

Summary
Soluble Aβ oligomers (oAβs) contribute importantly to synaptotoxicity in Alzheimer disease (AD), but the mechanisms related to heterogeneity of synaptic functions at local circuits remain elusive. Nearly all studies of the effects of oAβs on hippocampal synaptic plasticity have only examined homosynaptic plasticity. Here we stimulated the Schaffer collaterals and then simultaneously recorded in stratum radiatum (apical dendrites) and stratum oriens (basal dendrites) of CA1 neurons. We found that the apical dendrites are significantly more vulnerable to oAβ-mediated synaptic dysfunction: the heterosynaptic basal dendritic long-term potentiation (LTP) remained unchanged, whereas the homosynaptic apical LTP was impaired. However, the heterosynaptic basal dendritic plasticity induced by either spaced 10-Hz bursts or low-frequency (1-Hz) stimulation was disrupted by oAβs in a mGluR5-dependent manner. These results suggest that different firing patterns in the same neurons may be selectively altered by soluble oAβs in an early phase of AD, before frank neurodegeneration.
Subject Areas: Neuroscience, Cellular Neuroscience, Disease
Graphical Abstract
Highlights
-
•
Soluble Aβ oligomers have little effect on heterodendritic basal dendritic LTP
-
•
Soluble Aβ oligomers facilitate both basal and apical dendritic LTD induction
-
•
Stimulation timing determines the oAβ impairment of heterosynaptic basal LTP
-
•
Basal dendrites are less sensitive to Aβ oligomer-mediated synaptotoxicity
Neuroscience; Cellular Neuroscience; Disease
Introduction
Alzheimer disease (AD), the most common neurodegenerative disorder, is characterized by the initial subtle impairment of episodic memory followed by an insidious cognitive decline and devastating neurodegeneration. The typical histopathology of AD includes the accumulation of amyloid-β peptide (Aβ) in extracellular diffuse and fibrillar plaques, hyper-phosphorylated tau in intracellular neurofibrillary tangles, and the loss of neurons in the hippocampus, amygdala, and association cerebral cortices (Braak and Braak, 1991, Musiek and Holtzman, 2015). It has been demonstrated that Aβ accumulation in amyloid plaques may begin at least 1–2 decades before significant cortical tau pathology and the onset of initial clinical symptoms (Bateman et al., 2012, Maruyama et al., 2013). Accumulating evidence also supports the concept that Aβ acts in a common pathway for various molecular precipitants of AD (Eisele and Duyckaerts, 2016). Both postmortem examination and premortem positron emission tomography have shown that Aβ deposits may follow a pattern suggesting a “spread” to different brain regions over time (Villemagne et al., 2012), although a process of selective vulnerability of different classes of neurons cannot be ruled out (Walsh and Selkoe, 2016). The entorhinal cortex (EC) may be among the first regions affected by Aβ accumulation in the hippocampal formation (Harris et al., 2010). As for the neuronal loss, a histological study reported major losses in patients with terminal AD versus age-matched controls in CA1 (68%), subiculum (47%), and hilus (25%) (West et al., 1994, Bobinski et al., 1998, Rössler et al., 2002).
Consistent with the histopathological changes, clinical neuroimaging using high-resolution fMRI has shown that the hippocampus is already significantly damaged at the time of an AD (dementia) diagnosis: significant atrophy in all, or almost all, investigated subfields (La Joie et al., 2013, Li et al., 2013b, Boutet et al., 2014, Khan et al., 2015, de Flores et al., 2015), with the major atrophy generally located in CA1 (Mueller et al., 2010, La Joie et al., 2013, de Flores et al., 2015). Specifically, the apical dendrites of CA1 pyramidal neurons are first targeted, whereas the basal dendrites of CA1 neurons remain unchanged (Das et al., 2012, Engvig et al., 2012, Kerchner et al., 2012). In line with this clinicopathological evidence, several morphological studies show that soluble Aβ oligomers (oAβs) primarily affect the apical dendritic arbors, with no effect on basal dendrites of CA1 pyramidal neurons in AD model mice (Alpár et al., 2006, Steele et al., 2014, Price et al., 2014).
The hippocampus is a well-studied region in the vertebrate brain, due to its complex structure and importance to learning and memory. Hippocampal long-term potentiation (LTP) and long-term depression (LTD) serve as electrophysiological correlates of basic cellular mechanisms for learning and memory in mammals (Nicoll, 2017). Experimental findings suggest that distinct hippocampal subfields contribute to distinct aspects of the memory process (Stokes et al., 2015). For example, it has been shown that apical dendrites of CA1 help mediate spatial and working memory, whereas the basal dendrites are related to associative memory (Leuner et al., 2003, Mahmmoud et al., 2015). The hippocampal CA1 region is a major site for studying synaptic plasticity. Most data obtained from this region were recorded at apical dendrites of CA1 pyramidal neurons. Moreover, there are extensive studies on Aβ-impaired hippocampal LTP, but only a few have examined LTD induction, with inconsistent results. We previously demonstrated that soluble human oAβs applied to wild-type (wt) mouse hippocampal slices could enable a weak low frequency stimulation (LFS, 1 Hz for 5 min) that normally fails to induce LTD to elicit a significant LTD (Shankar et al., 2008; Li et al., 2009). This phenomenon has been replicated by several other groups (Ma et al., 2012, Olsen and Sheng, 2012, Chen et al., 2013, Hu et al., 2014, Salgado-Puga et al., 2017).
Most, if not all, studies of synaptic plasticity in the AD field have recorded input-specific homosynaptic plasticity that occurs only at the synapses that were active during the induction. However, the induction of plasticity at active synapses can also “spillover” to the neighboring synapses that were inactive during the plasticity induction, thereby producing changes in synaptic strength, referred to as heterosynaptic plasticity (Chistiakova et al., 2014). In addition to same-layer heterosynaptic plasticity, there are reports of neuron-wide heterosynaptic plasticity mediated by basal versus apical dendrites (Young and Nguyen, 2005, Hulme et al., 2012, Berberich et al., 2017). We could consider this cell-wide heterosynaptic plasticity as a current dipole change of the homosynaptic plasticity (Einevoll et al., 2013). Whether oAβs have any effect on the hippocampal heterosynaptic plasticity or current dipole has not been reported. That is to say, whether soluble oAβs have effects on the basal dendrites (stratum oriens) of CA1 and what the dynamic changes from apical to basal dendrites may be when the apical dendrite (in stratum radiatum) receives signal inputs have not been investigated. Here, by performing simultaneous recordings from basal and apical dendrites of CA1 in wt mice, we now report that LTP in basal dendrites remains unaffected when the LTP is impaired by oAβs in apical dendrites, whereas LTD was facilitated in both dendritic compartments. Interestingly, the basal dendritic LTP could be impaired by oAβs in a specific time-dependent manner.
