

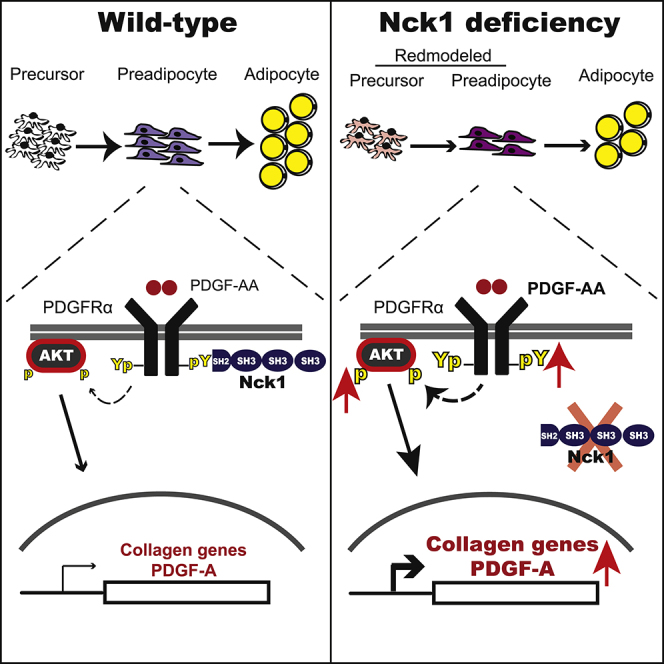
Summary
Obesity results from an excessive expansion of white adipose tissue (WAT), which is still poorly understood from an etiologic-mechanistic perspective. Here, we report that Nck1, a Src homology domain-containing adaptor, is upregulated during WAT expansion and in vitro adipogenesis. In agreement, Nck1 mRNA correlates positively with peroxisome proliferator-activated receptor (PPAR) γ and adiponectin mRNAs in the WAT of obese humans, whereas Nck1-deficient mice display smaller WAT depots with reduced number of adipocyte precursors and accumulation of extracellular matrix. Furthermore, silencing Nck1 in 3T3-L1 preadipocytes increases the proliferation and expression of genes encoding collagen, whereas it decreases the expression of adipogenic markers and impairs adipogenesis. Silencing Nck1 in 3T3-L1 preadipocytes also promotes the expression of platelet-derived growth factor (PDGF)-A and platelet-derived growth factor receptor (PDGFR) α activation and signaling. Preventing PDGFRα activation using imatinib, or through PDGF-A or PDGFRα deficiency, inhibits collagen expression in Nck1-deficient preadipocytes. Finally, imatinib rescues differentiation of Nck1-deficient preadipocytes. Altogether, our findings reveal that Nck1 modulates WAT development through PDGFRα-dependent remodeling of preadipocytes.
Subject Areas: Functional Aspects of Cell Biology, Pathophysiology
Graphical Abstract
Highlights
-
•
Nck1−/− mice display smaller WAT depots
-
•
Silencing Nck1 in preadipocyte impairs adipogenesis
-
•
Nck1 regulates PDGFRα activation and signaling in preadipocytes
-
•
Nck1 contributes to preadipocyte commitment
Functional Aspects of Cell Biology; Pathophysiology
Introduction
Obesity is characterized by an excessive accumulation of body fat from expansion of white adipose tissue (WAT). This remodeling process involves hypertrophy of pre-existing adipocytes and differentiation of committed precursor cells into adipocytes through adipogenesis. Adipocyte hypertrophy favors recruitment of inflammatory macrophages, adipose tissue dysfunction, and insulin resistance, whereas hyperplasia preserves WAT function and insulin sensitivity (Sun et al., 2011). However, mechanisms behind WAT expansion through hypertrophy and/or adipogenesis are still poorly understood and pharmacological approaches available to prevent the development of obesity are very few. In this context, identifying molecular targets regulating WAT expansion is promising for the design of more efficient strategies to treat obesity and its associated complications.
Adipogenesis is a complex process resulting from the coordinated temporal and spatial expression of specific transcriptional regulators (Lowe et al., 2011). In this perspective, CCAAT enhancer binding proteins (C/EBPs) serve as master initiators of the preadipocyte differentiation process (Tang and Lane, 2012). C/EBPβ and C/EBPδ are expressed early following the induction of adipogenesis. As a result, C/EBPα and peroxisome proliferator-activated receptor (PPAR) γ are induced and function together to promote the transcription of a large group of genes that govern the adipocyte phenotype and function (Rosen and MacDougald, 2006, Tang and Lane, 2012).
Our group has been at the forefront in unraveling the role of the Src homology (SH) domain-containing adaptor protein Nck2 (non-catalytic region of receptor tyrosine kinase 2) as a key player in limiting adipogenesis and adiposity, and regulating adipocyte function in mice (Dusseault et al., 2016, Haider et al., 2017). In agreement, we found that Nck2 downregulation in human visceral WAT correlates with severe obesity (Dusseault et al., 2016). At the mechanistic level, we demonstrated that Nck2 controls WAT homeostasis through the regulation of the endoplasmic reticulum (ER) transmembrane protein kinase R-like endoplasmic reticulum kinase (PERK), which plays a central role in the unfolded protein response (UPR) and critically contributes to lipogenesis and adipocyte differentiation (Han et al., 2013).
In mammals, Nck2 shares a high degree of identity with Nck1, a second member of the Nck family. Indeed, both lack catalytic activity and contain three SH3 and one SH2 domains that are particularly well conserved (Chen et al., 1998). These adaptor proteins facilitate the assembly of molecular complexes coupling cell surface and ER transmembrane receptor signaling to crucial cellular responses, such as proliferation, actin cytoskeleton reorganization, and ER stress-induced UPR (reviewed in Labelle-Cote and Larose (2011)). We previously reported that Nck1 is required to sustain ER stress-induced hepatic insulin resistance associated with impaired glucose homeostasis secondary to obesity (Latreille et al., 2011). However, a role for Nck1 in adipose tissue biology remains to be assessed.
Herein, using a combination of murine and human preadipocyte cell lines, as well as in vivo models, we demonstrate that, in contrast to Nck2 (Dusseault et al., 2016), Nck1 is required for in vivo WAT development as well as in vitro differentiation of preadipocytes into adipocytes. We determined that the level of Nck1 mRNA correlates positively with the mRNA levels of adipogenic markers PPARγ and adiponectin in adipose tissues of obese humans. From a mechanistic perspective, we demonstrate that Nck1 deficiency inhibits adipogenesis through remodeling of preadipocytes mediated by enhanced PDGFRα activation and signaling.
