

Abstract
We have isolated three Saccharomyces cerevisiae genes – CES1, CES2, and CES3 – that, when present in high copy, suppress the ts growth defect caused by mutations in the CEG1 gene encoding mRNA guanylyltransferase (capping enzyme). Molecular characterization of the capping enzyme supressor genes reveals the following. CES2 is identical to ESP1, a gene required for proper nuclear division. We show by deletion analysis that the 1573-amino acid ESP1 polypeptide is composed of distinct functional domains. The C-terminal portion of ESP1 is essential for cell growth, but dispensable for CES2 activity. The N-terminal half of ESP1, which is sufficient for CES2 function, displays local sequence similarity to the small subunit of the vaccinia virus RNA capping enzyme. This suggests a basis for suppression by physical or functional interaction between the CES2 domain of ESP1 and the yeast guanylyltransferase. CES1 encodes a novel hydrophilic 915-amino acid protein. The amino acid sequence of CES1 is uninformative, except for its extensive similarity to another yeast gene product of unknown function. The CES1 homologue (designated CES4) is also a multicopy suppressor of capping enzyme ts mutations. Neither CES1 nor CES4 is essential for cell growth, and a double deletion mutant is viable. CES3 corresponds to BUDS, which encodes a putative guanine nucleotide exchange factor. We hypothesize that CES1, CES4, and BUD5 may impact on RNA transactions downstream of cap synthesis that are cap dependent in vivo.
Keywords: Yeast, mRNA capping enzymes, Multicopy suppressor, Temperature-sensitive mutations
THE 5′ cap structure of eukaryotic mRNA consists of 7-methylguanosine linked to the end of the transcript via a 5′-5′ triphosphate bridge. Capping occurs by a series of three enzymatic reactions in which the 5′ triphosphate end of a primary transcript is hydrolyzed to a 5′ diphosphate by RNA triphosphatase, then capped with GMP by RNA guanylyltransferase, and methylated at N7 of guanine by RNA (guanine-7-)-methyl-transferase (23). Although the importance of the cap in mRNA function has been well documented in vitro (5,7,11,17), there is relatively little genetic evidence regarding specific roles for the cap in vivo (i.e., which RNA transactions are affected when cap synthesis is blocked). The most straight-forward way to address this issue is to genetically inactivate the enzymes involved in cap synthesis. This is feasible in the budding yeast Saccharomyces cerevisiae because the capping and methylating enzymes have been purified and the genes encoding them have been cloned (14,22).
Yeast mRNA guanylyltransferase (capping enzyme) is a 52-kDa protein encoded by the CEG1 gene (22). The yeast capping reaction resembles that of the vaccinia virus guanylyltransferase. In both cases, GTP reacts with the enzyme to form a covalent enzyme-GMP intermediate with concomitant release of pyrophosphate. The enzyme then donates the GMP to a 5' diphosphate-terminated RNA (23). Mechanistic similarities between the cellular and viral capping enzymes are echoed at the structural level by regional amino acid sequence conservation (4,24,26). Sequence homology has guided structure-function analyses of the yeast capping enzyme by targeted mutagenesis (6,21,24). Through this approach, the guanyl-yltransferase activity of CEG1 was shown to be essential for yeast cell growth (i.e., mutations of CEG1 that eliminated enzyme activity in vitro were invariably lethal in vivo).
In this article, we describe the isolation of a new collection of conditional lethal (ts) capping enzyme mutants and the use of this collection to identify multicopy suppressors of the ceg1-ts growth defect, the rationale being that such genes might encode proteins that either interact with CEG1 or impact on cap-dependent transactions in vivo. Three capping enzyme suppressor genes (CES1, CES2, CES3) were isolated by genetic selection from a multicopy genomic library. A fourth supressor gene, CES4, was identified by homology to CES1. A molecular characterization of the CES genes is presented.
