extensive astroglial activation in the spinal cord and brainstem (Fig. 1A). Astroglial immunoreactivity in the hippocampal formation in the forebrain did not show any obvious changes between the nontransgenic and JNPL3 mice (Fig. 1A). To further characterize the alterations in inflammatory transcriptome underlying such vivid astroglial immunoreactivity in JNPL3 mice, we used the NanoString nCounter inflammation gene array (24). The NanoString array has several advantages over PCR-based techniques, including no RNA amplification steps, direct sequence interrogation, and digital detection for absolute quantification. Using a combination of a capture probe and a fluorescent-labeled reporter probe that hybridize with specific nucleotide sequences of select genes, NanoString arrays directly measure gene expression in a multiplexed format. The hybridized probe-target complexes are then immobilized on a plastic cartridge. A charge-coupled device camera equipped with a microscope objective lens detects and counts these hybridized complexes based on a unique digital bar code comprised of a specific arrangement of fluorescent tags on the reporter probe corresponding to a particular gene of interest. Gene analysis results show that the detection limit for such reporter-probe complexes is as low as 0.1 fM with a linear dynamic range of >500-fold (24). Using the nCounter mouse inflammation array with 179 inflammation-related genes, we found that the brains and spinal cords of age-matched female nontransgenic mice and end-stage or asymptomatic homozygous JNPL3 mice have a very distinctive transcriptomic signature (Fig. 1B). There was a massive increase in expression of inflammatory chemokines related to chemotaxis (Ccl3, Ccl4, Ccl5, Cxcr4, and Cxcl10) in the spinal cords and brains of age-matched asymptomatic and end-stage JNPL3 mice. There was evidence of further activation of the innate immune system—expression of several TLRs (1, 2, 4, 6, and 7 in the spinal cord), complement system components (classic, alternative, and lectin pathways in the brain and spinal cord), and the transcription factor Stat1 (in the brain) were increased (Fig. 1B). Surprisingly, Hsbp1 transcription was up-regulated in the spinal cords of both asymptomatic and symptomatic JNPL3 mice, suggesting inhibition of the heat shock pathway. A subset of RNAs was also down-regulated or showed a lowering trend in transgenic JNPL3 mice compared with age-matched nontransgenic mice (Fig. 1B). These include major histocompatibility antigens (H2Ea and H2Eb), select basal/homeostatic chemokines (Cxcl5 and Ccl21b in the spinal cord and Ccl19 and Ccl21b in the brain), transcription factors (Myl2, Creb1, and Fos), and complement inhibitors (Cfd in the spinal cord).
Figure 1.

Inflammatory activation in asymptomatic and end-stage homozygous JNPL3 mice. A) Representative immunohistochemical images depict microglial (Iba-1) and astrocytic activation (GFAP) from cervical spinal cords and brains (hippocampus and brainstem) of 8-mo-old nontransgenic, asymptomatic, and symptomatic female homozygous JNPL3 mice (n = 3 females per group). B) Representative transcripts are significantly altered in the forebrains and whole spinal cords of 8-mo-old asymptomatic and end-stage (symptomatic) female JNPL3 mice. Data have been normalized to nontransgenic mice, which are depicted as the dotted line (n = 3 females per group). Multiple Student’s t test with a false discovery rate of 5% was performed. *P < 0.05; **P < 0.01.
IFN-γ expression does not alter τ expression or τ phosphorylation in WT primary neuroglial cultures
To explore how inflammatory signaling alters endogenous τ expression and phosphorylation, we transduced WT mouse primary neuroglial cultures with rAAV2/1–IFN-γ (Fig. 2). We used the empty rAAV2 vector packaged in serotype 1 as the control for these experiments (rAAV2/1-CTR0). rAAV2/1–IFN-γ transduction leads to increased levels of IFN-γ protein in the medium (6.04 ± 0.27 ng/ml) and increased intracellular Stat1 transcription factor compared with CTR0-transduced cultures following 4 d of infection (Fig. 2A–C). Immunohistochemistry with PHF1 antibody showed that IFN-γ expression does not significantly alter τ phosphorylation in these mixed cultures (Fig. 2D). Immunoblotting results confirm that following IFN-γ expression does not affect endogenous τ expression or PHF1 phospho τ levels in the soluble fraction (Tau5 and PHF1, Fig. 2E, F). In the insoluble fraction, we did not observe any PHF1-τ, whereas total τ levels remained in the IFN-γ-transduced cultures compared with controls (Fig. 2E, F).
Figure 2.

IFN-γ expression does not affect total τ or phospho τ levels in primary neuroglial cultures. A) WT mouse primary neuroglial cultures were transduced with rAAV2/1–IFN-γ or rAAV2/1-CTR0 (control) for 4 d, and medium was analyzed for IFN-γ protein using ELISA. B and C) WT primary neuroglia transduced with rAAV2/1–IFN-γ were lysed sequentially in TBS (pH 7.5) and 2% SDS. Cell lysates were analyzed for STAT1 and phosphorylated GSK3β (Ser9) protein by immunoblotting. Molecular mass markers are indicated on the left (kilodaltons). Data are normalized to an actin band that depicts the loading amount. D) Representative image depicts MAP2-stained neurons costained with antibodies against phospho τ (PHF1) antibody. DAPI represents nuclei. Magnification, ×200. E and F) Immunoblotting shows no change in total τ and PHF1 phospho τ in sequentially extracted soluble and insoluble cellular fractions of WT mouse neuroglial cultures transduced with rAAV2/1–IFN-γ or rAAV2/1-CTR0 (control). GAPDH depicts protein loading. Overexposing the PHF1/GAPDH-probed immunoblot containing insoluble cell lysates produced a faint unidentified band (E, asterisk). Image quantitation was done by normalizing the soluble τ levels to GAPDH, whereas normalization for insoluble τ levels was done to the wet weight of the starting material (F). Results are representative of 2 independent experiments. Data represent means ± sd with unpaired 2-tailed Student’s t test. **P < 0.01.
