

Abstract
Mutations of cystic fibrosis transmembrane conductance regulator (CFTR), an epithelial ligand-gated anion channel, are associated with the lethal genetic disease cystic fibrosis. The CFTR G551D mutation impairs ATP hydrolysis and thereby makes CFTR refractory to cAMP stimulation. Both wild-type (WT) and G551D CFTR have been implicated in regulatory volume decrease (RVD), but the underlying mechanism remains incompletely understood. Here, we show that the channel activity of both WT and G551D CFTR is directly stimulated by mechanical perturbation induced by cell swelling at the single-channel, cellular, and tissue levels. Hypotonicity activated CFTR single channels in cell-attached membrane patches and WT-CFTR-mediated short-circuit current (Isc) in Calu-3 cells, and this was independent of Ca2+ and cAMP/PKA signaling. Genetic suppression and ablation but not G551D mutation of CFTR suppressed the hypotonicity- and stretch-induced Isc in Calu-3 cells and mouse duodena. Moreover, ablation but not G551D mutation of the CFTR gene inhibited the RVD of crypts isolated from mouse intestine; more importantly, CFTR-specific blockers markedly suppressed RVD in both WT- and G551D CFTR mice, demonstrating for the first time that the channel activity of both WT and G551D CFTR is required for epithelial RVD. Our findings uncover a previously unrecognized mechanism underlying CFTR involvement in epithelial RVD and suggest that the mechanosensitivity of G551D CFTR might underlie the mild phenotypes resulting from this mutation.—Xie, C., Cao, X., Chen, X, Wang, D., Zhang, W. K., Sun, Y., Hu, W., Zhou, Z., Wang, Y., Huang, P. Mechanosensitivity of wild-type and G551D cystic fibrosis transmembrane conductance regulator (CFTR) controls regulatory volume decrease in simple epithelia.
Keywords: hypotonicity, ion channel, mechanosensitive, osmotic swelling
A cell’s volume is perturbed by fluctuations in extracellular or intracellular osmolarity. Mammalian cells face osmotic challenges even under normal physiologic conditions: whereas intracellular osmolarity might change as a result of transepithelial transport and accumulation of nutrients or metabolic waste products, extracellular osmolarity could vary because of exposure of intestinal epithelia to anisosmotic luminal fluids upon food/liquid ingestion, or prolonged experience by kidney medulla cells of rapid osmotic changes. Cell volumes also change under various pathologic conditions. Cells might swell during hypoxia/ischemia, hyponatremia, hypothermia, hyperkalemia, and intracellular acidosis/diabetic ketoacidosis. The constancy of cell volume is pivotal for cell function and survival: volume changes induced by a fluctuation in extracellular or intracellular osmolarity alter the hydration, structure, and membrane integrity of the cell. Specifically, enzymes concentrations/metabolic rates, intracellular ion concentrations/transporter activities, cell migration, proliferation, and differentiation, apoptosis, cell excitability/contraction, and hormone/transmitter release are all altered by cell volume shifts (1), which could, if not counterbalanced, damage and ultimately kill the cell. Cells have therefore developed strategies such as regulatory volume decrease (RVD) to withstand these perturbations.
Cystic fibrosis transmembrane conductance regulator (CFTR) is a critically important anion channel in various epithelia, including those in the lung, pancreas, bile duct, intestinal tract, and reproductive tract. Mutations of CFTR are associated with the lethal genetic disease cystic fibrosis (CF). CFTR is a multifunctional protein, and CFTR also regulates other transport proteins through mechanisms that remain incompletely elucidated. CFTR is traditionally viewed as an intracellular ligand-gated channel because its gating is subject to ATP hydrolysis and phosphorylation. Recently, we found that CFTR is also mechanosensitive and is directly activated by stretch (2, 3). However, the physiologic significance of CFTR mechanosensitivity remains largely unexplored.
Both wild-type (WT) and G551D mutant CFTR have been implicated in epithelial RVD (4–7). G551D CFTR is correctly processed to the plasmalemma but is refractory to cAMP stimulation because the mutation impairs ATP hydrolysis. CFTR was proposed to mediate RVD by modulating swelling-activated K+ conductance and thereby acting as a regulator of ATP release, rather than by conducting Cl− efflux as an ion channel (4–7). However, whether CFTR functioning as a channel can sense the mechanical perturbation induced by cell swelling has not been explored. Because CFTR is activated by stretch, we hypothesized that WT CFTR mediates epithelial RVD by directly responding to membrane stretch induced by hypotonicity. We further hypothesized that G551D CFTR functions similarly as WT CFTR because CFTR activation by stretch is independent of ATP binding and phosphorylation (2). Here, we show that both WT- and G551D CFTR channels are directly activated by mechanical perturbation induced by cell swelling and that they mediate Cl− efflux in epithelial RVD; this provides a previously unidentified mechanism underlying the involvement of CFTR in epithelial RVD. The preliminary account of this work has been presented at the second International Symposium of Mechanobiology (Okayma, Japan).
MATERIALS AND METHODS
Chemicals and reagents
All chemicals used in Ussing chamber and patch-clamp studies were purchased from Sigma-Aldrich (St. Louis, MO, USA), except for the CFTR-specific inhibitors CFTRi-172 and GlyH-101 (EMD Millipore, Billerica, MA, USA). Forskolin was prepared as a 1000-fold stock solution in ethanol, whereas all other chemicals were prepared as 1000-fold stock solutions in double-distilled water or DMSO, as appropriate. The vehicles at their final concentrations exerted no effects in any of the assays.
Cell culture, viral gene transfers, and CFTR mouse lines
Calu-3 cells were cultured as previously described for patch-clamp (8) and Ussing chamber (9) studies.
For knockdown experiments, double-stranded shRNA oligos targeting CFTR sequence 1 (GAAGTAGTGATGGAGAATGTA) and sequence 2 (AGAAGAAGAGGTGCAAGATAC) or a control sequence (ACGCATGCATGCTTGCTTT) unrelated to CFTR were cloned into the lentivirus transduction vector pLVTH (Addgene plasmid 12262). Green fluorescent protein (GFP), encoded in pLVTH under the control of a separate promoter, and was expressed as an indicator of shRNA expression. Lentivirus preparation, Calu-3 cell infection, and Western blot analysis procedures were as previously described (10, 11).
CFTR knockout (KO) mice (CFTRtm1Unc) (12), purchased from The Jackson Laboratory (Bar Harbor, ME, USA), and G551D CFTR mice, originally generated in B. Wainwright’s laboratory (13) (kindly provided by D. M. Bedwell, University of Alabama, Birmingham, AL, USA), and their WT controls were reared for 6 to 8 wk in the Animal and Plant Central Facility of the Hong Kong University of Science and Technology before experiments.
Patch-clamp recording
Experimental and data-acquisition/analysis procedures were the same as previously described (8). Both the bath and pipette solutions were initially an isotonic solution, containing 1 part of 300 mM mannitol and 2 parts of Tris-Cl buffer (in mM): 150 Tris-Cl, 2 MgCl2, 1 CaCl2, 5 HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 30 sucrose, 10 d-glucose; pH 7.4 was adjusted with Tris base. A 12% hypotonic challenge was induced by mixing 1 ml of the isotonic solution into the bath (331 mOsm/kg) with 1 ml of the hypotonic solution (252 mOsm/kg, the isotonic solution without mannitol). Negative pressures were applied to the back of the pipette manually through a 1 ml syringe attached to a 3-way stopcock (2); the pressures were monitored using a pressure transducer (PM01R; World Precision Instruments, Sarasota, FL, USA) and Axon pClamp9 software (Axon Instruments, Foster City, CA, USA).