Results
Expression of Nck1 Increases during Developmental and Obesogenic WAT Expansion
We previously reported that Nck1 is widely expressed in mouse tissues (Dusseault et al., 2016). However, further investigation of Nck1 expression in specific mouse adipose tissues reveals that epididymal (e) and subcutaneous (sc) WATs express similar levels, whereas a lower level of Nck1 is observed in brown adipose tissue (BAT) (Figure 1A). Furthermore, we found that Nck1 expression increases in eWAT and scWAT during aging (Figure 1B). To determine the cell type that contributes to an increase in Nck1 expression in WAT during development, we isolated adipocytes and adipocyte precursor cells (Lin−;CD29+;CD34+;Sca1+;PDGFRα+) following WAT collagenase digestion and fluorescence-activated cell sorting (FACS, Figure S1A) as previously described (Church et al., 2014). As expected, in both eWAT and scWAT, adipocytes show higher levels of Pparg compared with adipocyte precursor cells (Figure S1B). Similarly, Nck1 level appears higher in adipocytes than in preadipocytes, especially in older mice (Figure 1C). Interestingly, increased Nck1 at the protein level in eWAT and scWAT at week 16 post-weaning (Figure 1B) correlates at the same age with upregulation of Nck1 mRNA in adipocytes rather than in adipocyte precursor cells (Figure 1C). These results strongly suggest that during developmental WAT expansion, Nck1 expression mostly increases in adipocytes. Furthermore, Nck1 expression in eWAT from diet- and genetically induced obese mice (high-fat diet [HFD] and ob/ob) is also significantly increased (Figure 1D). Therefore, expression of Nck1 in WAT increases not only during normal development but also during WAT expansion associated with obesity. In agreement, we found that Nck1 mRNA positively correlates with PPARG and ADIPOQ mRNAs in omental (o) and subcutaneous (sc) WAT of obese humans (Figure 1E), further supporting the notion that increased expression of Nck1 relates to WAT expansion.
Figure 1.

Nck1 Is Induced during Developmental and Obesogenic WAT Expansion
(A) Relative expression of Nck1 in epididymal (e) and subcutaneous (sc) white adipose tissues (WAT) and brown adipose tissue (BAT) in mice as determined by western blot and densitometry of Nck1 relative to Hsp90 (n = 3/group).
(B) Relative expression of Nck1 in eWAT and scWAT during development (post-weaning week 0, 5, and 16) as determined by western blot and densitometry relative to β-actin (n = 3/group).
(C) Relative Nck1 mRNA levels in adipocytes and adipocyte precursors cells (Lin−;CD29+; CD34+;Sca1+;PDGFRα+) isolated from mice at weeks 5 and 16 post-weaning (n = 3–4/condition).
(D) Nck1 protein level in eWAT of mice fed a normal chow diet (NCD) (n = 6) or a high-fat diet (HFD) (n = 4) and Nck1 mRNA level in ob/ob mice eWAT (n = 4). Data are mean ± SEM. Statistical significance evaluated by unpaired Student's t test or one-way ANOVA is reported as *p ≤ 0.05, ***p ≤ 0.001.
(E) Correlation of Nck1 mRNA with PPARγ and adiponectin mRNA level in omental (o) and subcutaneous (sc) WAT of obese humans (n = 12). Statistical significance evaluated by two-tailed p value from analysis of Pearson correlation coefficient as reported in the figure.
Nck1 Is Required for WAT Development
To assess whether Nck1 contributes to postnatal WAT development in vivo, we characterized Nck1+/+ and Nck1−/− mice from 0 to 16 weeks after weaning. As we previously reported (Latreille et al., 2011), Nck1−/− mice display lower body weight early after weaning, but recover later on as shown by comparable body weight at week 16 (Nck1+/+: 38.83 g ± 1.26, Nck1−/−: 35.73 g ± 1.88; n = 9) (Figure 2A, inset). Under this time frame, eWAT and scWAT depots in Nck1−/− mice tend to be smaller, and this difference becomes significant at 16 weeks after weaning (Figure 2B), whereas BAT weight remains unchanged (Figure S2A). Taking body weight into account, eWAT and scWAT depots are still significantly reduced in Nck1−/− mice; excluding that smaller WAT depots at week 16 after weaning are due to lower overall body weight (Figures S2B and S2C). Interestingly, other organ weights normalized to body weight are not different between mice genotypes (Figure S2D), further highlighting a specific role for Nck1 in regulating WAT development in vivo.
Figure 2.

Impaired WAT Development in Nck1−/− Mice
(A) Body weight between 0 and 5 weeks and at 16 weeks (inset) after weaning (n = 6–15/group).
(B) Weight of eWAT and scWAT at 0, 5, and 16 weeks post-weaning (n = 5–16/group).
(C) H&E staining showing representative images at 10× magnification of indicated WAT and adipocyte area distribution with diameter measurements of 1000–5000 cells in Nck1+/+ and Nck1−/− mice at week 16 post-weaning (n = 3–4/group).
(D) FACS quantification of adipocyte precursor cells (Lin−;CD29+; CD34+;Sca1+;PDGFRα+) count relative to Lin− cells population or per gram of fat in indicated WAT depots from Nck1+/+ and Nck1−/− mice at week 5 post-weaning (n = 4/group).
(E and F) (E) Oil red O staining and quantification and (F) PPARγ and aP2 levels as determined by western blot and densitometry relative to Hsp90 at day 5 of differentiation in scWAT SVF isolated from Nck1+/+ and Nck1−/− mice at week 5 post-weaning (n = 3/group). Arrow represents PPARγ2 and * is a non-specific band in Nck1 western blot. Data are mean ± SEM.
Statistical significance evaluated by unpaired Student's t test or two-way ANOVA is defined as *p ≤ 0.05, **p ≤ 0.01, and ****p ≤ 0.0001.
Nck1+/+ and Nck1−/− mice display similar insulin and leptin serum levels, as well as fed blood triglycerides and glucose levels (Figure S2E). In addition, both mice genotypes were comparable in terms of metabolic and physical activities as shown by similar food and water intakes, energy expenditure, locomotor activity, O2 consumption and CO2 release, as well as respiratory exchange ratio (Figure S3). Therefore, decreased adiposity in Nck1−/− mice is not associated with obvious changes in metabolic or physical activity.