IFN-γ expression leads to increased phosphorylation of soluble τ in rTg4510 mice
We next investigated how expression affects the disease onset and severity in the rTg4510 mouse model of tauopathy (22). The rTg4510 mice, expressing tauopathy-associated human P301 mutant τ, exhibit age-progressive NFT formation, with pretangles first appearing in the neocortex at 3 mo of age and spreading to the hippocampus and limbic structures by 8.5 mo. We hypothesized that if IFN-γ signaling triggers tangle pathology, 3-mo-old rTg4510 mice expressing IFN-γ in the brain should have increased hyperphosphorylated insoluble τ and tangle pathology. For these experiments, we used rAAV2/1-EGFP mice as our control cohort, which by itself does not alter the inflammatory milieu (20). Analysis of these mouse brains showed that IFN-γ expression resulted in increased levels of IFN-γ protein, Stat1, GFAP, and CD68 (microglial activation marker) (Fig. 3A, B). Widespread glial and astrocytic activation was also indicated by the presence of morphologically activated Iba-1–immunoreactive microglia and GFAP-immunoreactive astrocytes in different areas of the brain of IFN-γ–expressing mice (Fig. 3C). Immunohistochemical staining with the phosphorylation-specific antibody PHF1 revealed increased levels of phospho τ in the cortex and hippocampus (forebrain; Fig. 4D, inset) of IFN-γ–expressing mice, which corresponded to increased gliosis in these brain areas (Fig. 4A). Thalamic neurons in the midbrain region of control mice did not show any PHF-1 immunoreactivity, whereas in the IFN-γ–expressing mice, few thalamic neurons were PHF1 immunoreactive (Fig. 4A, arrows). Another phospho τ–specific antibody, CP13, produced the same pattern of increased τ phosphorylation in IFN-γ–expressing mice, especially in the hippocampus (Fig. 4B, D). No changes in PHF1 or CP13 immunoreactivity were seen in the midbrain or pons region in IFN-γ–expressing mice compared with controls (Fig. 4D). A conformational epitope-specific antibody, Alz50, showed that IFN-γ–expressing mice had scattered Alz50-immunopositive neurons in the cortex and hippocampus, but not in the thalamus or pons (Fig. 4C). On the whole, Alz50 immunoreactivity did not increase significantly in the brains of IFN-γ–expressing mice, though a trend toward increased phosphorylation was noted in the cortex and hippocampus (forebrain) (Fig. 4C, D). To explore how IFN-γ–induced neuroinflammation impacts the relative abundance of τ species at different stages of self-assembly in these mice, we fractionated the brains into different fractions representing soluble τ (S1, S2, and S3) and insoluble τ (P3) (22) (Fig. 5A). Supernatant fractions of brain homogenates obtained in the first centrifugation steps (S1, S2, and S3) reflect τ assemblies at earlier stages of aggregation, whereas the P3 fraction contains enriched filamentous τ aggregates and is correlated to pathologic features of tauopathy, the NFTs. Biochemical analysis of sequentially extracted brains was done using 2 selected phosphorylated epitopes (CP13 and PHF1); albeit, these antibodies have been shown to be fairly representative and specific for pathologic τ by multiple groups (27). Analysis of sequentially extracted rTg4510 brain for the presence of PHF1 and CP13 phospho-epitopes showed that IFN-γ expression resulted in significantly increased levels of PHF1 phospho τ in the S1 fraction (Fig. 5B, C). An interesting observation was that total τ levels were lowered significantly in the S1 fraction of these mice, indicating differential expression of human τ, degeneration of τ-containing neurons, or more likely, that soluble S1 τ is being shifted into other fractions. In the S2 fraction, both of these phospho-epitopes showed a trend toward increased τ phosphorylation in IFN-γ–expressing mice compared with EGFP-expressing controls (Fig. 5B, C). There were no alterations in PHF1- or CP13-immunoreactive τ epitopes or total τ levels in the S3 fraction. In the sarkosyl-insoluble P3 fraction, the 64 kDa and ∼130 kDa (dimer) phosphorylated species were not selectively enriched in any of the cohorts (Fig. 5B). In order to compare the degree of epitope-specific phosphorylation, we also normalized the levels of each phospho τ epitope to the total τ levels in each cellular fraction (Fig. 5D). In the IFN-γ–expressing mice, the PHF1 epitope is significantly increased in both S1 and P3 fractions, whereas the CP13 epitope is enriched in the S1 fraction only when normalized to total τ levels (Fig. 5D).
Figure 3.

rAAV2/1–IFN-γ expression leads to immune activation in rTg4510 mice. A) TBS (pH 7.5)-extracted rTg4510 brain lysates were analyzed for IFN-γ protein by sandwich ELISA (n = 4 per group). Molecular mass markers are indicated on the left (kilodaltons). **P < 0.01, unpaired 2-tailed Student’s t test. B) TBS (pH 7.5)-extracted rTg4510 brain lysates were analyzed for STAT1, GFAP, and CD68. Data are normalized to an actin band that depicts the loading amount. **P < 0.01 and ***P < 0.001, unpaired 2-tailed Student’s t test. Data represent means ± sd. C) Representative fluorescent images of Iba-1 (green fluorophore) and GFAP (red fluorophore) immunoreactivity in different areas of the brain (cortex, hippocampus, and thalamus) of rAAV2/1–IFN-γ and rAAV2/1-EGFP (control) depict elevated gliosis in IFN-γ–expressing mice (n = 4–5 mice per group). DAPI represents nuclear staining. Magnification, ×400.
Figure 4.