Ussing chamber studies
Procedures used for recording the short-circuit current (Isc) and chloride current (ICl) in cultured Calu-3 cells and mouse duodena and applying negative pressures were the same as previously described (2), with a few modifications. For Isc experiments conducted without any hypotonic challenge in cultured cells and mouse duodenum tissues, both the mucosal and serosal baths contained Krebs–bicarbonate–Ringer (KBR) solution (in mM: 140 Na+, 120 Cl−, 5.2 K+, 25 HCO3−, 2.4 HPO42−, 0.4 HPO4−, 1.1 Ca2+, 1.2 Mg2+, 5.2 glucose; glucose was replaced with mannitol in the mucosal bath to avoid glucose-dependent Na absorption). The KBR solution was bubbled with 95% O2–5% CO2 at 37°C in the chambers and had a pH of 7.4. To assess apical ICl in isolation, the basolateral membrane was permeabilized by treating it for 20 to 30 min with 180 μg/ml nystatin, and a serosal-to-mucosal Cl− gradient was imposed by using a low-Cl− KBR (in mM: 140 Na+, 4.5 Cl−, 5.2 K+, 25 HCO3−, 2.4 HPO42−, 0.4 HPO4−, 1.1 Ca2+, 1.2 Mg2+, 115 gluconate, 5.2 mannitol) on the mucosal side and KBR on the serosal side. Complete permeabilization of the basolateral membrane was evidenced by recording a current consistent with the serosal-to-mucosal flow of negative charge (14). In Isc and ICl experiments involving hypotonic challenges, an isotonic bath solution (307 mOsm/kg), prepared by mixing 1 part of 300 mM mannitol and 2 parts of KBR (or low-Cl− KBR for ICl) (9), was added on both the mucosal and serosal sides. An 18.4% hypotonic challenge was introduced by replacing 2 out of 4 ml of the isotonic solution in the bath with the hypotonic buffer (194 mOsm/kg), which was prepared by omitting mannitol from the isotonic solution. The amplitudes of the pressure pulses used for Calu-3 cells and mouse duodena were 20 and 10 mmHg, respectively; the duration of the pressure pulses for Isc was 60 s; and the pressure pulse was released at the response plateau in all experiments. Test compounds were added to the apical side in all experiments unless indicated otherwise. In all experiments, peak current responses were measured and analyzed as previously described (2, 9).
RVD of Calu-3 and CHO cells
Volume measurements were obtained by using dissociated Calu-3 cells that had been plated 3 to 6 h earlier (15) and following the same procedure as previously described for T84 cells (16). The cells were bathed in an isotonic solution (in mM: 140 NaCl, 2.5 KCl, 0.5 MgCl2, 1.2 CaCl2, 10 HEPES, 5 glucose; pH 7.2, 302 mOsm/kg) and seeded onto coverslips, and 15 min later, the coverslips were mounted for imaging by using an LSM710 confocal microscope. The isotonic solution was replaced with a hypotonic solution (the isotonic solution without 50 mM NaCl; 220 mOsm/kg) and phase images of single cells were acquired at various intervals and analyzed by using ImageJ software (Image Processing and Analysis in Java; National Institutes of Health, Bethesda, MD, USA) with a plug-in for threshold color selection. The volume was calculated by assuming that each cell was a perfect sphere and then normalized to the zero time point (before the solution change).
For volume measurements of CHO cells, cells were transfected with pIRES–enhanced GFP (EGFP), pIRES-EGFP-CFTR, or pIRES-EGFP-G551D-CFTR. At 36 h after transfection, cells were digested with trypsin and resuspended in an isotonic buffer (in mM: 110 NaCl, 2.5 KCl, 1.2 CaCl2, 0.5 MgCl2, 5 glucose, 10 HEPES, 100 mannitol; pH 7.2, 345 mOsm/kg). Hypotonic shock was introduced by removing mannitol from the isotonic buffer (245 mOsm/kg). All other procedures were similar to those used for Calu-3 cells.
Crypt isolation and volume measurement
Small-intestinal crypts were isolated using procedures described previously (17), with slight modifications. Briefly, a 2-cm-long piece of duodenum was excised from 6- to 8-wk-old mice, washed through with ice-cold Hanks solution (in mM: 140 NaC1, 5 KC1, 1.3 CaC12, 0.5 MgCl2, 0.44 K2HPO4, 4.2 NaHCO3, 5.5 glucose, 10 HEPES; pH 7.2 with Tris at room temperature), and everted onto a 3-cm-long stainless steel rod (1.5 mm in diameter). The rod was incubated for 8.5 min in an EDTA (ethylenediaminetetraacetic acid)-Hanks bath (in mM: 30 Na-EDTA, 5 KC1, 60 HC1, 52 NaC1, 10 HEPES; pH 7.1 with Tris at room temperature), and then vibrated at 50 Hz in a Ca2+-free Hanks solution at 4°C; the isolated crypts were seeded into 6-well tissue culture plates. To ensure that the crypts adhered, the plate surface was precoated with 0.1% polyethyleneimine. Crypts were allowed to attach for 10 min in isotonic Hanks medium (2 parts of Hanks buffer mixed with 1 part of 300 mM mannitol; 332 mOsm/kg) and subsequently washed with 1 ml of the same solution.
To measure crypt volumes, crypts were examined under an inverted microscope (TE2000E-PFS; Nikon, Tokyo, Japan). Single cylindrical crypts were selected and photographed at 20 s intervals for 30 min. Hypotonicity was induced by replacing 1 part of mannitol with water in the isotonic Hanks medium in the bath (final osmolality: 257 mOsm/kg). Images of the crypts were analyzed for total area, and the volume of crypts was estimated, assuming a cylindrical shape, and normalized relative to that before the solution change.
Statistical analysis
Data are presented as means ± se. P values were determined by a 2-tailed Student's t test, and P < 0.05 was considered statistically significant.
RESULTS
Hypotonicity activated CFTR single channels in human airway Calu-3 epithelial cells
CFTR is activated by membrane stretch (2), but whether it is activated by hypotonicity is unknown; the response of CFTR to hypotonicity cannot be simply extrapolated from its stretch-activation and requires experimental testing, because osmotic stress and stretch are not the same stimulus (18, 19) and stretch-activated channels are not necessarily activated by cell swelling (20).
Similar to suction, mild (12%) hypotonicity in the bath solution robustly activated in cell-attached patches a channel that exhibited slow kinetics and a unitary conductance of ∼6 to 7 pS at a pipette potential of −60 mV, the same characteristics as those of CFTR channel (Fig. 1A, C) (2, 21). Moreover, this channel was sensitive to the CFTR-specific inhibitor GlyH-101 (Fig. 1A, B). These results suggest that hypotonicity activated CFTR single channels. Our previous study suggested that suction, particularly strong suction, also increased the unitary conductance of CFTR (2). However, 12% hypotonicity caused no significant changes in the unitary conductance of CFTR (Fig. 1C), which implies that this stimulus induced only mild mechanical stress on CFTR. In these experiments, no other types of hypotonicity-activated channels were detected.
Figure 1.
