

Abstract
The response of the skin to harmful environmental agents is shaped decisively by the status of the immune system. Keratinocytes constitutively express and secrete the chemokine-like mediator, macrophage migration inhibitory factor (MIF), more strongly than dermal fibroblasts, thereby creating a MIF gradient in skin. By using global and epidermis-restricted Mif-knockout (Mif−/− and K14-Cre+/tg; Miffl/fl) mice, we found that MIF both recruits and maintains antigen-presenting cells in the dermis/epidermis. The reduced presence of antigen-presenting cells in the absence of MIF was associated with accelerated and increased formation of nonmelanoma skin tumors during chemical carcinogenesis. Our results demonstrate that MIF is essential for maintaining innate immunity in skin. Loss of keratinocyte-derived MIF leads to a loss of control of epithelial skin tumor formation in chemical skin carcinogenesis, which highlights an unexpected tumor-suppressive activity of MIF in murine skin.—Brocks, T., Fedorchenko, O., Schliermann, N., Stein, A., Moll, U. M., Seegobin, S., Dewor, M., Hallek, M., Marquardt, Y., Fietkau, K., Heise, R., Huth, S., Pfister, H., Bernhagen, J., Bucala, R., Baron, J. M., Fingerle-Rowson, G. Macrophage migration inhibitory factor protects from nonmelanoma epidermal tumors by regulating the number of antigen-presenting cells in skin.
Keywords: skin carcinogenesis, chemokine, CD74, CD44, DMBA/TPA
The skin forms a thin but important barrier against a multitude of harmful external agents, such as infectious pathogens, allergens, or DNA-damaging agents. The functional unit of the skin contains keratinocytes and melanocytes in the epidermis as well as fibroblasts and endothelial cells in the underlying dermis. These cells provide the microenvironment that is required for mobile immune cells to perform their tasks of immunosurveillance and immune defense. Dendritic cells (DCs), macrophages, NK cells, and T and B cells are transported via the blood stream to small dermal vessels where they emigrate and patrol through the dermis. Some cells penetrate even the basal membrane and circulate through the epidermis. This directed migration is controlled by a number of chemotactic signals, typically provided by the chemokine family of mediators. Immigration of immature DCs into the skin is mediated via several different chemokine receptors, including CCR1, CCR2, CCR5, CCR6, CXCR1, and CXCR2 (1). In contrast, the homeostatic and inducible emigration of DCs from the skin to skin-draining lymph nodes has been shown to depend mainly on CCR7 and CXCR4 (2, 3). These low-molecular weight cytokines with chemotactic activity are classified as either constitutive or inducible. Constitutively expressed chemokines are generally implicated in the homeostasis of the immune system, whereas inducible chemokines are expressed mainly during inflammatory processes (4).
Recruitment of immune cells to the dermis and epidermis is highly relevant for the development of skin tumors. DCs/macrophages promote immune reactions to cutaneous antigens (5) and produce several immunoregulatory molecules, such as TNF-α, IL-1α, IL-23, prostaglandin E2, reactive oxygen species, and ornithine decarboxylase, all of which have been shown to be important regulators of DMBA (7,12-dimethylbenz[a]anthracene)/TPA-induced skin carcinogenesis (reviewed in ref. 6). These immune-activating features lead to antitumor immunity via cytotoxic T and NK cells, thereby suppressing skin tumorigenesis.
Nevertheless, immune cell recruitment to the epidermis may not always generate effective antitumor immunity and can even promote immune escape and tumor formation. DCs/macrophages may also secrete a number of survival-supporting cytokines, which may help tumor-initiated keratinocytes to overcome oncogene-induced senescence or apoptosis. In addition, immunosuppressive factors, such as IL-10 produced by DCs/macrophages, may help tumor cells to escape immune attack (7, 8). In chemical carcinogenesis protocols, immune cells have even been shown to add to the genotoxic impact of polycyclic carbohydrates, such as DMBA or benzo[α]pyrene (B[α]P), as their enzymes generate metabolic products with higher mutagenic potential (9).
Macrophage migration inhibitory factor (MIF) is a small homotrimeric (3 × 12.5 kDa) protein that was originally found to inhibit spontaneous random migration of macrophages out of capillary tubes in vitro (10). This is a sign of its macrophage-regulatory properties as macrophages stop their inherent migratory activity once they receive activating signals. More recently, MIF has been recognized as a pleiotropic proinflammatory and immunoregulatory mediator with chemokine-like functions that is secreted in a both constitutive and inducible fashion. MIF interacts with 3 surface receptors, CD74/CD44, CXCR2, and CXCR4 (11, 12). Within the immune system, MIF has been shown to activate macrophages and T and B cells and to prolong immune cell survival by inhibiting apoptosis (13, 14). MIF promotes inflammatory processes of acute and chronic conditions, such as infection, inflammation, and allergy (reviewed in refs. 15, 16). In atherosclerosis, which has features of chronic inflammation, MIF promotes recruitment of monocytes and T cells into the inflamed vessel wall (12). MIF displays chemotactic properties and binds to chemokine receptors, CXCR2 and CXCR4, with high affinity, yet lacks the N-terminal cysteine motif that is typical for classic chemokines. Accordingly, it is not considered a classic chemokine, but, rather, belongs to the class of chemokine-like function chemokines (12).
MIF expression in many tissues is ubiquitous and constitutive. High expression of MIF is found in monocytes/macrophages, epithelial cells, and keratinocytes. MIF is typically overexpressed in human and murine cancer cells compared with corresponding primary tissue. Several murine tumor models, such as myc- and TCL-1–induced lymphoma/leukemia, adenomatous polyposis coli-induced colon carcinoma, Her2-induced mammary carcinoma, and nitrosamine-induced bladder carcinoma, have demonstrated that MIF promotes tumor growth (17–21). Overexpressed MIF in malignant cells acts via multiple mechanisms to promote tumors. MIF is thought to be produced and released by tumor cells and to stimulate several survival pathways (e.g., MAPK, NF-κB, AKT) in a paracrine/autocrine manner, which leads to increased tumor cell proliferation and helps tumor cells escape from apoptosis (22–24). MIF exerts powerful actions within tumor cells that interfere with the 2 major tumor suppressor pathways, p53 and Rb-E2F, which are activated in response to oncogenic signaling (14, 25, 26).
Because MIF is highly expressed in normal epidermal keratinocytes, we hypothesized that MIF may promote skin tumorigenesis. Martin et al. (27) demonstrated that in Mif−/− BALB/c mice MIF deficiency reduces the incidence of UVB-induced nonmelanoma skin cancer by 45%. The authors associated this effect with reduced inflammation (as measured by myeloperoxidase activity), increased p53 activity, reduced VEGF levels in skin, and diminished angiogenesis (27). Here, we describe the unexpected finding that MIF suppresses epidermal tumorigenesis in mouse models of chemical skin carcinogenesis. We show that a MIF gradient from epidermis to dermis is essential for the recruitment of APCs to skin, which enables tumor suppression. Our results provide important insight into epidermal immunity and suggest a new mechanism of how MIF regulates the pathophysiology of skin inflammation and tumorigenesis.
MATERIALS AND METHODS
Animals
Mif−/− mice that were generated by Cre-mediated deletion of the entire Mif locus in the pure C57Bl/6 background (14) were from our laboratory. The same Mif−/− strain mice was outbred to the 129S1/SvImJ background for 6 generations and kindly provided to us by O. Petrenko (Stony Brook University, Stony Brook, NY, USA). Cd74−/− and Cd44−/− mice were obtained from R. Bucala and were outbred to the C57Bl/6 background for 6 generations. All animals were maintained in a homozygous state under virus-free conditions in a conventional animal facility. Experimental procedures in animals were approved by the Ethics Committee of Cologne University (8.87-50.10.37.09.273, 9.93.2.10.31.07.097, 9.93.2.10.31.07.098) and were performed in accordance with the Federation of European Laboratory Animal Science Associations/Society of Laboratory Animal Science (FELASA/GV-SOLAS) Guidelines.
Two-stage carcinogenesis with DMBA/TPA
Ten male Mif−/− and Mif+/+ littermates on the 129S1/SvImJ background were treated topically on their backs once with 25 µg DMBA (Sigma-Aldrich, St. Louis, MO, USA) in acetone after full completion of the first postnatal hair cycle (age 8 wk). After tumor initiation, mice were treated twice a week with 50 µg TPA (Sigma-Aldrich) in acetone for a total of 20 wk. Developing tumors with ≥1 mm3 were counted once weekly and measured in 3 dimensions with an electronic caliper.
One-stage carcinogenesis with B[α]P
Ten male and 10 female mice per group (Mif−/−, Miffl/fl K14-Cre+/tg; Miffl/fl, Cd74−/−, Cd44−/−), age 8 wk and on the C57Bl/6 background, were treated with 100 µg B[α]P (Sigma-Aldrich) in acetone twice a week on their shaved backs, and tumor formation was observed over a period of 32 wk. Mice were euthanized by cervical dislocation when tumor incidence reached 100% in one group. Skin tumors, B[α]P-treated skin adjacent to tumors, and untreated skin were excised for further histologic analysis after fixation in 4% buffered formalin (Roti Histofix; Carl Roth, Karlsruhe, Germany) or frozen for biochemical analysis.
Preparation of epidermal sheets
Both ear halves from 7 animals were placed on a drop of a 3.8% ammonium thiocyanate (Sigma-Aldrich) for 20 min at 37°C. After separation of the epidermis from the dermis with fine forceps, epidermal sheets were covered with ice-cold acetone for fixation and incubated for 15 min at 4°C with their inside up. The fixation step was followed by washing with PBS, then epidermal sheets were blocked with 0.5% bovine serum albumin (New England Biolabs, Ipswich, MA, USA) in PBS for 20 min, washed with PBS/Tween-20, and stained with Alexa Fluor 488 anti-mouse CD207 (Langerin) antibody (clone eBioRMUL.2, 1.100; eBioscience, San Diego, CA, USA) and DAPI (Sigma-Aldrich) for 30 min. For double immunofluorescence stainings, the solution contained both anti-CD207 and anti-MIF antibodies, and detection was performed with Alexa Fluor 546–conjugated secondary antibodies. After repeated washing steps with PBS, probes were mounted with Mowiol 4-88 (Calbiochem, San Diego, CA, USA) and analyzed under a Zeiss Axio microscope (Ziess, Jena, Germany).
Intraepidermal MIF rescue
The dorsal skin of 5 male mice age 8 wk was shaved, and 3 areas for injection were marked along the median of their backs. One site served for the intraepidermal injection of the buffer control (50 µl NaPP), one for recombinant murine MIF (100 ng/50 µl), and one for the structural mutant MIF protein D44A-MIF (100 ng/50 µl). The endotoxin content of wild-type (WT) and D44A-MIF was very low (<10 pg LPS/µg protein). Intraepidermal injections were administered using 32-gauge needles (B. Braun, Bethlehem, PA, USA), and successful injections were confirmed by blister formation at the injection sites. Animals were euthanized 5 d post-injection. Skins were taken, and the areas of injection were excised, divided into halves, and fixed in 4% buffered formaldehyde (Roti Histofix; Carl Roth) for 24 h. Samples were processed and embedded in paraffin, and sequential sections were cut. For analysis of the presence of immune cells at the injection sites, only tissue sections that were closest to the corresponding location were taken. The numbers of CD74+ and CD74− cells were counted within the whole epidermal or dermal compartments evaluated.
