

Abstract
Peroxisomal biogenesis disorders (PBDs) are autosomal recessive diseases caused by mutations in one of the 14 PEX genes described in the scientific literature.
All of these syndromes may be associated with different mutations in the PEX genes, the most frequent being PEX1 for patients with Zellweger syndrome (ZS).
In this paper, we present the case of a patient with a peculiar clinical history: evisceration of the left eye (LE) at 4 years of age because of a benign ocular teratoid medulloepithelioma and a progressive loss of visual acuity (VA) in the right eye (RE) beginning at 9 years of age, leading to the diagnosis of ZS. In addition, the patient presented a mutation in the PEX14 gene that has not been previously described in the literature.
This case broadens the spectrum of clinical expression in ZS patients because of not only the presence of a benign ocular teratoid medulloepithelioma at 4 years of age but also the late clinical expression of ZS (at 9 years of age).
Keywords: Zellweger syndrome, Ocular medulloepithelioma, Optic neuropathy, Leukodystrophy
Introduction
Peroxisomes play an important role in numerous metabolic processes, but primarily the metabolism of fatty acids, in humans.1 Therefore, any genetic mutation that leads to an alteration of the proteins that compose peroxisomes will result in the development of various peroxisomal disorders with severe metabolic disturbances. Notable disturbances include an absence of beta-oxidation of fatty acids with more than 22 carbons, alpha-oxidation of phytanic acid, and a lack of plasmalogen synthesis.1
Peroxisomal disorders are divided into two groups:
-
–
Peroxisomal biogenesis disorders (PBDs), in which a mutation in any of the peroxisome assembly (PEX) genes promotes a total absence of peroxisomal function. The Zellweger syndrome spectrum (ZSS) (Zellweger syndrome (ZS), neonatal adrenoleukodystrophy, and infantile Refsum) and rhizomelic chondrodysplasia punctata are found within this group.
-
–
Single peroxisomal enzyme deficiencies, which are syndromes that affect only one of the multiple peroxisomal functions. X-linked adrenoleukodystrophy, the adult form of Refsum, and pseudo-rhizomelic chondrodysplasia are found within this group.1
Among these syndromes, ZS is the most severe, causing death within the first year of life.1
PBDs are autosomal recessive diseases caused by mutations in one of the 14 PEX genes described in the scientific literature. These PEX genes encode peroxin proteins (denoted as PEX) that are involved in different peroxisomal functions.1
All of these syndromes may be associated with different mutations in PEX genes, the most frequent being PEX1 for patients with ZS.1, 2
In this paper, we present the case of a patient with a peculiar clinical history: evisceration of the left eye (LE) at 4 years of age because of a benign ocular teratoid medulloepithelioma and a progressive loss of visual acuity (VA) in the right eye (RE) beginning at 9 years of age, leading to the diagnosis of ZS. In addition, the patient presented a mutation in the PEX14 gene that has not been previously described in the literature.
Case report
The patient was a 4-year-old girl without a personal or family history of interest who presented with leukocoria in the LE in 2008. Examination revealed a retrolental mass that produced anterior displacement of the iris. Using ultrasound biomicroscopy (UBM) and a B-scan, the retrolental mass was detected in the ciliary body in the LE, with maximum dimensions of 7 × 3 mm. Cranial and orbital MRI was performed and showed that the tumor was confined to the ciliary body of the LE, without evidence of tumor extension. The rest of the brain scan was compatible with normal anatomy. As a retinoblastoma was suspected, enucleation of the LE was performed. The anatomo-pathology report described the existence of a translucent whitish mass in the ciliary body of the LE compatible with a benign teratoid medulloepithelioma (Fig. 1).
Fig. 1.