MATERIALS AND METHODS
PCR Mutagenesis and Selection of ceg1-ts Alleles
The 1.4-kbp CEG1 gene was amplified in vitro by Taq DNA polymerase using reaction conditions that promote nucleotide misincorporation (12). The standard PCR reaction mixture was modified to contain 0.5 mM MnCl2 and a reduced concentration of dATP (0.2 mM) relative to the other three dNTPs (each at 1 mM). The PCR product was digested with EcoRl (which cleaves at nucleotide position +102 relative to the ATG translation initiation codon) and Xhol (which cleaves at +1189). The mutagenized CEG1 DNA fragment was ligated into pGYCE-358 (CEN, TRP1, CEG1) that had been digested with EcoRl and Xhol and gel purified to separate the vector from the excised wild-type CEG1 sequence. The ligation mixture was transformed into E. coli. A pooled plasmid library was prepared from approximately 10,000 ampicillin-resistant colonies harvested directly from the agar plates. This DNA library was transformed into YBS2 (MATa, leu2, lys2, trpl, cegl::hisG, pGYCE-360), a yeast strain in which the chromosomal CEG1 gene had been deleted (21). Growth of YBS2 depends on an ex-trachromosomal copy of CEG1 on a CEN, URA3 plasmid (pGYCE-360). Trp+ transformants (1500) were selected and replica-plated at 25 °C on medium containing 5-FOA. Five percent of the isolates were unable to grow on 5-FOA. The surviving isolates, which had lost the CEG1, URA3 plasmid, were replica-plated at 25 °C (permissive temperature) and 37 °C (nonpermissive temperature). Plasmid DNA isolated from 19 candidate ts mutants was amplified in vivo in E. coli and retested by plasmid shuffle for the ts growth phenotype in YBS2. The ts mutations of nine cegl-ts plasmids were localized crudely by sub-cloning EcoRl/BamHl (nucleotides 102-804) and BamHl/Xhol (804-1189) fragments of the cegl-ts genes back into pGYCE-358 to replace the corresponding fragment of the wild-type gene with that of the ts gene. These plasmids were tested for ts function by plasmid shuffle. After determining which gene fragment conferred the ts phenotype, the relevant segment of each of the nine ceg-ts plasmids was sequenced to identify the molecular lesions.
Isolation of Multicopy Suppressors of ceg1-ts Mutations
Isogenic strains containing different ceg1-ts alleles on a CEN, TRP1 plasmid were transformed with a 2μ-URA3 genomic DNA library. An aliquot of the transformed cells was plated on Ura medium at 25 °C to gauge transformation efficiency. The rest of the cells (about 25,000 transformants) were plated on Ura medium at 37°C. For each ceg1-ts mutant, we picked 10 colonies that grew at 37°C. Yeast plasmid DNA was isolated and transformed into E. coli. Plasmids were prepared from cultures of individual ampicillin-resistant transformants. The DNAs were digested with EcoRl and Xhol to weed out 2μ plasmids containing a genomic insert of the wild-type CEG1 gene. Candidate suppressor clones were retested by transformation into the ceg1-ts strain in which they were originally selected. Three capping enzyme suppressor genes—CES1, CES2, and CES3—were identified in this screen.
Cloning and Deletion Analysis of CES2
Limited sequencing of the 9.5-kbp insert of the 2μ-CES2 isolate indicated that it contained the ESP1 gene (16). A subclone containing only ESP1 was constructed by excising a 5.6-kbp DNA fragment obtained by partial restriction endonuclease digestion of the CES2 clone with HindIII and EcoRI. This restriction fragment, which contained the entire ESP1 coding region plus 416 bp upstream of the start codon and 500 bp downstream of the stop codon (16), was inserted into pBlue-script KS+ to yield pKS-ESPl. The ESP1 insert was excised and inserted into YEP24 to yield pESPl. The C-terminal deletion clone pESPl(l-1033) was constructed by excising a 2121-bp SpeI restriction fragment from pKS-ESPl, and then cloning the truncated insert into YEP24. Deletion clone pESPl(1-887) was made by excising a 3071-bp Hindll! fragment of pKS-ESPl (containing ESP1 sequence from −307 to + 2655), cloning it back into pBluescript, and then transferring the insert into YEP24. Deletion clone pESPl(1–515) was made by deleting an internal BamHI fragment from pESPl. The deletion clones are numbered according to the amino acid coordinates of the truncated ESP1 proteins they encode.