To further characterize decreased adiposity in Nck1−/− mice, eWAT and scWAT sections from each mouse genotype were subjected to H&E staining and adipocyte area frequency distribution was quantified (Parlee et al., 2014). These analyses reveal no change in the size of adipocytes in eWAT between mice genotypes (Figure 2C, left panels). In contrast, it is clear that adipocytes are smaller and that the population of smaller adipocytes significantly increases in scWAT from Nck1−/− mice (Figure 2C, right panels). We then further investigated the relationship between average adipocyte volume and respective weight for each WAT depot (Figure S2F). For eWAT, this relationship is shifted to the left in Nck1−/− mice, suggesting that a decreased number of adipocytes could contribute to smaller eWAT. For scWAT, the relationship between adipocyte volume and tissue weight is shifted to the left and downward in Nck1−/− mice, suggesting that the smaller scWAT in these mice results from the lower number of adipocytes and decreased adipocyte size. To determine whether the decreased number of adipocytes in Nck1−/− mice WAT depots is related to a reduced number of adipocyte precursor cells, we compared the number of Lin−;CD29+;CD34+; Sca1+; PDGFRα+ adipocyte precursor cells in both WAT depots between mice genotypes using FACS analysis as reported above (Figure S1A). In agreement with the reduced number of adipocytes in eWAT and scWAT of Nck1−/− mice, the number of adipocyte precursor cells is significantly decreased in both WAT depots when compared with Nck1+/+ mice (Figure 2D). The decreased number of adipocyte precursor cells persists even after considering the weight of each WAT (Figure 2D), as well as the absolute count of precursors (Figure S2G). In addition, cells of stromal vascular fraction (SVF) isolated from Nck1−/− mice scWAT display impaired adipogenesis as shown by reduced oil red O staining at day 5 of differentiation compared with Nck1+/+ scWAT SVF cells similarly treated (Figure 2E). In agreement, western blot analysis of day 5 differentiated scWAT SVF shows significantly reduced levels of PPARγ and its downstream target aP2 in Nck1−/− mice compared with Nck1+/+ mice (Figure 2F). Therefore, impaired adipogenesis of SVF precursors isolated from scWAT of Nck1−/− mice could be attributed to a reduced number of adipocyte precursor cells. However, this approach does not exclude an intrinsic adipogenesis defect in Nck1−/− adipocyte precursor cells. Nonetheless, our findings demonstrate that Nck1 is required to maintain an adequate number of adipocyte precursor cells and WAT development.
Nck1 Is a Regulator of Adipogenesis
We observed that the expression of Nck1 at both mRNA and protein levels significantly increases during 3T3-L1 cells' differentiation (Figure 3A), a well-established model of in vitro differentiation of preadipocyte into adipocyte (Green and Meuth, 1974). To determine whether Nck1 regulates adipocyte differentiation in a cell-autonomous manner, we silenced Nck1 using specific small interfering RNA (siRNA) in 3T3-L1 preadipocytes. Indeed, we found that silencing Nck1 in 3T3-L1 preadipocytes strongly abrogates differentiation compared with cells transfected with control siRNA, as shown by the reduced accumulation of lipid droplets under light microscopy and quantification of oil red O staining (Figure 3B). In agreement, Pparg and Fabp4 (aP2) levels are significantly reduced during differentiation in Nck1-depleted 3T3-L1 cells (Figure 3C). Western blot analysis at day 6 of differentiation demonstrates that siRNA-mediated Nck1 silencing is efficient given that Nck1 protein levels remain low in differentiated cells (Figure 3D). In addition, silencing Nck1 significantly reduces the expression of the main adipogenic markers PPARγ2, aP2, C/EBPα, perilipin, and adiponectin at day 6 of differentiation (Figure 3D). Therefore, these results demonstrate that Nck1 is important to induce the differentiation of murine 3T3-L1 preadipocytes into adipocytes.
Figure 3.

Nck1 Is Required for 3T3-L1 Preadipocyte Differentiation
(A) Nck1 mRNA (n = 4) and protein (n = 3) levels during 3T3-L1 preadipocyte differentiation.
(B) Representative microscopic images (DIC, 10×) at days 0, 6, and 20 of differentiation and oil red O quantification in day 6 differentiated siControl and siNck1 3T3-L1 cells (n = 3/group).
(C) Relative Pparg and Fabp4 mRNA at indicated day of differentiation (n = 3/group).
(D) Expression of adipogenic markers at day 6 of differentiation as determined by western blot and densitometry relative to Hsp90 (representative of n = 3). Arrow represents PPARγ2. Data are mean ± SEM.
Statistical significance evaluated by one sample or unpaired Student's t test or two-way ANOVA is reported as *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001.
Similarly to murine 3T3-L1 preadipocytes, Nck1 expression increases during in vitro differentiation of human Simpson-Golabi-Behmel Syndrome (SGBS) preadipocytes into adipocytes (Figure S4A). In addition, silencing Nck1 in SGBS preadipocytes drastically reduces differentiation, as seen by both the lack of lipid droplet formation (light microscopy) and reduced lipid accumulation, revealed by decreased uptake of the lipophilic fluorescence dye, BODIPY, at day 12 of differentiation (Figure S4B). Consistently, the adipogenic markers PPARγ2, aP2, C/EBPα, perilipin, and adiponectin are significantly decreased in Nck1-deficient SGBS cells compared with control cells (Figure S4C). All these changes cannot be attributed to the recovery of Nck1 expression or compensation by Nck2 throughout the induction of differentiation in Nck1-depleted SGBS cells, as Nck1 expression remains low and Nck2 expression does not change upon silencing Nck1 in SGBS cells (Figure S4C). Therefore, Nck1 expression increases during in vitro adipocyte differentiation, and loss of function of Nck1 in preadipocytes strongly impairs mouse and human preadipocyte differentiation into adipocytes.
To determine whether gain of function of Nck1 affects adipogenesis, pools of 3T3-L1 preadipocytes stably transfected with a plasmid driving ectopic expression of mouse Nck1 tagged with a FLAG epitope (FLAG-Nck1) or an empty plasmid were generated (Figure 4A). Following induction of differentiation, we observed that 3T3-L1 preadipocytes overexpressing FLAG-Nck1 display increased formation of lipid droplets (light microscopy) and expression of the adipogenic markers Pparg and Fabp4 (Figures 4B and 4C). In addition, western blot analysis at day 6 of differentiation demonstrates that the expression of the main adipogenic markers PPARγ2, aP2, C/EBPα, perilipin, and adiponectin is increased in 3T3-L1 cells overexpressing FLAG-Nck1 when compared with control cells (Figure 4D). Overall, these results show that Nck1 loss and gain of function modulates in vitro adipocyte differentiation in a cell-autonomous manner.
Figure 4.

Overexpression of Nck1 Promotes Differentiation of 3T3-L1 Preadipocytes into Adipocytes
(A) Nck1 mRNA and protein levels in 3T3-L1 preadipocytes stably overexpressing murine FLAG-tagged Nck1.
(B) Representative microscopic images (DIC, 10×) of 3T3-L1 control and FLAG-Nck1 cells at days 0 and 6 of differentiation (n = 3/group).
(C) Relative expression of Pparg and Fabp4 in 3T3-L1 control and FLAG-Nck1 cells at day 6 of differentiation (n = 3/group).
(D) Western blots and quantification of adipogenic proteins in 3T3-L1 control and FLAG-Nck1 cells at day 6 of differentiation (representative of n = 3). Arrow represents PPARγ2. Data are mean ± SEM.