Results
Dipole-like Field Potentials Recorded from the Laminar Dendritic Trees of CA1
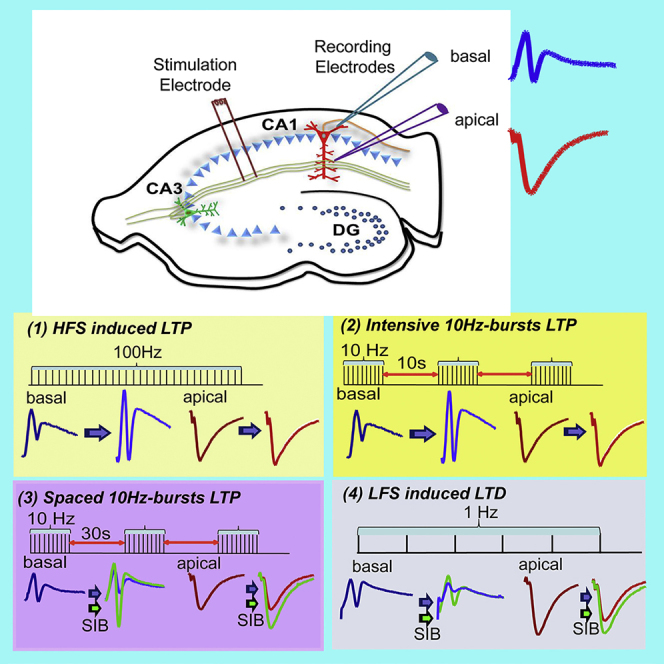
Synaptic potentials are generated as activated postsynaptic receptors enable current flow into neurons. At excitatory synapses, an EPSP (i.e. excitatory postsynaptic potentials, a postsynaptic potential changes caused by the flow of positively charged ions into the postsynaptic neuron) appears when positive ions flow intracellularly (active current sink) and exit the membrane at more distal locations (passive current source). As the hippocampal pyramidal neurons are arranged side by side in a columnar fashion, this structure could generate a dipole (equal but oppositely charged poles separated by a distance). To verify the basic electrical properties of the CA1 region in wt mouse hippocampus, we placed the recording electrodes in different positions of the laminar tree of CA1 pyramidal cells, with the stimulating electrode either on Schaffer collateral afferents (stratum radiatum) (Figure 1A left) or on basal dendrites (stratum oriens) (Figure 1A, right). The recording electrodes were placed on the distal (#1), middle (#2), and proximal (#3) basal dendrites (stratum oriens); pyramidal cell layer (#4); or proximal (#5), middle (#6), and distal (#7) apical dendrites (stratum radiatum) (Figure 1B, diagram on far left). We recorded the field EPSP (fEPSP) in three levels of stimulation intensities (15%–20% of maximum response as the low, 40%–50% as moderate, and 80% as the high). Our resultant recordings were consistent with such recordings performed in vivo (Kuo and Leung, 2017), in that negative extracellular potentials occur in apical dendrites and positive potentials occur in basal dendrites when the stimulus is delivered to the stratum radiatum, and positive extracellular potentials occur in apical dendrites and negative extracellular potentials occur in basal dendrites when the stimulus is delivered to the stratum oriens. To characterize the pattern of laminar synaptic plasticity in the hippocampal CA1 region, we chose the recording position #2 (middle; black star) for the basal and #6 (middle; black star) for the apical dendrites (Figure 1B).
Figure 1.

Basal and Apical Excitatory Postsynaptic Potentials Recorded in CA1
(A) Schematic diagram of a mouse hippocampal slice illustrating the placement of electrodes used in these experiments. The stimulating electrode was placed in either stratum radiatum (s. radiatum) (left) or stratum oriens (s. oriens) (right). Two simultaneous recording electrodes were placed in mid-CA1, one in s. radiatum to record apical dendritic field fEPSPs and one in s. oriens to record basal dendritic field EPSPs.
(B) Representative waveforms recorded from different positions along the laminar tree of CA1 pyramidal cells at three levels of stimulation intensities (low, moderate, high).
Soluble Aβ Oligomers Have No Effect on Heterosynaptic Basal Dendritic LTP
It is well documented that soluble oAβs impair evoked LTP by high-frequency tetanus of the Schaffer collateral afferents in the stratum radiatum (apical dendrites) of the CA1 region. We have also found that the LTP of population spikes, which are recorded immediately adjacent to the soma (i.e., in the pyramidal cell layer), is impaired by oAβs, similar to LTP impairment in the apical dendrites (Lei et al., 2016). To further explore the oAβ effect on the laminar compartments of the dendritic tree of CA1 neurons, we placed two recording electrodes on the basal dendrites (stratum oriens) and apical dendrites (stratum radiatum) simultaneously, to monitor synaptic activity in response to the stimulation of Schaffer collateral afferents (Figure 1A). We found that the magnitude of LTP of basal dendrites was significantly greater than that of apical dendrites (331% ± 44% versus 158% ± 6%, n = 8, p < 0.01) (Figures 2C versus 2A). This cell-wide heterosynaptic basal dendritic LTP is also N-methyl-D-aspartate receptor dependent, similar to the homosynaptic (apical) LTP (Figure S1). Interestingly, soluble oAβs derived from Tris-buffered saline (TBS)-soluble cortical extracts of typical AD brains (Shankar et al., 2008, Li et al., 2011) that were applied to the brain slices 30 min before high-frequency stimulation (HFS) had no significant effect on the basal dendrites, although they inhibited apical dendritic LTP in a manner consistent with our previous reports (Shankar et al., 2008, Li et al., 2011) (basal: 317% ± 29%; apical: 130% ± 4%, n = 8, p < 0.01) (Figure 2C versus 2A). To confirm this finding, we applied another source of oAβs: soluble oligomers present in the conditioned medium (CM) of 7PA2 cells (CHO cells stably expressing the hAPP-V717F AD mutant) (Podlisny et al., 1995). This oAβ-rich CM fully inhibited apical dendritic LTP in the wt hippocampal slices (123% ± 6%, n = 11, versus 157% ± 8%, n = 9) (p < 0.01), but the LTP from basal dendrites again remained the same as the control (CHO- CM) (275% ± 18%, n = 11, versus 265% ± 21%, p > 0.05) (Figures 2E and 2F). These two sources of soluble oAβs demonstrate that the cell-wide heterosynaptic basal dendritic LTP is insensitive to oAβ-mediated synaptic neurotoxicity.
Figure 2.

Soluble Aβ Oligomers Have No Effect on Heterosynaptic Basal Dendritic LTP
(A) Soluble oAβ-rich TBS extracts from AD brain inhibited apical dendritic LTP (red tracing) induced by high-frequency stimulation (HFS, arrow), whereas the control brain TBS extract has no effect on this homosynaptic LTP (black tracing).
(B) Time course and all individual recordings of apical dendritic LTP monitored by single-pulse stimuli (every 20 s) during AD-TBS treatment. LTP was induced by HFS (100 pulses at 100 Hz) at time 30 min and maintained over 60 min.
(C) Same treatments and stimulations as in (A) but with simultaneous recordings from the stratum oriens layer (basal dendrites); here, evoked heterosynaptic LTP remained unaffected.
(D) Time course and all individual recordings of basal dendritic LTP just as in (B).