Primary cell culture, MIF secretion, and skin equivalents
Primary keratinocytes and fibroblasts were prepared from sterile human skin samples and cultivated under regular culture conditions. For determination of MIF secretion, subconfluent primary cultures of keratinocytes or fibroblasts in 6-well dishes were washed with PBS twice and then cultured in either keratinocyte growth medium (Keratinocyte-SFM medium with l-glutamine, epidermal growth factor, and bovine pituitary extract) or fibroblast growth medium for 48 h. Supernatants were harvested and analyzed by a commercially available hMIF ELISA (DuoSet ELISA Development System; R&D Systems, Minneapolis, MN, USA).
Organic skin equivalents were constructed as previously described (28). DCs were prepared, cultivated as described (29), and seeded into the middle of a layer of human keratinocytes on top of a layer of dermal fibroblasts in 6-well plates. Dermal skin equivalents were covered with Keratinocyte Growth Medium (KGM; Lonza, Basel, Switzerland), DMEM (Thermo Fisher Scientific, Waltham, MA, USA) that contained 10% fetal calf serum and RPMI 1640 (Thermo Fisher Scientific) that was supplemented with 2% heat-inactivated autologous human serum (all media in a ratio of 1:1:1; 10 ml per well), 800 U/ml granulocyte-macrophage colony-stimulating factor, and 1000 U/ml IL-4 (R&D Systems). After 1 d of culturing, skin equivalents were lifted to the air–liquid interphase, and MIF (100 ng in 1 ml) or MIF plus anti-MIF antibody (100 ng/500 ng in 1 ml) were applied on the top of the model for a total of 7 or 14 d. Untreated models were maintained as controls. The reconstructs were harvested after 7 or 14 d, cut into pieces, and embedded in Tissue Tek optimum cutting temperature (OCT) compound (Sakura Finetek, Torrance, CA, USA) for cryosectioning.
MIF receptor expression on human primary keratinocytes
Primary keratinocytes were prepared from sterile human skin samples and cultivated under regular culture conditions. Keratinocytes were seeded at a density of 5 × 104 cells per 12-well plate and incubated for 24 h. Then, cells were stimulated with IFN-γ (20 ng/ml; PeproTech, Rocky Hill, NJ, USA) for 48 h, whereas control cells remained untreated. CXCR2 and CXCR4 receptor expression was detected by flow cytometry using FITC anti-CXCR2 and PE anti-CXCR4 mAbs and respective isotype controls (Becton Dickinson, Brea, CA, USA).
DC migration assay
Transwell cell migration experiments were carried out to prove whether CXCR2 mediates the chemotactic effect of hMIF in immature human DCs used in the 3-dimensional skin culture model mentioned above. Immature human DCs (2.5 × 105) were placed in the insert of Transwell cell migration chamber and preincubated with 100 nM of the selective CXCR2 inhibitor SB225002 (Calbiochem). Controls remained untreated. After 30 min, 100 ng/ml recombinant human MIF was added to the lower chamber. The percentage of cells that migrated to the lower chamber was determined after 6 h by manual counting using a Neubauer chamber.
Histology, immunohistochemistry, and immunofluorescence
Tissues were either fixed with 4% buffered formalin ON, then processed, embedded in paraffin or embedded in Tissue Tek OCT compound, and shock-frozen in isopentane and liquid nitrogen. Paraffin sections (4 μm) were dewaxed with xylene and rehydrated via a graded series of isopropanol and ethanol.
For determination of tumor types, tumor sections were stained with hematoxylin and eosin and evaluated by a blinded pathologist according to the current World Health Organization Classification Skin Tumors edition (http://www.iarc.fr/en/publications/pdfs-online/pat-gen/bb6/).
For immunohistochemistry, 1% H2O2 in methanol was used to block endogenous peroxidase activity, and citric acid antigen retrieval was performed for all antibodies—except for anti-MIF—by heating in CitraPlus (BioGenex, Fremont, CA, USA) for 1 min at 600 W in a microwave (Siemens, Washington, DC, USA).
Primary antibodies against B220 (clone RA3-6B2; 1:200; Becton Dickinson), CD44 (clone IM7; 1:400; Becton Dickinson), CD68 (clone KP1; 1:800; Novus Biologicals, Littleton, CO, USA), CD74 (clone In-1; 1:200, Becton Dickinson), CD163 (clone 10D6; 1:400; Thermo Fisher Scientific), CXCR2 (clone 5E8; 1:400; Novus Biologicals), JAB1 (clone 42; 1:200; Becton Dickinson), and MIF (clone FL-115; 1:400; Santa Cruz Biotechnology, Santa Cruz, CA, USA) were detected by using biotin-labeled secondary antibodies (clone E0433: goat anti-mouse-IgG/biotinylated; clone E0432: goat anti-rabbit IgG/biotinylated; or clone E0468: rabbit anti-rat IgG/biotinylated; Dako, Carpinteria, CA, USA) in conjunction with streptavidin peroxidase (in 25% of the concentration of the secondary antibodies; Zymed, South San Francisco, CA, USA) and a colorimetric read-out based on either 3-amino-9-ethylcarbazole or 3,3′-diaminobenzidine (Dako). Ready-to-use background-reducing antibody diluent solution (Dako) was used as solvent for all antibodies. Counterstaining of nuclei was done using Meyer’s hematoxylin (Sigma-Aldrich).
For immunofluorescent detection of proteins in paraffin-embedded material, the following antibodies were used in a 1:50 dilution: anti-CD1a (clone CBT6; Novus Biologicals), anti-CD3 ζ (AnaSpec), anti-CD19 (clone LS-C112467; LifeSpan), F4/80 (clone CI:A3-1; Abcam, Cambridge, MA, USA), and anti-p27 (clone 57; Becton Dickinson). In accordance with the intended color combinations, different Alexa Fluor–coupled secondary antibodies were combined and DAPI (Sigma-Aldrich) was used as nuclear counterstain. All secondary antibodies for immunofluorescent detection were purchased from Thermo Fisher Scientific.
For immunofluorescent detection of proteins, 4-µm cryosections were thawed to room temperature, fixed in ice-cold acetone at 4°C for 10 min, washed with PBS, and incubated with primary antibodies in a dilution of 1:50 at room temperature for 30 min. After staining with secondary antibodies/DAPI for 30 min and washing, samples were mounted in Mowiol 4-88. For immunofluorescence of the 3-dimensional skin models, 4-µm cryosections were processed as previously described (28). Sections were stained with antibodies against CD1a (clone BL6, 1:25; Abnova) and CD80 (clone MAB 104; 1:20; Abnova). For control stainings, either isotype mAbs were used or the primary antibody was omitted as indicated.
Statistical analysis
Tumor incidence during skin carcinogenesis experiments was analyzed using Cox proportional hazards regression models and log-rank statistics. Tumors per mouse over time were analyzed using repeated-measures ANOVA adjusted for multiple comparisons by the Tukey-Kramer procedure. Other statistical analyses for given time points were done using a Student’s t test or the Pearson’s χ2 test as indicated. A value of P < 0.05 was considered significant.
RESULTS
The epidermis: an abundant source of MIF
The epidermis is a known source of MIF production (30–32). Immunohistochemical detection of MIF in normal murine and human skin samples revealed that keratinocytes of the epidermis strongly express MIF in a constitutive fashion. MIF expression in both mice and humans was most prominent in the stratum basale and granulosum and became weaker with increasing keratinization of the cells in the stratum spinosum and corneum (Fig. 1A). MIF was also expressed in the epithelial component of skin appendices, such as hair follicles and sebaceous glands (Fig. 1B). Mif-deficient mice do not show any abnormality of skin or hair structure and the thickness of Mif−/− epidermis was not different from that of Mif+/+ mice. In both mice and humans, skin tumors diagnosed as actinic keratoses, keratoakanthomas (both premalignant lesions), and squamous cell carcinomas also strongly express MIF (Fig. 1D). Thus, expression of MIF in murine epidermis and epithelial tumors paralleled the pattern observed in humans. In human and murine skin, the epidermal layer stains much more intensely for MIF than the dermal layer by immunohistochemistry (IHC) and immunofluorescence, and constitutive expression of MIF in keratinocytes is higher than in fibroblasts (Fig. 1A, B). In subconfluent cultures of human primary keratinocytes and dermal fibroblasts, more MIF is secreted into the supernatant from keratinocytes than fibroblasts (MIF concentration in supernatant: mean ± sd: keratinocytes 1363 ± 3 ng/ml vs. dermal fibroblasts 725 ± 9 ng/ml; Fig. 1C). The difference between the secretory activity of keratinocytes and fibroblasts suggests that there is a gradient of MIF secretion from the epidermis (MIFhigh) to the dermis (MIFlow) in normal skin.
Figure 1.

MIF expression in human and murine skin. A) MIF is constitutively expressed in untreated normal human skin. Representative section stained by IHC [3-amino-9-ethylcarbazole (AEC), reddish brown]; original magnification, ×400. Inset shows negative control staining with anti-MIF antibody omitted. Epidermal keratinocytes show more pronounced MIF staining than dermal fibroblasts. B) MIF is constitutively expressed in untreated normal murine skin. Representative section of murine skin stained by immunofluorescence. MIF expression is much stronger in epidermis and epithelial skin appendices, such as hair follicles, compared with dermal structures. MIF visualized by Alexa Fluor 488 (green), nuclei counterstained by DAPI (blue); original magnification, ×100. C) MIF secretion in growth medium by subconfluent primary human dermal fibroblasts or keratinocytes after 48 h. Keratinocytes secrete twice as much MIF as dermal fibroblasts into the supernatant. D) IHC for MIF (AEC, reddish brown) in human and murine nonmelanoma skin tumors. Top: human tumors; bottom: murine tumors; left: keratosis (precancerous lesion); middle: keratoakanthoma (benign tumor); right: squamous cell carcinomas (SCC; malignant tumor). Insets: control without anti-MIF mAb. Counterstain with hematoxylin; original magnification, ×100.
Mif-deficient mice are more sensitive to tumor formation in chemical skin carcinogenesis
Because MIF was present at all stages of human and murine skin carcinogenesis, we hypothesized that this molecule potentially promoted skin carcinogenesis. We therefore tested the functional consequences of Mif deletion by using Mif-knockout (Mif−/−) mice, in which the entire Mif gene had been deleted by Cre-mediated excision in C57Bl/6 embryonic stem (ES) cells (14). As the C57Bl/6 background is not sensitive to the tumorigenic action of DMBA/TPA, we backcrossed these mice for 6 generations to the more tumor-sensitive 129S1/SvImJ background. DNA damage in murine epidermis was initiated in age- and sex-matched Mif+/+ and Mif−/− mice after the first postnatal hair cycle by a single application of the aromatic polycyclic carcinogen DMBA, followed by tumor promotion during 20 wk with the phorbol ester TPA.