Statistical significance evaluated by one sample or unpaired Student's t test is reported as *p ≤ 0.05 and **p ≤ 0.01.
Nck1 Deficiency Remodels Preadipocyte Signature
To explain how silencing Nck1 in preadipocytes impairs in vitro differentiation into adipocytes, we hypothesize that Nck1 is required to maintain preadipocyte commitment. Indeed, we found that silencing Nck1 affects preadipocyte function, as shown by reduced oleate-induced lipid uptake monitored by BODIPY FL C16 (Figure 5A). Accordingly, this correlates with lower mRNA and protein levels of the lipid transporter CD36 in Nck1-deficient 3T3-L1 preadipocytes (Figure 5B). On the other hand, silencing Nck1 in 3T3-L1 preadipocytes significantly enhances proliferation as determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and bromodeoxyuridine (BrdU) incorporation (Figure 5C). Therefore, these results suggest that Nck1 regulates preadipocyte function. However, propidium iodide (PI) staining to monitor cell cycle progression in 2-day post-confluent preadipocytes and through the first phase of mitotic clonal expansion (MCE) induced by the differentiation cocktail (DMI) is comparable between Nck1-deficient and control 3T3-L1 preadipocytes (Figure S5A). These data rule out that increased proliferation in Nck1-deficient preadipocytes impairs differentiation by altering MCE. Finally, Nck1-deficient preadipocytes show normal DMI-induced signaling, as demonstrated by comparable DMI-induced pAKT and pERK1/2 with control cells (Figure S5B) Altogether, these results suggest that Nck1 deficiency more importantly affects preadipocyte fate than events related to the induction of differentiation into adipocyte.
Figure 5.

Silencing Nck1 Remodels 3T3-L1 Preadipocyte Signature
(A) BODIPY FL C16 uptake during 48 hr with DAPI-stained siControl and siNck1 3T3-L1 preadipocytes (n = 3/group).
(B) Relative expression of CD36 mRNA and protein in siControl and siNck1 3T3-L1 preadipocytes (representative of n = 3).
(C) Preadipocyte proliferation assessed using MTT assay and quantification of BrdU-positive cells (n = 3/group).
(D) Relative mRNA expression of adipogenic master genes in growing and at day 0 of differentiation (n = 3/group).
(E) PPARγ expression at day 0 of differentiation as determined by western blots. Arrow indicates PPARγ2 (representative of n = 3). Data are mean ± SEM.
Statistical significance evaluated by unpaired Student's t test or two-way ANOVA is reported as *p ≤ 0.05, ***p ≤ 0.001, and ****p ≤ 0.0001.
To get more insight into the potential mechanism underlying Nck1-dependent regulation of preadipocyte function, we compared the expression levels of adipogenic markers in control and Nck1-deficient 3T3-L1 preadipocytes. Interestingly, growing Nck1-deficient preadipocytes only show a significant slight decrease in Pparg, whereas the level of Cebps is significantly unchanged (Figure 5D). In contrast, Pparg and Cebps levels are significantly reduced at day 0 of differentiation when preadipocytes are growth arrested (Figure 5D). In agreement, PPARγ protein levels are significantly reduced upon silencing Nck1 in 3T3-L1 preadipocytes (Figure 5E). These results suggest that impaired adipocyte differentiation of Nck1-depleted cells resides in preadipocytes per se and potentially results from reduced levels of critical transcription factors essential for adipogenesis. Altogether, our findings demonstrate that silencing Nck1 modulates 3T3-L1 preadipocyte functions and gene signatures.
Silencing Nck1 in 3T3-L1 Preadipocytes Promotes PDGFRα Activation and Signaling
Activation of PDGFRα inhibits WAT development by shifting precursor cells toward a stromal fibroblastic lineage secreting collagen rather than a preadipocyte lineage (Iwayama et al., 2015, Marcelin et al., 2017, Sun et al., 2017). In pull-down assays, we found that Nck1 interacts with PDGFRα through its SH2 domain in pervanadate (PV)-treated 3T3-L1 preadipocyte lysate (Figure S6A). We also detected increased PDGFRα phosphorylation on pY754 activation site in PV-treated Nck1-deficient 3T3-L1 preadipocytes compared with similarly treated control cells (Figure S6B, short exposure). Increased phosphorylation of PDGFRα on Y754 is also seen in the absence of PV treatment in Nck1-deficient 3T3-L1 preadipocytes (Figure S6B, long exposure). Therefore, increased phosphorylation of PDGFRα on Y754 suggests enhanced basal activation of PDGFRα in Nck1-deficient 3T3-L1 preadipocytes. Consistently, basal pAKT, p-p70S6 kinase, cyclin D1, and cytosolic FOXO1 were all increased in serum-starved Nck1-deficient 3T3-L1 preadipocytes (Figure S6C), suggesting that Nck1 limits PDGFRα activation and signaling in preadipocytes. Importantly, we found that PDGF-A-induced PDGFRα activation and signaling is also increased in Nck1-deficient 3T3-L1 preadipocytes as shown by increased phosphorylation of PDGFRα on Y754, phosphorylation of AKT on both activation sites T308 and S473, and phosphorylation of ERK1/2 to a lower extent (Figures 6A and S6D). Therefore, increased PDGRα activation and signaling could contribute to impair adipocyte differentiation in 3T3-L1 preadipocytes depleted of Nck1. On the other hand, we eliminated that enhanced differentiation in 3T3-L1 preadipocytes stably overexpressing FLAG-Nck1 (Figure 4B) clearly relies on attenuated PDGFRα activation and signaling, given that PDGF-A-induced PDGFRα pY754, pAKT, and pERK are mostly comparable to control 3T3-L1 preadipocytes (Figure S7A). Finally, we excluded that Nck1 loss and gain of function in 3T3-L1 preadipocytes affect adipocyte differentiation by modulating PDGFRα expression level (Figure S7B).
Figure 6.

Nck1 Deficiency Promotes PDGFRα Activation and Signaling in 3T3-L1 Preadipocytes and Enhances Collagen Deposition in WAT
(A) Western blots analysis of PDGF-A (5 ng/μL)-induced PDGFRα activation and signaling in control and Nck1-depleted 3T3-L1 preadipocytes (representative of n = 3).
(B) Relative expression of collagen genes in control and Nck1-depleted 3T3-L1 preadipocytes (n = 3/group).
(C) Picrosirius red and Masson's trichrome staining of eWAT and scWAT from Nck1+/+ and Nck1−/− mice at week 16 post-weaning (n = 3/group). Representative images were taken at 20× magnification.
(D) Hydroxyproline content in eWAT from Nck1+/+ and Nck1−/− mice at week 16 post- weaning (n = 3–5/group).
(E and F) (E) Relative expression of PDGF-A and PDGF-C in control and Nck-1-depleted 3T3-L1 preadipocytes (n = 3/group) (F) Relative expression of PDGF-A in scWAT of Nck1+/+ and Nck1−/− at week 5 post-weaning (n = 3–4).