Gene Disruptions
The CES1 gene was disrupted by insertion of a MsG-URA3-hisG cassette (1). We first constructed plasmid pAcesl, in which the hisG-URA3-hisG casssete was flanked by a 500-bp PCR fragment derived from the 5′ end of the CES1 gene and a 1058-bp Xbal/Spel fragment from the 3′ portion of the gene. Linearized pΔcesl was transformed into the diploid strain YPH274 (MATa/MATα, trp1Δ1/ trp1Δ1, his3Δ200/his3Δ200, ura3−52/ ura3−52, ade2−101/ade2-101, lys2-801/lys2-801, leu2-Δ1/ leu2-Δ1). Insertion of the hisG-URA3-hisG cassette at one CES1 locus resulted in deletion of amino acids 13-712 of the CES1 protein; correct insertion was confirmed by Southern blotting of DNA from Ura+ transformants. The dip-loids were then sporulated. All four spores of each tetrad were viable and URA3 segregated 2:2. CES1 disuption was also performed in a separate experiment by introduction of pAcesl into the haploid strain TR2 (MATa>, trpl, his3, ura3, ade2, lys2). Correct insertion into the CES1 locus was confirmed by Southern blotting.
The entire CES4 gene was deleted by replacing the sequence from nucleotides −13 to +2318 (relative to the ATG intiation codon) with the hisG-URA3-hisG cassette. To do this, we constructed plasmid pΔces4, in which the hisG-URA3-hisG casssete was flanked by a 929-bp EcoRI/DraI 5′ of CES4 and a PCR-amplified 1-kbp fragment beginning downstream of the CES4 stop codon. Linearized pΔces4 DNA was transformed into strain YPH399 (MATa, trp1Δ1, his3Δ200, ura3−52, ade2−101, lys2−801, leu2-Δ1) and into strain YBS10 (MATa, trp1, his3, ura3, ade2, lys2, cesl:: hisG) in which the chromosomal copy of CES1 had been deleted (see above). Correct insertions were confirmed by Southern blotting.
ESP1 was disrupted by insertion of a LEU2marker. To do this we constructed plasmid pAespl in which a 1673-bp EcoRV fragment of ESP1 from nucleotide positions + 414 to + 2087 (relative to the translation initiation codon) was replaced by a 2.2-kbp DNA fragment containing the LEU2 gene. Linearized pΔespl was introduced into the diploid strain YPH274. Corrected insertion was confirmed by Southern blot analysis of DNA from Leu+ transformants.
RESULTS
Isolation of Temperature-Sensitive ceg1 Mutants
PCR amplification of the CEG1 gene was performed using reaction conditions that promote nucleotide misincorporation (12). The PCR products were cloned into a CEN, TRP1, CEG1 plasmid, to replace the wild-type CEG1 gene fragment with the PCR-mutagenized DNA. After amplification in vivo in E. coli, a pooled plasmid library was transformed into a yeast Δceg1 null strain containing wild-type CEG1 on a URA3 plasmid. Selection for Trp+ growth at 25 °C was followed by counterselection on 5-FOA to eliminate the wild-type allele. Survivors were screened for growth at 37°C, defined hereafter as the nonpermissive condition. Nine ceg1-ts isolates that formed colonies at 25°C, but not at 37°C, were selected for further analysis. Plasmid DNA was recovered from the ts isolates and the relevant mutations were localized within the ceg-ts genes by replacing wild-type CEG1 DNA with restriction fragments from each ts mutant clone, followed by rescreening for the conditional growth phenotype. The ceg-ts mutations were then mapped at the nucleotide level by DNA sequencing (Table 1). In every case, the thermosensitive phenotype was attributable to missense mutations. Several of the isolates had more than one amino acid substitution; no effort was made to assign the phenotype to a single missense change, as we were not interested in drawing inferences about structure-function relationships from conditional mutations. Each missense change was encountered only once, indicating that our initial round of selection and mapping did not exhaust the available pool of ts mutations.