(E) CM of 7PA2 cells that is rich in soluble oAβs inhibited LTP (red tracing) induced by high-frequency stimulation (HFS, arrow) in the apical dendrites.
(F) Heterosynaptic LTP recorded simultaneously from basal dendrites was not affected by the same treatment. The recording electrodes (see Figure 1) placed the positions of “2” (basal) and “6” (apical) dendrites, whereas the stimulation electrode aims to Schaffer collaterals. Inset traces are typical fEPSPs recorded before (gray) and after (black or red) HFS for each condition. Horizontal calibration bars: 10 ms; vertical bars: 0.5 mV.
Soluble Aβ Oligomers Facilitate both Basal Dendritic and Apical Dendritic LTD Induction
Since the oAβs impaired the homosynaptic LTP but did not noticeably affect the heterodendritic LTP, we sought to determine whether the process of synaptic depression was altered. To investigate whether basal dendritic LTD has any difference from the apical dendritic LTD, we first verified the synaptic responses after a weak LFS (300 Hz, 1 Hz) in each compartment (Li et al., 2009). Consistent with previous reports, we found that a weak LFS failed to induce significant LTD in apical dendrites in control slice perfusate (Figure 3A, black). Likewise, the same stimulus did not induce synaptic depression in basal dendrites (Figure 3B, black). When soluble oAβ extracted from AD brain was added to the slices, the same weak LFS protocol induced significant LTDs in both dendritic compartments (apical: 88% ± 4%, n = 8, versus 101% ± 3%, n = 7, p < 0.01, Figure 3A; basal: 80% ± 5% versus 100% ± 6%, p < 0.01, Figure 3B). Similarly, the other source of soluble human oAβs obtained from cell-secreted medium (7PA2 CM) had the same effects on both compartments of the CA1 dendritic tree (apical: 77% ± 3%, n = 8, versus 102% ± 2%, n = 6, p < 0.0, Figure 3C; basal: 79% ± 3%, versus 99% ± 2%, p < 0.01, Figure 3D). As regards the mechanism by which oAβs facilitated LTD, we (Shankar et al., 2008) and others (Hu et al., 2014) have demonstrated that this is metabotropic glutamate receptor (mGluR) dependent, so we applied the mGluR5 antagonist, SIB 1757 (3 μM) to the brain slices 10 min before 7PA2 CM administration. The oAβ-facilitated LTD was fully blocked in both CA1 subregions (apical: 97% ± 3%, n = 7, Figure 3E; basal: 103% ± 3%, n = 7, Figure 3F). In addition to such evidence that mGluRs help regulate synaptic depression, synaptic localization of the GluA2R-lacking, calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (CPAMPARs) may also be important for the expression of hippocampal LTD (Isaac et al., 2007, Sanderson et al., 2016). We used the CP-AMPAR selective antagonist Philanthotoxin 74 (10 μM) to block the GluA1 and GluA3 AMPAR activities. However, this antagonist failed to prevent oAβ-enhanced LTD in CA1 (Figure S2), suggesting that CP-AMPARs are not significantly involved in oAβ-mediated synaptic depression in CA1.
Figure 3.

Soluble Aβ Oligomers Facilitate Both Homosynaptic and Heterosynaptic Hippocampal LTD
(A) A train of 300 single pulses at 1 Hz (5 min; small gray bar) did not induce LTD in acute mouse hippocampal slices in the presence of control brain TBS extract (black diamonds, n = 7) but did so in the presence of oAβ-rich AD-TBS (red circles, n = 7), as recorded from apical dendrites in CA1 region.
(B) A simultaneous recording from stratum oriens (basal dendrites) in CA1 also showed the same results with the respective treatments and stimulations (black: control TBS, red: AD-TBS).
(C) The train of 300 single pulses at 1 Hz (5 min; small gray bar) did not induce LTD in hippocampal slices in the presence of CHO- CM (black diamonds, n = 7) but induced a significant LTD in the presence of oAβ-rich 7PA2 CM (red circles, n = 7).
(D) The simultaneous recording from the stratum oriens (basal dendrites) also showed a significant LTD with 7PA2 CM (red circles), but not the CHO- CM (black diamonds), under the same stimulation.
(E) Homosynaptic apical LTD induced by the 300-pulse protocol (gray bar) in the presence of 7PA2 CM was blocked upon pre-administration of the highly selective mGluR5 antagonist, SIB 1757 (3 μM, blue circles, n = 8).
(F) Heterosynaptic basal dendritic LTD induced by the 300-pulse protocol in the presence of 7PA2 CM was similarly blocked by SIB 1757 (3 μM, blue circles). The electrode placement is the same as in Figure 2.
Aβ Oligomers Decrease Neurotransmission at Strong Intensities, Decrease PPF, and Delay the Latency of Peak Positive Responses in Basal Dendrites
Soluble oAβs appear to have no significant effect on basal dendritic LTP, but they can still facilitate LTD in that subregion and disrupt the homosynaptic (apical) LTP as well as the LTP at somata (population spikes). To further explore these oAβ-mediated dipolar heterodendritic changes, we next measured the basal neurotransmission by input/output curve in basal dendrites compared with apical dendrites. Although basal neurotransmission did not change significantly in the apical dendrites (Figure 4A), the recorded fEPSPs from basal dendrites were significantly reduced in response to strong stimulus intensities when recorded 30 min after exposure to 7PA2 CM (Figure 4B), suggesting that the soluble oAβ can disrupt basal but not apical dendritic activity in response to strong stimulation.
Figure 4.

Soluble Aβ Oligomers Disrupt Neuronal Network Integration
(A and B) The input-output (I/O) curve in the pathways recorded from apical dendrites (A) and basal dendrites (B) during stimulation of Schaffer collaterals in the presence of control CHO- CM (black circles, n = 12) and oAβ-rich 7PA2 CM (red circles, n = 14).
(C and D) Paired-pulse facilitation (PPF) in the two compartments (C: apical dendrites, D: basal dendrites) was measured by varying the intervals (20, 40, 60, 100, 200, and 500 ms) between pairs of stimuli (interstimulus interval; ISI) 30 min after applying CHO- CM (black circles, n = 14) or 7PA2 CM (red circles, n = 12) treatments.
(E) Difference between the onset of first EPSP response recorded from apical dendrites (blue) and basal dendrites (red) (illustrated on the right as indicated by two vertical lines) under the oAβ-rich (red circles, n = 7) and control CM (black circles, n = 8) treatments.
Short-term forms of synaptic plasticity are crucial for regulating the temporal code and information processing between neurons in a network (Tsodyks and Markram, 1997). Accordingly, we recorded paired-pulse facilitation (PPF) in the apical and basal dendrites of CA1 simultaneously. The second pulse-evoked response increased significantly at every interstimulus interval (ISI) tested (20–200 ms, p < 0.01, n = 14) compared with the first pulse-evoked response, as expected. The PPF ratios did not show significant differences between the apical and basal dendrites (Figures 4C and 4D, black). In line with previous reports from our and other laboratories (Shankar et al., 2008, Schmid et al., 2008, Li et al., 2009, Cerpa et al., 2010, Talantova et al., 2013), the apical PPF did not change after soluble oAβ exposure (Figure 4C). However, the PPF in the basal dendrites was significantly lower in oAβ-rich 7PA2 CM than in the CHO- CM (ISI 60 ms, p < 0.05, or ISI 100 and 200 ms, p < 0.01, Figure 4D).