(G) Western blot analysis of indicated proteins in eWAT from Nck1+/+ and Nck1−/− at week 16 post-weaning (n = 4). Data are mean ± SEM. Statistical significance evaluated by unpaired Student t test is reported as *p≤0.05.
Increased PDGFRα activation in adipocyte precursor cells is known to promote collagen gene expression and deposition of extra-cellular matrix (ECM) leading to adipose tissue fibrosis in adult mice (Iwayama et al., 2015, Marcelin et al., 2017, Sun et al., 2017). In agreement, increased PDGFRα activation and signaling in Nck1-deficient 3T3-L1 preadipocytes correlates with higher expression of various collagen genes, as well as Tgfb1, a potent mediator of collagen gene expression in various cell types (Meng et al., 2016) (Figure 6B). These findings are supported by in vivo data showing increased picrosirius red and Masson's trichrome staining, revealing enhanced collagen deposition in both eWAT and scWAT in Nck1−/− mice (Figure 6C). Increased collagen deposition is further confirmed by increased hydroxyproline content in eWAT of Nck1−/− mice (Figure 6D). In addition, the pro-fibrosis cytokines, transforming growth factor β1 and fibronectin, appear elevated in eWAT of Nck1−/− mice (Figure S7D). Overall, our findings suggest that lack of Nck1 impairs adipogenesis by shifting the preadipocyte phenotype toward a less committed cell fate with reduced preadipocyte lineage markers, enhanced proliferation, and increased collagen production.
To understand how Nck1 deficiency leads to increased PDGFRα activation and signaling, we assessed the expression of various PDGF ligands in 3T3-L1 preadipocytes. In contrast to PDGF-A and PDGF-C mRNAs, 3T3-L1 preadipocytes expressed very low levels of PDGF-B and PDGF-D mRNAs. Nonetheless, in Nck1-deficient 3T3-L1 preadipocytes, we found that the PDGF-A mRNA level is upregulated, whereas PDGF-C mRNA level is not changed (Figure 6E). Our findings suggest that increased expression of PDGF-A by silencing Nck1 in 3T3-L1 preadipocytes could contribute to further induce PDGFRα activation, leading to enhanced PDGFRα signaling. On the other hand, PDGF-A mRNA is not changed in FLAG-Nck1 3T3-L1 preadipocytes (Figure S7C), consistent with no change in PDGF-A-induced PDGFRα signaling (Figure S7A). Interestingly, in WAT of Nck1−/− mice, PDGF-A mRNA and protein levels tend to be higher and positively correlate with increased cyclin D1 expression, whereas the expression of PDGFRα remains unchanged (Figures 6F and 6G). These results further support that Nck1 deficiency promotes PDGFRα activation and signaling, leading to increased expression of collagen genes in preadipocytes and ECM accumulation in WAT.
PDGFRα Mediates the Effects of Silencing Nck1 in 3T3-L1 Preadipocytes
To determine whether PDGFRα mediates the effects of silencing Nck1 in preadipocytes, we treated Nck1-depleted and control 3T3-L1 preadipocytes with imatinib, a potent PDGFR inhibitor (Fitter et al., 2012). In this context, we found that induction of collagen genes in Nck1-depleted 3T3-L1 preadipocytes is prevented by overnight treatment with imatinib (Figure 7A). Furthermore, we next used specific inhibitors to determine which PDGFRα pathway is involved in inducing collagen genes in Nck1-depleted 3T3-L1 preadipocytes. Treatment of Nck1-depleted preadipocytes with an Akt inhibitor (Akti) completely prevents the induction of Col6A3, Col3A1, and Col1A1. Interestingly, impeding ERK activation using a MAPK/ERK Kinase (MEK) inhibitor (U0129) has no effect on the induction of Col6A3 and Col3A1 in Nck1-depleted 3T3-L1 preadipocytes, whereas it prevents Col1A1 induction (Figure 7A), suggesting differential regulation of collagen genes in Nck1-depleted 3T3-L1 preadipocytes.
Figure 7.

PDGFRα Mediates the Effects of Silencing Nck1 on Collagen Genes Expression and Differentiation in 3T3-L1 Preadipocytes
(A) Effect of imatinib (1 μM), Akti (10 μM), and U0126 (10 μM) and downregulation of either PDGFRα or PDGF-A on indicated collagen gene expression in siControl and siNck1 3T3-L1 preadipocytes (n = 3–7/condition).
(B) Effect of imatinib on differentiation of siControl and siNck1 3T3-L1 preadipocytes (n = 3/group). Differentiation is assessed at day 6 using light microscopy images (10× magnification) showing lipid droplets formation, lipid accumulation by oil red O quantification, and relative Pparg expression by qPCR (n = 3). Data are mean ± SEM.
Statistical significance evaluated by unpaired Student's t test or two-way ANOVA is reported as *p ≤ 0.05 and **p ≤ 0.01.
Imatinib is reported to be a potent inhibitor of PDGFRα and β, as well as Bcr-Abl, Abl, and c-kit tyrosine kinases. To our knowledge, Bcr-Abl, Abl, and c-kit are not expressed in 3T3-L1 preadipocytes. However, to discriminate between PDGFR isoforms mediating the effects of silencing Nck1 in 3T3-L1 preadipocytes on collagen gene expression, we simultaneously silenced PDGFRα and Nck1 through co-transfection of specific siRNAs. In parallel, we also determined the effects of silencing PDGF-A ligand expression in Nck1-depleted preadipocytes, given we found that deficiency in Nck1 induces PDGF-A mRNA in preadipocytes (Figure 6E) as well as PDGF-A ligand in WAT (Figure 6F). As expected, PDGFRα and PDGF-A siRNAs were specific in downregulating their respective mRNA (Figures S8A and S8B). Interestingly, silencing Nck1 in combination with PDGFRα or PDGF-A prevents induction of PDGF-A mRNA compared with Nck1-depleted preadipocytes (Figure S7E). In addition, depletion of PDGFRα or PDGF-A prevents the effects of silencing Nck1 on collagen gene expression in 3T3-L1 preadipocytes (Figure 7A), demonstrating that the effects of silencing Nck1 in preadipocytes are mediated by PDGFRα.