TABLE 1.
| cegl-ts Allele | CEG1 Protein Mutations | ||
|---|---|---|---|
| cegl-J | Phe(197)Tyr | Met(201)Thr | |
| cegl-3 | Phe(51)Tyr | Asn(190)Tyr | Tyr(226)Phe |
| cegl-5 | Ser(216)Arg | ||
| cegl-6 | Val(289)Ala | Glu(364)Gly | |
| cegl-7 | Asp(300)Gly | ||
| cegl-13 | Ser(168)Pro | Asp(205)Gly | Lys(215)Asn |
| cegl-17 | Leu(123)Pro | Ile(135)Thr | Tyr(181)Ser |
| cegl-25 | Asn(283)Tyr | Asp(370)Ala | Thr(378)Ser |
| cegl-27 | Phe(195)Leu | Tyr(208)Asn | |
| cegl-95 | Asp(95)Ala | Arg(96)Ala | Glu(97)Ala |
The nine ceg1-ts mutants grew as well as the wild-type strain at 25°C (Fig. 1). Six mutants (ceg1−3, ceg1−5, ceg1−6, ceg1−13, ceg1−17, and ceg1−25) failed to form colonies at 37°C. Three mutants (ceg1−1, cegl−7, and cegl−27) were leaky and formed small colonies at the nonpermissive temperature (Fig. 1). We analyzed in parallel a 10th ts allele, cegl−95, that we had identified during charge-cluster to alanine mutagenesis of CEG1 (24). The molecular lesion of cegl−95 is a triple alanine substitution at residues Asp95, Arg96, and Glu97. The ceg1−95 mutant cells grew as well as wild-type cells at 25 °C, but did not form colonies at 37°C (Fig. 1). All of the ceg1-ts alleles were recessive to wild-type CEG1 (not shown).
FIG. 1.
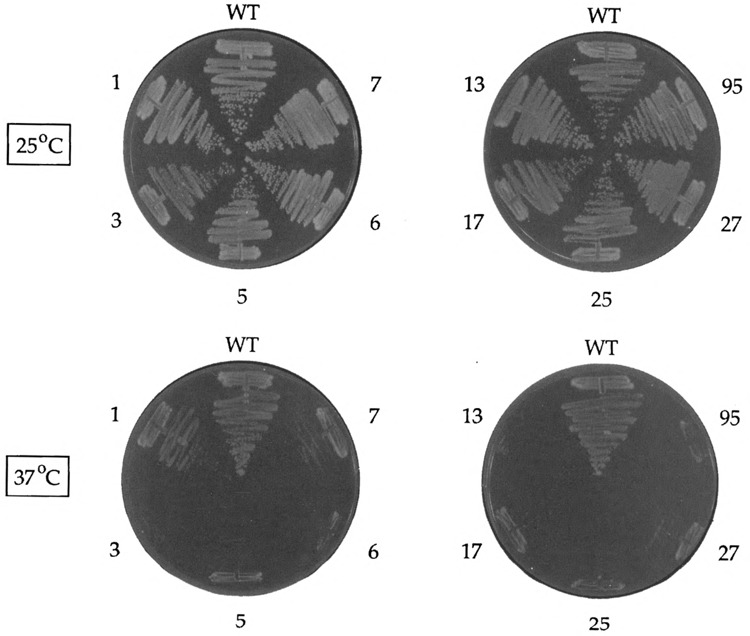
Conditional growth phenotype of ceg1-ts mutants. Cells carrying the wild-type CEG1 gene or ceg1-ts mutants (denoted by the ts allele number; see Table 1) on a CEN plasmid were plated on YPD medium and incubated for 3 days at either 25 °C or 37°C. Photographs of the plates are shown.