Tumor incidence induced by DMBA/TPA was accelerated in Mif−/− mice compared with WT controls (first tumor in Mif−/− in wk 7 vs. Mif+/+ in wk 11; 50% tumor incidence in Mif−/− in wk 11 vs. Mif+/+ in wk 14; 100% tumor incidence Mif−/− in wk 14 vs. Mif+/+ in wk 20; Fig. 2A, upper left). In statistical terms, this corresponds to a 2-fold-higher likelihood of Mif−/− mice to develop a tumor compared with Mif+/+ mice (hazard ratio, 2.4; CI, 0.9–6.2; P < 0.036). Regarding tumors per mouse, both groups were similar until wk 12, but starting at wk 13 until 20, Mif−/− mice exhibited a statistically significant increase in number of tumors compared with Mif+/+ (tumors per mouse mean ± sd: Mif−/− 11 ± 5 vs. Mif+/+ 5 ± 4; P < 0.0001; Fig. 2A, upper right). Tumor size was not different between genotypes (data not shown). Absence of MIF also altered the spectrum of resulting epithelial tumors: whereas most tumors were histologically classified as low-grade neoplasias (keratosis, akanthosis, akanthoma, papilloma) and appeared in both genotypes, Mif−/− mice developed more keratoses and benign tumors arising from the hair follicle. Pilar tumors and trichoadenomas were only observed in Mif−/− mice (Table 1). The observation that genetic deletion of MIF leads to increased tumor formation was unexpected, as all other cancer mouse models studied to date had shown that MIF promotes tumor formation and metastasis (17–21). To ensure that this observation was not a result of specific features of the 129S1/SvImJ background, we sought to confirm this phenotype in another model of chemical skin carcinogenesis.
Figure 2.

Mif deficiency leads to increased tumor formation in chemical and HPV8-induced murine skin carcinogenesis. A) Chemical skin carcinogenesis with DMBA/TPA or B[α]P. Tumor incidence (left) and tumors per mouse (right) in Mif+/+ and Mif−/− mice (n = 10) topically treated with 25 µg DMBA on d 1 and with 50 µg TPA biweekly for 20 wk. Mif−/− mice developed skin tumors significantly earlier and in increased number (top). Tumor incidence (left) and tumors per mouse (right) in Mif+/+ and Mif−/− mice (n = 20) topically treated with 100 µg B[α]P twice per week. Mif−/− mice developed skin tumors significantly earlier and in increased number (bottom). B) HPV8-induced skin carcinogenesis. K14-HPV8-CER+/tg; Mif−/− mice developed tumors at a faster rate than did K14-HPV8-CER+/tg; Mif+/+ controls [hazard ratio (CI), 3.971 (0.868–18.168)]; however, this result was not yet statistically significant. P = 0.0543.
TABLE 1.
Histologic classification of epithelial lesions induced by chemical carcinogenesis in Mif-deficient and control mice
| Histopathologic diagnosis | DMBA/TPA |
B[α]P |
||
|---|---|---|---|---|
| Mif+/+ | Mif−/− | Mif+/+ | Mif−/− | |
| Basal cell carcinoma | — | — | — | 9 |
| Squamous cell carcinoma | 1 | — | 7 | 27 |
| Keratosis | 9 | 23 | 1 | 8 |
| Keratoakanthoma | 1 | — | 3 | 13 |
| Akanthosis | 3 | 4 | 1 | 3 |
| Papilloma | 3 | 2 | 1 | 2 |
| Cyst | 2 | 3 | — | 2 |
| Dysplasia | — | — | 1 | — |
| Pila tumor | — | 5 | — | — |
| Trichoadenoma | — | 4 | — | — |
| Hair follicle hyperplasia | — | 3 | — | — |
| Sebaceous hyperplasia | 2 | 3 | — | — |
| Total number of findings | 21 | 47 | 14 | 65 |
Skin condition spectra according to World Health Organization classification of epithelial skin tumors in the DMBA/TPA and the B[α]P chemical carcinogenesis models as assessed by a pathologist blinded to genotype. In the DMBA/TPA experiment, incidence of keratoses and tumors arising from hair follicles (pilar tumor, trichoadenoma) in Mif−/− mice was significantly increased compared with Mif+/+ mice (χ2 test). P < 0.0001.
Benzo[α]pyrene (B[α]P), an aromatic polycyclic carbohydrate found in tobacco smoke and exhaust fumes, has a higher mutagenic potential than DMBA and is therefore suitable for inducing tumors in the C57Bl/6 strain. The metabolized epoxide of B[α]P not only induces Ras but also p53 mutations and subsequently leads to development of both benign and malignant tumors (33). Mif+/+ and Mif−/− mice age 6–8 wk were treated weekly with topical B[α]P on their shaved backs, and formation of skin tumors was monitored over a period of 32 wk. Similar to the DMBA/TPA experiment in the 129S1/SvImJ strain, incidence of epithelial skin tumors was strongly enhanced in Mif-deficient mice compared with WT controls (first tumor in Mif−/− in wk 17 vs. Mif+/+ in wk 26; 50% tumor incidence Mif−/− in wk 26 vs. Mif+/+ not reached), and Mif−/− mice developed 10-fold more tumors than did respective controls (tumors per mouse mean ± sd: Mif−/− 5 ± 6 vs. Mif+/+ 0.4 ± 0.9; Fig. 2A, bottom). In statistical terms, these data indicate that in the B[α]P 1-stage carcinogenesis experiment, C57Bl/6 Mif−/− mice are more than 6 times more likely to develop a tumor than are Mif+/+ controls (hazard ratio, 6.4; CI 2.3–17.7; P < 0.001). Again, tumor size was not different between genotypes (data not shown).
Mif deficiency also accelerates epithelial skin tumor formation in Keratin14-HPV8-CER transgenic mice
To exclude the possibility that the apparent tumor-suppressive action of MIF is limited to chemical carcinogenesis, we next tested the impact of Mif gene deletion in a second model of skin carcinogenesis. In the K14-HPV8-CER mouse model, epithelial tumors are induced by transgenic overexpression of the complete early region of the HPV8 genome, with the oncogenes E2, E6, and E7 under control of the cytokeratin 14 promoter 129S1/SvImJ (34). Mif−/− mice were crossed with epidermis-specific Keratin14-HPV8-CER-transgenic mice, and K14-HPV8-CER+/tg; Mif+/+ and K14-HPV8-CER+/tg; Mif−/− offspring in the F2 generation were monitored with respect to spontaneous development of skin tumors over a period of 52 wk. In keeping with our findings in chemical carcinogenesis, animals of the K14-HPV8-CER+/tg; Mif−/− cohort developed epithelial tumors at a significantly faster rate than did K14-HPV8-CER+/tg; Mif+/+ controls (first tumor K14-HPV8-CER+/tg; Mif−/− in wk 15 vs. K14-HPV8-CER+/tg; Mif+/+ in wk 51; (P = 0.0003; Fig. 2B). In Mif-deficient mice, we observed both benign papillomas and malignant squamous cell carcinomas. Because we only had 2 tumors in the K14-HPV8-CER+/tg; Mif+/+ control group, a formal comparison of the histology was not performed.
Taken together, all in vivo carcinogenesis experiments using chemical carcinogenesis with 2 different carcinogens and transgenic overexpression of the HPV8 oncogenes consistently demonstrated that MIF exhibits tumor suppressor activity in nonmelanoma skin cancer. We concluded that the tumor-suppressor properties of MIF are not likely to be dependent on the model, but rather a result of MIF biology in the epidermis. Subsequently, we chose the chemical carcinogenesis model to further elucidate the mechanism of action of MIF in skin tumor formation.
MIF receptors CD74, CXCR2, and CXCR4 are not expressed on keratinocytes
Secreted MIF transmits its extracellular signal via surface receptors, such as the high-affinity CD74/CD44 complex (11, 35) or CXCR2/CXCR4 (12). Next, we tested whether all components of the MIF signaling pathway were present in epidermal cells. Although the MIF coreceptor CD44 is present constitutively on keratinocytes (Fig. 3A), it cannot transmit a MIF signal without the MIF-binding partner CD74. However, CD74 was not detectable on keratinocytes but was only expressed on immune cells, such as Langerhans cells (LCs), macrophages, and lymphocytes (Fig. 3B–D). Although it has been previously reported that in vitro stimulation with IFN-γ may lead to weak expression of CD74 on keratinocytes (36), we were not able to observe detectable CD74 in keratinocytes by immunohistochemistry in our samples of normal skin or malignant tumors. Even irritation of the epidermis via topical application of TPA once or over several days did not lead to any de novo expression of CD74 in keratinocytes (data not shown). Flow cytometry studies showed that the alternative MIF receptors CXCR2 and CXCR4 were also not expressed constitutively on neonatal human epidermal keratinocytes. Upon stimulation with IFN-γ, CXCR2 remained absent and CXCR4 was only detectable at a very low level (Fig. 3F). Given the almost complete absence of MIF receptor expression on keratinocytes, CD74+ immune cells—which also are reported to express higher levels of the alternative MIF receptors—seem to be the primary target cells for the effects of keratinocyte-derived MIF.
Figure 3.
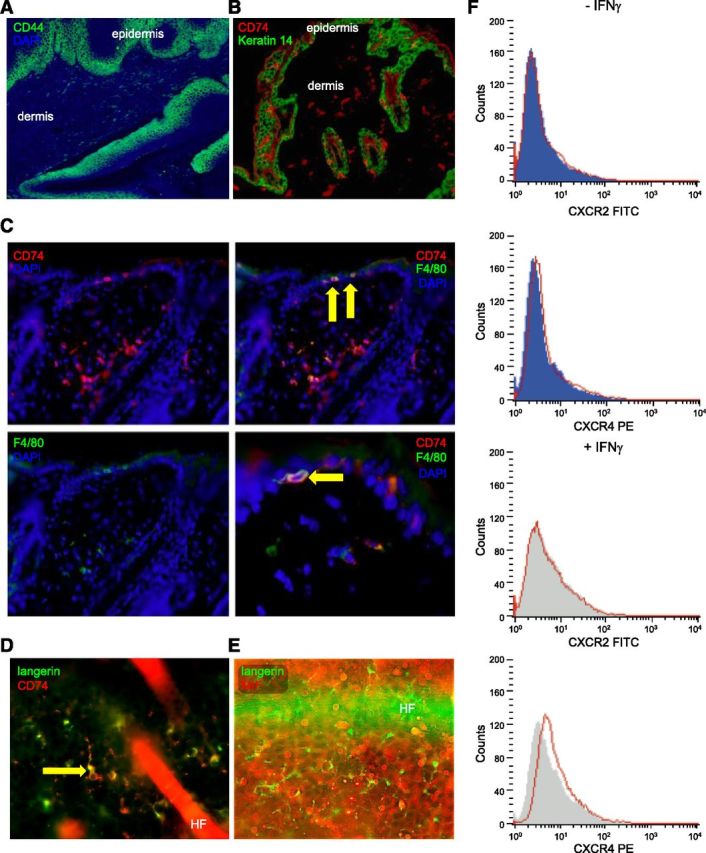
Mif receptor expression and association of MIF deficiency with reduced epidermal infiltration by immune cells. A) Representative immunofluorescence for CD44 (Alexa Fluor 488, green) with DAPI nuclear counterstain (blue) demonstrating strong and constitutive expression of CD44 in the basal layers of untreated WT murine epidermis; original magnification, ×100. B) Representative IHC for expression of CD74 (Alexa Fluor 546, red) and Keratin 14 (Alexa Fluor 488, green) in skin of Mif+/+ mice. CD74 is absent on keratinocytes, but clearly expressed on immune cells infiltrating epidermis and dermis; original magnification, ×100. C) Representative double-IHC for expression of CD74 (Alexa Fluor 546, red) and F4/80 (Alexa Fluor 488, green) in skin of Mif+/+ mice. DAPI was used as nuclear counterstain (blue). Arrows show intraepidermal F4/80+ cells with coexpression of CD74. Original magnification, ×200 for all panels but lower right, which is magnified ×400. D) Immunofluorescence for langerin (Alexa Fluor 488, green) and CD74 (Alexa Fluor 546, red) with DAPI counterstain in murine epidermis demonstrating that LCs coexpress CD74. Original magnification, ×400. E) Double-immunofluorescence for langerin (Alexa Fluor 488, green) and MIF (Alexa Fluor 546, red) in untreated murine epidermis demonstrating that langerin+ cells do not express MIF. Original magnification, ×400. F) Expression of CXCR2 and CXCR4 by flow cytometry on primary normal human keratinocytes in vitro with and without prior stimulation by IFN-γ (20 ng/ml, 48 h). No expression of CXCR2 and CXCR4 is seen on keratinocytes.