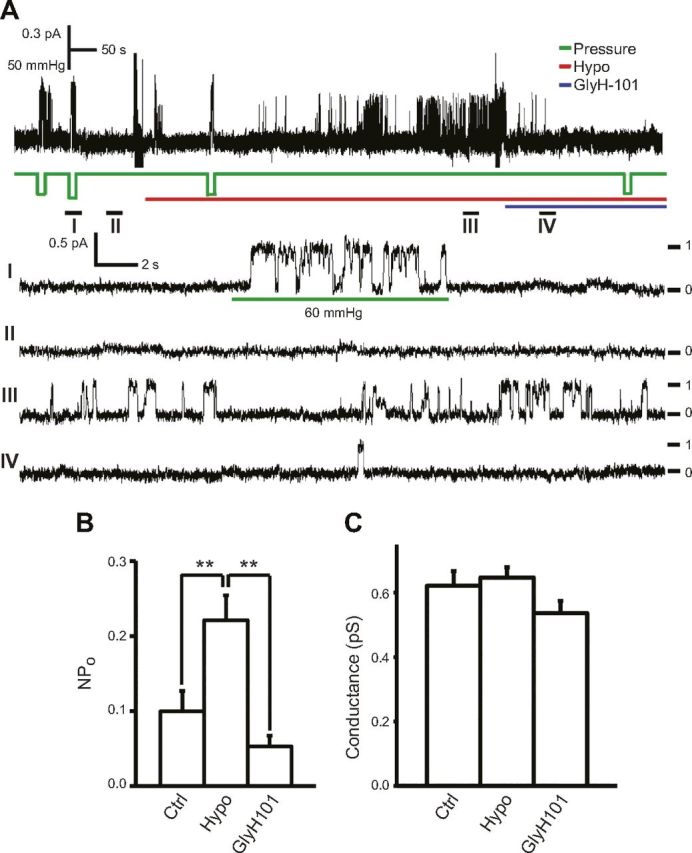
Hypotonicity activates CFTR single channels in cell-attached patches of Calu-3 cells. A, upper) representative trace of CFTR single-channel activation in response to suction applied to membrane patch (pressure, green line) and 12% hypotonicity (Hypo, red line) before and after 40 μM GlyH-101 (blue line) at −60 mV pipette potential. Bottom: time expansion of segments I to IV; numbers and dashes indicate number of active channels. Unitary conductance of CFTR activated by suction (60 mmHg) here is slightly larger than that of basal CFTR channel, as previously reported (2). B, C) Summary data of NPo (B) and unitary conductance (C) of CFTR channels in panel (n = 10). NPo of Ctrl, Hypo, and GlyH101 were calculated from 50 to 100 s recording before hypotonic challenge (Ctrl) and from 100 s segment (of 300 s) showing highest NPo after hypotonic challenge before (Hypo)/after (GlyH101) GlyH-101, respectively. **P = 0.004 and 0.002 for Hypo vs. Ctrl and GlyH101, respectively.
Mucosal hypotonicity activated apical CFTR in Calu-3 epithelial monolayers
The patch-clamp technique is a powerful method for studying the detailed kinetics of hypotonicity-activated channels. However, in single-channel studies, the process of forming a giga-seal might drastically perturb the native mechanical state of the membrane patches and change the behavior of resident channels (22–24); moreover, in whole-cell studies, dilution of the cytoplasm by the artificial pipette buffer might alter the physiologic state of ion channels. To avoid these problems, we examined hypotonicity-induced CFTR activation by using the Ussing chamber technique, which is comparatively less invasive to the cell membrane and is most physiologically relevant in terms of maintaining the mechanical state of the membrane and the intracellular environment.
First, we applied hypotonicity mucosally in order to imitate mucosal osmotic changes encountered by simple epithelial cells in a physiologic or therapeutic setting (25–27). Mucosal hypotonicity of 18.4% elicited a robust and sustained rise in the Isc (ΔIsc), with the current amplitudes and channel-inhibitor sensitivities being similar to those in ΔIsc activated by forskolin, a classic activator of CFTR (Fig. 2A–D). Among the panel of inhibitors used, CFTRinh-172 (CFTRi) is a CFTR-specific inhibitor that does not affect the volume-sensitive outwardly rectifying chloride channel (VSOR) (28), which was recently suggested to be composed of LRRC8 family proteins (29, 30); glybenclamide blocks both CFTR and VSOR (31, 32); and DIDS (4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid) and tamoxifen are classic VSOR inhibitors that exert little effect on CFTR (33, 34) (Fig. 2C). Interestingly, as little as 3% mucosal hypotonicity triggered a small yet statistically significant ΔIsc, and the dose-response curve was largely linear up to 20% hypotonicity (Fig. 2E); this suggests that the channel or channels underlying the current responds to osmolarity changes with high sensitivity and over a wide detection range.
Figure 2.

Mucosal hypotonicity activates CFTR-mediated ΔIsc and ΔICl in Calu-3 cells. A) Representative traces of ΔIsc induced by 18.4% mucosal hypotonicity (Hypo) with/without 20 μM CFTRinh-172 (CFTRi) in intact Calu-3 epithelial monolayers. B) Summary data of panel [CFTRi, n = 6 (Ctrl), 6 (treatment)] and similar experiments performed with 300 μM glybenclamide (GLY, n = 7, 7), 100 μM DIDS (n = 18, 22), 10 μM tamoxifen (TAMO, n = 19, 19), 20 μM H89 (n = 6, 8), and 100 μM BAPTA-AM (n = 5, 8). C) Representative traces of ΔIsc induced by 10 µM forskolin (Fsk) under same conditions with/without 20 μM CFTRi in intact Calu-3 epithelial monolayers. D) Summary data of (C) (CFTRi, n = 7, 7) and similar experiments with 300 μM glybenclamide (GLY, n = 5, 4), 100 μM DIDS (n = 6, 6), and 20 μM H89 (n = 10, 8). E) Dose–response curve of hypotonicity-induced ΔIsc in intact Calu-3 epithelial monolayers (n = 8 independent experiments for 1, 3, and 5% hypotonicity; n = 12 independent experiments for 10, 15, and 20% hypotonicity). F) Representative traces of ΔICl induced by 18.4% mucosal hypotonicity (Hypo) with/without 20 μM CFTRi, followed by addition of 10 μM Fsk, in basolaterally nystatin-permeabilized Calu-3 cells. G) Summary data of F; n = 8 (control) and 9 (CFTRi). Except for nystatin, all tested compounds shown in figure were added mucosally; different from control or 0% hypotonicity: *P ≤ 0.039; **P ≤ 0.01; ***P ≤ 0.001.
Hypotonicity-induced ΔIsc was sensitive to neither the PKA inhibitor H89 nor the intracellular Ca2+ chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester (BAPTA-AM), which excludes the involvement of PKA and Ca2+ signaling (Fig. 2B). By contrast, H89 markedly attenuated forskolin-activated ΔIsc as expected (Fig. 2D). Because ΔIsc was sensitive to Cl-channel blockers and Calu-3 cells lack of pathways for electrogenic Na+ absorption (35, 36) (glucose-dependent Na+ absorption was eliminated by replacing glucose with mannitol in the bath), the positive (mucosal-to-serosal) ΔIsc represented either transepithelial anion secretion, consistent with the activation of an apical Cl channel or channels (37, 38), or activation of a basolateral K channel or channels, which would increase the driving force for anion exit through an apical Cl channel (37, 39).
To further examine whether mucosal hypotonicity activates apical CFTR channels, we assessed the effect of mucosal hypotonicity on apical CFTR-mediated ICl in isolation by using basolaterally permeabilized Calu-3 epithelial monolayers. Previous work has suggested that even nystatin-permeabilized cells retain a partial response to hypotonicity (40). Like forskolin, mucosal hypotonicity elicited an apical, CFTRi-sensitive ΔICl (Fig. 2F–G). These results indicate that mucosal hypotonicity activates CFTR, the predominant apical Cl channel in Calu-3 cells (21), directly (through lipid-bilayer/cytoskeletal proteins) or indirectly [through volume-sensitive signaling molecules other than PKA and Ca2+ (Fig. 2B)]. In contrast to the similar magnitudes of ΔIsc measured in response to mucosal hypotonicity and forskolin in intact Calu-3 epithelial monolayers (Fig. 2A–C), hypotonicity-induced apical ΔICl was markedly smaller than forskolin-induced ΔICl in basolaterally permeabilized Calu-3 epithelial monolayers (Fig. 2F, G); this presumably reflects a considerable loss of the cells’ ability to swell after nystatin treatment (40, 41).