The longer ISI between the second pulse-induced and the first pulse-induced fEPSP decrease in the basal but not apical dendrites suggested that the oAβs may interrupt the excitatory conduction from apical to basal dendrites. To assess this, we measured the latent period (latency) of fEPSP (i.e., time from stimulus onset to onset of fEPSP) in the apical and basal dendrites and found that the latency difference between apical and basal dendrites was significantly longer in 7PA2 CM-treated group at all stimulation intensities (Figure 4E). These results suggest that soluble oAβs disrupt neuronal dipolar features in the hippocampus.
Stimulation Time, Not Frequency, Determines the oAβ Impairment of Heterosynaptic Basal LTP
Because oAβs impaired 100-Hz-induced LTP and facilitated 1-Hz-induced LTD, we sought to test whether oAβ has any effect on the modification threshold or sliding threshold (θm) frequency of 10 Hz, which represents the point of crossover between LTP and LTD (or LTP threshold) in frequency-response experiments (Bear, 1996, Hulme et al., 2012). Also, the 10-Hz frequency band of electroencephalogram (EEG) (α waves) was reported to be decreased in patients with mild cognitive impairment (MCI) and AD (Moretti., 2015), and it is an important signal in the understanding of cognitive processes (Başar and Güntekin, 2012). Previous reports using a single train of 900 pulses at 10 Hz did not alter the synaptic efficacy (Bear, 1996, Heynen et al., 1996). Here we used 10 trains of 10 pulse bursts in 10-Hz intensive (10-s interval between the trains) stimulation, and it induced a small but significant LTP in the apical dendrites (10 Hz-10 s: 134% ± 7% versus 100 Hz: 157% ± 8%, p < 0.05) but no difference in the basal dendrites (10 Hz-10 s: 290% ± 26% versus 100 Hz: 265% ± 25%, p > 0.05) (contrast Figures 5A and 5B versus 2E and 2F). Consistent with the regular 100-Hz HFS-induced LTP, soluble oAβs (7PA2 CM) did not inhibit the 10Hz-10s basal dendritic LTP (296% ± 27%, n = 7) but only inhibited the apical dendritic LTP (115% ± 4%, n = 7) (Figure 5A). To further explore this apparent temporal integration of the stimulation, we used the same 10-Hz and 10-pulse bursts for each train but increased the interval from 10 s to 30 s (to mimic the LTD protocol timing). We still obtained significant LTPs from both compartments of the CA1 dendritic tree. Interestingly, the heterodendritic basal LTPs were significantly impaired after 7PA2 CM treatment in this protocol (apical: 113% ± 4%, n = 9 versus 129% ± 7%, n = 17, p < 0.01, Figure 5C; basal: 143% ± 11%, n = 9 versus 317% ± 21%, n = 17, p < 0.01, Figure 5D). To assess if the 10-Hz-30-s-interval-induced LTP required mGluR5 activation like LTD did, we added the mGluR5 selective antagonist SIB 1757 (3 μM) to the perfusion buffer 10 min before 7PA2 CM. Interestingly, this also reversed the oAβ effect on LTP in both dendritic compartments (apical: SIB + CHO- CM 120% ± 4%, n = 6 versus SIB+7PA2 CM 116% ± 4%, n = 7, p > 0.05, Figure 5E; basal: 263% ± 34%, n = 6 versus 245% ± 34%, n = 7, p > 0.05, Figure 5F). Certain other receptor antagonists such as a GABAB receptor antagonist (CGP 35348, 10 μM) or an H-channel blocker (ZD7288, 5 μM) could not affect the spaced 10-Hz burst-induced heterodendritic LTP (Figure S3). Taken together, these results confirm that oAβs inhibit heterodendritic LTP and facilitate LTD in a way that requires activation of mGluR5.
Figure 5.

Time, Not Frequency, Determines the Aβ Oligomer Impairment of Heterosynaptic Basal LTP
(A–F) Homosynaptic apical dendritic LTP (A) and heterosynaptic basal dendritic LTP (B) induced by 10-Hz burst stimulation (each burst interval is 10 s for 10 trains [arrows]) produced different responses to the oAβ-rich 7PA2 CM treatment (red circles, n = 10). Both apical (C) and basal (D) dendritic LTPs were inhibited by the 7PA2 CM (red circles, n = 12) when the same 10-Hz burst interval was spaced to 30 s instead of 10 s. Homosynaptic apical (E) and heterodendritic basal (F) 30-s-spaced 10-Hz burst LTPs were compared with and without the mGluR5 selective antagonist, SIB 1757 (3 μM) combined with either CHO- CM (black) or 7PA2 CM (red). The electrode placement is the same as Figure 2. Inset traces are typical fEPSPs recorded before (gray) and after (black or red) 10-Hz burst stimulations in each condition. Horizontal bars: 10 ms; vertical bars: 0.5 mV.
Basal Dendrites Are less Sensitive to Aβ Oligomer-Mediated Synaptotoxicity
The heterodendritic LTP recorded from basal dendrites by stimulation of Schaffer collateral afferents may reflect the electrical properties of a neuron in that the hippocampal pyramidal cell has a dipole nature (Einevoll et al., 2013). To further confirm that basal dendrites are resistant to soluble oAβs, we placed the stimulation electrode in the same layer (stratum oriens) to record the homosynaptic basal dendritic LTP. In this experiment, we first verified that the oAβ-rich TBS extract from an AD brain decreased the conventional apical LTP. Indeed, consistent with our prior work (above), these soluble oAβs partially blocked homosynaptic apical dendritic LTP (158% ± 8%, n = 7, versus 134% ± 4%, n = 7, p < 0.05) (Figure 6A), whereas they had no effect on the heterodendritic basal LTP (270% ± 25%, versus 289% ± 20%, p < 0.05, Figure 6B). Interestingly, this same batch of AD brain oAβs had no effect on the homosynaptic basal dendritic LTP upon stimulation in the stratum oriens (190% ± 6%, n = 7, versus 181% ± 9%, n = 8, p > 0.05) (Figure 6C). Here, the heterodendritic apical dendritic LTP was not decreased (316% ± 28%, versus 290% ± 31%, p > 0.05) (Figure 6D). Consistent with previous reports (Haley et al., 1996, Sajikumar et al., 2007, Fan and Fu, 2014), we found that the magnitude of homosynaptic basal dendritic LTP (190%–217%; Figure 6C) was larger than that of homosynaptic apical LTP (157%–158%; Figures 2A, 2E, and 6A).
Figure 6.