Mif deficiency leads to reduced accumulation of APCs in skin
Skin is highly exposed to external pathogens and harmful environmental agents, and, therefore, a specialized system of immune surveillance is required to maintain immunologic protection. Immune cells, such as DCs/macrophages and T and B cells, have the ability to patrol through the epidermis and dermis and to return to lymphoid organs. Specific to the epidermis are LCs, a specialized subset of DCs that form a network within the epidermis that can be visualized by immunofluorescent staining of langerin (CD207), a type II transmembrane cell surface receptor. LCs are not the only cellular components of innate immunity in skin: macrophages (langerin negative) are also present in the dermis adjacent to the epidermal-dermal junction. Chemical carcinogenesis with DMBA/TPA or B[α]P induces a pronounced inflammatory reaction, and the number of inflammatory cells is known to be strongly increased in the epidermis and epidermal-dermal junction during chemical carcinogenesis experiments (37).
MIF receptor CD74 is expressed on most immune cell types and its presence suggests that cells may be MIF responsive. Therefore, we investigated whether loss of MIF in Mif−/− mice had an impact on the presence of CD74+ cells at the end of the chemical carcinogenesis experiment. CD74 was chosen as an indicator for the presence of MIF receptor–bearing cells as its expression was readily detectable by IHC. In all skin compartments analyzed (epidermis and dermis, epithelial tumor and stroma of tumor), the number of CD74+ cells was significantly reduced in Mif−/− animals (Fig. 4, top). Next, we sought to determine what type of immune cell was affected by the absence of MIF. Double IHC stainings revealed that many of the CD74+ cells were F4/80+, thus qualifying them as macrophages/DCs or LCs if located within the epidermis (Fig. 3C). On the basis of IHC, we quantified the presence of F4/80+ (macrophages/DCs/LCs), langerin+ cells (CD207), B220+ (all B cells) and CD3+ (all T cells) in skin/tumor sections of the DMBA/TPA experiment. Loss of MIF was associated with a 3- to 10-fold numerical reduction of F4/80+ cells both in treated, nontumor-bearing dermis and in epithelial parts of tumors, whereas cell numbers in the epidermis did not differ significantly (F4/80+ cells/mm2 in nontumor-bearing dermis Mif+/+ 58 ± 12 vs. Mif−/− 20 ± 11; P = 0.0012; in epithelial parts of tumor Mif+/+ 12 ± 4 vs. Mif−/− 1 ± 1; P = 0.002; Fig. 4, second row from top). Whereas the number of langerin+ (CD207) DCs within the nontumor-bearing epidermis was significantly reduced in Mif−/− mice and a similar trend was observed for squamous tumors, there was a nonsignificant converse trend toward higher numbers of langerin+ cells in the dermis of Mif−/− mice, which suggests that these cells may lack the ability to cross the basal membrane in the absence of MIF (nontumor-bearing epidermis: Mif+/+ 16 ± 2 vs. Mif−/− 9 ± 4 langerin+ cells/mm2; normal dermis: Mif+/+ 2 ± 2 vs. Mif−/− 7 ± 2 langerin+ cells/mm2; epithelial part of tumor: Mif+/+ 11 ± 4 vs. Mif−/− 2 ± 2 langerin+ cells/mm2; stromal part of tumor: Mif+/+ 30 ± 8 vs. Mif−/− 24 ± 4 langerin+ cells/mm2; Fig. 4, middle row).
Figure 4.
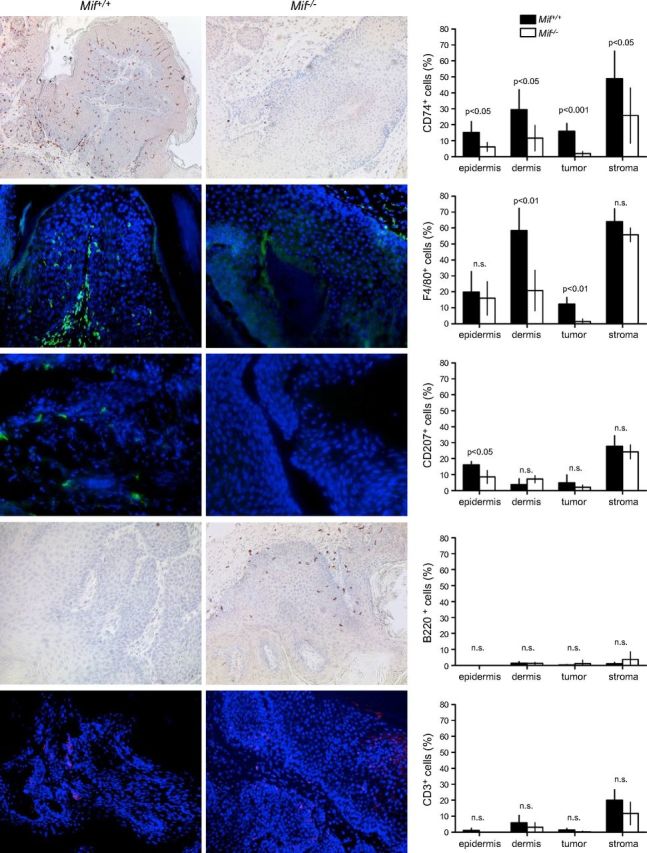
IHC analysis of immune cell infiltrates in DMBA/TPA-treated murine skin and DMBA/TPA-induced murine keratosis and corresponding statistics for each marker (right). Pictures of Mif+/+ control mouse tissues (left). Pictures of Mif−/− mice (middle). Original magnification, ×100. Epidermis, DMBA/TPA-treated, but nontumor-bearing epidermis; dermis, DMBA/TPA-treated, but nontumor-bearing dermis; tumor, epithelial part of the DMBA/TPA-induced keratosis; stroma, stromal part of the DMBA/TPA-induced keratosis. Mif+/+ indicated by solid bars; Mif−/− indicated by open bars. Percentage of positive cells was calculated by number of positive cells divided by total number of cells. For every tumor and tissue, total available cells were counted. HF, hair follicle; n.s., not significant.
CD3+ T cells were rare in the nontumor-bearing epidermis/dermis and the epithelial portion of the tumors (< 5%), but were frequently present at the junction between epithelial tumor and stroma (Mif+/+ 20 ± 4 vs. Mif−/− 12 ± 4 CD3+ cells/mm2; Fig. 4, bottom row). The number of B220+ B cells was negligible in the epidermis and epithelial parts of tumors. Dermal and stromal parts of tumors contained slightly more B cells, but there was no significant difference between the skin of Mif+/+ and Mif−/− animals (Fig. 4, second row from bottom).
The lower density of langerin+ cells in the epidermis of Mif−/− mice was independently confirmed in epithelial sheets taken from ears of untreated mice. Mif−/− mice showed a significantly reduced number of langerin+ LCs compared with WT controls (Mif+/+ 38 ± 23 vs. Mif−/− 17 ± 10 LCs per 20 high-power fields; P < 0.001), and the network of langerin+ cells in the epidermis was rarified in the absence of MIF. Although macrophages are known to produce MIF, we could not observe detectable levels of MIF expression in langerin+ cells by IHC (Fig. 3E). Thus, we conclude that MIF production and secretion in skin is mostly a result of synthesis of MIF in keratinocytes, not in immune cells.
Taken together, our data indicate that loss of MIF is associated with a reduction in APCs in the epithelial parts of murine skin and tumors.
MIF acts as a chemokine for dendritic cells in vitro and in vivo
Establishment of 3-dimensional skin culture models offers a unique opportunity to study the effects of a chemotactic molecule. As 3-dimensional skin models have been established for human, but not murine cells, we chose to study the chemotactic effects of MIF on human DCs in a human 3-dimensional skin model. Human peripheral blood mononuclear cells were differentiated ex vivo to CD1ahigh, CD80intermediate, CD86low, CD83–, CXCR2intermediate, CXCR4–, CD74high, and CD14– DCs and were seeded into the middle of a layer of human keratinocytes on top of a layer of dermal fibroblasts. Once the epidermal layer had stratified, daily stimulation with recombinant human MIF (100 ng in 1 ml of PBS), MIF plus anti-MIF antibody NIHIII.D.9 (100 ng/500 ng in 1 ml), or PBS (control) was applied on top of the epidermal layer for a total of 7 or 14 d. When treated with control or MIF/anti-MIF, DCs migrated from the epidermal to the dermal layer and successively disappeared from the model by day 14. In contrast, stimulation with recombinant hMIF alone stopped emigration of DCs from the organotypic model. On d 7, and even on d 14, the CD1a+/CD80+ population of DCs remained clearly present at the epidermal-dermal junction and in the dermal layer (Fig. 5A).
Figure 5.