We next assessed whether neutralizing PDGFRα activity during differentiation would restore adipogenesis in Nck1-deficient 3T3-L1 preadipocytes. For this, we decided to introduce imatinib during differentiation rather than to address this point using Nck1-deficient 3T3-L1 preadipocytes co-transfected with PDGFRα or PDGF-A siRNAs. This is based on the rationale that imatinib completely blocks PDGFRα activation, whereas siRNA PDGFRα only partially decreases PDGFRα activity due to incomplete downregulation of PDGFRα expression. Similarly, downregulating PDGF-A still allows serum-induced PDGFRα activation. Therefore, their effects on restoring adipocyte differentiation of Nck-1-depleted 3T3-L1 preadipocytes could be mitigated. In this perspective, we observed that adding imatinib to the differentiation cocktail improves differentiation of Nck1-depleted 3T3-L1 preadipocytes, as shown by comparable levels of lipid droplet formation (light microscopy) and quantification of oil red O between Nck1-depleted and control 3T3-L1 preadipocytes. Although imatinib has been reported to promote adipocyte differentiation of primary human mesenchymal stromal cells (Fitter et al., 2012), it has no significant effect on lipid droplet formation and oil red O content in control 3T3-L1 preadipocytes (Figure 7B). Recovery of adipocyte differentiation by imatinib in Nck1-deficient 3T3-L1 preadipocytes is further supported by the induction of Pparg to the level found in control cells similarly treated with imatinib (Figure 7B). Altogether, these findings support that enhanced PDGFRα activation and signaling mediate the effects of silencing Nck1 in 3T3-L1 preadipocytes and could contribute to impair adipogenesis in vivo.
Discussion
SH domain-containing adaptor proteins of the Nck family, Nck1 and Nck2, play an important role in mediating cell surface receptor signaling that orchestrates crucial cellular responses such as cell adhesion, migration and invasion (Dubrac et al., 2016, Labelle-Cote et al., 2011, Morris et al., 2017), and low-affinity antigen response (Borroto et al., 2016). However, the implication of Nck in intracellular signaling pathways governing WAT biology has never previously been investigated. In this perspective, our work addressing the role of both Ncks in WAT unveils insights on mechanisms regulating adipogenesis. Indeed, we have previously demonstrated that Nck2 limits adiposity in mice and adipocyte differentiation in vitro (Dusseault et al., 2016, Haider et al., 2017). Mechanistically, we provided evidence that Nck2 regulates adipogenesis by controlling activation of PERK during preadipocyte to adipocyte transition. Herein, we report that Nck1 is required for normal WAT development. Indeed, Nck1 deficiency in mice results in smaller WAT depots and specific deletion of Nck1 abrogates the ability of murine and human preadipocytes to differentiate into adipocytes in vitro. Moreover, we identified that Nck1 interacts with PDGFRα and Nck1 deficiency in preadipocytes leads to enhanced PDGFRα activation and signaling. It is well established that increased PDGFRα activity causes adipose tissue fibrosis in mice by inhibiting the formation of adipocytes in favor of promoting collagen-expressing fibroblasts (Iwayama et al., 2015, Marcelin et al., 2017, Sun et al., 2017). Accordingly, Nck1-deficient 3T3-L1 preadipocytes display enhanced PDGFRα activation and signaling, leading to greater collagen expression. Furthermore, WAT depots in Nck1−/− mice present increased collagen accumulation that correlates with a reduced population of adipocyte precursor cells. Nck1 has been identified in an activated PDGFRβ complex (Chen et al., 2000, Nishimura et al., 1993, Park and Rhee, 1992), whereas its recruitment to PDGFRα has never been reported. Therefore, Nck1 interaction with activated PDGFRα in preadipocytes represents a mechanism by which PDGFRα activation and signaling is controlled to maintain preadipocytes in an adequate committed state indispensable for their transition to adipocytes. In addition, we reported enhanced proliferation of Nck1-deficient 3T3-L1 cells that correlates with increased PDGFRα activation and signaling. In Nck1−/− mice, the cell population that is sensitive to PDGFRα stimulation might not be the specific adipogenic precursor cells that we analyzed (Lin−;CD29+;CD34+;Sca1+;PDGFRα+), but rather the profibrotic precursors (Lin−;GP38+; PDGFRα+) as reported by others (Marcelin et al., 2017). Further investigation is required to identify adipocyte precursor population responsive to PDGFRα activation in Nck1−/− mice that could contribute to alter the fate of adipocyte precursors in Nck1−/− mice. Nonetheless, altogether our findings suggest that an imbalance between collagen producing fibroblasts and committed preadipocytes rather than differentiation per se is responsible for the reduced adiposity in Nck1−/− mice. However, the fact that mice lacking Nck1 are not lipodystrophic suggests adipogenic precursor cells' heterogeneity, with a role for Nck1 only in a subset of adipocyte precursor cells.
In mammals, obesity has been associated with increased ECM deposition and fibrosis that affects progenitor adipogenic capacity leading to WAT dysfunction (Abdennour et al., 2014, Divoux et al., 2010, Khan et al., 2009, Marcelin et al., 2017). Our findings demonstrate that Nck1 expression is increased during obesogenic WAT expansion in mice and correlates with adipogenic markers in humans. Further investigations are required to demonstrate whether Nck1 plays a protective role in limiting ECM accumulation and fibrosis during WAT expansion associated with obesity. In this instance, WAT expansion and function in Nck1−/− mice fed an HFD should be explored further.
Several studies have shown that Nck1 and Nck2 are functionally redundant and share common interacting proteins (Buday et al., 2002, Labelle-Cote and Larose, 2011, Lettau et al., 2009). In agreement, individual Nck knockout mice are viable with no apparent phenotype, whereas the Nck1 and Nck2 double knockout mice are embryonically lethal (Bladt et al., 2003). However, increasing evidence supports specific functions and interacting partners for Nck1 and Nck2 (Mukherjee et al., 2014, Ngoenkam et al., 2014). In this perspective, our studies on the role of Nck adaptor proteins in WAT development reveal that both Ncks regulate adipogenesis, but in an opposite manner and at different stages during this process. Nck2 is involved in regulating the transition of preadipocyte into adipocyte by regulating PERK activation and signaling (Dusseault et al., 2016, Haider et al., 2017), whereas our findings reveal that Nck1 controls preadipocyte commitment by modulating PDGFRα activation and signaling, and is required to maintain an adequate number of adipocyte precursor cells in WAT. Analysis of Nck1 and Nck2 interactome profiles in the preadipocyte and adipocyte might provide an understanding of how these two highly identical adaptor proteins differentially regulate WAT homeostasis.