An alternative explanation for these data is that mucosal hypotonicity does not activate CFTR, and that the basal channel activity of CFTR as a regulator is permissive for hypotonicity-induced activation of another apical Cl channel. This channel, if present, must be insensitive to DIDS, tamoxifen, and PKA/Ca2+ signaling (Fig. 2B), but it could be a VSOR-like channel, because there are exceptions to the general DIDS sensitivity of VSOR-like channels (31, 32). However, this possibility contradicts our observation that other hypotonicity-activated channels are absent in the cell-attached patches.
Bilateral hypotonicity triggered a transient ΔIsc
Next, we applied hypotonic challenges to both the mucosal and serosal sides in order to mimic bilateral or intracellular osmotic changes under physiologic or pathologic conditions (42, 43). Interestingly, bilateral hypotonicity elicited a transient ΔIsc, with CFTR-blocker sensitivities being similar to those in the case of forskolin-activated ΔIsc (Fig. 3A, B); this reinforces the notion that CFTR channel activity is involved in hypotonicity-induced anion secretion. Moreover, mucosal DIDS application exerted a negligible effect on bilateral hypotonicity-induced ΔIsc (Fig. 3C), which agrees with the results in Fig. 2 and further verifies the finding that apical VSOR does not contribute to hypotonicity-induced ΔIsc.
Figure 3.

Bilateral hypotonicity activates CFTR-mediated ΔIsc in Calu-3 cells. A) Representative traces of ΔIsc induced by 18.4% bilateral hypotonicity (Hypo) and mucosal 10 μM forskolin (Fsk) in absence or presence of 20 μM mucosal CFTRi. B–E) Summary data of (B), and similar experiments performed with 100 μM mucosal (C) or serosal (D) DIDS, and 10 μM serosal tamoxifen (E). In (B), n = 3 (Ctrl) and 6 (CFTRi); in (C), 6 (Ctrl) and 7 (DIDS); in (D), 12 (Ctrl) and 11 (DIDS); in (E), 8 (ctrl) and 8 (Tamo). Ctrl, control; Tamo, tamoxifen; M, mucosal; S, serosal. Different from control: *P ≤ 0.043; **P ≤ 0.01; ***P = 0.001. F) Representative traces of ΔIsc induced by basolateral 18.4% hypotonicity (Hypo), with or without 20 μM mucosal CFTRi.
Bilateral hypotonicity-induced ΔIsc was transient, which is in marked contrast to the sustained ΔIsc induced by mucosal hypotonicity (Fig. 2); thus, we speculated that serosal hypotonicity activates a negative (serosal-to-mucosal) current in the basolateral domain and thereby partially offsets the sustained positive (mucosal-to-serosal) ΔIsc induced by mucosal hypotonicity in the apical domain. To determine whether any VSOR in the basolateral domain is activated by bilateral hypotonicity and thus contributes to a negative (serosal-to-mucosal) current, we tested the effect of serosal DIDS application. Interestingly, serosal DIDS suppressed bilateral hypotonicity-induced ΔIsc (Fig. 3D). However, the inhibition does not appear to result from blocking VSOR because 1) serosal application of tamoxifen, another VSOR blocker, exerted no effect (Fig. 3E); and 2) serosal DIDS also decreased the forskolin response (Fig. 3D), probably because DIDS imitates its analog 4,4′-dinitro stilbene-2,2′-disulfonate (DNDS) and inhibits basolateral Na/HCO3 transporter (44) and thereby suppresses the forskolin-induced current that predominately contains HCO3− (9, 14, 45). These results also imply that, like forskolin, bilateral hypotonicity opened CFTRs and induced a ΔIsc that, at least partially, contains HCO3−.
To further verify that serosal hypotonicity activated a negative current, we examined the effect of serosal hypotonicity on ΔIsc. Indeed, serosal hypotonicity evoked a moderate, negative current that was insensitive to mucosal CFTRi (Fig. 3F; ΔIsc, −5.05 ± 1.74 µA/cm2 for control and −5.28 ± 1.06 µA/cm2 for CFTRi, n = 4 for both the control and CFTRi; P = 0.916 for the difference between control and CFTRi). This current could be mediated by a basolateral K channel or channels or a DIDS/tamoxifen-insensitive volume-sensitive Cl channel or channels (43, 46). However, simply adding serosal hypotonicity-induced ΔIsc to mucosal hypotonicity-induced ΔIsc could only partly but not completely account for the transient characteristics of ΔIsc induced by bilateral hypotonicity, suggesting that the effects of mucosal and serosal hypotonicty on ΔIsc are not completely additive or independent.
Collectively, our results show that hypotonicity stimulated CFTR channel activity in Calu-3 cells, which contrasts previous studies suggesting that CFTR channel activity is not involved in epithelial RVD (5, 6) or that CFTR down-regulates swelling-activated ICl (47).
Genetic suppression and ablation of CFTR but not G551D mutation suppressed hypotonicity-induced Isc
We next knocked down the expression of endogenous CFTR in Calu-3 cells by means of RNA interference performed using a lentiviral vector (Fig. 4A). Knocking down CFTR markedly reduced mucosal hypotonicity-induced ΔIsc (Fig. 4A–C), which strengthened the notion that CFTR mediated the hypotonicity-induced current. To examine hypotonicity-dependent activation of CFTR in situ, we investigated the effect of CFTR gene ablation on hypotonicity-induced Isc in mouse tissue. Hypotonicity-induced ΔIsc was significantly lower in duodena of mice lacking the CFTR gene than in duodena of WT mice (P = 0.031), which verified that CFTR, either as a channel or as a nonchannel regulator, mediates hypotonicity-induced ΔIsc. Moreover, the CFTR blocker GlyH-101 lowered the ΔIsc in the duodena of WT mice but exerted a negligible effect on the ΔIsc in CFTR KO mice (Fig. 4D, E). These results suggest that at the concentration used (40 µM), GlyH-101 specifically blocks CFTR channel activity in mouse duodena and that CFTR channel activity is involved in hypotonicity-induced ΔIsc.
Figure 4.

Genetic suppression or ablation of CFTR but not G551D mutation reduces hypotonicity-induced ΔIsc. A) Western blot analysis of CFTR after treatment with control-shRNA (CTRL) and 2 CFTR-shRNAs (CFTR siRNA-1 and -2) in polarized Calu-3 cells grown on permeable support. GAPDH, loading control. B, C) Representative traces (B) and summary data (C) of mucosal hypotonicity-induced ΔIsc in Calu-3 cells treated with control-shRNA (CTRL, n = 6) or CFTR-shRNA (KD, n = 4). Different from control: *P = 0.047. D, E) Representative traces (D) and summary data (E) of ΔIsc induced by 18.4% mucosal hypotonicity with or without 40 μM GlyH-101 pretreatment in duodena in WT-CFTR (WT), G551D CFTR (G551D), and CFTR KO (KO) mice. Ctrl, control; n (Ctrl and GlyH101, respectively) = 5 and 5 for WT, 5 and 5 for G551D, and 3 and 3 for KO; different from control: **P ≤ 0.01. F, G) Representative trace (F) and summary data (G) of ΔIsc induced by stretch (10 mmHg) before and after 20 μM CFTRi treatment in mouse duodena; n = 5 (WT), 6 (G551D), and 3 (KO); different from control: *P ≤ 0.029; **P < 0.007.
Interestingly, the duodena in mice expressing G551D CFTR showed normal hypotonicity-induced ΔIsc (Fig. 4E; P = 0.982 relative to WT-CFTR mice), and, notably, the hypotonicity-induced ΔIsc was lowered by GlyH-101. These results suggest that hypotonicity stimulates the channel activity of G551D CFTR to the same level as that of WT CFTR, and that the G551D mutation has little impact on the mechanosensitivity of CFTR, even though it abolishes the channel’s cAMP response (48, 49).