Apical Dendrites Are More Vulnerable to Soluble Aβ Oligomers
(A–D) Homosynaptic basal dendritic LTP could be recorded from stratum oriens layers when the stimulation electrode was placed in the same layer. Consistent with Figure 2 (above), AD-TBS extract inhibited homosynaptic apical dendritic LTP (A) and did not affect the heterosynaptic basal dendritic LTP (B) induced by 100-Hz HFS (arrows) delivered to stratum radiatum, as before. The same batch of AD-TBS extract did not produce a significant effect on the homosynaptic basal dendritic LTP (C) and heterosynaptic apical dendritic LTP (D) by stimulation in stratum oriens. Inset traces are typical fEPSPs recorded before (gray) and after (black or red) 10-Hz burst stimulations for each condition. Horizontal bars: 10 ms; vertical bars: 0.5 mV.
To further confirm whether the heterodendritic LTD could be recorded by stimulation of basal dendrites, we recorded the homosynaptic LTD from stratum oriens and heterodendritic LTD from stratum radiatum. In line with the stimulation of Schaffer collateral afferents, both the homosynaptic basal dendritic LTD and heterosynaptic apical LTD were also facilitated by 7PA2 CM treatment upon stimulation in stratum oriens (basal: 76% ± 6%, n = 7, in 7PA2 CM, versus 99% ± 2%, n = 6, in CHO- CM, p < 0.01; apical: 72% ± 4%, n = 7, in 7PA2 CM, versus 103% ± 2%, n = 6, in CHO- CM, p < 0.01) (Figure S4). These results further support our above-mentioned findings that oAβs significantly facilitate both homosynaptic and heterosynaptic LTDs.
Discussion
AD is characterized by memory loss, cognitive decline, and devastating neurodegeneration, not only as a result of the extracellular accumulation of Aβ and intracellular accumulation of tau but also as a consequence of a multifactorial dysfunction and loss of synapses. Recent high-resolution human fMRI studies demonstrated that hippocampal subfields are specialized in different learning processes and can undergo selective damage in early stages of AD in patients. Specifically, the apical dendrites of CA1 pyramidal neurons are first targeted, whereas the basal dendrites of CA1 neurons remain unchanged (Das et al., 2012, Engvig et al., 2012, Kerchner et al., 2012). Several studies also report that soluble oAβ primarily affects the apical dendritic arbors, with little or no effect on basal dendrites of CA1 pyramidal neurons in AD-like mouse models of Aβ accumulation (Alpár et al., 2006, Steele et al., 2014, Price et al., 2014). The present study uses dual recording to assess oAβ effects on synaptic plasticity and dynamic changes in network interactions in the dendritic trees of CA1 neurons. Our results show that soluble oAβ extracted from human AD brain has no effect on the heterodendritic basal dendritic LTP but still facilitates the heterodendritic basal dendritic LTD. The apical dendrites are more vulnerable to oAβ-mediated synaptotoxicity. Using a 10-Hz burst stimulation, we found that the oAβs impair the heterodendritic basal dendritic LTP at a spaced interval (30 s), not at an intensive interval (10 s). Mechanistically, soluble oAβ impairment of heterodendritic plasticity is mediated in part by the activation of mGluRs.
Most LTP recordings examine homosynaptic LTP that occurs at synapses that were active during the induction. However, certain synapses that are not directly active by the afferent stimulation during the induction could be active by “spillover,” inducing a heterosynaptic plasticity. Such plasticity is usually recorded from the same layer of hippocampus but with different afferent input; however, several reports defined the neuron-wide heterosynaptic plasticity as heterodendritic plasticity (Young and Nguyen, 2005, Hulme et al., 2012). Heterosynaptic plasticity has a strong stabilizing effect on synaptic weights (the amount of influence the firing of one neuron has on another) and neuronal circuits. It helps preserve the ability of a neuron with plastic synapses for further learning. A possible signal that may trigger cell-wide heterosynaptic plasticity is an increase of intracellular calcium concentration caused by back-propagating action potentials. Due to the dipole nature of hippocampal pyramidal neurons as to their field potentials (Einevoll et al., 2013), it is likely that when the apical dendrites are depolarized, the basal dendrites will be hyperpolarized, resulting from the current flow. The present study also demonstrates that the latency of onset of basal dendritic fEPSP/PS (population spike) is longer after oAβ exposure, suggesting that oAβ can interrupt the current propagation, or otherwise interfere with membrane electrical properties. The result showed that the oAβs cause the heterosynaptic response delay (Figure 4E).
LTP in apical (stratum radiatum) and basal (stratum oriens) dendrites of hippocampal CA1 pyramidal neurons are known to differ in induction and maintenance (Kramar and Lynch, 2003, Sajikumar et al., 2007, Navakkode et al., 2012, Fan, 2013). Several reported molecular mechanisms may differ in the distribution and/or fine tone of synaptic plasticity between apical and basal dendritic spines (Brzdak et al., 2017). Another reason may be the patterns of innervation, e.g., CA1 stratum oriens receives more input from ipsilateral CA2 and contralateral CA3, whereas CA1 stratum radiatum receives more input from the ipsilateral CA3 and small portions of local CA2 (Shinohara et al., 2012). In our slice study, the innervations from the contralateral hippocampus are cut off, meaning that a larger proportion of inputs to stratum oriens are missing, when compared with stratum radiatum, so we considered our basal dendritic LTP to be a cell-wide heterosynaptic LTP. The basal dendrites being shorter and less branched permit for less attenuation of signal (Henze et al., 1996) and thus can be induced to have a greater LTP and are more resistant to the oAβ-mediated synaptotoxicity.
Using human AD brain extracts rich in oAβs, we found that the basal dendritic homosynaptic LTP is unaffected, whereas the apical homosynaptic LTP is partially impaired, suggesting that basal dendrites are less sensitive to the Aβ-mediated synaptic dysfunction when compared with the apical dendrites. We have seen no reports on oAβ effects on neuron-wide heterosynaptic LTPs in vitro, but a similar report can be found in vivo (Hu et al., 2009). In contrast to the effect of soluble oAβs on the basal dendritic LTP, a facilitated LTD could be recorded in both heterosynaptic basal dendritic LTD (Schaffer collateral stimulation) and homosynaptic basal dendritic LTD (stratum oriens stimulation) after oAβ administration. Similarly, using 10-Hz burst stimulation, where the total pulses were the same and only the train intervals were 10 or 30 s, we found that oAβs only impaired the more widely spaced (30 s) burst-induced heterosynaptic LTP. The oAβ-disrupted heterosynaptic plasticities by both longer timing conditioning stimulations (i.e., LFS lasts for 5 min, spaced 10-Hz burst lasts for 4.5 min) were also mGluR5 dependent. The longer stimulation triggered synaptic currents may influence escape of charged glutamate from the cleft (Sylantyev et al., 2008), therefore consistently activates the postsynaptic mGluRs and mGluR-dependent activation of the MAPK cascade can lead to AMPAR internalization (Casimiro et al., 2011).