Nevertheless, in this study we clearly established in mouse 3T3-L1 and human SGBS preadipocytes that Nck1 regulates adipogenesis in a cell-autonomous manner. Furthermore, we provide strong evidence that Nck1 regulation of adipogenesis involves a tight control of collagen expression through activation of PDGFRα in preadipocytes. Indeed, treatment with imatinib, a PDGFR inhibitor, and Akti, an Akt inhibitor, both prevented increased collagen expression in Nck1-deficient 3T3-L1 preadipocytes. Downregulation of PDGFRα also prevents increased collagen expression in Nck1-depleted preadipocytes, suggesting that PDGFRα mediates the preadipocyte phenotype upon Nck1 depletion. More importantly, imatinib maintained during differentiation restores induction of PPARγ and improves adipogenesis in Nck1-deficient 3T3-L1 cells. Although the exact mechanism by which Nck1 regulates PDGFRα activity in preadipocytes remains to be elucidated, our findings show that increased production of the PDGF-A ligand by silencing Nck1 in 3T3-L1 cells could contribute to higher activation of PDGFRα. Indeed, PDGFRα-mediated induction of collagen genes is prevented upon downregulation of PDGF-A in Nck1-depleted 3T3-L1 preadipocytes, suggesting that enhanced production of PDGF-A contributes to further increase PDGFRα activation and signaling. In addition, Nck1 interaction with the activated PDGFRα could mediate the recruitment of a phosphatase or a kinase involved in attenuating PDGFRα activation and signaling. For instance, we recently demonstrated that Nck1, which interacts with the tyrosine phosphatase PTP1B through its SH3 domains (Clemens et al., 1996, Li et al., 2014, Wu et al., 2011), regulates PTP1B expression and controls receptor tyrosine kinases-induced activation of the PI3K/AKT pathway (Li et al., 2014). On the other hand, we have previously shown that Nck1 interacts with the isoform γ2 casein kinase I (CKIγ2) through its SH3 domains (Lussier and Larose, 1997) and that CKIγ2 negatively regulates PDGFRβ activation by increasing its phosphorylation on serine residues (Bioukar et al., 1999). Therefore, silencing Nck1 in 3T3-L1 preadipocytes could potentially prevent PTP1B or CKIγ2 translocation in close proximity of PDGFRα, thus enhancing its activation and signaling. More investigation is needed to further explore these avenues.
Overall, our study provides insight on the role of Nck1 in regulating adipocyte precursor cells and preadipocytes through modulation of PDGFRα activation. We have identified Nck1 as a regulator of adipose tissue biology in mouse and humans through various in vitro and in vivo approaches. We believe that Nck1 regulation of WAT development could potentially highlight a powerful avenue to overcome or treat obesity.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank Dr. T. Pawson (Mount Sinai Hospital, Toronto, ON, Canada) for providing Nck1+/− mice several years ago. We also thank Dr. Bing Li for providing data on ob/ob mice and the MUHC-RI immunophenotyping platform for flow cytometry expertise. Finally, the authors acknowledge the surgery team, bariatric surgeons, and Biobank staff of the IUCPQ. During this study, N.H. was supported by studentships from the MUHC-RI and Fonds de la Recherche du Québec en Santé (FRQS). Work in this study was supported by a grant from the Canadian Institutes of Health Research to L.L. (MOP-115045) and a grant from the Zavalkoff Foundation. No potential conflicts of interest relevant to this article were reported.
Author Contributions
N.H. contributed to experimental design, data collection, interpretation and analysis, preparation of figures, and writing of the manuscript. J.D. contributed to experimental design, data analysis, and critical reading of the manuscript. L.L. contributed to study design and final editing of the manuscript. L.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Declaration of Interests
The authors declare no competing interests.
Published: August 31, 2018
Footnotes
Supplemental Information includes Transparent Methods and eight figures and can be found with this article online at https://doi.org/10.1016/j.isci.2018.07.010.
Supplemental Information
References
- Abdennour M., Reggio S., Le Naour G., Liu Y., Poitou C., Aron-Wisnewsky J., Charlotte F., Bouillot J.L., Torcivia A., Sasso M. Association of adipose tissue and liver fibrosis with tissue stiffness in morbid obesity: links with diabetes and BMI loss after gastric bypass. J. Clin. Endocrinol. Metab. 2014;99:898–907. doi: 10.1210/jc.2013-3253. [DOI] [PubMed] [Google Scholar]
- Bioukar E.B., Marricco N.C., Zuo D., Larose L. Serine phosphorylation of the ligand-activated beta-platelet-derived growth factor receptor by casein kinase I-gamma2 inhibits the receptor's autophosphorylating activity. J. Biol. Chem. 1999;274:21457–21463. doi: 10.1074/jbc.274.30.21457. [DOI] [PubMed] [Google Scholar]
- Bladt F., Aippersbach E., Gelkop S., Strasser G.A., Nash P., Tafuri A., Gertler F.B., Pawson T. The murine Nck SH2/SH3 adaptors are important for the development of mesoderm-derived embryonic structures and for regulating the cellular actin network. Mol. Cell. Biol. 2003;23:4586–4597. doi: 10.1128/MCB.23.13.4586-4597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroto A., Reyes-Garau D., Jimenez M.A., Carrasco E., Moreno B., Martinez-Pasamar S., Cortes J.R., Perona A., Abia D., Blanco S. First-in-class inhibitor of the T cell receptor for the treatment of autoimmune diseases. Sci. Transl. Med. 2016;8:370ra184. doi: 10.1126/scitranslmed.aaf2140. [DOI] [PubMed] [Google Scholar]
- Buday L., Wunderlich L., Tamas P. The Nck family of adapter proteins: regulators of actin cytoskeleton. Cell Signal. 2002;14:723–731. doi: 10.1016/s0898-6568(02)00027-x. [DOI] [PubMed] [Google Scholar]
- Chen M., She H., Davis E.M., Spicer C.M., Kim L., Ren R., Le Beau M.M., Li W. Identification of Nck family genes, chromosomal localization, expression, and signaling specificity. J. Biol. Chem. 1998;273:25171–25178. doi: 10.1074/jbc.273.39.25171. [DOI] [PubMed] [Google Scholar]
- Chen M., She H., Kim A., Woodley D.T., Li W. Nckbeta adapter regulates actin polymerization in NIH 3T3 fibroblasts in response to platelet-derived growth factor bb. Mol. Cell. Biol. 2000;20:7867–7880. doi: 10.1128/mcb.20.21.7867-7880.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church C.D., Berry R., Rodeheffer M.S. Isolation and study of adipocyte precursors. Methods Enzymol. 2014;537:31–46. doi: 10.1016/B978-0-12-411619-1.00003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens J.C., Ursuliak Z., Clemens K.K., Price J.V., Dixon J.E. A Drosophila protein-tyrosine phosphatase associates with an adapter protein required for axonal guidance. J. Biol. Chem. 1996;271:17002–17005. doi: 10.1074/jbc.271.29.17002. [DOI] [PubMed] [Google Scholar]