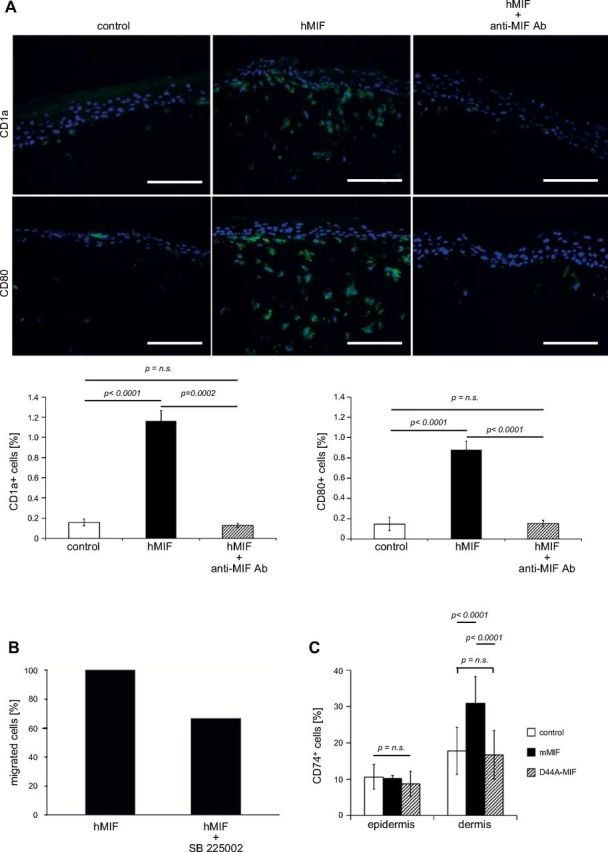
MIF acts as chemokine for DCs in human 3-dimensional skin equivalents. A) Normal human epidermal keratinocytes, fibroblasts, and DCs were grown as 3-dimensional organotypic skin equivalents and stimulated with MIF (100 ng/ml) and MIF plus anti-MIF antibody (500 ng/ml) for a total of 7 d. Untreated models were maintained as control. Skin equivalents were harvested after 7 d and histologic sections were stained for CD1a and CD80 by immunofluorescence. The number of DCs in histologic sections was analyzed semiquantitatively for frequency of CD1a+ and CD80+ signals using the multifluorescence imaging and processing software Cell F (Olympus, Tokyo, Japan). Representative example from 3 independent experiments is shown. DCs spontaneously migrate out of the skin equivalent, but are retained in the presence of hMIF on top of the epidermal layer. Neutralizing anti-MIF mAb abrogates the effect of hMIF. Original magnification, ×200. Scale bar, 100 µm. B) In vitro chemotaxis experiments using Transwell cell migration chambers: 2 × 105 immature human DCs were placed in the insert of the Transwell system and preincubated with 100 nM SB225002. Controls remained untreated. After 30 min, 100 ng/ml recombinant hMIF was added to the lower chamber. The percentage of cells migrated to the lower chamber was determined after 6 h. Human MIF-mediated DC migration (set as 100%) is reduced by preincubation with the CXCR2 chemokine receptor antagonist SB225002 by 34%. C) Mif−/− mice (n = 5) were each injected intraepidermally in 3 separate locations with either 50 µl that contained mMIF (100 ng/ml), 50 µl that contained mD44A-MIF (100 ng/ml), or 50 µl NaPP buffer as vehicle control. After 72 h, mice were euthanized and the skin part of injections excised. In sequential sections through the sites of injection, the relative proportion of CD74+ cells of all cells were counted. The injection of mMIF, but not of the mutant D44A-MIF, resulted in an accumulation of CD74+ cells. n.s., not significant. P < 0.001.
It is known that human tumors use the chemokine receptor CXCR2 to inhibit DCs from migrating out of the tumor (38). Therefore, in vitro chemotaxis experiments using Transwell cell migration chambers were carried out to prove whether CXCR2 mediates the chemotactic retention effect of human MIF on the same immature human DCs used in the 3-dimensional skin culture model above. DCs were placed in the insert of the Transwell system and migration was stimulated by addition of human MIF (100 ng/ml). After 6 h, 24% of DCs had migrated in response to MIF. Of note, preincubation of DCs with the selective, competitive CXCR2 inhibitor SB225002 (100 nM) reduced this MIF-induced cell migration by 34% (Fig. 5B).
To test whether CXCR2-mediated inhibition of immune cell migration was also operative in the mouse in vivo, we intraepidermally injected recombinant murine MIF (2 μg/ml), the murine D44A-MIF mutant (mD44A-MIF; 2 μg/ml), or vehicle control into the back skin of Mif−/− mice (n = 5). The D44A mutation lacks binding activity for CXCR2 (39). Whereas mD44A-MIF or control-injected mice failed to significantly increase the number of CD74+ cells in the dermal area of injected mice, only WT MIF was able to lead to accumulation of CD74+ cells (P < 0.0001; Fig. 5C). These results support the concept that MIF attracts APCs and also demonstrate that CXCR2 may be operative in MIF-mediated signaling for macrophage recruitment in vivo.
Generation of an epidermis-specific Mif-knockout mouse
Next, we sought to determine more definitively whether the phenotype we observed in the complete MIF knockout was a result of loss of MIF produced in the epidermis. The Mif gene locus was targeted in C57Bl/6 ES cells by using a targeting vector in which the entire Mif gene was flanked by 2 loxP sites (promoter plus all exons/introns and poly A; Fig. 6A) (14). After successful selection of homologous integrants by neomycin selection/thymidine kinase enrichment, the selection cassette was removed by transient transfection with a Cre-expressing plasmid in vitro. ES cell clones in which only the loxP flanked neomycin resistance marker, but not the Mif locus was removed, were selected, verified by Southern blotting (Fig. 6B), and finally injected into BALB/c blastocysts. Resulting chimeric mice were bred with C57Bl/6 WT mice until germline transmission from ES cell clones was achieved. Miffl/fl mice (genotyped by PCR; Fig. 6C) were phenotypically normal and showed normal fertility and behavior. MIF mRNA and protein expression in skin and organs of Miffl/fl mice was comparable to that of WT C57Bl/6 mice (Fig. 6D, E). To generate an epidermis-specific Mif knockout, we bred homozygous C57Bl/6 Miffl/fl mice with female K14-Cre transgenic mice (40) on a C57Bl/6 background. K14 (cytokeratin 14) promoter leads to expression of Cre recombinase in cells of the basal layer of the epidermis, thereby effectively deleting the Mif gene in all cells of the epidermis (Fig. 6A), which can be detected by genotyping PCR (Fig. 6F). As expected, K14-Cre+/tg; Miffl/fl showed a complete loss of MIF protein by IHC and immunofluorescence in the epidermal layer, whereas MIF expression remained unaffected in immune cells and dermis (Fig. 7A). The epidermis, however, was structurally unaffected by loss of MIF and showed a thickness and differentiation pattern that was comparable to the nontransgenic Miffl/fl controls.
Figure 6.
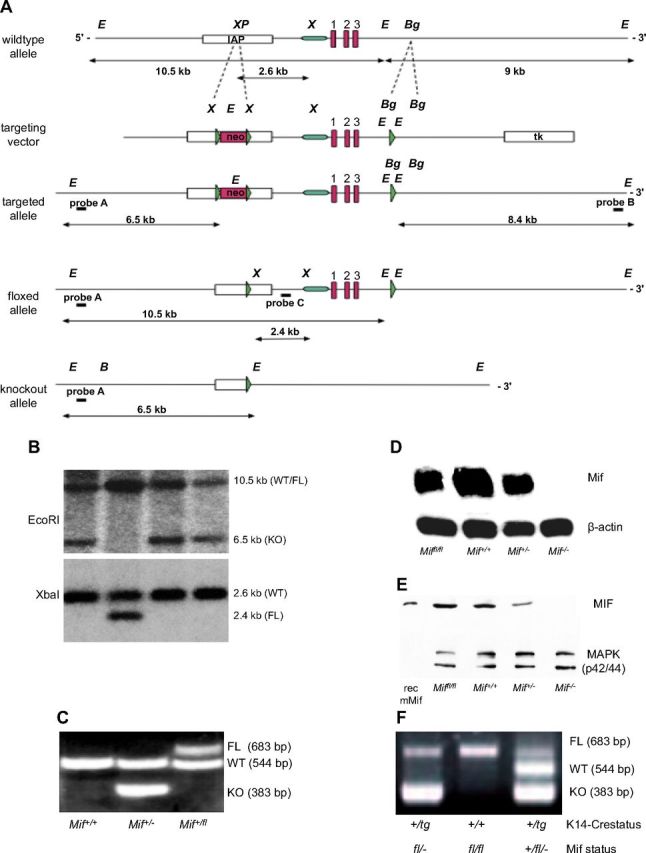
Absence of epidermal MIF, but not of MIF receptors CD74 and CD44, reproduces the phenotype of the complete Mif knockout. A) Design and initial targeting of C57Bl/6 ES cells is reported in the previous work (14). After transient Cre-expression in targeted ES cells, in vitro clones, ES cell clones with floxed Mif (FL), and deletion of the neo selection cassette were identified by sensitivity to GA101. Neo-deleted, floxed MIF ES cells were injected into BALB/c blastocysts and resulting chimeras were bred to C57Bl/6 mice to obtain pure C57Bl/6 Miffl/+ mice. Homozygous Miffl/fl females were crossed with K14-Cre transgenic males to achieve epidermis-specific K14-Cre+/tg; Mif−/− mice. B) Selection of Miffl/fl ES cells after transient transfection with a Cre-expressing plasmid by restriction fragment length polymorphism of EcoRI (top) and XbaI (bottom) digested genomic DNA and southern blotting with an external/internal probe. EcoRI-digest and external probe A in G418-sensitive clones: 10.5-kb band indicates the WT or floxed allele, 6.5-kb indicates deletion of both Mif and Neo. XbaI-digest and internal probe C: 2.6-kb band indicates WT allele, 2.4-kb band corresponds to floxed allele. C) Representative example of the genotyping PCR differentiating Mif+/+, Mif+/−, and Mif+/fl mice. D) Northern blotting for MIF mRNA from skin of Miffl/fl, Mif+/+, Mif+/−, and Mif−/− mice. β-Actin mRNA used as loading control. E) Western blotting for MIF protein from skin of Miffl/fl, Mif+/+, Mif+/−, and Mif−/− mice. MAPK used as loading control. F) Representative example of the genotyping PCR from tail DNA in K14-Cre transgenic and K14-Cre-negative Miffl/fl mice. Presence of the K14-driven Cre recombinase leads to deletion of the mif gene in keratinocytes visualized by the presence of the 383-bp band.
Figure 7.
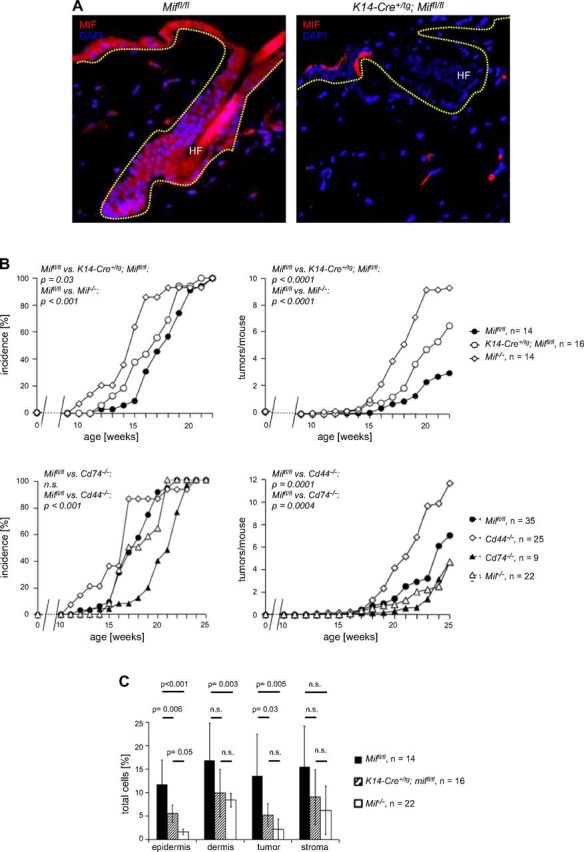
A) Immunofluorescence for MIF (Alexa Fluor 546, red) in skin from a Miffl/fl (top) and an epidermis-specific Mif-knockout mouse (K14-Cre+/tg; Miffl/fl) (bottom). Magnification, ×100. B) Epidermal deletion of Mif, but not deletion of the MIF receptor complex Cd74/Cd44, partially recapitulates increased tumorigenesis seen in the complete Mif knockout. Tumor incidence and tumors per mouse in B[α]P-induced 1-stage carcinogenesis experiments. Comparison of Miffl/fl (control), K14-Cre+/tg; Miffl/fl (epidermis-specific Mif knockout), and Mif−/− mice (top). Comparison of Miffl/fl (control) with Mif−/−, Cd74−/−, and Cd44−/− mice (bottom). C) Statistical evaluation of the IHC analysis of F4/80+ immune cell infiltrate in DMBA/TPA-treated murine skin and DMBA/TPA-induced murine keratosis. Epidermis, DMBA/TPA-treated, but nontumor-bearing epidermis; dermis, DMBA/TPA-treated, but nontumor-bearing dermis; tumor, epithelial part of the DMBA/TPA-induced keratosis; stroma, stromal part of the DMBA/TPA-induced keratosis. Miffl/fl (control) indicated by solid bars; K14-Cre+/tg Miffl/fl (epidermis-specific Mif knockout) indicated by striped bars; Mif−/− (complete knockout) indicated by open bars. Percentage of positive cells was calculated by number of positive cells divided by number of all cells (>1000 cells counted). n.s., not significant.