Learning-induced LTP is a heterosynaptic phenomenon that requires inputs from other neural structures (Zhu et al., 2011). Mechanistically, it has been shown that activation of β-adrenergic receptors generates long-lasting enhancements of heterosynaptic plasticity (Connor et al., 2011). In this regard, we previously reported that an enriched environment (EE) in wt mice significantly protects hippocampal LTP against the effects of soluble oAβs via activation of β-adrenergic receptors (Li et al., 2013a). In accordance, EE may delay the onset of AD in humans and can ameliorate the memory deficits of AD model animals (Li et al., 2013a). This could be due to EE enhancing both homosynaptic and heterosynaptic plasticities. Our present results suggest that heterosynaptic basal dendritic LTP is resistant to oAβ-induced synaptotoxicity, similar to the resistance to oAβ-induced toxicity of homosynaptic apical LTP in EE mice. It has been reported that EE counteracts the age-dependent shift of EEG spectral power toward slow oscillations (δ and θ rhythms) (Mainardi et al., 2014). Compared with age-matched healthy control subjects, patients with MCI and AD exhibit an increase in the relative power of slow oscillations (δ and θ rhythms) associated with a decrease in relative power of fast oscillations (α, β, and γ rhythms) (Moretti, 2015). The alpha activity is significantly decreased in patients with cognitive impairment and AD, suggesting that soluble oAβ interferes with the spontaneous neuronal network that generates the alpha oscillations (Başar and Güntekin, 2012, Moretti, 2015). Therefore, the frequency and time-dependent heterosynaptic plasticity impairment induced by soluble oAβs suggest that certain neuronal firing patterns could be affected by soluble oAβ or in the earlier phase of AD.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Drs. Nikolai Otmakhov and Zemin Wang for their expert advice. This work was supported by NIH grant RF1 AG006173 (D.J.S.); Alzheimer's Association (S.L.); the project for the Disciplinary group of Psychology and Neuroscience, Xinxiang Medical University, China (J.Z.); Henan Natural Science Foundation (182300410389) (J.Z.); and Scientific and Technological Project of Health and Family Planning Commission, Henan Province, China 201303105 (J.Z.).
Author Contributions
J.Z, A.L., and S.L. performed experiments and analyzed the data; M.R. prepared the 7PA2 CM and CHO- CM; Y.D. prepared the human AD brain and control brain extracts; S.L. designed the experiments and wrote the paper; and D.J.S. advised the experimental design and edited the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: August 31, 2018
Footnotes
Supplemental Information includes Transparent Methods and four figures and can be found with this article online at https://doi.org/10.1016/j.isci.2018.07.018.
Supplemental Information
References
- Alpár A., Ueberham U., Brückner M.K., Seeger G., Arendt T., Gärtner U. Different dendrite and dendritic spine alterations in basal and apical arbors in mutant human amyloid precursor protein transgenic mice. Brain Res. 2006;1099:189–198. doi: 10.1016/j.brainres.2006.04.109. [DOI] [PubMed] [Google Scholar]
- Başar E., Güntekin B. A short review of alpha activity in cognitive processes and in cognitive impairment. Int. J. Psychophysiol. 2012;86:25–38. doi: 10.1016/j.ijpsycho.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Bateman R.J., Xiong C., Benzinger T.L., Fagan A.M., Goate A., Fox N.C., Marcus D.S., Cairns N.J., Xie X., Blazey T.M. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear M.F. A synaptic basis for memory storage in the cerebral cortex. Proc. Natl. Acad. Sci. USA. 1996;93:13453–13459. doi: 10.1073/pnas.93.24.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberich S., Pohle J., Pollard M., Barroso-Flores J., Köhr G. Interplay between global and pathway-specific synaptic plasticity in CA1 pyramidal cells. Sci. Rep. 2017;7:17040. doi: 10.1038/s41598-017-17161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobinski M., de Leon M.J., Tarnawski M., Wegiel J., Reisberg B., Miller D.C., Wisniewski H.M. Neuronal and volume loss in CA1 of the hippocampal formation uniquely predicts duration and severity of Alzheimer disease. Brain Res. 1998;805:267–269. doi: 10.1016/s0006-8993(98)00759-8. [DOI] [PubMed] [Google Scholar]
- Boutet C., Chupin M., Lehéricy S., Marrakchi-Kacem L., Epelbaum S., Poupon C., Wiggins C., Vignaud A., Hasboun D., Defontaines B. Detection of volume loss in hippocampal layers in Alzheimer's disease using 7 T MRI: a feasibility study. Neuroimage Clin. 2014;5:341–348. doi: 10.1016/j.nicl.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H., Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- Brzdak P., Wójcicka O., Zareba-Koziol M., Minge D., Henneberger C., Wlodarczyk J., Mozrzymas J.W., Wójtowicz T. Synaptic potentiation at basal and apical dendrites of hippocampal pyramidal neurons involves activation of a distinct set of extracellular and intracellular molecular cues. Cereb. Cortex. 2017 doi: 10.1093/cercor/bhx324. [DOI] [PubMed] [Google Scholar]
- Casimiro T.M., Sossa K.G., Uzunova G., Beattie J.B., Marsden K.C., Carroll R.C. mGluR and NMDAR activation internalize distinct populations of AMPARs. Mol. Cell. Neurosci. 2011;48:161–170. doi: 10.1016/j.mcn.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerpa W., Farías G.G., Godoy J.A., Fuenzalida M., Bonansco C., Inestrosa N.C. Wnt-5a occludes Abeta oligomer-induced depression of glutamatergic transmission in hippocampal neurons. Mol. Neurodegener. 2010;5:3. doi: 10.1186/1750-1326-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Lin R., Chang L., Xu S., Wei X., Zhang J., Wang C., Anwyl R., Wang Q. Enhancement of long-term depression by soluble amyloid β protein in rat hippocampus is mediated by metabotropic glutamate receptor and involves activation of p38MAPK, STEP and caspase-3. Neuroscience. 2013;253:435–443. doi: 10.1016/j.neuroscience.2013.08.054. [DOI] [PubMed] [Google Scholar]
- Chistiakova M., Bannon N.M., Bazhenov M., Volgushev M. Heterosynaptic plasticity: multiple mechanisms and multiple roles. Neuroscientist. 2014;20:483–498. doi: 10.1177/1073858414529829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor S.A., Wang Y.T., Nguyen P.V. Activation of {beta}-adrenergic receptors facilitates heterosynaptic translation-dependent long-term potentiation. J. Physiol. 2011;589:4321–4340. doi: 10.1113/jphysiol.2011.209379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S.R., Avants B.B., Pluta J., Wang H., Suh J.W., Weiner M.W., Mueller S.G., Yushkevich P.A. Measuring longitudinal change in the hippocampal formation from in vivo high-resolution T2-weighted MRI. Neuroimage. 2012;60:1266–1279. doi: 10.1016/j.neuroimage.2012.01.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Flores R., La Joie R., Landeau B., Perrotin A., Mézenge F., de La Sayette V., Eustache F., Desgranges B., Chételat G. Effects of age and Alzheimer's disease on hippocampal subfields: comparison between manual and FreeSurfer volumetry. Hum. Brain Mapp. 2015;36:463–474. doi: 10.1002/hbm.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele Y.S., Duyckaerts C. Propagation of Aß pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016;131:5–25. doi: 10.1007/s00401-015-1516-y. [DOI] [PubMed] [Google Scholar]