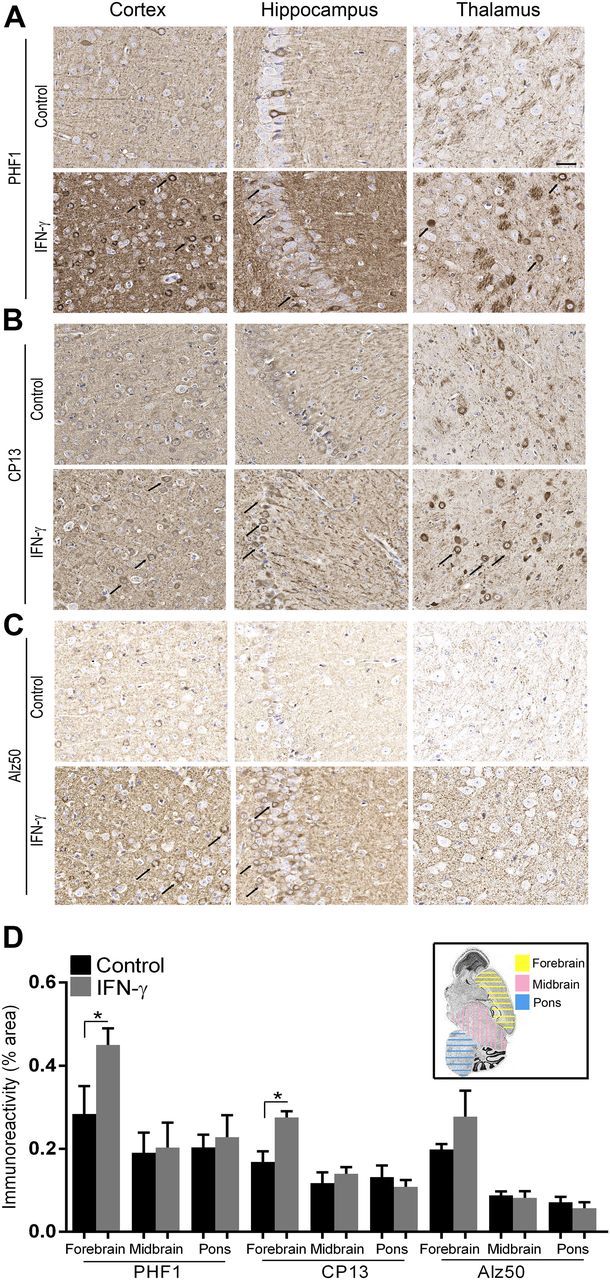
IFN-γ expression increases τ phosphorylation in rTg4510 mice. Representative images depict increased levels of phospho τ epitopes [PHF1 (A), CP13 (B), and Alz50 (C)] in IFN-γ–injected rTg4510 compared with controls (arrows in cortex, hippocampus, and thalamus). Phospho τ immunoreactivity (mean ± sem) in areas of interest [depicted in inset in (D)] has been quantified using the Positive Pixel Count program (Aperio) (n = 4 mice per cohort.). Scale bar, 30 μm.
Figure 5.

IFN-γ expression increases phosphorylation of soluble τ in rTg4510 mice. A) Schematic depiction describes biochemical extraction of τ from mouse brain. B–D) Tau levels were evaluated using phospho τ (PHF1 and CP13) and total τ antibodies from sequentially extracted brain lysates (A) of 3-mo-old rTg4510 mice expressing IFN-γ or EGFP (control) (B). Molecular mass markers are indicated on the left (kilodaltons). Asterisks denote presumed dimer and higher oligomeric τ. ND, not done; p-tau, phosphorylated τ. Data (means ± sd) from soluble fractions (S1–S3) are normalized to an actin band that depicts the loading amount (C). C) P3 data are depicted following normalization to the dry weight of the hemibrain. D) Analysis of the phospho τ relative to total τ levels is depicted (n = 4 mice per group). *P < 0.05 and **P < 0.01, unpaired 2-tailed Student’s t test.
Abnormal intracellular proteostasis in AD and other dementias is characterized by aggregation of misfolded and hyperphosphorylated τ protein into tangle-like structures in the somatodendritic compartment (1). Accumulation of such intracellular NFTs is an inherent feature of the neurodegenerative process in these dementias. To determine whether IFN-γ-driven τ phosphorylation promotes NFT-type pathology, we immunostained for ubiquitin and directly evaluated NFT pathology by silver staining (Supplemental Figs. 1A and 2A). Ubiquitin and silver staining showed that 3-mo-old mice have few pretangle structures in the hippocampus and cortex (Supplemental Figs. 1A and 2A). Our observations indicate that IFN-γ expression did not result in any obvious alterations in ubiquitin-immunopositive or silver-impregnated pretangle structures, consistent with unchanged biochemical τ burden in the P3 fraction (Supplemental Figs. 1A and 2A).
IFN-γ expression increases phosphorylation of soluble τ in P301L JNPL3 mice
We next investigated whether IFN-γ expression affected disease progression in a second mouse model of tauopathy, the hemizygous JNPL3 mice (23). This mouse model also expresses the human P301L τ gene but under the prion promoter and has a comparatively protracted pathology progression compared with the rTg4510 model. In this model, τ tangles are commonly seen at ∼9 mo of age in the amygdala, midbrain, and spinal cord, with hindlimb paresis phenotype most consistently noticed in females. Heterozygous JNPL3 mice were injected as neonates, and female transgenics were analyzed at 12 mo of age. Mice were injected similarly with rAAV2/1-EGFP as a control cohort. IFN-γ expression resulted in increased expression of IFN-γ protein, Stat1, GFAP, and CD68, indicating activation of the IFN-γ signaling pathway and widespread astrogliosis (Fig. 6A, B). Widespread GFAP immunoreactivity was noted in all the brain regions examined, whereas Iba-1 immunoreactivity was more prominent in the cortex (Fig. 6C). We found that IFN-γ–expressing mice showed increased parenchymal immunoreactivity of the PHF1 τ epitope in the cortex, hippocampus (forebrain), and pons, though PHF1 levels were not altered in the cell bodies (Fig. 7A, D). On the other hand, CP13 immunostaining revealed robustly increased intraneuronal phospho τ levels in forebrain and pons of IFN-γ–expressing mice, corresponding to increased gliosis patterns (Fig. 7B, D). Intraneuronal staining with Alz50 was increased in few neurons in the pons area of IFN-γ–expressing mice, whereas other brain areas showed minimal Alz50 immunopositivity (Fig. 7C, D). CNS expression of IFN-γ did not affect gliosis or CP13-τ immunoreactivity in the white matter or gray matter of spinal cords of these mice (Supplemental Fig. 3).
Figure 6.

