

Abstract
The health risks of a dysregulated immune response during spaceflight are important to understand as plans emerge for humans to embark on long-term space travel to Mars. In this first-of-its-kind study, we used adoptive transfer of T-cell receptor transgenic OT-II CD4 T cells to track an in vivo antigen-specific immune response that was induced during the course of spaceflight. Experimental mice destined for spaceflight and mice that remained on the ground received transferred OT-II cells and cognate peptide stimulation with ovalbumin (OVA) 323-339 plus the inflammatory adjuvant, monophosphoryl lipid A. Control mice in both flight and ground cohorts received monophosphoryl lipid A alone without additional OVA stimulation. Numbers of OT-II cells in flight mice treated with OVA were significantly increased by 2-fold compared with ground mice treated with OVA, suggesting that tolerance induction was impaired by spaceflight. Production of proinflammatory cytokines were significantly increased in flight compared with ground mice, including a 5-fold increase in IFN-γ and a 10-fold increase in IL-17. This study is the first to show that immune tolerance may be impaired in spaceflight, leading to excessive inflammatory responses.—Chang, T. T., Spurlock, S. M, Candelario, T. L. T., Grenon, S. M., Hughes-Fulford, M. Spaceflight impairs antigen-specific tolerance induction in vivo and increases inflammatory cytokines.
Keywords: inflammation, microgravity, T-cell memory, T-cell tolerance, transgenic model
Immune dysregulation in microgravity, defined as 10−3 to 10−6 of normal gravity near Earth’s surface (1g), is predicted to be a significant challenge to human health during long-term space travel and habitation (1, 2). Evidence indicates that dysregulated CD4 T-cell–mediated immunity is a major mechanism of spaceflight-associated immune dysfunction. The response of T cells isolated from humans postflight to phytohemagglutinin mitogen stimulation was significantly reduced compared with preflight (3). Importantly, delayed-type hypersensitivity reactions mediated by CD4 T cells were significantly reduced in astronauts when antigenic challenge was administered during flight compared with preflight (4). Spaceflight in vitro studies showed that microgravity directly disrupted T-cell activation independently of systemic factors such as the stress response. Cultures of leukocytes isolated from human peripheral blood that were stimulated during spaceflight with the T-cell mitogen concanavalin A (ConA) demonstrated profoundly suppressed T-cell proliferation compared with ground control animals (5). T cells activated with either anti-CD3 alone or anti-CD3 plus anti-CD28 demonstrated significantly decreased surface expression of CD25 (high-affinity IL-2 receptor or IL-2Rα) in microgravity comparison to 1g-centrifuge controls in flight (6). Expression of IL-2, CD25, and numerous cytokines and chemokines were significantly decreased in T cells activated with ConA in ground-based simulated microgravity compared with 1g (7–9). In addition, differential expression analysis of immediate early genes revealed that Rel/NF-κB signal transduction was likely a major pathway affected by microgravity in T-cell activation compared with 1g-centrifuge controls in flight (10). These findings indicated that spaceflight altered CD4 T-cell responses independently of the systemic stress response and that microgravity inhibited T-cell activation by mitogens at the cellular level.
What remained unknown after these studies was what would happen to humans exposed to infectious agents during long-term spaceflight, such as on a voyage to Mars, or if stationed for a prolonged period on an outpost in space. Critical to answering this question was to determine the effect of spaceflight on the response of CD4 T cells to cognate peptide–major histocompatibility complex stimulation in vivo. Although it is possible that human space travelers may encounter novel extraterrestrial microbes, a more likely scenario is that they would continue to contend with hitchhiking Earth-derived microorganisms to which there was immunologic experience and established immune memory. Earth-derived bacteria, fungi, and viruses are brought into space through normal human skin, oral, and gastrointestinal flora. Studies have shown that numerous species of common Earth bacteria and fungi could be cultured from the environment of the Mir and International Space Stations and that the degree of microbial contamination increased with time (11, 12). In addition, latent viral infections have been shown to reactivate in astronauts during spaceflight (13). Memory CD4 T-cell responses to these bacteria, fungi, and viruses may be altered during spaceflight because normal T-cell activation is inhibited by microgravity. An easily treatable infection on Earth may have serious consequences if acquired in space because of the associated immune dysregulation.
In order to test and track a physiologic antigen-specific response in vivo during the course of spaceflight, we used the well-vetted adoptive transfer model of T-cell receptor (TCR) transgenic T cells in mice (14). In our chosen experimental system, small numbers of T cells from CD45.2-expressing OT-II transgenic mice, in which almost all T cells expressed a single TCR specific to ovalbumin (OVA) peptide 323-339 (15), were transferred into congenic CD45.1 C57BL/6 mice with a normal immune repertoire. Memory CD4 T cells have been shown to rapidly generate from effector CD4 T cells after in vitro activation (16, 17). Therefore, we first activated OT-II transgenic T cells with OVA peptide in vitro and then adoptively transferred them into recipients just before launch in order to test the development and maintenance of CD4 T-cell memory during spaceflight. Experimental mice were rechallenged in vivo during spaceflight with OVA peptide in an inflammatory adjuvant to determine the likely outcome of a memory CD4 T-cell response to infection. The OVA-specific memory CD4 T-cell response was specifically tracked using the congenic surface marker CD45.2 expressed on transferred OT-II T cells.
As a result of limitations of the spaceflight hardware and crew time availability, our experiment was designed to be self-contained and internally delivered the dose protocol during the 15 d flight mission on Space Transport System (STS)-131 without requirement of astronaut time. Antigen delivery to recipient mice during spaceflight was achieved through minipumps preloaded with treatment and surgically implanted subcutaneously into mice before flight. The minipumps delivered 2 burst-releases of antigen several days into the flight mission in order to test the immune system after mice have acclimatized to spaceflight and a few days of in vivo quiescence in the absence of antigen. OVA peptide was delivered mixed with the inflammatory adjuvant, monophosphoryl lipid A (MPLA), in order to simulate the context of an infection in vivo. MPLA is a low-toxicity derivative of LPS, a component of the cell membranes of gram-negative bacteria. MPLA has been used successfully as an adjuvant in vaccines instead of LPS because, like LPS, it is a TLR4 agonist (18). The reduced toxicity of MPLA is related to reduced potency at producing inflammation compared with LPS. Because infectious pathogens and toxic materials like LPS were not allowed on flight missions, use of MPLA as an adjuvant was the best choice to test the CD4 T-cell memory response to infection in vivo during spaceflight.
Our experimental design examined 4 groups of mice in order to comprehensively characterize the effect of spaceflight on the antigen-specific CD4 T-cell response in vivo using the internal in situ delivery of adjuvant/antigen: ground mice stimulated with MPLA alone (ground control), ground mice stimulated with MPLA plus OVA peptide (ground OVA), flight mice stimulated with MPLA alone (flight control), and flight mice stimulated with MPLA plus OVA peptide (flight OVA). Unexpectedly, we found that our experimental protocol led to impaired tolerance induction in mice that received OVA challenge during spaceflight. Although not the original intention of the experimental design, these results allowed us to discover a unique effect of spaceflight on in vivo tolerance induction. This study is the first detailed analysis of an antigen-specific immune response upon antigenic challenge in the mouse during the course of spaceflight. The findings are an important advance in our understanding of the potential impact of spaceflight immune dysregulation on human space travelers and provide additional insight on the mechanisms of immune tolerance here on Earth.