Approximately 50% of the hypotonicity-induced ΔIsc persisted in the presence of GlyH-101 in both WT and CFTR KO mice (Fig. 4D, E). However, this residual ΔIsc was insensitive to mucosal DIDS (not shown), which excludes the possibility that it is mediated by apical DIDS-sensitive channels such as VSOR. The molecular identify of the channel underlying this residual ΔIsc is unclear, but it is possible that either another hypotonicity-activated Cl channel is present in the apical membrane, or a hypotonicity-activated K channel is present in the basolateral membrane. Similarly, a residual stretch-activated ΔIsc was observed in CFTR KO mice in the previous study (2) and also here (Fig. 4F, G). Whether the same channel underlies the residual hypotonicity- and stretch-activated ΔIsc warrants further examination.
To further confirm that G551D CFTR is mechanosensitive, the stretch response of G551D CFTR was tested. In the duodena of WT mice, a mild apical suction elicited a ΔIsc that was sensitive to the CFTR inhibitor CFTRi (Fig. 4F, G), as previously reported (2). Notably, the CFTRi-sensitive fraction of the ΔIsc was abolished when CFTR was knocked out (Fig. 4G); this suggests that 20 µM CFTRi specifically blocked CFTR channel activity in the mouse duodenum. Moreover, in the duodena of G551D CFTR mice, we detected a suction-activated ΔIsc featuring an amplitude and CFTRi sensitivity similar to those in the duodena of WT mice (P = 0.84 relative to WT CFTR) (Fig. 4G). This finding suggests that G551D CFTR channel activity can also be activated by stretch and further bolsters the notion that the G551D mutation has no impact on the mechanosensitivity of CFTR.
Epithelial RVD depends on the channel activity of WT and G551D CFTR
Because both WT and G551D CFTR channel activities were stimulated by hypotonicity, we determined their role in epithelial RVD. In Calu-3 cells, CFTRi potently suppressed the RVD (Fig. 5A), which indicated that CFTR channel activity is essential for RVD in these cells, as reported (15). In another set of experiments, ectopic expression of either WT or G551D CFTR by using the vector pIRES-EGFP equally triggered a clear RVD (Fig. 5B, top) that is normally absent in CHO cells (50); this agrees with the previous observation that both WT and G551D CFTR can mediate RVD in epithelia (4–7). More importantly, the effects of these channels on RVD were eliminated by GlyH-101 (Fig. 5B, top) and glybenclamide (data not shown), which supports the notion that the channel activity—rather than a nonchannel regulatory function—of WT and G551D CFTR is essential for RVD. Here, Western blot analysis revealed no difference in the expression level of WT and G551D CFTR (Fig. 5B, bottom), as previously reported (49).
Figure 5.

Epithelial RVD depends on channel activity of WT and G551D CFTR. Volume change shown here is normalized relative to cell or crypt volume before applying hypotonicity. A) Volume of Calu-3 cells at 5 min after 27.2% hypotonic challenge is significantly different from that at 21 min in control group (P = 0.0027, n = 7) but not in group treated with 40 μM CFTRi (P = 0.73, n = 5), which indicates CFTRi-sensitive RVD in Calu-3 cells. Moreover, at 21 min, cell volume in control is significantly different from that in CFTRi group (P = 0.023). B) CHO cells were transfected with pIRES-EGFP (GFP), pIRES-EGFP-CFTR (CFTR), or pIRES-EGFP-G551D-CFTR (G551D). Top: volume of CHO cells at 20 min after 30.0% hypotonic challenge in GFP group is significantly different from that in WT-CFTR group (P = 0.029, n = 5) and G551D CFTR group (P = 0.046, n = 5), and addition of 40 μM GlyH-101 eliminated these differences (n = 4–5; P = 0.0114 and 0.022, control vs. WT CFTR and G551D CFTR, respectively). Bottom: Western blot analysis of WT and G551D CFTR expressed in CHO cells. GFP, pIRES-EGFP alone; GAPDH, loading control. C) Volumes of intestinal crypts at 25 min after 22.6% hypotonic challenge in control group differed significantly from corresponding volumes in GlyH-101 (GlyH) group in case of crypts from WT CFTR mice (WT) or G551D CFTR mice (G551D) but not CFTR KO mice (KO). P = 0.02 (WT), 0.002 (G551D), and 0.38 (KO); n (control and GlyH, respectively) = 8 and 5 (WT), 10 and 6 (G551D), 3 and 3 (KO).
Last, we examined the role of WT and G551D CFTR in crypts isolated from mouse duodena in order to determine the channels’ in vivo function. In accord with previous studies (5, 6), knocking out CFTR inhibited RVD, but the G551D mutation did not alter the RVD relative to that in crypts from WT-CFTR mice (Fig. 5C). Moreover, GlyH-101 markedly suppressed RVD in both WT- and G551D CFTR mice. The concentration of GlyH-101 seems to specifically block CFTR as well (Fig. 4D). These results further verify that the channel activity of WT and G55D CFTR is crucial for RVD in intestinal epithelial cells and thus argue against the previously suggested mechanisms (5, 6).
DISCUSSION
During RVD in animal cells, cell swelling activates transport pathways that mediate the net efflux of K+, Cl−, small organic osmolytes, and osmotically obligated water. K+ and Cl− are released from cells primarily by KCl cotransporters or separate K+ and anion channels. Cell swelling has been documented to activate or up-regulate several anion channels, including ClC-2, ClC-3, ICln, dBest1, maxi-anion channel, and TMEM16A (Ano1). Among such anion channels, VSOR has been suggested to be the most prominently activated and ubiquitously expressed. The current consensus is that in whole-cell recordings, VSOR typically exhibits outward rectification, voltage-dependent inactivation, low-field-strength anion selectivity, and DIDS sensitivity (1–4). Although the biophysical and pharmacological properties of VSOR had been characterized in detail, the channel’s molecular identity remained unelucidated and contentious until recently. In 2014, 2 independent studies suggested that LRRC8 family proteins form components of VSOR, if not the channel pore per se (29, 30). However, it is not yet known how ubiquitous and critical a role LRRC8 family proteins play in cell volume regulation; this is because only several cells lines were tested in previous studies (29, 30) and LRRC8 mutations or KO in humans and mice displayed less severe phenotypes than expected considering the importance of volume-regulated anion channels (51–53). Other chloride channels such as TMEM16 and bestrophin1, besides LRRC8 family proteins, have also been implicated in the volume regulation of certain cell types and tissues (53, 54).
CFTR has previously been implicated in RVD. Chan et al. (55, 56) reported that an anti-CFTR antibody inhibited both cAMP-activated and swelling-induced VSOR-like whole-cell Cl conductance in T84 cells, and further that the swelling-activated ICl was smaller in CF epithelia than in non-CF cells. Moreover, CFTR KO was reported to impair a VSOR-like current and RVD in primary cultures of mouse nephrons due to the loss of impaired CFTR-dependent ATP release (57). Conversely, ectopic expression of CFTR down-regulated a swelling-induced, VSOR-like whole-cell current in calf endothelial cells and COS cells (58). CFTR expression also suppressed VSOR-like currents in HEK293T cells, and this depended on CFTR NBD2 (47). However, whether these results were an artifact of CFTR overexpression remains unclear. By contrast, several groups have suggested that CFTR enhances swelling-activated K+ conductance and thereby RVD, possibly by promoting ATP release (4, 5, 7).