Loss of epidermal MIF partially recapitulates the phenotype of increased tumor formation and reduced macrophage accumulation in chemical skin carcinogenesis
Next, we tested whether epidermal MIF was required for macrophage accumulation in epidermis and dermis in vivo. Whereas K14-Cre+/tg; Miffl/fl mice had normal numbers of monocytes/macrophages in peripheral blood and spleen, IHC using either CD74 or F4/80 showed that the number of macrophages in epidermis and dermis was reduced significantly compared with nontransgenic control Miffl/fl mice (data not shown). We subjected C57Bl/6 keratinocyte-specific (K14-Cre+/tg; Miffl/fl) and complete (Mif−/−) knockout mice and Miffl/fl controls to the B[α]P 1-stage carcinogenesis protocol and determined the incidence, number, and size of tumors over time. Tumor incidence was accelerated in complete and epidermis-specific Mif-knockout mice compared with Miffl/fl controls (time to first tumor: Miffl/fl in wk 12 vs. K14-Cre+/tg; Miffl/fl in wk 12 vs. Mif−/− in wk 10; 50% tumor incidence Miffl/fl in wk 18 vs. K14-Cre+/tg; Miffl/fl in wk 17 vs. Mif−/− in wk 15; P = 0.03; Fig. 7B, upper left). Tumors per mouse were significantly increased in the epidermis-specific Mif knockout compared with Miffl/fl controls (P < 0.0001), but not quite as strongly as in Mif−/− mice (tumors per mouse at 22 wk: Miffl/fl 3 ± 2, K14-Cre+/tg; Mif−/− 7 ± 3, Mif−/− 9 ± 5; Fig. 7B, upper right). As already observed in complete Mif−/−, the average size of resulting tumors in keratinocyte-specific Mif knockouts was not significantly different from floxed Mif controls.
In all dermal compartments, both the keratinocyte-specific and the complete Mif knockout showed reduced numbers of CD74+ and F4/80+ cells compared with Miffl/fl controls at the end of the B[α]P chemical carcinogenesis experiment. Reductions of cell numbers exceeded factor two and were generally more pronounced in the complete Mif−/− compared with keratinocyte-specific Mif knockout. Of interest, as already observed in the DMBA/TPA tumorigenesis experiments (Fig. 4), absence of MIF had a stronger effect within the epidermis and epithelial parts of the tumors and remained a nonsignificant trend in dermis and stromal parts of the tumors (Fig. 7C). Taken together, these results demonstrated that keratinocyte-derived MIF and not only MIF in immune cells is an essential mediator for migration of immune cells to skin.
Epidermal deletion of Mif, but not deletion of the MIF receptor complex Cd74/Cd44, recapitulates increased tumorigenesis seen in the complete Mif knockout
The MIF receptor complex CD74/CD44 is present on immune cells and potentially also on IFN-γ–activated keratinocytes. MIF secreted by epidermal keratinocytes may mediate immune cell activation via CD74 and thereby strengthen immune surveillance of skin tumor formation. If immune cell activation via the MIF-CD74/CD44 axis was the relevant mechanism, genetic deletion of Cd74 or Cd44 should produce a phenotype similar to the Mif complete knockout. To test this hypothesis, we subjected C57Bl/6 Cd74- and Cd44-knockout mice to B[α]P-induced skin carcinogenesis in comparison with Mif−/− mice and Miffl/fl control mice. Cd74−/− mice were very similar to Miffl/fl control mice with respect to tumor incidence (first tumor Cd74−/− in wk 15 vs. Miffl/fl in wk 12; 50% tumor incidence in Miffl/fl and Cd74−/− in wk 17; 100% tumor incidence Cd74−/− in wk 20 vs. Miffl/fl in wk 21; P = 0.92; Fig. 7B, bottom left), tumors per mouse (in wk 22: Cd74−/− 2 ± 0.7 vs. Miffl/fl 3 ± 2; P = 0.04; Fig. 7B, lower right). Cd44−/− mice showed an opposite phenotype with delayed tumor formation (first tumor Miffl/fl in week 12 vs. Cd44−/− in wk 13, 50% tumor incidence Miffl/fl in wl 17 vs. Cd44−/− in wk 21 and 100% tumor incidence Miffl/fl in wk 21 vs. Cd44−/− in week 24; P < 0.001; Fig. 7B, lower left) and reduced number of tumors per mouse (wk 25: Miffl/fl 3 ± 2 vs. Cd44−/− 1.3 ± 1; P = 0.0001; Fig. 7B, lower right). These results suggest that the effects of MIF in chemical skin tumorigenesis are not mediated by the MIF receptor CD74 or its coreceptor CD44. It was not possible to test CXCR2 and CXCR4 in a similar fashion as genetic deletion of Cxcr4 is embryonically lethal, and deletion of Cxcr2 leads to very ill mice, which cannot be bred any further. Conditional-knockout models for these chemokines are not yet available.
Overall, our results demonstrate that epidermal keratinocytes produce chemotactic MIF, which is important for recruitment of APCs to the dermis and epidermis (model in Fig. 8). Increased nonmelanoma skin tumor formation in mice deficient in epidermal MIF suggests that immune cells recruited to skin with the help of MIF exhibit an important role in antitumor immunity within the epidermis.
Figure 8.

Model of the action of MIF as chemokine in skin tumorigenesis. Keratinocytes in the epidermis produce and secrete chemotactic MIF, which helps to attract cells of the innate system to the dermis and epidermis.
DISCUSSION
The skin-resident immune system plays an important role in skin pathology, such as allergy, inflammation, infection, and tumor formation. Although it has been known for quite some time that keratinocytes strongly and constitutively express MIF and this is associated with inflammatory disorders (reviewed in ref. 41), the mode of action of MIF and its role for skin homeostasis and pathophysiology is largely unknown. We used global and epidermis-specific Mif-knockout mice in the setting of chemically induced skin tumorigenesis to investigate the biologic role of MIF in skin. Our results classify MIF as a functional tumor suppressor in skin. This finding was unexpected given that all other nonskin tumor models had shown that MIF increases malignant transformation, tumor growth, and metastatic potential (42). Moreover, in UVB-induced skin tumorigenesis, a 45% reduced tumor incidence in BALB/c Mif−/− mice and a corresponding increased tumor incidence in MIF transgenic mice had been reported (27, 43). Such alternative findings can be excellent opportunities to understand the critical actions of a given mediator in greater detail, such as in the case of TGF-β.
Several possible mechanisms for the mode of action of MIF in tumorigenesis have been described, such as inhibition of p53 transcriptional activity (44), stabilization of the cyclin-dependent kinase inhibitor p27Kip1 (45), activation of survival-promoting signaling pathways, neoangiogenesis, and several others (46). Given our results, none of these possible modes of action provides an explanation of how MIF deficiency in keratinocytes can result in increased nonmelanoma skin tumor formation. LCs convert DMBA into the mutagenic 3,4-diole, and LC-deficient skin has been shown to be resistant to DMBA-induced tumorigenesis (9). As we observe reduced numbers of LCs in the epidermis of MIF-deficient mice, this is also not a likely explanation for our phenotype of enhanced tumor formation in the absence of MIF.
However, there is wide consensus that MIF promotes inflammatory responses, and this has been confirmed in numerous publications that have explored many different experimental settings and tissues. In skin, the effect of MIF on inflammation was measured by various means, such as simple cell counts in hematoxylin and eosin–stained sections, myeloperoxidase activity, dermal neutrophil or eosinophil influx, or edematous response, each supporting the validity of this concept in skin (27, 43, 47). The functional association of MIF with macrophage migration is also well established. Regulation of macrophage migration by lymphocyte-derived MIF led to the initial functional characterization of this mediator (10). MIF is an essential factor for delayed type hypersensitivity reactions (48) and for regulating neutrophil and eosinophil migration (47, 49). In atherosclerosis, MIF is required for monocyte/macrophage migration into atherosclerotic plaques (12).
Our work provides genetic proof that MIF, secreted by keratinocytes, acts as a regulator of APC numbers in dermis and epidermis under conditions of both normal skin homeostasis and chemically induced tumorigenesis. Our human 3-dimensional skin culture model supports this conclusion, as application of rMIF on top of the epidermal layer leads to retention of DCs seeded into the epidermal layer, and neutralizing anti-MIF antibodies revert to this phenotype. We also rescued dermal macrophage numbers to normal levels present in control mice by intraepidermally reinjecting MIF into the skin of Mif−/− mice. Hence, MIF is involved in the control of epidermal immune cell pools. On the basis of these findings, we propose the concept that MIF, in its function as a chemokine, recruits cells of the innate immune system to replenish the skin with APCs.
As IHC and in vitro experiments identify keratinocytes as main producers of MIF in skin, our results suggest that the spatial structure of skin generates a chemotactic MIF gradient from the epidermis to the dermis, which contributes to regulation of APC numbers in the skin. If keratinocytes are stimulated to produce and/or secrete more or less MIF, this gradient may be modulated to support an adaptive immune response toward the external challenge.
How does MIF recruit APCs? Several cytokines/chemokines are produced by epidermal keratinocytes and were shown to regulate trafficking of APCs under steady-state conditions or during inflammation (reviewed in ref. 1, 50): CXCL-14 (BRAK, breast and kidney expressed chemokine) and CCL-20 (MIP-3α) are both constitutively expressed in basal keratinocytes and support steady-state recruitment of DCs from blood. Under conditions of inflammation, MCP-1/CCL2 and IL-8/CXCL8 trigger transendothelial migration of monocyte subsets (51). It is known that MIF secretion leads to CXCL-8 production in B cells (24) and that CXCL-8 acts to retain DCs in skin or in malignant lesions (38). MIF also is known to induce the expression of cyclooxygenase-2 and to regulate prostaglandin E2 production, which is a key factor for CCR7 surface expression and migration of monocyte-derived DCs (52). Presence of MIF may also enhance the secretion of other chemokines as recently shown by us for the migration of B cells (21).
The biology of HPV8 also provides us with important clues about the relevance of chemokines for skin carcinogenesis: by suppressing CCL-20 expression in keratinocytes, HPV8 effectively inhibits LC migration to the epidermis and thereby creates a microenvironment favorable for tumorigenesis on the basis of disturbed innate immune control and viral persistence (53).