IFN-γ expression induces immune activation in JNPL3 mice. A) TBS (pH 7.5)-extracted JNPL3 brain lysates were analyzed for IFN-γ protein by sandwich ELISA (n = 4 per group). Molecular mass markers are indicated on the left (kilodaltons). B) TBS (pH 7.5)-extracted JNPL3 brain lysates were analyzed for STAT1, GFAP, and CD68. Data are normalized to an actin band that depicts the loading amount. Data represent means ± sd with unpaired 2-tailed Student’s t test. *P < 0.05; **P < 0.01. C) Representative images of Iba-1 (green fluorophore) and GFAP (red fluorophore) immunoreactivity in different areas of the brain (cortex, hippocampus, and pons) of rAAV2/1–IFN-γ and rAAV2/1-EGFP (control) depict elevated gliosis in IFN-γ–expressing mice. DAPI represents nuclear staining (n = 4–5 mice per group). Magnification, ×400.
Figure 7.
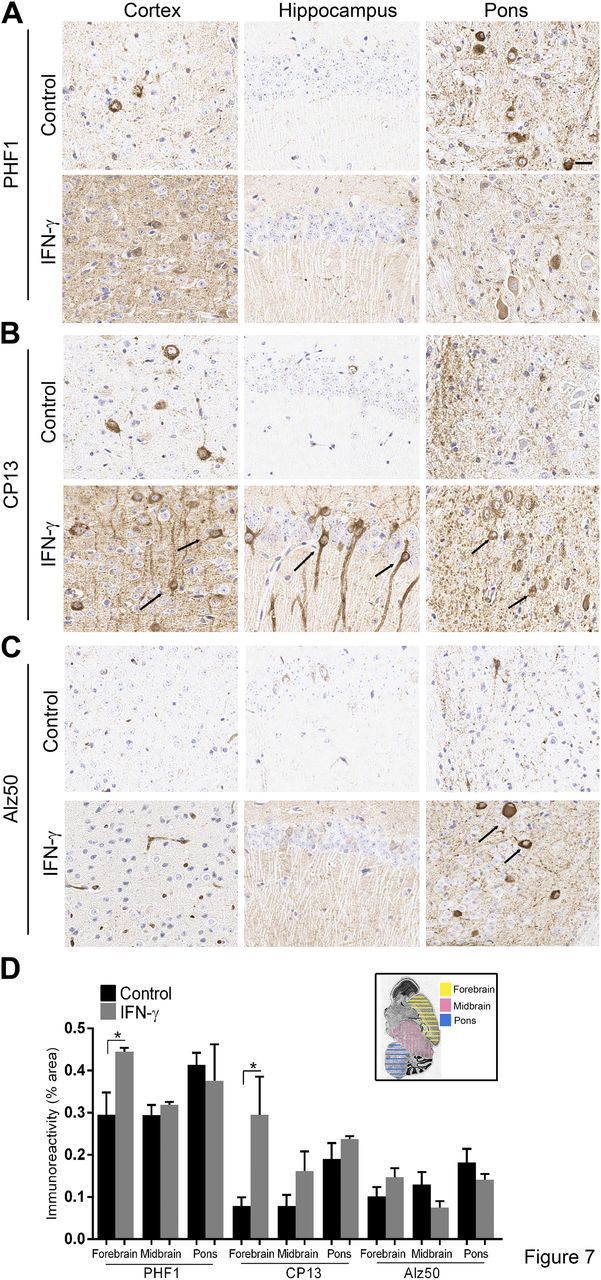
IFN-γ expression increases τ phosphorylation in JNPL3 mice. Representative images depict increased levels of phospho τ epitopes [PHF1 (A), CP13 (B), and Alz50 (C)] in IFN-γ–expressing JNPL3 compared with controls (arrows in cortex, hippocampus, and thalamus). Phospho τ immunoreactivity (mean ± sem) in areas of interest [depicted in inset in (D)] has been quantified using the Positive Pixel Count program (n = 4 mice per cohort). *P < 0.05, unpaired 2-tailed Student’s t test.
Biochemical analysis of sequentially extracted brains demonstrated that there was a robust increase in soluble hyperphosphorylated τ species in the S1 fraction of IFN-γ–expressing mice compared with controls (Fig. 8). Specifically, PHF1 τ was most robustly increased in the S1 fraction of IFN-γ–expressing animals, whereas the CP13 epitope showed a trend toward increased phosphorylation in the S1, S2, and S3 fractions. However, no change in either PHF1 or CP13 phospho-epitopes was noted in the sarkosyl-insoluble P3 fraction in the IFN-γ–expressing mice compared with control mice (Fig. 8A, B). No change in total τ was observed in any of these cellular fractions (Fig. 8A, B). Analysis of the ratio of phospho τ epitopes and total τ showed increased PHF1 levels in the S1 fraction of IFN-γ–expressing mice compared with control mice (Fig. 8C).
Figure 8.

IFN-γ expression increases phosphorylation of soluble τ in JNPL3 mice. Tau levels were evaluated using phospho τ (PHF1 and CP13) and total τ antibodies from sequentially extracted brain lysates of 12-mo-old JNPL3 mice expressing IFN-γ or EGFP (control) (A). Asterisks denote presumed dimer or higher oligomeric τ. ND, not done; p-tau, phosphorylated τ. Data from soluble fractions (S1–S3) are presented following normalization to an actin band that depicts the loading amount (B). B) P3 data are depicted following normalization to the dry weight of the hemibrain. C) Analysis of the phospho τ relative to total τ levels is depicted (n = 4 female mice per group). Data represent means ± sd with unpaired 2-tailed Student’s t test. **P < 0.01; ****P < 0.001.
Given that we did not observe any changes in sarkosyl-insoluble τ levels, we wanted to further determine whether increased τ phosphorylation in IFN-γ mice leads to increased NFTs. We immunostained for ubiquitin and performed Gallyas silver staining (Supplemental Figs. 1B and 2B). Immunostaining revealed that there were very occasional ubiquitinated structures in the hippocampus and the pons of IFN-γ mice compared with controls (Supplemental Fig. 1B). On the whole, ubiquitin immunoreactivity was unaltered between the IFN-γ–expressing and control cohorts (Supplemental Fig. 1B). Similar results were obtained with silver staining, indicating that IFN-γ–induced inflammatory signaling does not drive NFT formation (Supplemental Fig. 2B).