Although the aforementioned studies implicated CFTR in RVD, discrepancies and unresolved issues remain. One key question is whether CFTR channel activity is required for RVD. Valverde et al. (5) suggested that CFTR per se is not a volume-sensitive channel for Cl− efflux, because they found that in intestinal crypt epithelia in mice, RVD was sensitive to CFTR KO but not to the CFTR blocker glybenclamide. This could be considered unexpected, unless the explanation is that the activity of neither CFTR nor VSOR is involved in the RVD, because glybenclamide also blocks VSOR (31). However, most of the previous studies did not address whether CFTR channel activity is involved in RVD, partly because the studies were performed before the discovery of CFTR-specific inhibitors (28). After the discovery of CFTR-specific inhibitors, 3 studies addressed this question. In one study, a single representative trace without any indication of the sample size showed that 5 µM CFTRi did not block hypotonicity-activated ICl in FRT epithelial cells expressing CFTR (59); in another study, CFTRi at an unreported concentration failed to block swelling-induced anion currents in mouse ventricular myocytes in 4 experiments (60). By contrast, one study reported that the RVD in Calu-3 cells was sensitive to 10 μM CFTRi but not 250 μM DIDS (15), which agrees with our results (Figs. 2 and 5). In this study, we used multiple approaches together with several CFTR blockers in both cultured cells and mouse tissues, and our results clearly demonstrated that CFTR channel activity is stimulated by cell swelling and is involved in epithelial RVD.
The possibility that CFTR contributes to cell volume regulation through ATP release has previously been intensively studied and remains a viable alternative to CFTR channel activity per se (4, 5, 7). Although our study demonstrated that swelling-activated CFTR channel activity mediates anion efflux in RVD, it did not exclude the possibility that the regulatory function of CFTR (with or without channel activity) might mediate ATP release and thereby enhance swelling-activated K+ conductance in epithelial RVD.
Braunstein et al. (61) reported a loss of the regulatory ability of G551D CFTR in RVD; conversely, another study suggested that mice carrying the G551D CFTR mutant exhibited normal RVD (6). In accord with the second study, we found that G551D CFTR mice maintain a normal RVD (Fig. 5C); more importantly, our results showed that G551D CFTR is activated by mechanical stimuli, including hypotonicity and stretch (Fig. 4E, G), and that the mechanosensitive channel activity of G551D CFTR is required for the RVD (Fig. 5C). Because G551D CFTR shows impaired ATP binding and is refractory to cAMP/PKA stimulation, the mechanosensitivity of G551D CFTR (i.e., hypotonicity- and stretch-activated channel activities) must be independent of ATP binding and activation of any mechanosensitive cAMP/PKA signaling. Similarly, our data on WT CFTR here (Fig. 2) and in a previous study (2) indicate that hypotonicity- and stretch-induced activation of WT CFTR is also independent of ATP and cAMP/PKA-dependent phosphorylation.
Because of its impaired ATP binding, G551D CFTR was estimated to exhibit only ∼ 4% of WT-CFTR activity (48, 49), which might reflect ATP-independent gating (49). However, the G551D CFTR mutation phenotypes in patients or mice are milder than those of CFTR KO or ΔF508 CFTR mutation (48, 62, 63), and these mild phenotypes were attributed to the ∼ 4% residual activity (48). Interestingly, Valverde et al. (6) provided an alternative explanation: the G551D mutant might retain the WT-CFTR function as a regulator and thus contribute to the milder phenotypes, because G551D CF mice maintain a normal RVD; however, in the study, the exact mechanism underlying the normal RVD of G551D mice was not examined.
Sustained cell swelling leads to necrosis, and in this process, volume regulatory anion channels rescue cells by ensuring RVD. Conversely, normotonic activation of these anion channels leads to cell death by triggering apoptotic volume decrease upon stimulation with apoptosis inducers (64, 65). Here, we found that CFTR channel activity is involved in epithelial volume regulation, and thus CFTR mutations such as ΔF508 could result in abnormal volume regulation. This abnormal volume regulation would lead to necrosis upon cell swelling under physiologic/pathologic conditions and to deficient apoptotic volume decrease/apoptosis of epithelial, immune, and other cells; both of these processes would contribute to the pathology of CF (66, 67), in addition to the pathology associated with the defective cAMP/PKA response of ΔF508 CFTR. By contrast, the G551D mutant retains the response to cell swelling, and this could, at least partially, account for the mild phenotypes. Similarly, G551D and WT CFTR are equally stretch-sensitive, and, consequently, G551D CFTR would retain residual channel activity under basal or elevated mechanical tension of the cell membrane [e.g., during maximal exercise (2) or food ingestion]. Therefore, the mechanoresponses of G551D CFTR—its hypotonicity- and stretch-induced responses together—might explain the mild pathology resulting from G551D CFTR mutation.
Acknowledgments
The authors are grateful to B. Wainwright (University of Queensland, Brisbane, QLD, Australia) and D. M. Bedwell (University of Alabama at Birmingham, Birmingham, AL, USA) for generating and providing G551D CFTR mice, and to T. C. Hwang (University of Missouri, Columbia, MO, USA) for providing the cDNA of G551D CFTR. The authors thank K.-L. So for technical support. This work was supported by Hong Kong Research Grants Council (Grants GRF660913 and GRF16102415 to P.H.). The authors declare no conflicts of interest.
Glossary
- BAPTA-AM
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- CFTRi
CFTR inhibitor 172
- DIDS
4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid
- DNDS
4,4′-dinitro stilbene-2,2′-disulfonate
- EDTA
ethylenediaminetetraacetic acid
- EGFP
enhanced green fluorescent protein
- GFP
green fluorescent protein
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- ICl
chloride circuit
- Isc
short-circuit current
- KBR
Krebs–bicarbonate–Ringer
- KO
knockout
- RVD
regulatory volume decrease
- VSOR
volume-sensitive outwardly rectifying chloride channel
- WT
wild-type
REFERENCES
- 1.Sardini A., Amey J. S., Weylandt K. H., Nobles M., Valverde M. A., Higgins C. F. (2003) Cell volume regulation and swelling-activated chloride channels. Biochim. Biophys. Acta , 153–162 [DOI] [PubMed] [Google Scholar]
- 2.Zhang W. K., Wang D., Duan Y., Loy M. M., Chan H. C., Huang P. (2010) Mechanosensitive gating of CFTR. Nat. Cell Biol. , 507–512 [DOI] [PubMed] [Google Scholar]
- 3.Gray M. A. (2010) CFTR is a mechanosensitive anion channel: a real stretch? Cellscience , 1–7 [PMC free article] [PubMed] [Google Scholar]
- 4.Belfodil R., Barrière H., Rubera I., Tauc M., Poujeol C., Bidet M., Poujeol P. (2003) CFTR-dependent and -independent swelling-activated K+ currents in primary cultures of mouse nephron. Am. J. Physiol. Renal Physiol. , F812–F828 [DOI] [PubMed] [Google Scholar]
- 5.Valverde M. A., O’Brien J. A., Sepúlveda F. V., Ratcliff R. A., Evans M. J., Colledge W. H. (1995) Impaired cell volume regulation in intestinal crypt epithelia of cystic fibrosis mice. Proc. Natl. Acad. Sci. USA , 9038–9041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valverde M. A., Vázquez E., Muñoz F. J., Nobles M., Delaney S. J., Wainwright B. J., Colledge W. H., Sheppard D. N. (2000) Murine CFTR channel and its role in regulatory volume decrease of small intestine crypts. Cell. Physiol. Biochem. , 321–328 [DOI] [PubMed] [Google Scholar]