- Engvig A., Fjell A.M., Westlye L.T., Skaane N.V., Sundseth Ø., Walhovd K.B. Hippocampal subfield volumes correlate with memory training benefit in subjective memory impairment. Neuroimage. 2012;61:188–194. doi: 10.1016/j.neuroimage.2012.02.072. [DOI] [PubMed] [Google Scholar]
- Einevoll G.T., Kayser C., Logothetis N.K., Panzeri S. Modelling and analysis of local field potentials for studying the function of cortical circuits. Nat. Rev. Neurosci. 2013;14:770–785. doi: 10.1038/nrn3599. [DOI] [PubMed] [Google Scholar]
- Fan W. Group I metabotropic glutamate receptors modulate late phase long-term potentiation in hippocampal CA1 pyramidal neurons: comparison of apical and basal dendrites. Neurosci. Lett. 2013;553:132–137. doi: 10.1016/j.neulet.2013.08.030. [DOI] [PubMed] [Google Scholar]
- Fan W., Fu T. Somatostatin modulates LTP in hippocampal CA1 pyramidal neurons: differential activation conditions in apical and basal dendrites. Neurosci. Lett. 2014;561:1–6. doi: 10.1016/j.neulet.2013.12.025. [DOI] [PubMed] [Google Scholar]
- Harris J.A., Devidze N., Verret L., Ho K., Halabisky B., Thwin M.T., Kim D., Hamto P., Lo I., Yu G.Q. Transsynaptic progression of amyloid-β-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron. 2010;68:428–441. doi: 10.1016/j.neuron.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley J.E., Schaible E., Pavlidis P., Murdock A., Madison D.V. Basal and apical synapses of CA1 pyramidal cells employ different LTP induction mechanisms. Learn Mem. 1996;3:289–295. doi: 10.1101/lm.3.4.289. [DOI] [PubMed] [Google Scholar]
- Henze D.A., Cameron W.E., Barrionuevo G. Dendritic morphology and its effects on the amplitude and rise-time of synaptic signals in hippocampal CA3 pyramidal cells. J. Comp. Neurol. 1996;369:331–344. doi: 10.1002/(SICI)1096-9861(19960603)369:3<331::AID-CNE1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Heynen A.J., Abraham W.C., Bear M.F. Bidirectional modification of CA1 synapses in the adult hippocampus in vivo. Nature. 1996;381:163–166. doi: 10.1038/381163a0. [DOI] [PubMed] [Google Scholar]
- Hu N.W., Klyubin I., Anwyl R., Rowan M.J. GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc. Natl. Acad. Sci. USA. 2009;106:20504–20509. doi: 10.1073/pnas.0908083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu N.W., Nicoll A.J., Zhang D., Mably A.J., O'Malley T., Purro S.A., Terry C., Collinge J., Walsh D.M., Rowan M.J. mGlu5 receptors and cellular prion protein mediate amyloid-β-facilitated synaptic long-term depression in vivo. Nat. Commun. 2014;5:3374. doi: 10.1038/ncomms4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme S.R., Jones O.D., Ireland D.R., Abraham W.C. Calcium-dependent but action potential-independent BCM-like metaplasticity in the hippocampus. J. Neurosci. 2012;32:6785–6794. doi: 10.1523/JNEUROSCI.0634-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac J.T., Ashby M.C., McBain C.J. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Kerchner G.A., Deutsch G.K., Zeineh M., Dougherty R.F., Saranathan M., Rutt B.K. Hippocampal CA1 apical neuropil atrophy and memory performance in Alzheimer's disease. Neuroimage. 2012;63:194–202. doi: 10.1016/j.neuroimage.2012.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan W., Westman E., Jones N., Wahlund L.O., Mecocci P., Vellas B., Tsolaki M., Kłoszewska I., Soininen H., Spenger C., AddNeuroMed consortium and for the Alzheimer’s Disease Neuroimaging Initiative Automated hippocampal subfield measures as predictors of conversion from mild cognitive impairment to Alzheimer's disease in two independent cohorts. Brain Topogr. 2015;28:746–759. doi: 10.1007/s10548-014-0415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramar E.A., Lynch G. Developmental and regional differences in the consolidation of long-term potentiation. Neuroscience. 2003;118:387–398. doi: 10.1016/s0306-4522(02)00916-8. [DOI] [PubMed] [Google Scholar]
- Kuo M.C., Leung L.S. Disruption of hippocampal multisynaptic networks by general anesthetics. Anesthesiology. 2017 doi: 10.1097/ALN.0000000000001861. [DOI] [PubMed] [Google Scholar]
- La Joie R., Perrotin A., de La Sayette V., Egret S., Doeuvre L., Belliard S., Eustache F., Desgranges B., Chételat G. Hippocampal subfield volumetry in mild cognitive impairment, Alzheimer's disease and semantic dementia. Neuroimage Clin. 2013;3:155–162. doi: 10.1016/j.nicl.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M., Xu H., Li Z., Wang Z., O'Malley T.T., Zhang D., Walsh D.M., Xu P., Selkoe D.J., Li S. Soluble Aβ oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol. Dis. 2016;85:111–121. doi: 10.1016/j.nbd.2015.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner B., Falduto J., Shors T.J. Associative memory formation increases the observation of dendritic spines in the hippocampus. J. Neurosci. 2003;23:659–665. doi: 10.1523/JNEUROSCI.23-02-00659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Hong S., Shepardson N.E., Walsh D.M., Shankar G.M., Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Jin M., Koeglsperger T., Shepardson N.E., Shankar G.M., Selkoe D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Jin M., Zhang D., Yang T., Koeglsperger T., Fu H., Selkoe D.J. Environmental novelty activates β2-adrenergic signaling to prevent the impairment of hippocampal LTP by Aβ oligomers. Neuron. 2013;77:929–941. doi: 10.1016/j.neuron.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.D., Dong H.B., Xie G.M., Zhang L.J. Discriminative analysis of mild Alzheimer's disease and normal aging using volume of hippocampal subfields and hippocampal mean diffusivity: an in vivo magnetic resonance imaging study. Am. J. Alzheimers Dis. Other Demen. 2013;28:627–633. doi: 10.1177/1533317513494452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T., Du X., Pick J.E., Sui G., Brownlee M., Klann E. Glucagon-like peptide-1 cleavage product GLP-1(9-36) amide rescues synaptic plasticity and memory deficits in Alzheimer's disease model mice. J. Neurosci. 