MATERIALS AND METHODS
Mice
Female wild-type, CD45.1 congenic (B6.SJL-PtprcaPepcb/BoyJ), and OT-II C57BL/6 mice [B6.Cg-Tg(TcraTcrb)425Cbn/J] were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and transferred directly to the Kennedy Space Center Space Life Sciences Laboratory specific-pathogen-free animal care facility at least 30 d before the flight experiment. Six- to 8-wk-old wild-type C57BL/6 mice were used as sources of antigen-presenting cells and OT-II mice as sources of transgenic OT-II T cells. CD45.1 congenic mice >18 wk old at the time of launch and between 22 and 24 g in weight were used as recipient animals. Three weeks before launch, CD45.1 mice were acclimated to the modified water delivery Lixit that mimicked those they would use during spaceflight and placed on a diet of U.S. National Aeronautics and Space Administration (NASA) Rodent Foodbars in preparation for flight conditions. All animal procedures were performed in accordance with a protocol approved by the NASA Institutional Animal Care and Use Committee.
Flight hardware
Through the duration of the spaceflight experiment, flight and ground mice were housed in animal enclosure modules (AEMs) (19). The AEM is a self-contained enclosed rodent habitat that provided ventilation at a rate of 14 ft3/min, 12 h light/dark lighting cycle, and waste collection through an exhaust filter. Each AEM housed 7–8 mice and conformed to the housing space guidelines in the Guide for the Care and Use of Laboratory Animals. During the experiment, rodents were periodically observed through the clear plastic cover, but there was no access availability to the crew.
Preparation and adoptive transfer of OT-II memory cells
CD4 T cells were purified from the spleens of naive OT-II mice using magnetic Dynabead negative isolation per the manufacturer’s instructions (Life Technologies, Grand Island, NY, USA). Purified OT-II CD4 T cells were combined with mitomycin C (Sigma-Aldrich, St. Louis, MO, USA)-treated splenocytes from wild-type C57BL/6 mice as antigen-presenting cells at a ratio of 1:2.5 to a final cell density of 8 × 106 cell/ml. Cells were cultured in complete medium consisting of RPMI 1640 medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Hyclone), 1% l-glutamine (Mediatech, Manassas, VA, USA), 1% antibiotics/antimycotic (Mediatech), 1% sodium pyruvate (Hyclone), and 1.2% 1 M HEPES (Sigma-Aldrich). To generate activated OT-II effector T cells, cultures were stimulated with 20 μg/ml OVA peptide and 30 U/ml of recombinant mouse IL-2 (R&D Systems, Minneapolis, MN, USA). After 2 or 3 d of culture, medium was exchanged with fresh complete medium supplemented with 10 μg/ml OVA peptide and 15 U/ml IL-2. Cells were collected on d 4 of culture, washed, and resuspended in PBS for adoptive transfer. Five million cells per mouse were adoptively transferred into CD45.1 congenic mice by intravenous tail-vein injection.
Antigen and adjuvant delivery by subcutaneous minipump
Alzet osmotic minipumps (model 2002, 3 cm length, 0.7 cm diameter; Durect, Cupertino, CA, USA) with a reservoir volume of 200 μl and release rate of 0.5 μl/h over 2 wk were modified with a coiled external polyethylene catheter (PE-60; Durect) to deliver 200 μg OVA 323-339 peptide (Anaspec, San Jose, CA, USA) with 30 μg MPLA (InvivoGen, San Diego, CA, USA) or 30 μg MPLA alone in 2 separate timed burst-releases. MPLA was initially dissolved in cyclodextran and then combined 1:1 with OVA peptide dissolved in ddH2O to final concentration. In mice that received MPLA alone, MPLA in cyclodextran was combined 1:1 with ddH2O. Timed burst-release of antigen/adjuvant was achieved by filling the catheter with saline spacers that were separated from the antigen/adjuvant solution by mineral oil to prevent mixing and dilution. The antigen/adjuvant mixture was delivered in 10 μl over 20 h of release time on flight d 6 and 10 so that mice had time to acclimatize to spaceflight and transferred OT-II cells had time to develop into memory CD4 T cells before being restimulated with antigen in vivo (Fig. 1A). The combined volume of the loaded solutions (including saline, mineral oil, and antigen/adjuvant mixture) was 167 μl, which required 36.6 cm of catheter and was released over 334 h (14 d). The minipumps were sterilely prepared by first weighing and filling the pumps with saline per the manufacturer’s instructions. The coiled catheter was separately filled with saline, mineral oil, and the antigen/adjuvant mixture sequentially using gel loading tips. The distal delivery end of the catheter was attached to the flow modulator of the minipump. The pumps were then primed by submersion in a beaker of sterile saline overnight at 37°C with the catheter extended outside of the saline. Immediately before implantation, the filled coiled catheter was pushed over the body of the pump to reduce to overall size of the implant.
Figure 1.

Experimental design. A) Schematic of minipump modified with catheter extension used to deliver antigen in vivo. B) Timeline of experimental procedures. Procedural events for flight cohort are shown above timeline and events for ground cohort below timeline. C) Experimental groups. Four experimental groups and corresponding sample sizes are represented.
For implantation, CD45.1 mice were anesthetized with isoflurane. Using aseptic technique, a small 1 cm transverse incision was made on the upper back of mice, and a subcutaneous pocket just large enough for the pump was developed by dissection. Prepared minipumps were placed into the subcutaneous pocket with the catheter opening toward the neck of the mice. The incision was closed with several interrupted permanent sutures. The mice were then treated with buprenorphine for pain relief and allowed to recover.
Experiment procedural timeline
Mice intended for the flight cohort were implanted with minipumps 3 d before the launch of STS-131 Space Shuttle Discovery on April 5, 2010 (Fig. 1B). Activated OT-II cells were adoptively transferred into these mice 1 d later. Mice were placed into the AEM and integrated into the space shuttle 24 h before launch. Flight AEMs remained in the middeck of the space shuttle for the duration of spaceflight. After a 15 d mission, STS-131 landed back in Kennedy Space Center on April 20, 2010. Mice were received by the science team 2 h after landing, and tissue dissection began immediately. Cell suspensions from cervical lymph nodes and spleens were made and stained immediately for flow cytometry analysis. Splenocytes were also restimulated in vitro for 3 d for additional analysis.
To minimize environmental factors other than spaceflight that may affect the immune response, ground mice were housed in identical AEM flight hardware within an orbital environment simulator that mimicked the temperature, humidity, and carbon dioxide concentration of the space shuttle middeck. To accomplish this simulation and to accommodate for potential changes in the flight plan, synchronous ground cohort mice were generated 48 h after flight mice were launched into space per standard NASA protocol. A separate fresh preparation of memory OT-II T cells was used in the adoptive transfer of ground mice. All postflight analyses, including staining for flow cytometry, were also performed synchronously 48 h later for the ground cohort compared with flight mice. All reagents and protocols were kept identical between flight and ground mice to minimize variability. For flow cytometry, although cells from flight and ground mice were stained on different days, flow cytometer analysis was performed on the same day using the same acquisition settings to minimize variability.
Experimental groups and power analysis
Ground and flight mice were either implanted with minipumps containing MPLA alone or with minipumps containing OVA peptide and MPLA. Therefore, a total of 4 experimental groups were generated: ground mice stimulated with MPLA alone (ground control), ground mice stimulated with MPLA plus OVA peptide (ground OVA), flight mice stimulated with MPLA alone (flight control), and flight mice stimulated with MPLA plus OVA peptide (flight OVA) (Fig. 1C). The sample size of each group was 4 in order to detect a 30% difference between groups with a 20% sd, α of 0.05, and power of 0.85. Just before integration with the space shuttle, one mouse in the flight control group was disqualified for flight because of an incompletely healed wound at the minipump implantation site. The sample size for the flight control group was reduced to 3, and therefore any comparisons to this group had decreased experimental power.