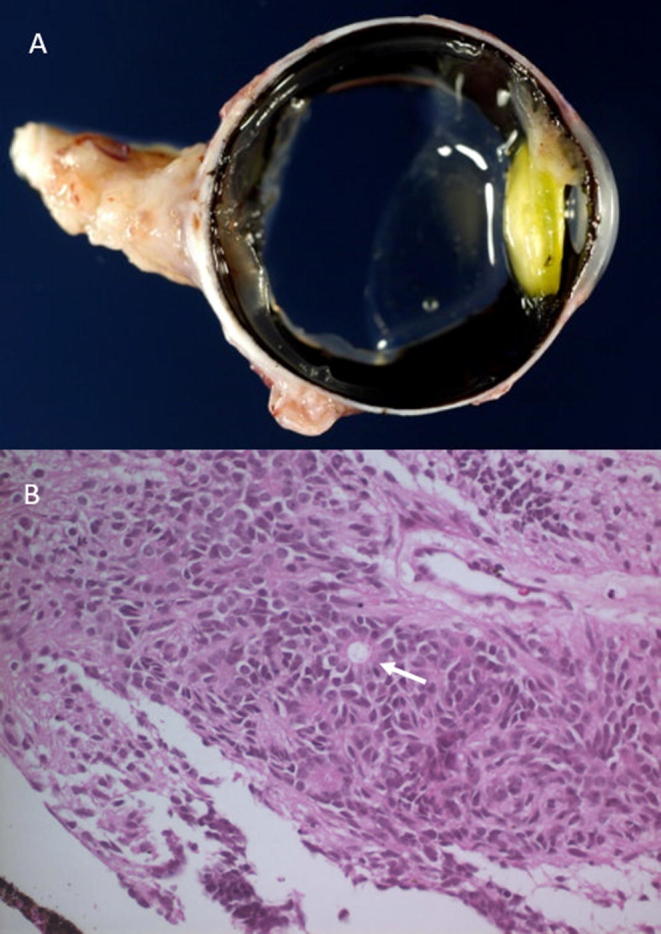
Anatomopathological aspect of the tumor in the LE. (A) Macroscopic aspect of the retrolental mass after LE enucleation. (B) Microscopic cross-sectional view of the tumor (hematoxylin and eosin staining, ×400) showing typical cells and rosettes (arrow) in the benign teratoid medulloepithelioma.
Subsequently, the patient had a stable course until 9 years of age, when she began to exhibit a progressive loss of VA in the RE, accompanied by progressive bilateral hypoacusis and a decline in school performance.
Clinical examination at that time was compatible with VA RE 20/400 (with an ocular prosthesis in the LE). The pupil of the RE was reactive to light and accommodation. The fundus of the RE showed temporal pallor of the optic nerve and a normal retina (Fig. 2). There was bilateral hypoacusis, conserved and symmetrical muscular balance, and a lack of dysmetria. Additionally, examination revealed a normal gait.
Fig. 2.
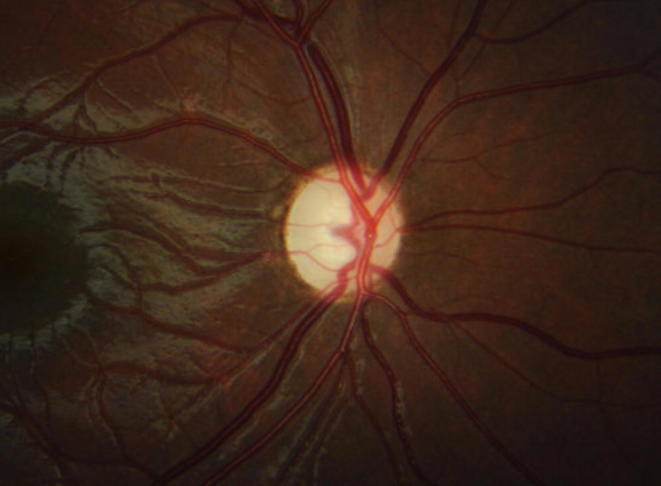
Funduscopic aspect of the RE, showing temporal pallor of the optic nerve and a normal retina.
At that time, a hemogram; measurement of vitamin B12 and folic acid levels, ANA levels, anti-DNA levels, and ENA levels; infectious serology (RPR, TPHA, Brucella, Bartonella, Borrelia, HIV, herpes virus, and TORCH serology testing); thrombotic risk profile measurement; a QuantiFERON assay; and genetic testing to rule out dominant optic atrophy (OPA1) were requested, and all results were within the normal range.
In subsequent visits, the patient presented a progressive deterioration of vision in the RE and progressive gait instability. The parents also detected marked cognitive deterioration, with an alarming decline in school performance.