Effect of IFN-γ expression on synaptic proteins
Because soluble τ species have been shown to be neurotoxic (28), we investigated whether IFN-γ–induced elevation in soluble phospho τ affects synaptic function. Immunoblotting for synaptophysin, a major vesicle-associated synaptic protein, showed slightly lower levels in IFN-γ–expressing rTg4510 or JNPL3 mice compared with controls, suggesting subtle synaptic abnormalities (Fig. 9A). Similarly, there is a lowering trend in the levels of PSD95, a membrane-associated synaptic protein, in IFN-γ–expressing rTg4510 or JNPL3 mice compared with control mice (Fig. 9B).
Figure 9.

IFN-γ expression modestly reduces synaptophysin and PSD95 protein levels. Synaptophysin (A) and PSD95 (B) levels in 3-mo-old rTg4510 mice and 12-mo-old JNPL3 are presented. All blots were reprobed with actin antibody to depict the loading amount. Intensity analysis (mean ± sem) of immunoreactive bands of interest was normalized to actin (n = 4 mice per group).
IFN-γ increases GSK3β activation without affecting p38MAPK or cell division cycle kinase 5 levels
Dysfunction in cellular kinase activities results in an imbalance in τ phosphorylation and dephosphorylation leading to NFT pathology. Overactivity of cellular kinases, such as GSK3β, p38MAPK, and cell division cycle kinase 5 (cdk5), increases τ inclusion formation (29). In particular, GSK3β phosphorylation accounts for at least a third of the pathologic phosphorylation events and colocalizes with NFTs in AD. Phosphorylation at Ser9 of GSK3β decreases its active site availability and thus inhibits its kinase activity (30). We found that IFN-γ expression decreases phospho-Ser9 levels in both rTg4510 and JNPL3 mice without significantly affecting total GSK3β levels, indicating that IFN-γ–induced τ hyperphosphorylation is mediated by the release of the inhibitory effect of GSK3β activity (Fig. 10). In primary neuroglial cultures, however, we failed to notice any changes in phosphorylation levels of GSK3β in response to IFN-γ (Fig. 2B, C). p38MAPK is involved in abnormal τ phosphorylation and is increased in AD brains (29). Immunoblotting revealed no major changes in p38MAPK levels in either rTg4510 or JNPL3 cohorts (Fig. 10A, B). Cleavage of cdk5, another major τ kinase, into p35 and p25 fragments is necessary for formation of the potent cdk5/p25 hyperactive complex and is correlated with neurodegenerative tauopathies (29). In the rTg4510 and JNPL3 mice, we did not observe any significant differences in the absolute levels of either p35 and p25 proteins or p25:p35 ratio following IFN-γ expression, indicating that IFN-γ does not affect this pathway (Fig. 10A, B).
Figure 10.
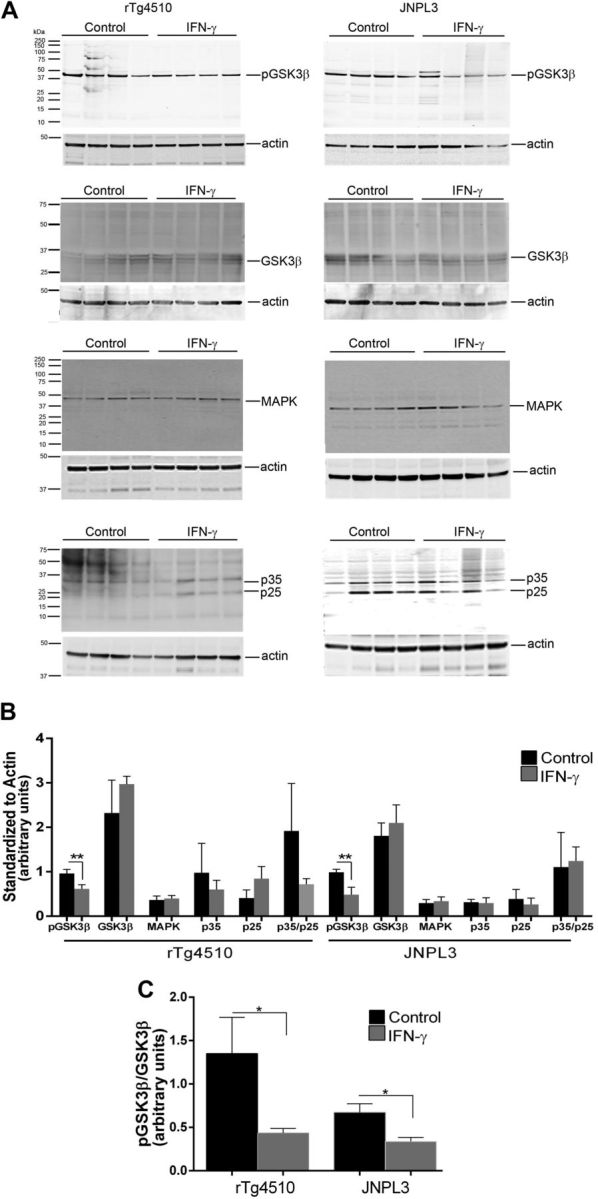
GSK3β activation mediates IFN-γ–induced τ phosphorylation in vivo. A) Representative immunoblot shows levels of pSer9GSK3β, total GSK3β, p38MAPK, and p35/p25 in IFN-γ–expressing 3-mo-old rTg4510 and 12-mo-old JNPL3 mice. Molecular mass markers are indicated on the left (kilodaltons). B) Data (means ± sem) are normalized to an actin band that depicts the loading amount. C) pSer9GSK3β signal has been normalized to the averaged signal of total GSK3β protein (n = 4 mice per group). **P < 0.05, unpaired 2-tailed Student’s t test.