Flow cytometry
Red blood cells were lysed with Pharm Lyse buffer (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions. Cell suspensions were washed and incubated in PBS supplemented with 1% bovine serum albumin (BSA; Sigma-Aldrich), 1% mouse serum (Life Technologies), and 10 μg/ml purified anti-CD16/CD32 (2.4G2; BD Biosciences) to block nonspecific binding. Cells were then stained with antibodies in 1% BSA/1% mouse serum/PBS for 45 min on ice. Antibodies and dilutions used were: anti-CD3-PE (145-2C11; 1:20), anti-CD4-FITC (GK1.5; 1:100), anti-B220-PerCP-Cy5.5 (RA3-62B; 1:100), anti-CD45.2 PerCP-Cy5.5 (104; 1:20), anti-CD62L-PE (MEL-14; 1:20) anti-CD25-PE (7D4; 1:20), anti-CD44-PE (IM7; 1:20). Isotype controls were used at the same dilution as the corresponding antibody—mouse IgG2a-PerCP-Cy5.5 for anti-CD45.2, rat IgG2a-PE for anti-CD62L, rat IgM for anti-CD25-PE, and rat IgG2b-PE for anti-CD44 (BD Biosciences). Cells were washed and fixed in 4% paraformaldehyde for 10 min in room temperature. Fixative was washed off and cells were stored in 1% BSA/1% mouse serum/PBS in 4°C protected from light until analysis. Flow cytometry data acquisition was performed using a BD FACSCalibur (BD Biosciences) located at the Sanford-Burnham Medical Research Institute (Orlando, FL, USA). A minimum of 10,000 CD4+CD45.2+ cells were counted for analysis of transferred OT-II T cells. Analysis was performed by FlowJo, v10 software (FlowJo, Ashland, OR, USA).
In vitro restimulation cultures
Spleens were mechanically dissociated into single cell suspensions and red blood cells lysed with Pharm Lyse buffer. Cells were washed, resuspended in complete medium, and plated in 12-well plates at a density of 1 × 107 cells/well. Cells were cultured with either medium alone, 20 μg/ml OVA peptide, 20 μg/ml OVA peptide plus 5 U/ml IL-2, or 5 μg/ml ConA (Sigma-Aldrich) plus 4 μg/ml anti-CD28 antibody (BD Biosciences) for 3 d. Cells were then collected for flow cytometry or preserved for RNA isolation.
Carboxyfluorescein succinimidyl ester (CFSE) labeling and proliferation analysis
Splenocytes were resuspended in 0.1% BSA/PBS at 1 × 106 cells/ml and incubated with 1.5 μM CFSE (Life Technologies) for 10 min in room temperature. Cell labeling was quenched by adding an equal volume of ice-cold fetal bovine serum and incubation on ice for 5 min. Cells were resuspended in complete medium, washed, and plated in 96-well U-bottom plates at 1 × 106 cells/well. Cells were cultured with medium alone, OVA peptide, OVA peptide plus IL-2, or ConA and anti-CD28 for 3 d. On d 3, cells were stained with anti-CD3-PE and anti-CD45.2-PerCP-Cy5.5 for flow cytometry analysis. Expansion index of CD45.2+ cells for each sample (number of CD45.2+ cells at end of culture/number of CD45.2+ cells at start of culture) was calculated by the Proliferation Platform of FlowJo v9.8.1 (FlowJo).
Multiplex cytokine analysis
Cell culture supernatant from cultures restimulated in vitro with ConA and anti-CD28 for 2 d or restimulated with medium alone or OVA peptide for 3 d were collected, frozen, and stored at −80°C until analysis. Supernatant samples were assayed using a mouse cytokine 20-plex assay (Life Technologies) following the manufacturer’s procedures to detect basic fibroblast growth factor, granulocyte macrophage colony-stimulating factor (GM-CSF), IFN-γ, IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12 (p40/p70), IL-13, IL-17, IP-10, KC, MCP-1, MIG, MIP-1α, TNF-α, and VEGF-A. Multiplex results were acquired on a Labscan 200 analyzer (Luminex, Austin, TX, USA) using Bio-Plex manager software v6.1 (Bio-Rad, Hercules, CA, USA). A 5-point logistic regression curve was used to calculate the concentration from the fluorescence intensity of the bead measurements. Samples that were below the level of detection were assigned half the lowest detectable value for that analyte.
RNA isolation and quantitative real-time reverse transcription PCR
Cells were preserved in RNAprotect Cell Reagent (Qiagen, Germantown, MD, USA) and stored at −80°C until processing. RNA was isolated using the RNeasy Mini kit (Qiagen) according to the manufacturer’s protocol. RNA quantity and purity were measured by a Nanodrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and 260/280 ratios for all samples were 1.8 to 2.0. Reverse transcription was carried out with 300 ng of RNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) per the manufacturer’s instructions. One microliter of the resulting cDNA was added to a final 20 μl mixture containing 10 μl of VeriQuest SYBR Green Master Mix (Affymetrix, Santa Clara, CA, USA) and 12 pmol oligonucleotide primers. PCRs were carried out in a 7300 Real-Time PCR System (Applied Biosystems). Primer sequences used were: Faslg (12597287a1), forward 5′-GCCCATGAATTACCCATGTCC-3′, reverse 5′-ACAGATTTGTGTTGTGGTCCTT-3′; Fas (226443052c1), forward 5′- GCGGGTTCGTGAAACTGATAA-3′, reverse 5′- GCAAAATGGGCCTCCTTGATA-3′; Foxp3 (16905075a1), forward 5′- CCCATCCCCAGGAGTCTTG-3′, reverse 5′- ACCATGACTAGGGGCACTGTA-3′; 18S, forward 5′-GTGGAGCGATTTGTCTGGTT-3′, reverse 5′-CGCTGAGCCAGTCAGTGTAG-3′. For primer sequences obtained from the Harvard Primer Bank (http://pga.mgh.harvard.edu/primerbank/citation.html), the PrimerBank IDs were provided in parentheses (20). Additional primers were designed by using the Web-based Primer3 primer design program. The thermal profile was 50°C for 2 min and 95°C for 10 min, followed by 40 amplification cycles consisting of 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s. Samples were normalized to rRNA 18S internal standard. Relative quantification of gene expression was calculated by the 2ΔΔCt equation.
DNA fragmentation assay
A portion of cells preserved in RNAprotect were used for DNA isolation and fragmentation assay according to a previously described protocol (21). Briefly, cells were lysed with 5 mM Tris-HCl, pH 8.0, 20 mM EDTA, and 0.1% Triton X-100 solution. An equal volume of 5% polyethylene glycol in 2 M NaCl solution was added to precipitate nonfragmented DNA. Samples were incubated on ice for 10 min and then centrifuged at 16,000 g for 10 min. The supernatants were collected as the fragmented DNA fraction. The pellets were redissolved in an equal volume of Tris EDTA buffer and represented the nonfragmented DNA fraction. A 100 μl of supernatant and pellet samples were combined 1:1 with 1 μg/ml Hoechst 33342 dye (Life Technologies). Fluorescence was read at 355 nm excitation/460 nm emission to measure DNA content. Percentage DNA fragmentation for each sample = [fluorescence intensity of supernatant/(fluorescence intensity of supernatant + fluorescence intensity of pellet)].
Statistical analysis
Two-tailed Student’s t tests and 2-way ANOVA analyses were performed by Prism v5.0 software (GraphPad Software, La Jolla, CA, USA).