Cranial and orbital MRI was performed at that time, showing a white-matter hyperintensity predominantly in the occipital lobe (with bilateral thalamic involvement) and in the splenium of the corpus callosum in T2 and FLAIR sequences (Fig. 3).
Fig. 3.

MRI of the brain and orbits, with axial T2 (A) and axial FLAIR (B, C, D) sequences, showing white-matter hyperintensity predominantly in the occipital lobe (with bilateral thalamic involvement) and in the splenium of the corpus callosum. The enucleation of the LE is also visible.
Because leukodystrophy was suspected, a determination of very-long-chain fatty acid (VLCFA) levels was requested, which demonstrated increases in phytanic acid, pristanic acid, and cerotic acid (C26: 0) levels, compatible with a possible peroxisomal disorder.
A biochemical analysis with a hepatic profile (aspartate aminotransferase, alanine aminotransferase, gamma-glutamyl transferase, and total bilirubin) was performed, which yielded normal results.
Genetic testing was performed by next-generation sequencing, revealing that in exon 8 of the PEX14 gene, the patient carried a homozygous missense mutation that yielded a non-conservative substitution of an evolutionarily highly conserved amino acid (p.Arg226Gln) (c.677G > A) in the PEX14 gene. To the best of our knowledge, this mutation has not been described in the literature.
On the last visit, the patient displayed marked cognitive deterioration. In particular, she was incapable of speaking or walking and required a wheelchair.
Discussion
In this paper, we present the case of a patient with a peculiar clinical history: LE evisceration at 4 years of age because of a benign ocular teratoid medulloepithelioma and a progressive loss of VA in the RE beginning at 9 years of age, leading to the diagnosis of ZS. In addition, the patient presented a mutation in the PEX14 gene that has not been previously described in the literature.
ZS was called hepatorenal syndrome in its first clinical descriptions. This syndrome is characterized by the following craniofacial abnormalities: hypoplastic supraorbital ridges, midface hypoplasia, and an epicanthal fold.3, 4, 5
Ophthalmologically, patients may present corneal opacity, cataracts, glaucoma, retinal alteration with poor responses on ERG, and optic atrophy.6, 7
At the level of the central nervous system, there is typically a neuronal migration defect with polymicrogyria. In certain patients, leukodystrophy develops, with degeneration of the myelin, causing deterioration of acquired skills and spasticity. The demyelination has a predilection for the occipital area and the brain stem.7
Patients with ZS typically exhibit general, progressive deterioration, with death during the first year of life secondary to apnea and respiratory failure.5
Our patient had a peculiar clinical presentation because of not only the presence of a benign ocular teratoid medulloepithelioma at 4 years of age but also the late clinical expression of ZS (at 9 years of age). Moreover, her ensuing clinical course was relatively rapid, with swift cognitive deterioration within a few months.
Craniofacial alterations were very mild in our patient, with only very mild hypoplastic supraorbital ridges. Ophthalmologically, the existence of marked pallor of the optic nerve was noteworthy. Corneal opacity, cataract, and glaucoma, which are anomalies described in other patients with ZS in the literature, were not detected.
MRI of our patient’s brain revealed a normal appearance for the age of 4 years (except for the presence of the benign teratoid medulloepithelioma in the LE). However, when the patient began to have symptoms of loss of VA and cognitive deterioration at 9 years of age, the brain MRI clearly showed alterations of the white-matter signal at the level of the occipital lobe, the splenium of the corpus callosum, and the bilateral thalamus. These alterations are similar to those described in other patients with ZS.
There is no current treatment for ZS and related disorders; we can only offer patients supportive therapy for the different clinical manifestations. Oral bile administration has been performed in certain patients with ZS, improving hepatobiliary function.8, 9 In patients with ZS, supplements of fat-soluble vitamins (A, D, E, and K) are recommended due to the lipid malabsorption present in this syndrome.10, 11
The metabolic alterations in PBDs consist of the accumulation of VLCFAs, pristanic acid, phytanic acid, and other lipids, as in the deficiency of certain macromolecules metabolized by peroxisomes, such as plasmalogens.1, 4, 12 These were precisely the alterations found in our patient, with increases in the levels of phytanic acid, pristanic acid, and cerotic acid (C26: 0), compatible with a possible peroxisomal disorder.