DISCUSSION
Emerging data suggest that dysregulated inflammatory signaling precedes and accompanies neurodegenerative proteinopathy in AD-type dementias (2). Indeed, we show here that compared with nontransgenic mice, asymptomatic and end-stage symptomatic τ mice have elevated levels of immune gene expression, clearly implying that accumulating τ pathology is accompanied by inflammatory changes, preceding the appearance of the end-stage paralysis phenotype. This is consistent with epidemiologic and postmortem data showing that microglial activation closely mirrors the development of the τ lesions observed in human tauopathies and in rodent models of tauopathies (26, 31, 32). However, whether immune signaling is a trigger for maturation of intracellular τ to frank NFTs remains a contentious issue. Here, we demonstrate that expression of IFN-γ, a master regulator of inflammatory activation, causes increased τ hyperphosphorylation at AD-relevant epitopes (PHF1 and CP13) in 2 mouse models of tauopathy. Our data suggest that such an increase in phosphorylated pathologic τ epitopes is mediated via activation of GSK3β, though other kinases may have an additive or synergistic role. The hyperphosphorylated τ was localized biochemically to soluble cellular fractions, and insoluble NFT τ levels remained unchanged. Our data show that IFN-γ–mediated immune signaling increases τ phosphorylation in multiple models of tauopathies but does not lead to increased NFTs.
Given the complexity of the neuroglial architecture and variability in the context-specific and type of inflammatory signaling, the net effect of immune activation on tangle formation and neurodegeneration can often display a double-edged sword phenotype (2). Collective data from various independent groups have consistently demonstrated that neuroinflammation increases disease-associated phosphorylation of τ epitopes. Intraperitoneal injection of bacterial LPS or direct hepatitis virus infection in the 3xTg mouse model resulted in exacerbated tauopathy by activating GSK3β signaling cascade (12, 14). Notably, intraperitoneal LPS administration in WT mice also promotes endogenous τ hyperphosphorylation, which is abrogated in TLR4 or IL-1 receptor 1 knockout mice (33). Antibody-mediated blockage of IL-1β signaling also attenuates τ pathology in the 3xTg mice by reducing τ kinase activity (34). In another study, using this same mouse model, hippocampal overexpression of rAAV–IL-1β resulted in GSK3β and p38MAPK-induced τ hyperphosphorylation (11). Direct intracranial injection of LPS into rTg4510 brains also resulted in increased soluble hyperphosphorylated τ and closely recapitulates our own observations (13). In some of these studies, inflammatory signaling pathways resulted in elevated levels of insoluble NFTs (14, 33), though other studies did not find any changes in sarkosyl-insoluble phospho τ levels (11–13). In our study, we found that despite robust accumulation of PHF1- and CP13-immunopositive τ-bearing cell bodies and increased biochemical levels of soluble phospho τ, sarkosyl-insoluble τ or argyrophilic τ deposits were not induced. Additionally, we also did not observe any behavioral phenotype in hemizygous JNPL3 mice, suggesting that IFN-γ does not promote pathologic τ-associated neurologic deficits (hindlimb weakness or paralysis). Taken together, these observations suggest that type II IFN signaling can enhance τ hyperphosphorylation but has limited impact on disease onset, τ tangle formation, or neurologic deficits.
In addition to its effect on NFTs, innate immune activation also modulates Aβ deposition, another cardinal feature of AD [reviewed in Heppner et al. (2)]. Genome-wide association study and accompanying biochemical data implicate many inflammation-related genes in modulating Aβ pathology in patients with late-onset AD (35). Although initially it was hypothesized that inflammatory signaling may trigger and subsequently exacerbate Aβ deposition (36) and thereby potentially lead to τ dysfunction (37), the current data are much less irreconcilable with a straightforward hypothesis. Some preclinical studies suggest that inflammatory signaling directly contributes to Aβ pathogenesis (38, 39), whereas other studies suggest that innate immune activation may be beneficial (40, 41).
Much of the available data, along with ours, are harder to reconcile with the observations of Mastrangelo et al. (42), who showed that hippocampal expression of adeno-associated virus (AAV) IFN-γ in adult 3xTg mice results in an unexpected reduction in phospho τ burden. The discrepancy between these observations and ours may be explained by the type of τ transgenic model used and the strength and spatiotemporal distribution of IFN-γ signaling. Whereas we have utilized mouse models of pure tauopathy, this study used a much more complex transgenic model that overexpresses mutant human APP, mutant human τ, and presenilin (43). Given that IFN-γ signaling modulates Aβ plaques in APP mice models (20) and inflammatory signaling may further affect astroglial fate via presenilins (44), it is unclear how these changes would finally alter τ pathology in the presence of mutant APP and presenilin overexpression. In addition, IFN-γ signaling resulted in both elevated astrocytosis and microgliosis in all of the brain areas in our study, whereas astrocyte activation was minimally observed in the study by Mastrangelo et al. (42), which may suggest that this hippocampal paradigm could not recapitulate the full inflammatory spectrum. Such divergent data underscore the need for more rigorous and thorough testing of pathway-based manipulations in τ transgenic models.
Phosphorylation of τ can be mediated by various kinases; the main cellular τ kinases are GSK3β, p38MAPK, and cdk5 (29). In previous studies on IL-1β–induced τ phosphorylation, both GSK3β and p38MAPK were shown to be involved (11, 45). On the other hand, LPS administration exacerbated τ pathology by cdk5/p25 activation (12). We did not observe any significant changes in p38MAPK or the p35:p25 ratio in these mice, though the role of other τ kinases (e.g., casein kinase, Erk1/2, and Jnk) cannot be ruled out. We observed that GSK3β from IFN-γ–expressing τ mice had decreased phospho-Ser9 epitope immunoreactivity, suggesting that IFN-γ signaling increases GSK3β activity. Previous studies have shown that GSK3β overexpression alone is accompanied by gliosis and τ hyperphosphorylation (46). In addition, IFN-γ can directly activate GSK3β in macrophages and RAW264.7 cells (47). Further work needs to be done to clarify the exact mode of action of IFN-γ on GSK3β-induced τ phosphorylation in the brain and its relevance to disease progression.