- 7.Vázquez E., Nobles M., Valverde M. A. (2001) Defective regulatory volume decrease in human cystic fibrosis tracheal cells because of altered regulation of intermediate conductance Ca2+-dependent potassium channels. Proc. Natl. Acad. Sci. USA , 5329–5334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y., Lam C. S., Wu F., Wang W., Duan Y., Huang P. (2005) Regulation of CFTR channels by HCO(3)-sensitive soluble adenylyl cyclase in human airway epithelial cells. Am. J. Physiol. Cell Physiol. , C1145–C1151 [DOI] [PubMed] [Google Scholar]
- 9.Wang D., Sun Y., Zhang W., Huang P. (2008) Apical adenosine regulates basolateral Ca2+-activated potassium channels in human airway Calu-3 epithelial cells. Am. J. Physiol. Cell Physiol. , C1443–C1453 [DOI] [PubMed] [Google Scholar]
- 10.Duan Y., Sun Y., Zhang F., Zhang W. K., Wang D., Wang Y., Cao X., Hu W., Xie C., Cuppoletti J., Magin T. M., Wang H., Wu Z., Li N., Huang P. (2012) Keratin K18 increases cystic fibrosis transmembrane conductance regulator (CFTR) surface expression by binding to its C-terminal hydrophobic patch. J. Biol. Chem. , 40547–40559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun Y., Duan Y., Eisenstein A. S., Hu W., Quintana A., Lam W. K., Wang Y., Wu Z., Ravid K., Huang P. (2012) A novel mechanism of control of NFκB activation and inflammation involving A2B adenosine receptors. J. Cell Sci. , 4507–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snouwaert J. N., Brigman K. K., Latour A. M., Malouf N. N., Boucher R. C., Smithies O., Koller B. H. (1992) An animal model for cystic fibrosis made by gene targeting. Science , 1083–1088 [DOI] [PubMed] [Google Scholar]
- 13.Oceandy D., McMorran B. J., Smith S. N., Schreiber R., Kunzelmann K., Alton E. W., Hume D. A., Wainwright B. J. (2002) Gene complementation of airway epithelium in the cystic fibrosis mouse is necessary and sufficient to correct the pathogen clearance and inflammatory abnormalities. Hum. Mol. Genet. , 1059–1067 [DOI] [PubMed] [Google Scholar]
- 14.Devor D. C., Singh A. K., Lambert L. C., DeLuca A., Frizzell R. A., Bridges R. J. (1999) Bicarbonate and chloride secretion in Calu-3 human airway epithelial cells. J. Gen. Physiol. , 743–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harron S. A., Clarke C. M., Jones C. L., Babin-Muise D., Cowley E. A. (2009) Volume regulation in the human airway epithelial cell line Calu-3. Can. J. Physiol. Pharmacol. , 337–346 [DOI] [PubMed] [Google Scholar]
- 16.Bond T. D., Ambikapathy S., Mohammad S., Valverde M. A. (1998) Osmosensitive C1− currents and their relevance to regulatory volume decrease in human intestinal T84 cells: outwardly vs. inwardly rectifying currents. J. Physiol. , 45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valverde M. A., O’Brien J. A., Sepúlveda F. V., Ratcliff R., Evans M. J., Colledge W. H. (1993) Inactivation of the murine cftr gene abolishes cAMP-mediated but not Ca(2+)-mediated secretagogue-induced volume decrease in small-intestinal crypts. Pflugers Arch. , 434–438 [DOI] [PubMed] [Google Scholar]
- 18.Voets T., Droogmans G., Raskin G., Eggermont J., Nilius B. (1999) Reduced intracellular ionic strength as the initial trigger for activation of endothelial volume-regulated anion channels. Proc. Natl. Acad. Sci. USA , 5298–5303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sachs F. Stretch-activated ion channels: what are they? Physiology , 50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamill O. P., Maroto R. (2007) TRPCs as MS channels. In Mechanosensitive Ion Channel, Part B (Hamill O. P., ed), pp. 191–231, Academic Press, San Diego/London: [DOI] [PubMed] [Google Scholar]
- 21.Haws C., Finkbeiner W. E., Widdicombe J. H., Wine J. J. (1994) CFTR in Calu-3 human airway cells: channel properties and role in cAMP-activated Cl− conductance. Am. J. Physiol. , L502–L512 [DOI] [PubMed] [Google Scholar]
- 22.Morris C. E., Sigurdson W. J. (1989) Stretch-inactivated ion channels coexist with stretch-activated ion channels. Science , 807–809 [DOI] [PubMed] [Google Scholar]
- 23.Small D. L., Morris C. E. (1994) Delayed activation of single mechanosensitive channels in Lymnaea neurons. Am. J. Physiol. , C598–C606 [DOI] [PubMed] [Google Scholar]
- 24.Wan X., Juranka P., Morris C. E. (1999) Activation of mechanosensitive currents in traumatized membrane. Am. J. Physiol. , C318–C327 [DOI] [PubMed] [Google Scholar]
- 25.Gisolfi C. V., Summers R. W., Lambert G. P., Xia T. (1998) Effect of beverage osmolality on intestinal fluid absorption during exercise. J. Appl. Physiol. , 1941–1948 [DOI] [PubMed] [Google Scholar]
- 26.Lemoine J. L., Farley R., Huang L. (2005) Mechanism of efficient transfection of the nasal airway epithelium by hypotonic shock. Gene Ther. , 1275–1282 [DOI] [PubMed] [Google Scholar]
- 27.Dagenais A., Tessier M. C., Tatur S., Brochiero E., Grygorczyk R., Berthiaume Y. (2013) Hypotonic shock modulates Na(+) current via a Cl(−) and Ca(2+)/calmodulin dependent mechanism in alveolar epithelial cells. PLoS One , e74565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma T., Thiagarajah J. R., Yang H., Sonawane N. D., Folli C., Galietta L. J., Verkman A. S. (2002) Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin–induced intestinal fluid secretion. J. Clin. Invest. , 1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiu Z., Dubin A. E., Mathur J., Tu B., Reddy K., Miraglia L. J., Reinhardt J., Orth A. P., Patapoutian A. (2014) SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell , 447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voss F. K., Ullrich F., Münch J., Lazarow K., Lutter D., Mah N., Andrade-Navarro M. A., von Kries J. P., Stauber T., Jentsch T. J. (2014) Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science , 634–638 [DOI] [PubMed] [Google Scholar]
- 31.Liu Y., Oiki S., Tsumura T., Shimizu T., Okada Y. (1998) Glibenclamide blocks volume-sensitive Cl− channels by dual mechanisms. Am. J. Physiol. , C343–C351 [DOI] [PubMed] [Google Scholar]
- 32.Schultz B. D., DeRoos A. D., Venglarik C. J., Singh A. K., Frizzell R. A., Bridges R. J. (1996) Glibenclamide blockade of CFTR chloride channels. Am. J. Physiol. , L192–L200 [DOI] [PubMed] [Google Scholar]
- 33.Schwiebert E. M., Morales M. M., Devidas S., Egan M. E., Guggino W. B. (1998) Chloride channel and chloride conductance regulator domains of CFTR, the cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA , 2674–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valverde M. A., Mintenig G. M., Sepúlveda F. V. (1993) Differential effects of tamoxifen and I− on three distinguishable chloride currents activated in T84 intestinal cells. Pflugers Arch. , 552–554 [DOI] [PubMed] [Google Scholar]
- 35.MacVinish L. J., Cope G., Ropenga A., Cuthbert A. W. (2007) Chloride transporting capability of Calu-3 epithelia following persistent knockdown of the cystic fibrosis transmembrane conductance regulator, CFTR. Br. J. Pharmacol. , 1055–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen B. Q., Finkbeiner W. E., Wine J. J., Mrsny R. J., Widdicombe J. H. (1994) Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl− secretion. Am. J. Physiol. , L493–L501 [DOI] [PubMed] [Google Scholar]