RESULTS
Antigen-specific challenge in vivo during spaceflight resulted in greater numbers of antigen-specific T cells compared with ground-based responses
Draining lymph nodes and spleens from flight mice were collected within 4 h of landing for immediate analysis, and the same procedure was followed for ground mice. Consistent with previously published findings, flight mice demonstrated a significant overall decrease in the proportion of T cells in the spleen compared with ground mice. Lower T-cell numbers in flight mice were due to decreased numbers of CD4 T cells, whereas the CD8 T-cell compartment was relatively unaffected (Supplemental Fig. 1A) (22). Two-color flow cytometry for CD4 and CD45.2 expression was used to identify the adoptively transferred OT-II memory CD4 T cells. Unexpectedly, the number of CD45.2+CD4+ T cells decreased significantly in flight OVA mice compared with flight control mice in both lymph node and spleen, suggesting that in vivo exposure to OVA led to some sort of T-cell tolerance or deletion (Fig. 2). CD45.2+CD4+ T cells tended to decrease in ground OVA compared with ground control mice, suggesting that induction of T-cell tolerance or deletion was due to our experimental system and not specific to the spaceflight condition. Importantly, for both the lymph nodes and spleen, the numbers of CD45.2+CD4+ T cells were significantly greater in flight OVA mice than in ground OVA mice, indicating that OVA-specific T-cell tolerance or deletion was less effective in spaceflight. Furthermore, numbers of CD45.2+CD4+ T cells tended to increase in flight control compared with ground control mice, suggesting that even in the absence of in vivo OVA restimulation, CD45.2+CD4+ T cells had a slight proliferative or survival advantage in the flight condition. These findings were maintained even after we normalized for baseline alterations in the overall CD4 T-cell numbers between flight and ground mice (Supplemental Fig. 1B). Taken together, these results indicated that: 1) spaceflight conditions likely offered a growth or survival advantage to OVA-specific memory T cells compared with ground conditions, 2) in vivo restimulation with OVA using minipump delivery in our system led to a dominant T-cell tolerance or deletion response, and 3) induced T-cell tolerance or deletion was less effective in flight mice than in ground mice.
Figure 2.

Flight mice demonstrated more transferred OVA-specific T cells in vivo than ground mice. A) Fluorescence-activated cell sorting (FACS) plots show CD45.2+CD4+ T cells from draining lymph nodes of 1 representative mouse from each experimental group. Bar graph shows average frequency of CD45.2+CD4+ T cells as percentage of total CD4 cells for entire experimental group (n = 4, except flight control n = 3). Error bars represent se. B) Similar analysis of CD4+CD45.2+ cell frequency in spleen. G, ground; F, flight. *P < 0.05 (2-tailed Student’s t test).
In vivo OVA peptide plus MPLA stimulation by minipump delivery led to induction of antigen-specific tolerance
One potential explanation for the higher numbers of CD45.2+CD4+ T cells detected directly ex vivo from flight mice was that deletional tolerance mechanisms, such as activation-induced cell death, were impaired in spaceflight. However, Fas expression did not differ among the 4 experimental groups, and expression of Fas ligand was higher in flight mice than in ground mice (Supplemental Fig. 2A). Corresponding to Fas ligand expression, there were more apoptotic cells in flight compared with ground cultures restimulated with OVA peptide in vitro (Supplemental Fig. 2B), suggesting that spaceflight did not induce any global defects in Fas/Fas ligand expression or resistance to activation-induced cell death after antigen-specific stimulation. Expression of other molecules that modulate T-cell apoptosis, including Bcl2 and Bcl-xL, were not significantly changed between the 4 experimental conditions (data not shown).
To further examine whether antigen-specific tolerance was induced in transferred OT-II cells by implanted minipumps that delivered antigen in vivo, we determined the proliferative response of CD45.2+CD4+ T cells restimulated in vitro in medium alone, OVA peptide, OVA peptide plus IL-2, or ConA and anti-CD28. We found that CD45.2+CD4+ T-cell proliferation to OVA peptide restimulation significantly decreased in cultures from mice that received OVA compared with control in vivo, indicating that our experimental protocol resulted in tolerance induction in mice that received OVA peptide by minipump (Fig. 3A). Interestingly, CD45.2+CD4+ T cells from flight mice tended to proliferate slightly more than those from ground mice, again suggesting that tolerance induction was less robust in flight mice. Corresponding to proliferation, OVA-specific IL-2 production from in vivo OVA-stimulated mice was decreased compared with that in control mice for both flight and ground cohorts, a finding that supports the idea that antigen-specific tolerance was induced (Fig. 3B). When IL-2 was added to OVA in the in vitro restimulation cultures, the proliferation of CD45.2+CD4+ T cells did not increase significantly (Fig. 3A), suggesting that the mechanism of tolerance was not classic T-cell anergy.
Figure 3.

In vivo OVA plus MPLA stimulation by minipump inhibited antigen-specific proliferation and IL-2 production. A) Splenocytes were labeled with CFSE and restimulated in vitro with medium alone, OVA peptide, OVA peptide plus IL-2, or ConA and anti-CD28. After 3 d, cultured cells were collected, stained with CD45.2, and analyzed by FACS for proliferation. *P < 0.05 by 2-way ANOVA. B) Antigen-specific IL-2 production was measured by restimulating splenocytes with OVA peptide for 3 d and subtracting IL-2 level from parallel cultures stimulated with medium alone. IL-2 production resulting from mitogen activation was measured from culture supernatants after 2 d of ConA and anti-CD28 stimulation. *P < 0.05 (2-tailed Student’s t test). G, ground; F, flight. All groups n = 4, except for flight control n = 3. Error bars represent se.
In contrast to the OVA-specific response, flight cultures produced significantly less IL-2 than ground cultures upon stimulation with ConA and anti-CD28, regardless of whether mice received control or OVA in vivo (Fig. 3B). This finding indicated that IL-2 production in response to mitogen stimulus was highly dependent on the gravity condition and confirmed previously published results that showed significantly reduced IL-2 expression from mitogen-activated T cells exposed to simulated microgravity (8, 9). Proliferation of CD45.2+CD4+ T cells in response to ConA and anti-CD28 stimulation was similar in all 4 experimental groups, suggesting there are compensatory mechanisms that maintain T-cell proliferation despite significantly reduced IL-2 production in flight cultures.
Spaceflight had differential effects on CD62L, CD25, and CD44 expression on endogenous and transferred CD4 T cells
To determine the expression of CD62L, CD25, and CD44 on endogenous (CD45.2−CD4+) and transferred OT-II (CD45.2+CD4+) T cells in draining lymph nodes, we performed 3-color flow cytometry. Endogenous CD4 T cells represented the normal CD4 T-cell repertoire of the mouse as altered by the ground vs. flight conditions, including a small fraction that might have been activated by the in vivo administration of OVA peptide. Transferred OT-II cells were the OVA-specific memory CD4 T cells. As expected, the majority of endogenous CD4 T cells in ground and flight mice were nonactivated CD62Lhi cells, regardless of in vivo treatment (Fig. 4). Upon closer inspection, endogenous CD4 T cells in flight mice consistently showed a higher proportion of CD62Llo cells, reflecting an overall lower median fluorescence intensity (MFI), compared with ground mice. This finding suggested that a small percentage of endogenous CD4 T cells in flight conditions had a more activated phenotype compared with ground control mice. Although most of the transferred OT-II cells from ground control and flight control mice also showed a CD62Lhi phenotype, MFI did not differ between the 2 groups. Importantly, transferred OT-II cells from ground control and flight OVA mice significantly down-regulated CD62L expression by comparable magnitudes, suggesting that initial activation of these cells by in vivo OVA challenge was equally effective in flight and ground conditions.
Figure 4.