- Divoux A., Tordjman J., Lacasa D., Veyrie N., Hugol D., Aissat A., Basdevant A., Guerre-Millo M., Poitou C., Zucker J.D. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes. 2010;59:2817–2825. doi: 10.2337/db10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrac A., Genet G., Ola R., Zhang F., Pibouin-Fragner L., Han J., Zhang J., Thomas J.L., Chedotal A., Schwartz M.A. Targeting NCK-mediated endothelial cell front-rear polarity inhibits neovascularization. Circulation. 2016;133:409–421. doi: 10.1161/CIRCULATIONAHA.115.017537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusseault J., Li B., Haider N., Goyette M.A., Cote J.F., Larose L. Nck2 deficiency in mice results in increased adiposity associated with adipocyte hypertrophy and enhanced adipogenesis. Diabetes. 2016;65:2652–2666. doi: 10.2337/db15-1559. [DOI] [PubMed] [Google Scholar]
- Fitter S., Vandyke K., Gronthos S., Zannettino A.C. Suppression of PDGF-induced PI3 kinase activity by imatinib promotes adipogenesis and adiponectin secretion. J. Mol. Endocrinol. 2012;48:229–240. doi: 10.1530/JME-12-0003. [DOI] [PubMed] [Google Scholar]
- Green H., Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell. 1974;3:127–133. doi: 10.1016/0092-8674(74)90116-0. [DOI] [PubMed] [Google Scholar]
- Haider N., Dusseault J., Rudich A., Larose L. Nck2, an unexpected regulator of adipogenesis. Adipocyte. 2017;6:154–160. doi: 10.1080/21623945.2017.1291102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Murthy R., Wood B., Song B., Wang S., Sun B., Malhi H., Kaufman R.J. ER stress signalling through eIF2alpha and CHOP, but not IRE1alpha, attenuates adipogenesis in mice. Diabetologia. 2013;56:911–924. doi: 10.1007/s00125-012-2809-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwayama T., Steele C., Yao L., Dozmorov M.G., Karamichos D., Wren J.D., Olson L.E. PDGFRalpha signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev. 2015;29:1106–1119. doi: 10.1101/gad.260554.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T., Muise E.S., Iyengar P., Wang Z.V., Chandalia M., Abate N., Zhang B.B., Bonaldo P., Chua S., Scherer P.E. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol. Cell. Biol. 2009;29:1575–1591. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labelle-Cote M., Dusseault J., Ismail S., Picard-Cloutier A., Siegel P.M., Larose L. Nck2 promotes human melanoma cell proliferation, migration and invasion in vitro and primary melanoma-derived tumor growth in vivo. BMC Cancer. 2011;11:443. doi: 10.1186/1471-2407-11-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labelle-Cote M., Larose L. A uNick protein. Med. Sci. (Paris) 2011;27:746–752. doi: 10.1051/medsci/2011278017. [DOI] [PubMed] [Google Scholar]
- Latreille M., Laberge M.K., Bourret G., Yamani L., Larose L. Deletion of Nck1 attenuates hepatic ER stress signaling and improves glucose tolerance and insulin signaling in liver of obese mice. Am. J. Physiol. Endocrinol. Metab. 2011;300:E423–E434. doi: 10.1152/ajpendo.00088.2010. [DOI] [PubMed] [Google Scholar]
- Lettau M., Pieper J., Janssen O. Nck adapter proteins: functional versatility in T cells. Cell Commun. Signal. 2009;7:1. doi: 10.1186/1478-811X-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Dusseault J., Larose L. Nck1 depletion induces activation of the PI3K/Akt pathway by attenuating PTP1B protein expression. Cell Commun. Signal. 2014;12:71. doi: 10.1186/s12964-014-0071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe C.E., O'Rahilly S., Rochford J.J. Adipogenesis at a glance. J. Cell Sci. 2011;124:2681–2686. doi: 10.1242/jcs.079699. [DOI] [PubMed] [Google Scholar]
- Lussier G., Larose L. A casein kinase I activity is constitutively associated with Nck. J. Biol. Chem. 1997;272:2688–2694. doi: 10.1074/jbc.272.5.2688. [DOI] [PubMed] [Google Scholar]
- Marcelin G., Ferreira A., Liu Y., Atlan M., Aron-Wisnewsky J., Pelloux V., Botbol Y., Ambrosini M., Fradet M., Rouault C. A PDGFRalpha-mediated switch toward CD9high adipocyte progenitors controls obesity-induced adipose tissue fibrosis. Cell Metab. 2017;25:673–685. doi: 10.1016/j.cmet.2017.01.010. [DOI] [PubMed] [Google Scholar]
- Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-beta: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- Morris D.C., Popp J.L., Tang L.K., Gibbs H.C., Schmitt E., Chaki S.P., Bywaters B.C., Yeh A.T., Porter W.W., Burghardt R.C. Nck deficiency is associated with delayed breast carcinoma progression and reduced metastasis. Mol. Biol. Cell. 2017;28:3500–3516. doi: 10.1091/mbc.E17-02-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee C., Bakthavachalu B., Schoenberg D.R. The cytoplasmic capping complex assembles on adapter protein nck1 bound to the proline-rich C-terminus of Mammalian capping enzyme. PLoS Biol. 2014;12:e1001933. doi: 10.1371/journal.pbio.1001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngoenkam J., Paensuwan P., Preechanukul K., Khamsri B., Yiemwattana I., Beck-Garcia E., Minguet S., Schamel W.W., Pongcharoen S. Non-overlapping functions of Nck1 and Nck2 adaptor proteins in T cell activation. Cell Commun. Signal. 2014;12:21. doi: 10.1186/1478-811X-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura R., Li W., Kashishian A., Mondino A., Zhou M., Cooper J., Schlessinger J. Two signaling molecules share a phosphotyrosine-containing binding site in the platelet-derived growth factor receptor. Mol. Cell. Biol. 1993;13:6889–6896. doi: 10.1128/mcb.13.11.6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D., Rhee S.G. Phosphorylation of Nck in response to a variety of receptors, phorbol myristate acetate, and cyclic AMP. Mol. Cell. Biol. 1992;12:5816–5823. doi: 10.1128/mcb.12.12.5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlee S.D., Lentz S.I., Mori H., MacDougald O.A. Quantifying size and number of adipocytes in adipose tissue. Methods Enzymol. 2014;537:93–122. doi: 10.1016/B978-0-12-411619-1.00006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen E.D., MacDougald O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- Sun C., Berry W.L., Olson L.E. PDGFRalpha controls the balance of stromal and adipogenic cells during adipose tissue organogenesis. Development. 2017;144:83–94. doi: 10.1242/dev.135962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K., Kusminski C.M., Scherer P.E. Adipose tissue remodeling and obesity. J. Clin. Invest. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q.Q., Lane M.D. Adipogenesis: from stem cell to adipocyte. Annu. Rev. Biochem. 2012;81:715–736. doi: 10.1146/annurev-biochem-052110-115718. [DOI] [PubMed] [Google Scholar]
- Wu C.L., Buszard B., Teng C.H., Chen W.L., Warr C.G., Tiganis T., Meng T.C. Dock/Nck facilitates PTP61F/PTP1B regulation of insulin signalling. Biochem. J. 2011;439:151–159. doi: 10.1042/BJ20110799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.