In conclusion, we show that type II IFN signaling increases soluble phospho τ but does not necessarily trigger sarkosyl-insoluble τ. Future experiments will focus on how inflammatory pathways affect disease progression in adult mice with existing τ pathology. Understanding how innate immune activation states regulate τ pathology and τ-induced neurodegeneration could reveal optimal therapeutic approaches for tauopathies, including AD.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
Acknowledgments
This work was supported by funding from the Alzheimer's Association (to P.C.) and BrightFocus Foundation (to P.C.). Tau antibodies (PHF1, CP13, and Alz50) were a kind gift from Peter Davies (Feinstein Institute for Medical Research, Manhasset, NY, USA). Homozygous P301L τ transgenics were obtained from Karen Duff (Columbia University, New York, NY, USA). J.L. is an inventor and intellectual property holder for the JNPL3 and rTg4510 mice, who receives royalties for these inventions. P.C. conceived the idea, planned the experiments, and wrote the manuscript. A.L. performed immunoblotting and enzyme-linked immunosorbent assays. C.C.-D. prepared viruses, primary neuronal culture experiments, immunoblotting, and NanoString analysis. N.D. performed immunohistochemistry. Y.L. and P.E.C. assisted in mouse experiments. J.L. provided transgenic mice and critical input. T.E.G. provided overall support. The authors declare no conflicts of interest.
Glossary
- AAV
adeno-associated virus
- Aβ
amyloid β
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- Cdk5
cell division cycle kinase 5
- EGFP
enhanced green fluorescent protein
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GFAP
glial fibrillary acidic protein
- GSK3β
glycogen synthase kinase 3β
- Iba-1
ionized calcium binding adapter protein 1
- MAP2
microtubule-associated protein 2
- MAPT
microtubule-associated protein τ
- NFT
neurofibrillary tangle
- P38MAPK
microtubule-associated protein kinase p38
- PHF
paired helical filament
- phospho τ
phosphorylated τ
- PSD95
postsynaptic density protein 95
- rAAV
recombinant adeno-associated virus
- rAAV2/1
recombinant adeno-associated virus 2 plasmid packaged in serotype 1
- SDS
sodium dodecyl sulfate
- STAT1
signal transducer and activator of transcription 1
- TBS
Tris-buffered saline
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Spillantini M. G., Goedert M. (2013) Tau pathology and neurodegeneration. Lancet Neurol. , 609–622 [DOI] [PubMed] [Google Scholar]
- 2.Heppner F. L., Ransohoff R. M., Becher B. (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. , 358–372 [DOI] [PubMed] [Google Scholar]
- 3.Carter S. F., Schöll M., Almkvist O., Wall A., Engler H., Långström B., Nordberg A. (2012) Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. , 37–46 [DOI] [PubMed] [Google Scholar]
- 4.Santillo A. F., Gambini J. P., Lannfelt L., Långström B., Ulla-Marja L., Kilander L., Engler H. (2011) In vivo imaging of astrocytosis in Alzheimer’s disease: an ¹¹C-L-deuteriodeprenyl and PIB PET study. Eur. J. Nucl. Med. Mol. Imaging , 2202–2208 [DOI] [PubMed] [Google Scholar]
- 5.Komori T. (1999) Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol. , 663–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaworski T., Lechat B., Demedts D., Gielis L., Devijver H., Borghgraef P., Duimel H., Verheyen F., Kügler S., Van Leuven F. (2011) Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am. J. Pathol. , 2001–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanger D. P., Lau D. H., Phillips E. C., Bondulich M. K., Guo T., Woodward B. W., Pooler A. M., Noble W. (2014) Intracellular and extracellular roles for tau in neurodegenerative disease. J. Alzheimers Dis. (Suppl 1), S37–S45 [DOI] [PubMed] [Google Scholar]
- 8.Zilka N., Stozicka Z., Kovac A., Pilipcinec E., Bugos O., Novak M. (2009) Human misfolded truncated tau protein promotes activation of microglia and leukocyte infiltration in the transgenic rat model of tauopathy. J. Neuroimmunol. , 16–25 [DOI] [PubMed] [Google Scholar]
- 9.Sheng J. G., Zhu S. G., Jones R. A., Griffin W. S., Mrak R. E. (2000) Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Exp. Neurol. , 388–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noble W., Garwood C., Stephenson J., Kinsey A. M., Hanger D. P., Anderton B. H. (2009) Minocycline reduces the development of abnormal tau species in models of Alzheimer’s disease. FASEB J. , 739–750 [DOI] [PubMed] [Google Scholar]
- 11.Ghosh S., Wu M. D., Shaftel S. S., Kyrkanides S., LaFerla F. M., Olschowka J. A., O’Banion M. K. (2013) Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. , 5053–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitazawa M., Oddo S., Yamasaki T. R., Green K. N., LaFerla F. M. (2005) Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. , 8843–8853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee D. C., Rizer J., Selenica M. L., Reid P., Kraft C., Johnson A., Blair L., Gordon M. N., Dickey C. A., Morgan D. (2010) LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J. Neuroinflammation , 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sy M., Kitazawa M., Medeiros R., Whitman L., Cheng D., Lane T. E., Laferla F. M. (2011) Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am. J. Pathol. , 2811–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schroder K., Hertzog P. J., Ravasi T., Hume D. A. (2004) Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. , 163–189 [DOI] [PubMed] [Google Scholar]
- 16.Colangelo V., Schurr J., Ball M. J., Pelaez R. P., Bazan N. G., Lukiw W. J. (2002) Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J. Neurosci. Res. , 462–473 [DOI] [PubMed] [Google Scholar]
- 17.Ricciarelli R., d’Abramo C., Massone S., Marinari U., Pronzato M., Tabaton M. (2004) Microarray analysis in Alzheimer’s disease and normal aging. IUBMB Life , 349–354 [DOI] [PubMed] [Google Scholar]
- 18.Abbas N., Bednar I., Mix E., Marie S., Paterson D., Ljungberg A., Morris C., Winblad B., Nordberg A., Zhu J. (2002) Up-regulation of the inflammatory cytokines IFN-gamma and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APP(SWE) transgenic mice. J. Neuroimmunol. , 50–57 [DOI] [PubMed] [Google Scholar]