Expression of CD62L, CD25, and CD44 on endogenous and transferred CD4 T cells from flight and ground mice differed. Draining lymph nodes were collected and single cell suspensions stained with CD4, CD45.2, and 1 of indicated T-cell markers or its corresponding isotype control. Endogenous CD4 T cells were identified as CD45.2−CD4+ cells, and transferred OT-II T cells were identified as CD45.2+CD4+ cells. FACS histograms show expression profile of CD62L, CD25, or CD44 on endogenous or OT-II cells of 1 representative mouse per group. Gray lines within histograms show gating between cells expressing high vs. low levels of indicated surface marker. Bar graphs show MFI for each T-cell marker averaged across all mice for entire experimental group (n = 4, except flight control n = 3). MFI of surface markers was calculated for entire indicated T-cell population (endogenous or OT-II), except for CD25 on endogenous cells, which represent MFI of gated CD25hi population. G, ground; F, flight. *P < 0.05 and **P < 0.005 (2-tailed Student’s t test). Error bars represent se.
Interpretation of CD25 expression was more complex than that of other T cell activation markers because it could also serve as a marker for regulatory T (Treg)cells. Approximately 20 to 25% of the endogenous CD4 T cells in ground and flight mice expressed CD25. Among CD25-expressing endogenous CD4 T cells, the level of CD25 expression was significantly lower in flight mice compared with ground mice as manifested by a significantly lower MFI (Fig. 4). Not surprisingly, the entire population of transferred OT-II memory T cells demonstrated up-regulated expression of CD25 in ground control and flight control mice, which was further up-regulated upon in vivo OVA challenge in ground control and flight OVA mice. Similarly to endogenous CD4 T cells, transferred OT-II cells in flight control mice expressed significantly lower levels of CD25 than did ground control animals. Although CD25 on OT-II cells was significantly up-regulated in ground OVA mice compared with ground control animals, up-regulation of CD25 on OT-II cells in flight OVA mice compared with flight control animals was less robust. In addition, expression of CD25 on OT-II cells tended to be reduced in flight OVA mice compared with ground OVA mice. Because appropriate down-regulation of CD62L indicated that T-cell activation was at least partially intact during spaceflight, these CD25 findings suggested that spaceflight caused targeted impairment of CD25 expression and/or up-regulation on both endogenous and transferred T cells.
Finally, high CD44 expression was used as a marker for memory T cell phenotype. Endogenous CD4 T cells in flight mice demonstrated a significantly lower proportion of CD44hi cells and overall lower CD44 MFI compared with ground mice (Fig. 4). Although transferred OT-II cells showed generally higher CD44 expression levels than endogenous cells, the proportion of CD44hi cells and CD44 MFI remained significantly lower in flight mice compared with ground mice regardless of whether there was in vivo OVA stimulation. Indeed, in vivo OVA stimulation did not significantly alter the level of CD44 expression within the ground or flight cohorts. These results indicated that spaceflight had a dominant effect in significantly reducing CD44 expression on both endogenous and OT-II memory CD4 T cells, regardless of in vivo antigen exposure, and that maintenance of the memory phenotype might be impaired.
Spaceflight differentially impaired subsequent expression of CD25, Foxp3, and CD44 upon antigen-specific restimulation in vitro
To characterize the effect of spaceflight on the antigen recall response after return to Earth, splenocytes from ground and flight mice were restimulated in vitro with medium alone, OVA peptide, OVA peptide plus IL-2, or ConA and anti-CD28. Upon OVA in vitro restimulation, expression of CD25 on OT-II (CD45.2+CD4+) T cells from flight control mice remained significantly reduced compared with ground control mice (Fig. 5A). CD25 expression tended to increase on OT-II cells from flight OVA compared with flight control mice, suggesting that in vivo exposure to OVA during spaceflight partially mitigated the defect in CD25 up-regulation. Adding IL-2 to OVA cultures significantly up-regulated CD25 expression on OT-II cells from flight OVA mice compared with flight control mice so that levels became comparable to those on OT-II cells from ground OVA mice. In contrast, upon ConA and anti-CD28 in vitro stimulation, expression of CD25 on OT-II cells was significantly reduced in flight compared with ground cultures regardless of prior in vivo treatment, confirming previously published results that showed impaired CD25 expression upon mitogen T-cell activation in spaceflight and simulated microgravity (6, 8). Together, these findings suggested that spaceflight induced a generalized defect in CD25 up-regulation. Antigen-specific exposure in vivo during spaceflight likely partially mitigated the deficiency in CD25 expression and additional exposure to exogenous IL-2 appeared to abrogate the defect.
Figure 5.
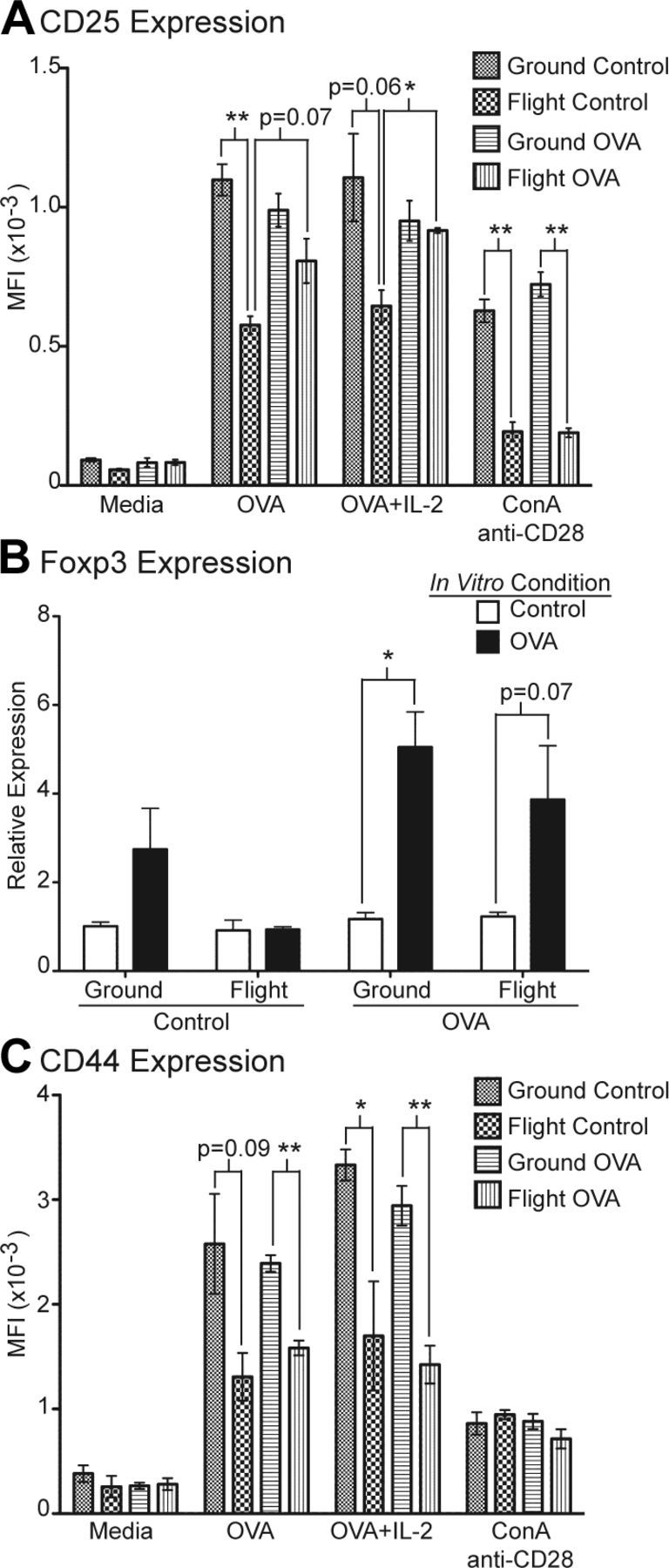
Transferred OT-II cells from flight mice demonstrated impaired expression of CD25, Foxp3, and CD44 upon OVA peptide restimulation in vitro compared with ground mice. A) Splenocytes were restimulated in vitro with medium alone, OVA peptide, OVA peptide plus IL-2, or ConA and anti-CD28. After 3 d, cultured cells were stained with CD4, CD45.2, and CD25 and analyzed by FACS. Data represent MFI of CD25 for gated CD45.2+CD4+ cells for each experimental group. *P < 0.005 and **P < 0.001. B) Quantitative real-time reverse transcription-PCR analysis of Foxp3 expression in splenocytes restimulated with OVA peptide for 3 d compared with freshly collected splenocytes as control. *P < 0.005. C) Analysis of CD44 expression by FACS on CD4+CD45.2+ cells after in vitro restimulation. *P < 0.05 and **P < 0.005. All groups n = 4, except for flight control n = 3. Error bars represent se. P values were calculated by 2-tailed Student’s t test.