Given the importance of skin-residing APCs in a variety of pathophysiologic conditions, our results imply that the status of the MIF chemotactic gradient may have profound implications for many skin conditions in which recruitment of immune cells is part of the pathophysiologic events. Whereas the effect of MIF in wound healing is controversial (41) and may depend on the model system studied, little doubt exists that MIF generally increases inflammatory processes in skin as in atopic dermatitis or contact hypersensitivity, and the up-regulation of MIF expression in these conditions has been described (54, 55).
A number of mechanisms may account for the differing results we observed in chemical skin carcinogenesis and those reported in UVB irradiation. First, UVB irradiation has immune-suppressive effects and leads to apoptosis of immune effector cells, such as lymphocytes, macrophages, and LCs (reviewed in ref. 56). Therefore, UV-irradiation administered 3 times/wk over a long period, as required for UVB-induced skin carcinogenesis, may make it impossible to detect differences in recruitment of immune cells. In contrast, chemical skin carcinogenesis, especially the DMBA/TPA model, increases leukocyte infiltration to the skin, thereby enabling the chemotactic effects of MIF to be observed more easily than in UV carcinogenesis protocols. Second, the choice of the BALB/c mouse strain may influence the outcome in the UVB-induced skin carcinogenesis model, as BALB/c mice harbor an inactivating mutation of the cyclin-dependent kinase inhibitor p16INK4a (57), thus exhausting epidermal stem cell pools and increasing risks of associated cell-cycle alterations in keratinocytes.
Nevertheless, the link between chronic inflammation and tumorigenesis is not simple because certain types of inflammatory processes may promote tumor development, whereas others serve a tumor-suppressing function. A prime example of this is tumor-associated macrophages, which may possess tumor promoting (M1) or tumor suppressive activity (M2). On the one hand, macrophages promote tumorigenesis via their ability to produce a variety of inflammatory cytokines and chemokines that lead to increased survival, proliferation, neoangiogenesis, accelerated malignant transformation of initiated epithelial cells, and suppressed local immunity toward the tumor (58). On the other hand, APCs may take up cutaneous tumor antigens and elicit a T-cell–based immune response against the tumor after migration to regional lymph nodes (59). Mice deficient in T cells are known to be much more susceptible to regimens of skin carcinogenesis (60).
The fact that MIF may use several surface receptors to trigger its biologic effects makes its functional evaluation complicated. Our finding that Cd74−/− and Cd44−/− receptor-knockout mice do not exhibit increased numbers of tumors in chemical skin carcinogenesis experiments rules out the possibility that the phenotype of MIF is mediated via this receptor complex on leukocytes. We speculate that the effects of MIF on recruitment of APCs will be mediated via CXCR2 or CXCR4, as these receptors are also expressed on immune cells. Our observation that macrophage recruitment is impaired when using the D44A-MIF mutant as chemoattractant implicates CXCR2 in the process of macrophage recruitment via MIF and suggests that similar mechanisms of MIF-mediated leukocyte recruitment could be active in skin as in the arterial atherosclerotic vessel wall (39). Because genetic deletion of Cxcr4 is embryonically lethal, mice with deletion of Cxcr2 cannot be bred. Corresponding cell type–specific knockout models are not yet publicly available. Thus, we were not able to functionally identify the MIF receptor responsible for immune cell migration to skin.
Taken together, our results identify MIF as a relevant tumor suppressor in chemically induced nonmelanoma skin cancer. Keratinocyte-produced MIF acts as a chemotactic factor for the regulation of APC pools in the skin. Therefore, constitutive and inducible MIF expression by keratinocytes confers an important contribution to the status of skin immunity.
ACKNOWLEDGMENTS
The authors thank Prof. A. Sharpe (Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA) for helping with BALB/c blastocyst injections and the generation of chimaeric conditional MIF mice. The K14-Cre transgenic mice were obtained from Drs. C. Niessen and A. Roers (University Hospital Cologne), and from Prof. Dr. I. Förster (University of Düsseldorf, Düsseldorf, Germany). This work was supported by the Deutsche Forschungsgemeinschaft (German Research Council) Grant Fi 712/2 (to G.F.-R.), Grant BA1803/7-1 (to J.M.B.), Grant BE1977/4-2 and SFB1123-A03 (to J.B.), Grant A16 of SFB 832 (to M.H.), Grant R06/20 of the German José Carreras Leukemia Foundation (to G.F.-R.), Grant AI042310 (to R.B.), and by the START programs of the Medical Faculty of the Rheinisch–Westfälische Technische Hochschule Aachen (to J.M.B., R.H., and J.B.). The authors declare no conflicts of interest.
Glossary
- APC
antigen-presenting cell
- B[α]P
benzo[α]pyrene
- DC
dendritic cell
- DMBA
7,12-dimethylbenz[a]anthracene
- IHC
immunohistochemistry
- LC
Langerhans cell
- MIF
macrophage migration inhibitory factor
- TPA
12-O-tetradecanoylphorbol-13-acetate
- WT
wild-type
AUTHOR CONTRIBUTIONS
T. Brocks designed and executed experiments, analyzed data, and designed the figures; O. Fedorchenko, N. Schliermann, U. M. Moll, M. Dewor, M. Hallek, Y. Marquardt, K. Fietkau, R. Heise, S. Huth, J. Bernhagen, R. Bucala, designed and executed experiments; A. Stein analyzed the pathologic sections; S. Seegobin performed the statistical calculations; R. Bucala funded the generation of the MIF-knockout mice; H. Pfister generated the K14-HPV8-CER+/tg mice and participated in their analysis of tumor formation; and T. Brocks, M. Hallek, J. M. Baron, and G. Fingerle-Rowson designed experiments, analyzed data, and wrote the manuscript.
REFERENCES
- 1.Koch S., Kohl K., Klein E., von Bubnoff D., Bieber T. (2006) Skin homing of Langerhans cell precursors: adhesion, chemotaxis, and migration. J. Allergy Clin. Immunol. , 163–168 [DOI] [PubMed] [Google Scholar]
- 2.Randolph G. J., Ochando J., Partida-Sánchez S. (2008) Migration of dendritic cell subsets and their precursors. Annu. Rev. Immunol. , 293–316 [DOI] [PubMed] [Google Scholar]
- 3.Kabashima K., Shiraishi N., Sugita K., Mori T., Onoue A., Kobayashi M., Sakabe J., Yoshiki R., Tamamura H., Fujii N., Inaba K., Tokura Y. (2007) CXCL12-CXCR4 engagement is required for migration of cutaneous dendritic cells. Am. J. Pathol. , 1249–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen S. J., Crown S. E., Handel T. M. (2007) Chemokine: receptor structure, interactions, and antagonism. Annu. Rev. Immunol. , 787–820 [DOI] [PubMed] [Google Scholar]
- 5.Wang L., Bursch L. S., Kissenpfennig A., Malissen B., Jameson S. C., Hogquist K. A. (2008) Langerin expressing cells promote skin immune responses under defined conditions. J. Immunol. , 4722–4727 [DOI] [PubMed] [Google Scholar]
- 6.Mueller M. M. (2006) Inflammation in epithelial skin tumours: old stories and new ideas. Eur. J. Cancer , 735–744 [DOI] [PubMed] [Google Scholar]
- 7.Lewis J., Filler R., Smith D. A., Golubets K., Girardi M. (2010) The contribution of Langerhans cells to cutaneous malignancy. Trends Immunol. , 460–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts S. J., Ng B. Y., Filler R. B., Lewis J., Glusac E. J., Hayday A. C., Tigelaar R. E., Girardi M. (2007) Characterizing tumor-promoting T cells in chemically induced cutaneous carcinogenesis. Proc. Natl. Acad. Sci. USA , 6770–6775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Modi B. G., Neustadter J., Binda E., Lewis J., Filler R. B., Roberts S. J., Kwong B. Y., Reddy S., Overton J. D., Galan A., Tigelaar R., Cai L., Fu P., Shlomchik M., Kaplan D. H., Hayday A., Girardi M. (2012) Langerhans cells facilitate epithelial DNA damage and squamous cell carcinoma. Science , 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.David J. R. (1966) Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc. Natl. Acad. Sci. USA , 72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leng L., Metz C. N., Fang Y., Xu J., Donnelly S., Baugh J., Delohery T., Chen Y., Mitchell R. A., Bucala R. (2003) MIF signal transduction initiated by binding to CD74. J. Exp. Med. , 1467–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernhagen J., Krohn R., Lue H., Gregory J. L., Zernecke A., Koenen R. R., Dewor M., Georgiev I., Schober A., Leng L., Kooistra T., Fingerle-Rowson G., Ghezzi P., Kleemann R., McColl S. R., Bucala R., Hickey M. J., Weber C. (2007) MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. , 587–596 [DOI] [PubMed] [Google Scholar]
- 13.Mitchell R. A., Liao H., Chesney J., Fingerle-Rowson G., Baugh J., David J., Bucala R. (2002) Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc. Natl. Acad. Sci. USA , 345–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fingerle-Rowson G., Petrenko O., Metz C. N., Forsthuber T. G., Mitchell R., Huss R., Moll U., Müller W., Bucala R. (2003) The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc. Natl. Acad. Sci. USA , 9354–9359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bucala R., Donnelly S. C. (2007) Macrophage migration inhibitory factor: a probable link between inflammation and cancer. Immunity , 281–285 [DOI] [PubMed] [Google Scholar]
- 16.Greven D., Leng L., Bucala R. (2010) Autoimmune diseases: MIF as a therapeutic target. Expert Opin. Ther. Targets , 253–264 [DOI] [PubMed] [Google Scholar]
- 17.Talos F., Mena P., Fingerle-Rowson G., Moll U., Petrenko O. (2005) MIF loss impairs Myc-induced lymphomagenesis. Cell Death Differ. , 1319–1328 [DOI] [PubMed] [Google Scholar]
- 18.Wilson J. M., Coletta P. L., Cuthbert R. J., Scott N., MacLennan K., Hawcroft G., Leng L., Lubetsky J. B., Jin K. K., Lolis E., Medina F., Brieva J. A., Poulsom R., Markham A. F., Bucala R., Hull M. A. (2005) Macrophage migration inhibitory factor promotes intestinal tumorigenesis. Gastroenterology , 1485–1503 [DOI] [PubMed] [Google Scholar]