Isolation of Multicopy Suppressors of cegl-ts Mutations
The suppressor screen entailed transformation of each of the ceg1-ts strains with a 2μ plasmid-based wild-type genomic DNA library and selection for Ura+ colonies that grew at 37 °C. We analyzed 10 positives for each of the ceg1-ts strains that were transformed with the 2μ library. Plasmid DNA was recovered from individual yeast colonies and transformed into E. coli. Diagnostic restriction enzyme digestion revealed whether wild-type CEG1 had been selected. Candidate suppressors that did not contain the CEG1 gene were retransformed into the ts strain from which they were originally isolated and tested for growth at 37 °C. Six genomic clones retested faithfully. Restriction mapping of the genomic inserts revealed that these six clones derived from three distinct genetic loci, which we named CES1, CES2, and CES3 (CES = capping enzyme suppressor). CES1 and CES2 were recovered as single isolates in cegl-25. CES3 was recovered four times, three times in cegl-5 and once in cegl-7.
A side-by-side comparison of efficacy of multicopy suppression of the cegl-25 and cegl-5 mutations by the three CES genes is shown in Fig. 2. ceg1-ts mutants transformed with a 2μ.-CEG1 plasmid served as a positive control for wild-type growth at 37°C. ceg1-ts strains transformed with the YEP24 vector served as negative controls (i.e., the vector transformants grew at 25 °C, but not at 37°C). CES1 and CES2 both restored growth of cegl−25 at 37°C. (This was expected given that CES1 and CES2 were both isolated in cegl-25.) The size of the CES1 and CES2 colonies at 37°C was slightly smaller than that of the CEG1 strain (Fig. 2). Defining the wild-type CEG1 colony size as three-plus (+ + +), we designated the extent of suppression of cegl-25 by CES1 and CES2 as two-plus (+ +). CES3 was less effective in suppressing cegl-25, as judged by the even smaller colony size at 37°C. We classified this degree of suppression as one-plus (+). A different hierarchy of suppression was observed with the cegl-5 mutation. Here, CES3 was the better suppressor (+ +), which was in keeping with the fact that CES3 was isolated in the cegl-5 strain. CES1 suppressed weakly ( + ), whereas CES2 did not suppress.
FIG. 2.
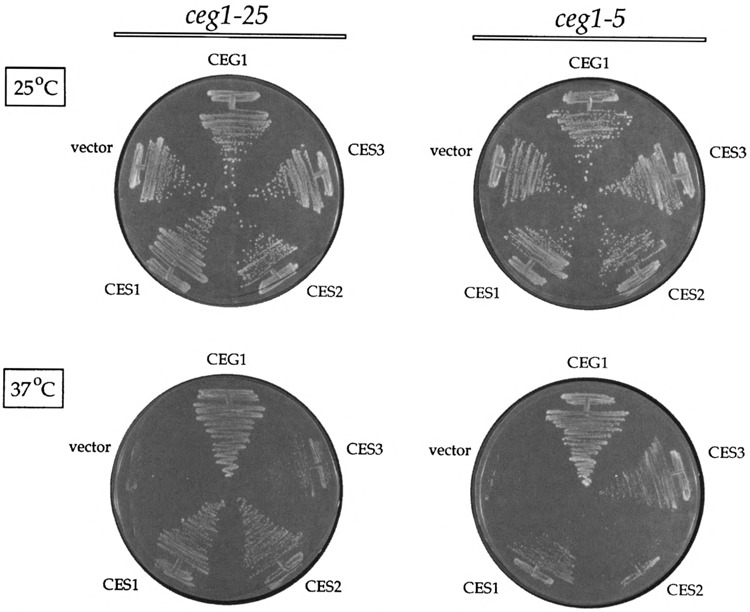
Suppression of the ts growth defect of ces1−25 and ces1−5. Strains ceg1−25 and cegl−5 were transformed with a 2μ-URA3 plasmid containing the wild-type CEG1 gene, with the 2 clones of CES1, CES2, and CES3 that were isolated in the multicopy suppressor screen, and with the YEP24 vector without any insert. Ura+ transformants were selected and streaked on plates lacking uracil. The plates were photographed after incubation for 4 days at either 25°C or 37°C, as indicated.