To determine whether differences in CD25 expression might be correlated with Treg cell development, we investigated the expression of Foxp3, a more specific marker for Treg cells. When restimulated with OVA in vitro, splenocytes from ground OVA mice significantly up-regulated Foxp3 expression compared with unstimulated controls (Fig. 5B). A similar trend was observed in flight OVA splenocytes cultures. In contrast, flight control splenocytes showed no appreciable up-regulation of Foxp3 when stimulated with OVA in vitro compared with unstimulated controls. These results, together with those for CD25 expression, suggested that reduced generation or maintenance of induced OVA-specific CD4+CD25+Foxp3+ Treg cells in flight mice might be a potential mechanism for impaired tolerance induction during spaceflight. Moreover, the deficiency in generating Treg cells appeared to persist upon antigen restimulation after return from spaceflight and might be partially mitigated if there was exposure to the specific antigen during spaceflight.
To further examine the generation and maintenance of memory T cells in ground and flight mice, we determined the expression of CD44 on OT-II cells upon in vitro restimulation. Similar to what was observed on freshly isolated cells, CD44 expression remained significantly reduced on OT-II cells from flight compared with ground cultures regardless of prior in vivo treatment (Fig. 5C). Addition of exogenous IL-2 did not increase CD44 expression on OT-II cells from flight mice. ConA and anti-CD28 stimulation induced comparably weak up-regulation of CD44 from all 4 experimental groups. These results suggested that exposure to spaceflight significantly impaired generation and maintenance of antigen-specific CD44hi memory T cells. Disrupted memory T-cell homeostasis was not mitigated by in vivo antigen-specific exposure during spaceflight, and impaired development of memory T cells persisted after return from spaceflight, at least in the short term.
Splenocyte cultures from mice exposed to spaceflight produced increased antigen-specific proinflammatory cytokines
Culture supernatants were collected from in vitro restimulation cultures to determine the cytokine production profile of splenocytes upon OVA-specific activation. Levels of each cytokine from OVA restimulated cultures were subtracted by the baseline level of that cytokine from parallel cultures with medium alone in order to determine its production due to antigen-specific stimulation. Overall, cultures from flight mice produced more antigen-specific proinflammatory cytokines than counterpart cultures from ground mice regardless of prior in vivo treatment (Fig. 6). Some of the increases in proinflammatory cytokine production were statistically significant (e.g., IL-1β and IL-17 for flight OVA vs. ground OVA; GM-CSF and IFN-γ for flight control vs. ground control animals). From the restimulation cultures in general, there was relatively less overall production of IL-10, a major anti-inflammatory cytokine product of T regulatory type 1 cells, and IL-4, the signature cytokine product of T helper 2 (Th2) cells. In contrast, production of IFN-γ and IL-17, the signature cytokines for Th1 and Th17 cells, respectively, were significantly increased in cultures from flight mice compared with ground mice. These results indicated that OVA-specific T cells from flight mice differentiated toward the Th1 and Th17 subsets more robustly and induced higher levels of proinflammatory cytokines than was true for ground mice. Importantly, these findings suggested that flight mice developed an excessive inflammatory response to the in vivo OVA challenge compared with ground mice. Production of TNF-α, GM-CSF, IFN-γ, and IL-17 tended to be lower in cultures from flight OVA compared with flight control mice, again suggesting that in vivo OVA treatment induced dominant T-cell tolerance that was somewhat less effective in flight compared with ground mice. There were no significant differences among the 4 experimental groups in the levels of these cytokines upon restimulation with ConA and anti-CD28 (Supplemental Fig. 3), indicating that in vivo treatment or gravity conditions did not induce any generalized defects in the ability to produce these cytokines. In addition, similar cytokine production profiles from ConA/anti-CD28 stimulated cultures indicated that differences in OVA-specific cytokine production were unlikely due to overall changes in T-cell numbers between flight and ground cultures.
Figure 6.

Cultures from flight mice produced increased proinflammatory cytokines to OVA peptide restimulation compared with ground mice. OVA-specific cytokine response was measured by restimulating splenocytes with OVA peptide for 3 d and subtracting level from parallel cultures stimulated with medium alone. Production of proinflammatory (IL-1β, IL-6, TNF-α, and GM-CSF) and T-cell subset cytokines (Th1 represented by IFN-γ, Th2 represented by IL-4, Th17 represented by IL-17, and T regulatory type 1 cells represented by IL-10) was measured by multiplex assay. G, ground; F, flight. *P < 0.05 (2-tailed Student’s t test). All groups n = 4, except for flight control n = 3. Error bars represent se.
DISCUSSION
This report represents the first detailed analysis of an in vivo antigen-specific CD4 T-cell response induced during spaceflight. We found that spaceflight disrupted induction of T-cell tolerance, impaired maintenance of memory T-cell homeostasis, and led to the development of a heightened inflammatory response.
Our experimental system, in which soluble OVA peptide was administered subcutaneously with MPLA by minipump, led to the development of tolerance in vivo. This was evidenced by decreased numbers of OT-II T cells in mice exposed to OVA plus MPLA compared with MPLA alone, as well as their decreased proliferation and IL-2 production upon restimulation. Continuous administration of subcutaneous soluble peptide by minipump has been shown to lead to Treg cell development (23). Although we attempted to avoid tolerance induction by giving 2 isolated bursts of antigen, the minipump infusion method of antigen delivery may still have favored Treg cell generation. In addition, MPLA, chosen for its lower toxicity and safety profile, might have skewed the immune response more toward Treg cells compared with highly inflammatory adjuvants such as LPS or complete Freund's adjuvant. The reduced toxicity of MPLA might be related to altered intracellular signaling that actively suppressed proinflammatory activity (18), thus favoring Treg cell generation. Although not our original intention, this experimental system allowed us to examine the effectiveness of tolerance induction during spaceflight. Importantly, our results suggested that the mechanism of impaired tolerance in spaceflight was not due to defects in clonal deletion or anergy, and instead might be related to impaired generation or maintenance of Treg cells.
Treg cells are critical for maintaining tolerance, preventing autoimmunity, limiting inflammatory responses, and preserving immune homeostasis. Natural Treg cells are differentiated within the thymus, whereas induced Treg cells may be derived from mature naive or memory CD4 T cells in the periphery (24). Rapidly dividing memory CD4 T cells are likely an important continued source of induced Treg cells later in life (25). Treg cells are CD4+CD25+Foxp3+ and mediate suppression of a wide range of immune cell types through a cell-contact-dependent mechanism (26). IL-2 is essential for the generation and expansion of Treg cells (27). Mice lacking expression of IL-2 or CD25 developed severe autoimmune lymphoproliferative diseases spontaneously (28, 29), indicating that the dominant nonredundant role of IL-2 in vivo was to maintain immune tolerance and homeostasis. Adoptive transfer of CD4+CD25+ T cells into mice with deficiencies in IL-2 receptor signaling rescued them from lethal autoimmunity (30, 31). Moreover, low-dose IL-2 increased Treg cell numbers and alleviated severity of autoimmune diseases (27), suggesting that IL-2 mediated its immunoregulatory effect through Treg cells. Our in vivo results confirmed previous findings that spaceflight and microgravity caused severe impairment of IL-2 and CD25 up-regulation when T cells were activated with mitogens in vitro (6–9). The dominant in vivo manifestation of decreased IL-2 and CD25 expression during spaceflight may be altered Treg cell homeostasis that leads to dysregulated inflammatory responses. On the basis of our in vivo results and findings from previous in vitro T-cell spaceflight experiments, we hypothesize that microgravity during spaceflight predominantly inhibited IL-2 and CD25 up-regulation during T-cell activation, leading to a disproportionate decrease in Treg cells and subsequent impairment in tolerance that is manifested as heightened inflammatory responses. Whether reduced antigen-specific expression of CD25 and Foxp3 caused by spaceflight represented transiently impaired up-regulation after T-cell activation or a lasting deficiency of the Treg cell lineage could not be definitively answered by our data. Nevertheless, our findings provided the first evidence that antigen-specific activation of T cells in vivo during spaceflight might lead to impaired generation of Treg cells and dysregulated inflammatory responses.