Regarding the mutations described in PBDs, it should be noted that the peroxisomes do not contain DNA and that all the proteins required for their operation are encoded by genes in the cell nucleus. After the proteins are synthesized, they are released into the cytosol, from where they are necessarily transported to the peroxisomes.1
The PEX1, PEX2, PEX5, PEX6, PEX10, PEX12, PEX13, PEX14, and PEX26 proteins intervene in the incorporation of cytosolic proteins into the peroxisomal matrix.1, 13 However, the PEX3, PEX16, and PEX19 proteins play a role in the incorporation of proteins from the cytosol into the peroxisomal membrane.1, 14
The ZSS can present great clinical variability. Several authors have tried to make a genotype-phenotype correlation. If a patient is homozygous for two severe mutations, the mutations will provoke severe clinical expression. However, patients heterozygous for a severe mutation and another milder mutation have milder clinical expression. Finally, the most benign clinical expression is that of patients who are homozygous for two mild mutations.1, 15
Currently, the fact that genetic testing is so affordable permits expansion of the clinical spectrum of ZS, allowing the diagnosis of older patients. Our patient carried the homozygous missense mutation p.Arg226Gln (c.677G > A) in exon 8 of the PEX14 gene. To the best of our knowledge, this mutation has not previously been described in the literature.
In the scientific literature, PEX14 mutations have been described as very rare in cases of ZS. In particular, there are currently only three published cases with these mutations.1, 16, 17, 18 The PEX14 gene encodes a peroxisomal membrane protein. Specifically, PEX14 is the port that receives proteins from the cytosol that have bound to PEX3 and PEX4 and initiates the proteins’ transport through the peroxisomal membrane and their incorporation into the peroxisomal matrix.1
The PEX14 gene is located on chromosome 1p36.22 and is composed of nine different exons. As already mentioned, three patients to date have been reported to have PEX14 mutations. We have found the following clinical data on two of them:
The first patient1, 17 died at 10 days of age and presented a mutation in exon 6 of PEX14 (c.553C > T; pQ185X).
The second patient1, 18 presented a genomic deletion leading to the deletion of exon 3 from the coding DNA (c.85-?_170+_del) and a concomitant change in the reading frame (p.(Ile 29_Lys56del; Gly57GlyfsX2)). This patient presented with jaundice, hepatomegaly, and axial hypotonia at 3 months of age and was still alive at 21 months of age.
The clinical course of our patient was quite different from those of these two patients. In particular, she presented a normal childhood until experiencing the first symptoms of ZS at 9 years of age. Therefore, this case demonstrates that the clinical expression of PEX14 mutations can be variable.
In conclusion, we present the case of a ZS patient with a new PEX14 mutation that has not been previously reported in the literature. This case broadens the spectrum of clinical expression in ZS patients because of not only the presence of a benign ocular teratoid medulloepithelioma at 4 years of age but also the late clinical expression of ZS (at 9 years of age).
Conflict of interest
The authors declared that there is no conflict of interest.
Footnotes
Peer review under responsibility of Saudi Ophthalmological Society, King Saud University.