- 19.Patel N. S., Paris D., Mathura V., Quadros A. N., Crawford F. C., Mullan M. J. (2005) Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflammation , 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakrabarty P., Ceballos-Diaz C., Beccard A., Janus C., Dickson D., Golde T. E., Das P. (2010b) IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J. Immunol. , 5333–5343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee D. C., Rizer J., Hunt J. B., Selenica M. L., Gordon M. N., Morgan D. (2013) Review: experimental manipulations of microglia in mouse models of Alzheimer’s pathology: activation reduces amyloid but hastens tau pathology. Neuropathol. Appl. Neurobiol. , 69–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E., Forster C., Yue M., Orne J., Janus C., Mariash A., Kuskowski M., Hyman B., Hutton M., Ashe K. H. (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science , 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis J., McGowan E., Rockwood J., Melrose H., Nacharaju P., Van Slegtenhorst M., Gwinn-Hardy K., Paul Murphy M., Baker M., Yu X., Duff K., Hardy J., Corral A., Lin W. L., Yen S. H., Dickson D. W., Davies P., Hutton M. (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. , 402–405 [DOI] [PubMed] [Google Scholar]
- 24.Geiss G. K., Bumgarner R. E., Birditt B., Dahl T., Dowidar N., Dunaway D. L., Fell H. P., Ferree S., George R. D., Grogan T., James J. J., Maysuria M., Mitton J. D., Oliveri P., Osborn J. L., Peng T., Ratcliffe A. L., Webster P. J., Davidson E. H., Hood L., Dimitrov K. (2008) Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. , 317–325 [DOI] [PubMed] [Google Scholar]
- 25.Hicks S. P. (1946) A rapid pyridine silver stain for nervous tissue and reticular fibers. J. Lab. Clin. Med. , 1375–1377 [PubMed] [Google Scholar]
- 26.Sheffield L. G., Marquis J. G., Berman N. E. (2000) Regional distribution of cortical microglia parallels that of neurofibrillary tangles in Alzheimer’s disease. Neurosci. Lett. , 165–168 [DOI] [PubMed] [Google Scholar]
- 27.Petry F. R., Pelletier J., Bretteville A., Morin F., Calon F., Hébert S. S., Whittington R. A., Planel E. (2014) Specificity of anti-tau antibodies when analyzing mice models of Alzheimer’s disease: problems and solutions. PLoS One , e94251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kopeikina K. J., Hyman B. T., Spires-Jones T. L. (2012) Soluble forms of tau are toxic in Alzheimer’s disease. Transl. Neurosci. , 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin L., Latypova X., Wilson C. M., Magnaudeix A., Perrin M. L., Yardin C., Terro F. (2013) Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res. Rev. , 289–309 [DOI] [PubMed] [Google Scholar]
- 30.Ali A., Hoeflich K. P., Woodgett J. R. (2001) Glycogen synthase kinase-3: properties, functions, and regulation. Chem. Rev. , 2527–2540 [DOI] [PubMed] [Google Scholar]
- 31.Sheng J. G., Mrak R. E., Griffin W. S. (1997) Glial-neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J. Neuropathol. Exp. Neurol. , 285–290 [PubMed] [Google Scholar]
- 32.Sasaki A., Kawarabayashi T., Murakami T., Matsubara E., Ikeda M., Hagiwara H., Westaway D., George-Hyslop P. S., Shoji M., Nakazato Y. (2008) Microglial activation in brain lesions with tau deposits: comparison of human tauopathies and tau transgenic mice TgTauP301L. Brain Res. , 159–168 [DOI] [PubMed] [Google Scholar]
- 33.Bhaskar K., Konerth M., Kokiko-Cochran O. N., Cardona A., Ransohoff R. M., Lamb B. T. (2010) Regulation of tau pathology by the microglial fractalkine receptor. Neuron , 19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kitazawa M., Cheng D., Tsukamoto M. R., Koike M. A., Wes P. D., Vasilevko V., Cribbs D. H., LaFerla F. M. (2011) Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. , 6539–6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tosto G., Reitz C. (2013) Genome-wide association studies in Alzheimer’s disease: a review. Curr. Neurol. Neurosci. Rep. , 381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffin W. S., Sheng J. G., Royston M. C., Gentleman S. M., McKenzie J. E., Graham D. I., Roberts G. W., Mrak R. E. (1998) Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. , 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oddo S., Caccamo A., Tran L., Lambert M. P., Glabe C. G., Klein W. L., LaFerla F. M. (2006) Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J. Biol. Chem. , 1599–1604 [DOI] [PubMed] [Google Scholar]
- 38.Heneka M. T., Kummer M. P., Stutz A., Delekate A., Schwartz S., Vieira-Saecker A., Griep A., Axt D., Remus A., Tzeng T. C., Gelpi E., Halle A., Korte M., Latz E., Golenbock D. T. (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature , 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vom Berg J., Prokop S., Miller K. R., Obst J., Kälin R. E., Lopategui-Cabezas I., Wegner A., Mair F., Schipke C. G., Peters O., Winter Y., Becher B., Heppner F. L. (2012) Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat. Med. , 1812–1819 [DOI] [PubMed] [Google Scholar]
- 40.Shaftel S. S., Kyrkanides S., Olschowka J. A., Miller J. N., Johnson R. E., O’Banion M. K. (2007) Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Invest. , 1595–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chakrabarty P., Jansen-West K., Beccard A., Ceballos-Diaz C., Levites Y., Verbeeck C., Zubair A. C., Dickson D., Golde T. E., Das P. (2010) Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. , 548–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mastrangelo M. A., Sudol K. L., Narrow W. C., Bowers W. J. (2009) Interferon-gamma differentially affects Alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. Am. J. Pathol. , 2076–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oddo S., Caccamo A., Shepherd J. D., Murphy M. P., Golde T. E., Kayed R., Metherate R., Mattson M. P., Akbari Y., LaFerla F. M. (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron , 409–421 [DOI] [PubMed] [Google Scholar]
- 44.Lee J., Chan S. L., Mattson M. P. (2002) Adverse effect of a presenilin-1 mutation in microglia results in enhanced nitric oxide and inflammatory cytokine responses to immune challenge in the brain. Neuromolecular Med. , 29–45 [DOI] [PubMed] [Google Scholar]
- 45.Li Y., Liu L., Barger S. W., Griffin W. S. (2003) Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. , 1605–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lucas J. J., Hernández F., Gómez-Ramos P., Morán M. A., Hen R., Avila J. (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. , 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu X., Paik P. K., Chen J., Yarilina A., Kockeritz L., Lu T. T., Woodgett J. R., Ivashkiv L. B. (2006) IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity , 563–574 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.