2012;32:13701–13708. doi: 10.1523/JNEUROSCI.2107-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmmoud R.R., Sase S., Aher Y.D., Sase A., Gröger M., Mokhtar M., Höger H., Lubec G. Spatial and working memory is linked to spine density and mushroom spines. PLoS One. 2015;10:e0139739. doi: 10.1371/journal.pone.0139739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi M., Di Garbo A., Caleo M., Berardi N., Sale A., Maffei L. Environmental enrichment strengthens corticocortical interactions and reduces amyloid-β oligomers in aged mice. Front. Aging Neurosci. 2014;6:1. doi: 10.3389/fnagi.2014.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama M., Shimada H., Suhara T., Shinotoh H., Ji B., Maeda J., Zhang M.R., Trojanowski J.Q., Lee V.M., Ono M. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094–1108. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti D.V. Theta and alpha EEG frequency interplay in subjects with mild cognitive impairment: evidence from EEG, MRI, and SPECT brain modifications. Front. Aging Neurosci. 2015;7:31. doi: 10.3389/fnagi.2015.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S.G., Schuff N., Yaffe K., Madison C., Miller B., Weiner M.W. Hippocampal atrophy patterns in mild cognitive impairment and Alzheimer's disease. Hum. Brain Mapp. 2010;31:1339–1347. doi: 10.1002/hbm.20934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek E.S., Holtzman D.M. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat. Neurosci. 2015;18:800–806. doi: 10.1038/nn.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navakkode S., Sajikumar S., Korte M., Soong T.W. Dopamine induces LTP differentially in apical and basal dendrites through BDNF and voltage-dependent calcium channels. Learn Mem. 2012;19:294–299. doi: 10.1101/lm.026203.112. [DOI] [PubMed] [Google Scholar]
- Nicoll R.A. A brief history of long-term potentiation. Neuron. 2017;93:281–290. doi: 10.1016/j.neuron.2016.12.015. [DOI] [PubMed] [Google Scholar]
- Olsen K.M., Sheng M. NMDA receptors and BAX are essential for Aβ impairment of LTP. Sci. Rep. 2012;2:225. doi: 10.1038/srep00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlisny M.B., Ostaszewski B.L., Squazzo S.L., Koo E.H., Rydell R.E., Teplow D.B., Selkoe D.J. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J. Biol. Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- Price K.A., Varghese M., Sowa A., Yuk F., Brautigam H., Ehrlich M.E., Dickstein D.L. Altered synaptic structure in the hippocampus in a mouse model of Alzheimer's disease with soluble amyloid-β oligomers and no plaque pathology. Mol. Neurodegener. 2014;9:41. doi: 10.1186/1750-1326-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rössler M., Zarski R., Bohl J., Ohm T.G. Stage-dependent and sector-specific neuronal loss in hippocampus during Alzheimer's disease. Acta Neuropathol. 2002;103:363–369. doi: 10.1007/s00401-001-0475-7. [DOI] [PubMed] [Google Scholar]
- Sajikumar S., Navakkode S., Frey J.U. Identification of compartment- and process-specific molecules required for “synaptic tagging” during long-term potentiation and long-term depression in hippocampal CA1. J. Neurosci. 2007;27:5068–5080. doi: 10.1523/JNEUROSCI.4940-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado-Puga K., Rodríguez-Colorado J., Prado-Alcalá R.A., Peña-Ortega F. Subclinical doses of ATP-sensitive potassium channel modulators prevent alterations in memory and synaptic plasticity induced by amyloid-β. J. Alzheimers Dis. 2017;57:205–226. doi: 10.3233/JAD-160543. [DOI] [PubMed] [Google Scholar]
- Sanderson J.L., Gorski J.A., Dell'Acqua M.L. NMDA receptor-dependent LTD requires transient synaptic incorporation of Ca2⁺-permeable AMPARs mediated by AKAP150-anchored PKA and calcineurin. Neuron. 2016;89:1000–1015. doi: 10.1016/j.neuron.2016.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid A.W., Freir D.B., Herron C.E. Inhibition of LTP in vivo by beta-amyloid peptide in different conformational states. Brain Res. 2008;1197:135–142. doi: 10.1016/j.brainres.2007.11.056. [DOI] [PubMed] [Google Scholar]
- Shankar G.M., Li S., Mehta T.H., Garcia-Munoz A., Shepardson N.E., Smith I., Brett F.M., Farrell M.A., Rowan M.J., Lemere C.A. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara Y., Hosoya A., Yahagi K., Ferecskó A.S., Yaguchi K., Sík A., Itakura M., Takahashi M., Hirase H. Hippocampal CA3 and CA2 have distinct bilateral innervation patterns to CA1 in rodents. Eur. J. Neurosci. 2012;35:702–710. doi: 10.1111/j.1460-9568.2012.07993.x. [DOI] [PubMed] [Google Scholar]
- Steele J.W., Brautigam H., Short J.A., Sowa A., Shi M., Yadav A., Weaver C.M., Westaway D., Fraser P.E., St George-Hyslop P.H. Early fear memory defects are associated with altered synaptic plasticity and molecular architecture in the TgCRND8 Alzheimer's disease mouse model. J. Comput. Neurol. 2014;522:2319–2335. doi: 10.1002/cne.23536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes J., Kyle C., Ekstrom A.D. Complementary roles of human hippocampal subfields in differentiation and integration of spatial context. J. Cogn. Neurosci. 2015;27:546–559. doi: 10.1162/jocn_a_00736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylantyev S., Savtchenko L.P., Niu Y.P., Ivanov A.I., Jensen T.P., Kullmann D.M., Xiao M.Y., Rusakov D.A. Electric fields due to synaptic currents sharpen excitatory transmission. Science. 2008;319:1845–1849. doi: 10.1126/science.1154330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantova M., Sanz-Blasco S., Zhang X., Xia P., Akhtar M.W., Okamoto S., Dziewczapolski G., Nakamura T., Cao G., Pratt A.E. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA. 2013;110:E2518–E2527. doi: 10.1073/pnas.1306832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsodyks M.V., Markram H. The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc. Natl. Acad. Sci. USA. 1997;94:719–723. doi: 10.1073/pnas.94.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne V.L., Mulligan R.S., Pejoska S., Ong K., Jones G., O'Keefe G., Chan J.G., Young K., Tochon-Danguy H., Masters C.L., Rowe C.C. Comparison of 11C-PiB and 18F-florbetaben for Aβ imaging in ageing and Alzheimer's disease. Eur. J. Nucl. Med. Mol. Imaging. 2012;39:983–989. doi: 10.1007/s00259-012-2088-x. [DOI] [PubMed] [Google Scholar]
- Walsh D.M., Selkoe D.J. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat. Rev. Neurosci. 2016;17:251–260. doi: 10.1038/nrn.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West M.J., Coleman P.D., Flood D.G., Troncoso J.C. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- Young J.Z., Nguyen P.V. Homosynaptic and heterosynaptic inhibition of synaptic tagging and capture of long-term potentiation by previous synaptic activity. J. Neurosci. 2005;25:7221–7231. doi: 10.1523/JNEUROSCI.0909-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., Sacco T., Strata P., Sacchetti B. Basolateral amygdala inactivation impairs learning-induced long-term potentiation in the cerebellar cortex. PLoS One. 2011;6:e16673. doi: 10.1371/journal.pone.0016673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.