References
- 1.Waterham H.R., Ebberink M.S. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochem Biophys Acta. 2012 Sep;1822(9):1430–1441. doi: 10.1016/j.bbadis.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 2.Fujiki Y. Peroxisome biogenesis and human peroxisome-deficiency disorders. Proc Jpn Acad Ser B Phys Biol Sci. 2016;92(10):463–477. doi: 10.2183/pjab.92.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinberg S.J., Raymond G.V., Braverman N.E., Moser A.B. Peroxisome biogenesis disorders, zellweger syndrome spectrum. In: Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., editors. GeneReviews® [Internet] University of Washington, Seattle; Seattle (WA): 2003. 1993–2015. [Google Scholar]
- 4.Wanders R.J., Klouwer F.C., Ferdinandusse S., Waterham H.R., Poll-Thé B.T. Clinical and laboratory diagnosis of peroxisomal disorders. Methods Mol Biol. 2017;1595:329–342. doi: 10.1007/978-1-4939-6937-1_30. [DOI] [PubMed] [Google Scholar]
- 5.Braverman N.E., Raymond G.V., Rizzo W.B., Moser A.B., Wilkinson M.E., Stone E.M. Peroxisome biogenesis disorders in the Zellweger spectrum: an overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. 2016;117(3):313–321. doi: 10.1016/j.ymgme.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crane D.I. Revisiting the neuropathogenesis of Zellweger syndrome. Neurochem Int. 2014;69:1–8. doi: 10.1016/j.neuint.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 7.Volpe J.J., Adams R.D. Cerebro-hepato-renal syndrome of Zellweger: an inherited disorder of neuronal migration. Acta Neuropathol. 1972;20(3):175–198. doi: 10.1007/BF00686900. [DOI] [PubMed] [Google Scholar]
- 8.Setchell K.D., Bragetti P., Zimmer-Nechemias L., Daugherty C., Pelli M.A., Vaccaro R. Oral bile acid treatment and the patient with Zellweger syndrome. Hepatology. 1992 Feb;15(2):198–207. doi: 10.1002/hep.1840150206. [DOI] [PubMed] [Google Scholar]
- 9.Setchell K.D., Heubi J.E., Bove K.E., O'Connell N.C., Brewsaugh T., Steinberg S.J. Liver disease caused by failure to racemize trihydroxycholestanoic acid: gene mutation and effect of bile acid therapy. Gastroenterology. 2003 Jan;124(1):217–232. doi: 10.1053/gast.2003.50017. [DOI] [PubMed] [Google Scholar]
- 10.Lee P.R., Raymond G.V. Child neurology: Zellweger syndrome. Neurology. 2013;80(20):e207–e210. doi: 10.1212/WNL.0b013e3182929f8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grayer J. Recognition of Zellweger syndrome in infancy. Adv Neonatal Care. 2005;5(1):5–13. doi: 10.1016/j.adnc.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Wanders R.J., Waterham H.R. Annu Rev Biochem. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329. Biochemistry of mammalian peroxisomes revisited. [DOI] [PubMed] [Google Scholar]
- 13.Carvalho A.F., Pinto M.P., Grou C.P., Alencastre I.S., Fransen M., Sá-Miranda C. Ubiquitination of mammalian Pex5p, the peroxisomal import receptor. J Biol Chem. 2007;282(43):31267–31272. doi: 10.1074/jbc.M706325200. [DOI] [PubMed] [Google Scholar]
- 14.Ebberink M.S., Csanyi B., Chong W.K., Denis S., Sharp P., Mooijer P.A. Identification of an unusual variant peroxisome biogenesis disorder caused by mutations in the PEX16 gene. J Med Genet. 2010 Sep;47(9):608–615. doi: 10.1136/jmg.2009.074302. [DOI] [PubMed] [Google Scholar]
- 15.Poll-The B.T., Gootjes J., Duran M., De Klerk J.B., Wenniger-Prick L.J., Admiraal R.J. Peroxisome biogenesis disorders with prolonged survival: phenotypic expression in a cohort of 31 patients. Am J Med Genet A. 2004 May 1;126A(4):333–338. doi: 10.1002/ajmg.a.20664. [DOI] [PubMed] [Google Scholar]
- 16.Ebberink M.S., Mooijer P.A., Gootjes J., Koster J., Wanders R.J., Waterham H.R. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Hum Mutat. 2011 Jan;32(1):59–69. doi: 10.1002/humu.21388. [DOI] [PubMed] [Google Scholar]
- 17.Huybrechts S.J., Van Veldhoven P.P., Hoffman I., Zeevaert R., de Vos R., Demaerel P. Identification of a novel PEX14 mutation in Zellweger syndrome. BMJ Case Rep. 2009;2009 doi: 10.1136/bcr.07.2008.0503. pii: bcr07.2008.0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimozawa N., Tsukamoto T., Nagase T., Takemoto Y., Koyama N., Suzuki Y. Identification of a new complementation group of the peroxisome biogenesis disorders and PEX14 as the mutated gene. Hum Mutat. 2004 Jun;23(6):552–558. doi: 10.1002/humu.20032. [DOI] [PubMed] [Google Scholar]