The relationship between Treg cells and memory CD4 T cells is interesting because memory CD4 T cells have been proposed to be an important source of induced Treg cells (25). Our results suggested that both Treg and memory CD4 T-cell numbers may be significantly reduced by spaceflight. However, Treg cells could be increased by antigen-specific stimulation in vivo during spaceflight, whereas memory CD4 T cells could not. If homeostasis of Treg and memory CD4 T cells were linked, our findings would suggest that spaceflight induced a profound and absolute inhibition of memory CD4 T-cell development and maintenance, and that a subset may be stimulated to become Treg cells if exposed to antigen during spaceflight. Because the prevailing functional outcome was increased inflammatory response in mice exposed to spaceflight, the deficiency of Treg cells had the dominant effect in our experimental system. Additional studies are required to determine whether spaceflight impairs unique memory T-cell functions, such as efficient homing to lymphoid organs, increased life span, and faster response kinetics (32), resulting in less effective immune responses in other contexts.
In our current study, a cohort of flight mice was compared with ground control mice that were housed in identical flight hardware enclosures and subjected to simulated ambient conditions experienced by the flight mice. Under these conditions, the spaceflight-related variables that remained different between the 2 groups were the presence of microgravity cosmic radiation, and systemic stress response related to launch and landing for the flight mice. In vitro spaceflight experiments using isolated human peripheral blood leukocytes demonstrated that spaceflight inhibited T-cell activation independent of systemic stress hormone responses (5, 6, 10). In addition, experiments that compared activation of T cells in spaceflight microgravity with an in-flight 1g centrifuge control demonstrated that suppressed T-cell activation can be attributed to effects of microgravity independent of other spaceflight-related environmental factors (6, 10). Cosmic radiation has been shown to have adverse effects on the immune system at high doses (2 to 3 Gy) which may occur during sporadic solar particle events and exposure risk is greatest for astronauts performing extravehicular activities (33, 34). It is very unlikely that flight mice in our study were exposed to these levels of radiation during a 15 d low-Earth-orbit mission while enclosed and protected by the space shuttle. Increased systemic stress hormones, as determine by spikes in urinary cortisol levels, have been documented immediately around the time of space shuttle launch and landing (35). However, how these changes in stress hormones may affect an immune response induced during the course of spaceflight remains unclear. Our current experiment was designed so that the in vivo antigen challenge was delivered on flight d 6 and 10 during a 15 d mission, remote from the acute neuroendocrine changes induced by launch and landing. As a result of this experimental design, the acute stress responses to launch and landing likely had minimal effect on the OVA-specific immune responses we measured. Although the possible effects of cosmic radiation and systemic stress hormones cannot be completely eliminated in our experiment, the most likely spaceflight-related factor that caused the immune dysregulation we observed remains to be microgravity.
Our study had some limitations. First, the sample size of 4 mice per experimental group was powered to detect a 30% difference between groups with up to a 20% sd. The flight control group had 3 animals because 1 mouse was disqualified for flight as a result of an incompletely healed wound at the minipump implant site. As a result of limited statistical power, there might have been significant differences between groups that could not be definitively determined. This was reflected by the fact that several consistent trends in our data did not reach statistical significance. Regardless of the limitations, we still detected several statistically significant effects of spaceflight on the immune response, indicating that the impact of spaceflight on the immune system was substantial and would likely pose considerable challenges for human health. Second, per standard NASA protocol, synchronous ground control mice were generated 48 h after flight mice in order to conform to the requirements of the Orbital Environment Simulator that mimicked space shuttle ambient conditions for the ground cohort. This minimized experimental variability that may be caused by differences in ambient conditions with the tradeoff that variability may be introduced by the adoptive transfer of separate preparations of activated OT-II T cells. Due diligence was followed to keep all experimental procedures and reagents the same between the flight and ground cohorts so that experimental differences may be attributable to exposure to spaceflight.
In conclusion, we showed that spaceflight caused significant impairment in tolerance induction and disrupted homeostasis of memory CD4 T cells, leading to increased inflammatory responses. There are examples in humans in which the host’s hyperstimulated inflammatory responses induce more overall damage than the inciting infectious agent or agents (e.g., sepsis.) Our results raise the possibility that humans who acquire an infection during long-term spaceflight, such as on an exploratory mission to Mars, may develop excessive inflammatory responses and associated adverse consequences. As a result of disrupted Treg cell homeostasis, autoimmune diseases may also develop during long-term spaceflight. Additional studies are required to assess the true risks of humans developing deleterious inflammatory responses and/or autoimmune manifestations during long-term space travel. With increased laboratory facilities and safety barriers on the International Space Station, performing such experiments using in vivo infectious and autoimmune disease models will be possible. Determining the nature of immune dysregulation during spaceflight and potential countermeasures will be critical for protecting of the health of humans who venture on long-term space voyages. Finally, understanding spaceflight immune dysregulation may reveal fundamental and previously unappreciated aspects of how the immune system functions under Earth’s gravity and provide critical insights into the mechanisms of immune dysfunction in various human diseases.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
Acknowledgments
This work was supported by U.S. National Aeronautics and Space Administration (NASA) Grant NNH07ZTT001N. The authors thank P. M. Dumars for science integration with NASA operations and S. Litherland (Sanford-Burnham Medical Research Institute, Orlando, FL, USA) for providing access their flow cytometer. The authors also thank S. Keating (Blood Systems Research Institute, San Francisco, CA, USA) for excellent technical assistance in processing the multiplex cytokine assays, Q. Tang and A. Abbas for thoughtful scientific feedback, and P. Derish for editorial work.