- 19.Schulz R., Marchenko N. D., Holembowski L., Fingerle-Rowson G., Pesic M., Zender L., Dobbelstein M., Moll U. M. (2012) Inhibiting the HSP90 chaperone destabilizes macrophage migration inhibitory factor and thereby inhibits breast tumor progression. J. Exp. Med. , 275–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor J. A. III, Kuchel G. A., Hegde P., Voznesensky O. S., Claffey K., Tsimikas J., Leng L., Bucala R., Pilbeam C. (2007) Null mutation for macrophage migration inhibitory factor (MIF) is associated with less aggressive bladder cancer in mice. BMC Cancer , 135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reinart N., Nguyen P. H., Boucas J., Rosen N., Kvasnicka H. M., Heukamp L., Rudolph C., Ristovska V., Velmans T., Mueller C., Reiners K. S., von Strandmann E. P., Krause G., Montesinos-Rongen M., Schlegelberger B., Herling M., Hallek M., Fingerle-Rowson G. (2013) Delayed development of chronic lymphocytic leukemia in the absence of macrophage migration inhibitory factor. Blood , 812–821 [DOI] [PubMed] [Google Scholar]
- 22.Lue H., Thiele M., Franz J., Dahl E., Speckgens S., Leng L., Fingerle-Rowson G., Bucala R., Lüscher B., Bernhagen J. (2007) Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene , 5046–5059 [DOI] [PubMed] [Google Scholar]
- 23.Lue H., Kapurniotu A., Fingerle-Rowson G., Roger T., Leng L., Thiele M., Calandra T., Bucala R., Bernhagen J. (2006) Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cell. Signal. , 688–703 [DOI] [PubMed] [Google Scholar]
- 24.Gore Y., Starlets D., Maharshak N., Becker-Herman S., Kaneyuki U., Leng L., Bucala R., Shachar I. (2008) Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J. Biol. Chem. , 2784–2792 [DOI] [PubMed] [Google Scholar]
- 25.Petrenko O., Fingerle-Rowson G., Peng T., Mitchell R. A., Metz C. N. (2003) Macrophage migration inhibitory factor deficiency is associated with altered cell growth and reduced susceptibility to Ras-mediated transformation. J. Biol. Chem. , 11078–11085 [DOI] [PubMed] [Google Scholar]
- 26.Petrenko O., Moll U. M. (2005) Macrophage migration inhibitory factor MIF interferes with the Rb-E2F pathway. Mol. Cell , 225–236 [DOI] [PubMed] [Google Scholar]
- 27.Martin J., Duncan F. J., Keiser T., Shin S., Kusewitt D. F., Oberyszyn T., Satoskar A. R., VanBuskirk A. M. (2009) Macrophage migration inhibitory factor (MIF) plays a critical role in pathogenesis of ultraviolet-B (UVB) -induced nonmelanoma skin cancer (NMSC). FASEB J. , 720–730 [DOI] [PubMed] [Google Scholar]
- 28.Neis M. M., Wendel A., Wiederholt T., Marquardt Y., Joussen S., Baron J. M., Merk H. F. (2010) Expression and induction of cytochrome p450 isoenzymes in human skin equivalents. Skin Pharmacol. Physiol. , 29–39 [DOI] [PubMed] [Google Scholar]
- 29.Cornelissen C., Brans R., Czaja K., Skazik C., Marquardt Y., Zwadlo-Klarwasser G., Kim A., Bickers D. R., Lüscher-Firzlaff J., Lüscher B., Baron J. M. (2011) Ultraviolet B radiation and reactive oxygen species modulate interleukin-31 expression in T lymphocytes, monocytes and dendritic cells. Br. J. Dermatol. , 966–975 [DOI] [PubMed] [Google Scholar]
- 30.Shimizu T., Abe R., Ohkawara A., Nishihira J. (1999) Ultraviolet B radiation upregulates the production of macrophage migration inhibitory factor (MIF) in human epidermal keratinocytes. J. Invest. Dermatol. , 210–215 [DOI] [PubMed] [Google Scholar]
- 31.Shimizu T., Ohkawara A., Nishihira J., Sakamoto W. (1996) Identification of macrophage migration inhibitory factor (MIF) in human skin and its immmunohistochemical localization. FEBS Lett. , 199–202 [DOI] [PubMed] [Google Scholar]
- 32.Fingerle-Rowson G., Koch P., Bikoff R., Lin X., Metz C. N., Dhabhar F. S., Meinhardt A., Bucala R. (2003) Regulation of macrophage migration inhibitory factor expression by glucocorticoids in vivo. Am. J. Pathol. , 47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruggeri B., DiRado M., Zhang S. Y., Bauer B., Goodrow T., Klein-Szanto A. J. (1993) Benzo[a]pyrene-induced murine skin tumors exhibit frequent and characteristic G to T mutations in the p53 gene. Proc. Natl. Acad. Sci. USA , 1013–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaper I. D., Marcuzzi G. P., Weissenborn S. J., Kasper H. U., Dries V., Smyth N., Fuchs P., Pfister H. (2005) Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res. , 1394–1400 [DOI] [PubMed] [Google Scholar]
- 35.Shi X., Leng L., Wang T., Wang W., Du X., Li J., McDonald C., Chen Z., Murphy J. W., Lolis E., Noble P., Knudson W., Bucala R. (2006) CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity , 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heise R., Vetter-Kauczok C. S., Skazik C., Czaja K., Marquardt Y., Lue H., Merk H. F., Bernhagen J., Baron J. M. (2012) Expression and function of macrophage migration inhibitory factor in the pathogenesis of UV-induced cutaneous nonmelanoma skin cancer. Photochem. Photobiol. , 1157–1164 [DOI] [PubMed] [Google Scholar]
- 37.Moon D. C., Nakayama J., Urabe A., Terao H., Kinoshita N., Hori Y. (1992) Immunohistochemical characterization of cellular infiltrates in epidermal tumors induced by two-stage and complete chemical carcinogenesis in mouse skin. J. Dermatol. , 146–152 [DOI] [PubMed] [Google Scholar]
- 38.Feijoo E., Alfaro C., Mazzolini G., Serra P., Penuelas I., Arina A., Huarte E., Tirapu I., Palencia B., Murillo O., Ruiz J., Sangro B., Richter J. A., Prieto J., Melero I. (2005) Dendritic cells delivered inside human carcinomas are sequestered by interleukin-8. Int. J. Cancer , 275–281 [DOI] [PubMed] [Google Scholar]
- 39.Weber C., Kraemer S., Drechsler M., Lue H., Koenen R. R., Kapurniotu A., Zernecke A., Bernhagen J. (2008) Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc. Natl. Acad. Sci. USA , 16278–16283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hafner M., Wenk J., Nenci A., Pasparakis M., Scharffetter-Kochanek K., Smyth N., Peters T., Kess D., Holtkötter O., Shephard P., Kudlow J. E., Smola H., Haase I., Schippers A., Krieg T., Müller W. (2004) Keratin 14 Cre transgenic mice authenticate keratin 14 as an oocyte-expressed protein. Genesis , 176–181 [DOI] [PubMed] [Google Scholar]
- 41.Gilliver S. C., Emmerson E., Bernhagen J., Hardman M. J. (2011) MIF: a key player in cutaneous biology and wound healing. Exp. Dermatol. , 1–6 [DOI] [PubMed] [Google Scholar]
- 42.Conroy H., Mawhinney L., Donnelly S. C. (2010) Inflammation and cancer: macrophage migration inhibitory factor (MIF)--the potential missing link. QJM , 831–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Honda A., Abe R., Yoshihisa Y., Makino T., Matsunaga K., Nishihira J., Shimizu H., Shimizu T. (2009) Deficient deletion of apoptotic cells by macrophage migration inhibitory factor (MIF) overexpression accelerates photocarcinogenesis. Carcinogenesis , 1597–1605 [DOI] [PubMed] [Google Scholar]
- 44.Hudson J. D., Shoaibi M. A., Maestro R., Carnero A., Hannon G. J., Beach D. H. (1999) A proinflammatory cytokine inhibits p53 tumor suppressor activity. J. Exp. Med. , 1375–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kleemann R., Hausser A., Geiger G., Mischke R., Burger-Kentischer A., Flieger O., Johannes F. J., Roger T., Calandra T., Kapurniotu A., Grell M., Finkelmeier D., Brunner H., Bernhagen J. (2000) Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature , 211–216 [DOI] [PubMed] [Google Scholar]
- 46.Bifulco C., McDaniel K., Leng L., Bucala R. (2008) Tumor growth-promoting properties of macrophage migration inhibitory factor. Curr. Pharm. Des. , 3790–3801 [DOI] [PubMed] [Google Scholar]
- 47.Yoshihisa Y., Makino T., Matsunaga K., Honda A., Norisugi O., Abe R., Shimizu H., Shimizu T. (2011) Macrophage migration inhibitory factor is essential for eosinophil recruitment in allergen-induced skin inflammation. J. Invest. Dermatol. , 925–931 [DOI] [PubMed] [Google Scholar]
- 48.Bernhagen J., Bacher M., Calandra T., Metz C. N., Doty S. B., Donnelly T., Bucala R. (1996) An essential role for macrophage migration inhibitory factor in the tuberculin delayed-type hypersensitivity reaction. J. Exp. Med. , 277–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santos L. L., Fan H., Hall P., Ngo D., Mackay C. R., Fingerle-Rowson G., Bucala R., Hickey M. J., Morand E. F. (2011) Macrophage migration inhibitory factor regulates neutrophil chemotactic responses in inflammatory arthritis in mice. Arthritis Rheum. , 960–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Imhof B. A., Aurrand-Lions M. (2004) Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. , 432–444 [DOI] [PubMed] [Google Scholar]
- 51.Muller W. A. (2001) New mechanisms and pathways for monocyte recruitment. J. Exp. Med. , F47–F51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scandella E., Men Y., Gillessen S., Förster R., Groettrup M. (2002) Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood , 1354–1361 [DOI] [PubMed] [Google Scholar]
- 53.Sperling T., Ołdak M., Walch-Rückheim B., Wickenhauser C., Doorbar J., Pfister H., Malejczyk M., Majewski S., Keates A. C., Smola S. (2012) Human papillomavirus type 8 interferes with a novel C/EBPβ-mediated mechanism of keratinocyte CCL20 chemokine expression and Langerhans cell migration. PLoS Pathog. , e1002833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimizu T. (2005) Role of macrophage migration inhibitory factor (MIF) in the skin. J. Dermatol. Sci. , 65–73 [DOI] [PubMed] [Google Scholar]
- 55.Shimizu T., Abe R., Nishihira J., Shibaki A., Watanabe H., Nakayama T., Taniguchi M., Ishibashi T., Shimizu H. (2003) Impaired contact hypersensitivity in macrophage migration inhibitory factor-deficient mice. Eur. J. Immunol. , 1478–1487 [DOI] [PubMed] [Google Scholar]
- 56.Timares L., Katiyar S. K., Elmets C. A. (2008) DNA damage, apoptosis and langerhans cells--activators of UV-induced immune tolerance. Photochem. Photobiol. , 422–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang S., Ramsay E. S., Mock B. A. (1998) Cdkn2a, the cyclin-dependent kinase inhibitor encoding p16INK4a and p19ARF, is a candidate for the plasmacytoma susceptibility locus, Pctr1. Proc. Natl. Acad. Sci. USA , 2429–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mantovani A., Allavena P., Sica A., Balkwill F. (2008) Cancer-related inflammation. Nature , 436–444 [DOI] [PubMed] [Google Scholar]
- 59.Vesely M. D., Kershaw M. H., Schreiber R. D., Smyth M. J. (2011) Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. , 235–271 [DOI] [PubMed] [Google Scholar]
- 60.Girardi M., Glusac E., Filler R. B., Roberts S. J., Propperova I., Lewis J., Tigelaar R. E., Hayday A. C. (2003) The distinct contributions of murine T cell receptor (TCR)gammadelta+ and TCRalphabeta+ T cells to different stages of chemically induced skin cancer. J. Exp. Med. , 747–755 [DOI] [PMC free article] [PubMed] [Google Scholar]