- 37.Diener M., Bertog M., Fromm M., Scharrer E. (1996) Segmental heterogeneity of swelling-induced Cl− transport in rat small intestine. Pflugers Arch. , 293–300 [DOI] [PubMed] [Google Scholar]
- 38.McCann J. D., Li M., Welsh M. J. (1989) Identification and regulation of whole-cell chloride currents in airway epithelium. J. Gen. Physiol. , 1015–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bajnath R. B., de Jonge H. R., Borgdorff A. J., Zuiderwijk M., Groot J. A. (1997) Characterization of swelling-induced ion transport in HT-29Cl.19A cells. Role of inorganic and organic osmolytes during regulatory volume decrease. Pflugers Arch. , 276–286 [DOI] [PubMed] [Google Scholar]
- 40.De Smet P., Li J., Van Driessche W. (1998) Hypotonicity activates a lanthanide-sensitive pathway for K+ release in A6 epithelia. Am. J. Physiol. , C189–C199 [DOI] [PubMed] [Google Scholar]
- 41.Joiner C. H., Rettig R. K., Jiang M., Risinger M., Franco R. S. (2007) Urea stimulation of KCl cotransport induces abnormal volume reduction in sickle reticulocytes. Blood , 1728–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fürst J., Gschwentner M., Ritter M., Bottà G., Jakab M., Mayer M., Garavaglia L., Bazzini C., Rodighiero S., Meyer G., Eichmüller S., Wöll E., Paulmichl M. (2002) Molecular and functional aspects of anionic channels activated during regulatory volume decrease in mammalian cells. Pflugers Arch. , 1–25 [DOI] [PubMed] [Google Scholar]
- 43.Hoffmann E. K., Lambert I. H., Pedersen S. F. (2009) Physiology of cell volume regulation in vertebrates. Physiol. Rev. , 193–277 [DOI] [PubMed] [Google Scholar]
- 44.Amlal H., Habo K., Soleimani M. (2000) Potassium deprivation upregulates expression of renal basolateral Na(+)-HCO(3)(−) cotransporter (NBC-1). Am. J. Physiol. Renal Physiol. , F532–F543 [DOI] [PubMed] [Google Scholar]
- 45.Krouse M. E., Talbott J. F., Lee M. M., Joo N. S., Wine J. J. (2004) Acid and base secretion in the Calu-3 model of human serous cells. Am. J. Physiol. Lung Cell. Mol. Physiol. , L1274–L1283 [DOI] [PubMed] [Google Scholar]
- 46.Pedersen S. F., Prenen J., Droogmans G., Hoffmann E. K., Nilius B. (1998) Separate swelling- and Ca2+-activated anion currents in Ehrlich ascites tumor cells. J. Membr. Biol. , 97–110 [DOI] [PubMed] [Google Scholar]
- 47.Ando-Akatsuka Y., Abdullaev I. F., Lee E. L., Okada Y., Sabirov R. Z. (2002) Down-regulation of volume-sensitive Cl− channels by CFTR is mediated by the second nucleotide-binding domain. Pflugers Arch. , 177–186 [DOI] [PubMed] [Google Scholar]
- 48.Delaney S. J., Alton E. W., Smith S. N., Lunn D. P., Farley R., Lovelock P. K., Thomson S. A., Hume D. A., Lamb D., Porteous D. J., Dorin J. R., Wainwright B. J. (1996) Cystic fibrosis mice carrying the missense mutation G551D replicate human genotype-phenotype correlations. EMBO J. , 955–963 [PMC free article] [PubMed] [Google Scholar]
- 49.Bompadre S. G., Sohma Y., Li M., Hwang T. C. (2007) G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J. Gen. Physiol. , 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Becker D., Blase C., Bereiter-Hahn J., Jendrach M. (2005) TRPV4 exhibits a functional role in cell-volume regulation. J. Cell Sci. , 2435–2440 [DOI] [PubMed] [Google Scholar]
- 51.Sawada A., Takihara Y., Kim J. Y., Matsuda-Hashii Y., Tokimasa S., Fujisaki H., Kubota K., Endo H., Onodera T., Ohta H., Ozono K., Hara J. (2003) A congenital mutation of the novel gene LRRC8 causes agammaglobulinemia in humans. J. Clin. Invest. , 1707–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar L., Chou J., Yee C. S., Borzutzky A., Vollmann E. H., von Andrian U. H., Park S. Y., Hollander G., Manis J. P., Poliani P. L., Geha R. S. (2014) Leucine-rich repeat containing 8A (LRRC8A) is essential for T lymphocyte development and function. J. Exp. Med. , 929–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kunzelmann K. (2015) TMEM16, LRRC8A, bestrophin: chloride channels controlled by Ca(2+) and cell volume. Trends Biochem. Sci. , 535–543 [DOI] [PubMed] [Google Scholar]
- 54.Milenkovic A., Brandl C., Milenkovic V. M., Jendryke T., Sirianant L., Wanitchakool P., Zimmermann S., Reiff C. M., Horling F., Schrewe H., Schreiber R., Kunzelmann K., Wetzel C. H., Weber B. H. (2015) Bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells. Proc. Natl. Acad. Sci. USA , E2630–E2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan H. C., Goldstein J., Nelson D. J. (1992) Alternate pathways for chloride conductance activation in normal and cystic fibrosis airway epithelial cells. Am. J. Physiol. , C1273–C1283 [DOI] [PubMed] [Google Scholar]
- 56.Chan H. C., Kaetzel M. A., Nelson D. J., Hazarika P., Dedman J. R. (1992) Antibody against a cystic fibrosis transmembrane conductance regulator-derived synthetic peptide inhibits anion currents in human colonic cell line T84. J. Biol. Chem. , 8411–8416 [PubMed] [Google Scholar]
- 57.Barrière H., Belfodil R., Rubera I., Tauc M., Poujeol C., Bidet M., Poujeol P. (2003) CFTR null mutation altered cAMP-sensitive and swelling-activated Cl− currents in primary cultures of mouse nephron. Am. J. Physiol. Renal Physiol. , F796–F811 [DOI] [PubMed] [Google Scholar]
- 58.Vennekens R., Trouet D., Vankeerberghen A., Voets T., Cuppens H., Eggermont J., Cassiman J. J., Droogmans G., Nilius B. (1999) Inhibition of volume-regulated anion channels by expression of the cystic fibrosis transmembrane conductance regulator. J. Physiol. , 75–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma T., Vetrivel L., Yang H., Pedemonte N., Zegarra-Moran O., Galietta L. J., Verkman A. S. (2002) High-affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high-throughput screening. J. Biol. Chem. , 37235–37241 [DOI] [PubMed] [Google Scholar]
- 60.Ichishima K., Yamamoto S., Iwamoto T., Ehara T. (2010) alpha-Adrenoceptor-mediated depletion of phosphatidylinositol 4,5-bisphosphate inhibits activation of volume-regulated anion channels in mouse ventricular myocytes. Br. J. Pharmacol. , 193–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Braunstein G. M., Zsembery A., Tucker T. A., Schwiebert E. M. (2004) Purinergic signaling underlies CFTR control of human airway epithelial cell volume. J. Cyst. Fibros. , 99–117 [DOI] [PubMed] [Google Scholar]
- 62.Parad R. B. (1996) Heterogeneity of phenotype in two cystic fibrosis patients homozygous for the CFTR exon 11 mutation G551D. J. Med. Genet. , 711–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Comer D. M., Ennis M., McDowell C., Beattie D., Rendall J., Hall V., Elborn J. S. (2009) Clinical phenotype of cystic fibrosis patients with the G551D mutation. QJM , 793–798 [DOI] [PubMed] [Google Scholar]
- 64.Okada Y., Maeno E., Shimizu T., Manabe K., Mori S., Nabekura T. (2004) Dual roles of plasmalemmal chloride channels in induction of cell death. Pflugers Arch. , 287–295 [DOI] [PubMed] [Google Scholar]
- 65.Okada Y., Sato K., Numata T. (2009) Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J. Physiol. , 2141–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gottlieb R. A., Dosanjh A. (1996) Mutant cystic fibrosis transmembrane conductance regulator inhibits acidification and apoptosis in C127 cells: possible relevance to cystic fibrosis. Proc. Natl. Acad. Sci. USA , 3587–3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watt A. P., Courtney J., Moore J., Ennis M., Elborn J. S. (2005) Neutrophil cell death, activation and bacterial infection in cystic fibrosis. Thorax , 659–664 [DOI] [PMC free article] [PubMed] [Google Scholar]