Glossary
- 1g
Earth’s gravity
- AEM
animal enclosure module
- BSA
bovine serum albumin
- CFSE
carboxyfluorescein succinimidyl ester
- ConA
concanavalin A
- FACS
fluorescence-activated cell sorting
- GM-CSF
granulocyte macrophage colony-stimulating factor
- MFI
median fluorescence intensity
- MPLA
monophosphoryl lipid A
- OVA
ovalbumin
- STS
Space Transport System
- TCR
T-cell receptor
- Th
T helper
- Treg
regulatory T
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Guéguinou N., Huin-Schohn C., Bascove M., Bueb J. L., Tschirhart E., Legrand-Frossi C., Frippiat J. P. (2009) Could spaceflight-associated immune system weakening preclude the expansion of human presence beyond Earth’s orbit? J. Leukoc. Biol. , 1027–1038 [DOI] [PubMed] [Google Scholar]
- 2.Hughes-Fulford M. (2011) To infinity…and beyond! Human spaceflight and life science. FASEB J. , 2858–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konstantinova I. V., Rykova M. P., Lesnyak A. T., Antropova E. A. (1993) Immune changes during long-duration missions. J. Leukoc. Biol. , 189–201 [DOI] [PubMed] [Google Scholar]
- 4.Taylor G. R., Janney R. P. (1992) In vivo testing confirms a blunting of the human cell–mediated immune mechanism during space flight. J. Leukoc. Biol. , 129–132 [DOI] [PubMed] [Google Scholar]
- 5.Cogoli A., Tschopp A., Fuchs-Bislin P. (1984) Cell sensitivity to gravity. Science , 228–230 [DOI] [PubMed] [Google Scholar]
- 6.Hashemi B. B., Penkala J. E., Vens C., Huls H., Cubbage M., Sams C. F. (1999) T cell activation responses are differentially regulated during clinorotation and in spaceflight. FASEB J. , 2071–2082 [DOI] [PubMed] [Google Scholar]
- 7.Cogoli A., Bechler B., Cogoli-Greuter M., Criswell S. B., Joller H., Joller P., Hunzinger E., Müller O. (1993) Mitogenic signal transduction in T lymphocytes in microgravity. J. Leukoc. Biol. , 569–575 [DOI] [PubMed] [Google Scholar]
- 8.Walther I., Pippia P., Meloni M. A., Turrini F., Mannu F., Cogoli A. (1998) Simulated microgravity inhibits the genetic expression of interleukin-2 and its receptor in mitogen-activated T lymphocytes. FEBS Lett. , 115–118 [DOI] [PubMed] [Google Scholar]
- 9.Boonyaratanakornkit J. B., Cogoli A., Li C. F., Schopper T., Pippia P., Galleri G., Meloni M. A., Hughes-Fulford M. (2005) Key gravity-sensitive signaling pathways drive T cell activation. FASEB J. , 2020–2022 [DOI] [PubMed] [Google Scholar]
- 10.Chang T. T., Walther I., Li C. F., Boonyaratanakornkit J., Galleri G., Meloni M. A., Pippia P., Cogoli A., Hughes-Fulford M. (2012) The Rel/NF-κB pathway and transcription of immediate early genes in T cell activation are inhibited by microgravity. J. Leukoc. Biol. , 1133–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castro V. A., Thrasher A. N., Healy M., Ott C. M., Pierson D. L. (2004) Microbial characterization during the early habitation of the International Space Station. Microb. Ecol. , 119–126 [DOI] [PubMed] [Google Scholar]
- 12.Ott C. M., Bruce R. J., Pierson D. L. (2004) Microbial characterization of free floating condensate aboard the Mir space station. Microb. Ecol. , 133–136 [DOI] [PubMed] [Google Scholar]
- 13.Mehta S. K., Laudenslager M. L., Stowe R. P., Crucian B. E., Sams C. F., Pierson D. L. (2014) Multiple latent viruses reactivate in astronauts during Space Shuttle missions. Brain Behav. Immun. , 210–217 [DOI] [PubMed] [Google Scholar]
- 14.Pape K. A., Kearney E. R., Khoruts A., Mondino A., Merica R., Chen Z. M., Ingulli E., White J., Johnson J. G., Jenkins M. K. (1997) Use of adoptive transfer of T-cell-antigen-receptor-transgenic T cell for the study of T-cell activation in vivo. Immunol. Rev. , 67–78 [DOI] [PubMed] [Google Scholar]
- 15.Barnden M. J., Allison J., Heath W. R., Carbone F. R. (1998) Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. , 34–40 [DOI] [PubMed] [Google Scholar]
- 16.Hu H., Huston G., Duso D., Lepak N., Roman E., Swain S. L. (2001) CD4(+) T cell effectors can become memory cells with high efficiency and without further division. Nat. Immunol. , 705–710 [DOI] [PubMed] [Google Scholar]
- 17.McKinstry K. K., Golech S., Lee W. H., Huston G., Weng N. P., Swain S. L. (2007) Rapid default transition of CD4 T cell effectors to functional memory cells. J. Exp. Med. , 2199–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mata-Haro V., Cekic C., Martin M., Chilton P. M., Casella C. R., Mitchell T. C. (2007) The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science , 1628–1632 [DOI] [PubMed] [Google Scholar]
- 19.Naidu S., Winget C. M., Jenner J. W., Mele G., Holley D. C. (1995) Effects of housing density on mouse physiology and behavior in the NASA animal enclosure module simulators. J. Gravit. Physiol. , P140. [PubMed] [Google Scholar]
- 20.Spandidos A., Wang X., Wang H., Seed B. (2010) PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. , D792–D799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ioannou Y. A., Chen F. W. (1996) Quantitation of DNA fragmentation in apoptosis. Nucleic Acids Res. , 992–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pecaut M. J., Nelson G. A., Peters L. L., Kostenuik P. J., Bateman T. A., Morony S., Stodieck L. S., Lacey D. L., Simske S. J., Gridley D. S. (2003) Genetic models in applied physiology: selected contribution: effects of spaceflight on immunity in the C57BL/6 mouse. I. Immune population distributions. J. Appl. Physiol. , 2085–2094 [DOI] [PubMed] [Google Scholar]
- 23.Apostolou I., von Boehmer H. (2004) In vivo instruction of suppressor commitment in naive T cells. J. Exp. Med. , 1401–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curotto de Lafaille M. A., Lafaille J. J. (2009) Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity , 626–635 [DOI] [PubMed] [Google Scholar]
- 25.Akbar A. N., Vukmanovic-Stejic M., Taams L. S., Macallan D. C. (2007) The dynamic co-evolution of memory and regulatory CD4+ T cells in the periphery. Nat. Rev. Immunol. , 231–237 [DOI] [PubMed] [Google Scholar]
- 26.Shevach E. M. (2009) Mechanisms of foxp3+ T regulatory cell–mediated suppression. Immunity , 636–645 [DOI] [PubMed] [Google Scholar]
- 27.Liao W., Lin J. X., Leonard W. J. (2013) Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity , 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadlack B., Merz H., Schorle H., Schimpl A., Feller A. C., Horak I. (1993) Ulcerative colitis–like disease in mice with a disrupted interleukin-2 gene. Cell , 253–261 [DOI] [PubMed] [Google Scholar]
- 29.Willerford D. M., Chen J., Ferry J. A., Davidson L., Ma A., Alt F. W. (1995) Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity , 521–530 [DOI] [PubMed] [Google Scholar]
- 30.Almeida A. R., Legrand N., Papiernik M., Freitas A. A. (2002) Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J. Immunol. , 4850–4860 [DOI] [PubMed] [Google Scholar]
- 31.Malek T. R., Yu A., Vincek V., Scibelli P., Kong L. (2002) CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity , 167–178 [DOI] [PubMed] [Google Scholar]
- 32.Dooms H., Abbas A. K. (2006) Control of CD4+ T-cell memory by cytokines and costimulators. Immunol. Rev. , 23–38 [DOI] [PubMed] [Google Scholar]
- 33.Gridley D. S., Dutta-Roy R., Andres M. L., Nelson G. A., Pecaut M. J. (2006) Acute effects of iron-particle radiation on immunity. Part II: leukocyte activation, cytokines and adhesion. Radiat. Res. , 78–87 [DOI] [PubMed] [Google Scholar]
- 34.Zhou Y., Ni H., Li M., Sanzari J. K., Diffenderfer E. S., Lin L., Kennedy A. R., Weissman D. (2012) Effect of solar particle event radiation and hindlimb suspension on gastrointestinal tract bacterial translocation and immune activation. PLoS ONE , e44329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stowe R. P., Yetman D. L., Storm W. F., Sams C. F., Pierson D. L. (2008) Neuroendocrine and immune responses to 16-day bed rest with realistic launch and landing G profiles. Aviat. Space Environ. Med. , 117–122 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.