The CES1, CES2, and CES3 multicopy plasmids were tested for their ability to rescue the conditional lethality of a GAL-CEG1 strain, in which the chromosomal CEG1 gene is deleted and a CEN, TRP1 plasmid carries a wildtype CEG1 gene under the control of a GAL10 promoter. Growth of the GAL-CEG1 strain is galactose dependent (21). Growth is precluded on medium containing glucose, which represses GAL-CEG1 expression. Growth on glucose can be restored by wild-type CEG1 expressed from a constitutive promoter, or by PCE1, a cDNA clone encoding the capping enzyme of Schizosaccharomyces pombe (24). We reasoned that if any of the multicopy capping enzyme suppressor genes were genetically bypassing CEG1, then the 2μ-CES plasmids should also permit growth of the GAL-CEG1 strain on glucose. Neither CES1, CES2, nor CES3 allowed any detectable growth of GAL-CEG1 on glucose (not shown). We conclude that the CES genes are not bypassing the requirement for cap or capping enzyme for yeast cell growth.
A llele Specificity of Multicopy Suppression
The 2μ-CES clones were tested for suppression of each of the 10 ceg-ts mutations in our collection (Fig. 3). A hierarchy of suppression was apparent in that some ts mutations (ceg1−1, cegl-7, ceg1−27, and ceg1−25) were suppressed by all three CES genes, whereas other mutations were not suppressed at all (e.g., ceg1−3, ces1−13). The former group includes the three ts mutants that display a partially leaky growth phenotype at 37°C (Fig. 1). We suspect that the least severe ts mutations were most prone to suppression because CEG1 activity hovers about some threshold level when these strains are plated at the nonpermissive temperature, whereas the most severe ceg1-ts mutants could not be suppressed because they are functionally null at 37°C. Several examples of allele-specific suppression were encountered; cegl-6 was suppressed by CES1 and CES2, but not by CES3, whereas cegl-17 and cegl-95 were suppressed only by CES3 (Fig. 3).
FIG. 3.
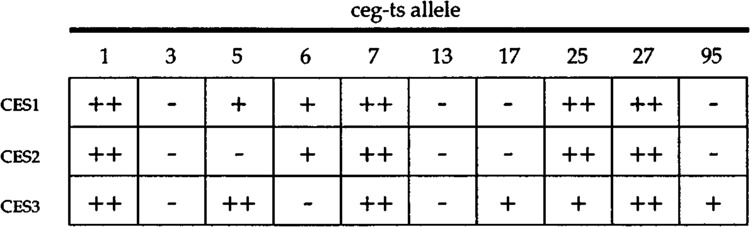
Test of allele specificity of suppression. Each of the ceg1-ts strains (see Table 1) was transformed with 2μ-URA3 clones of CES1, CES2, and CES3 in parallel with 2μ-CEG1 and YEP24 vector plasmids. Ura+ transformants were streaked to plates lacking uracil. The plates were incubated for 4 days at either 25°C or 37°C. The strength of suppression at 37°C was scored by comparison to the wild-type CEG1 transformant (+ + +) and the YEP24 vector (−), which were included as controls on every culture plate.
CES1 Encodes a Novel 103-kDa Protein
The 2μ-CES1 clone isolated in the suppressor screen contained a 9-kbp DNA insert. To narrow down the location of the CES1 gene, we screened the 2μ genomic library by colony hybridization using a labeled probe specific for the CES1 insert. We obtained a 2μ-CES1 plasmid with a 5-kbp insert that was as effective as the original isolate in suppressing the cegl-25 mutation (Figs. 4 and 5). A physical map of the insert is shown in Fig. 4. An internal deletion of a 1-kbp Bg/II fragment from the 5-kbp insert abolished CES1 function, whereas a 3.8-kbp HincII fragment including this Bg/II fragment was sufficient to suppress cegl-25 (Figs. 4 and 5). We completely sequenced both DNA strands of the insert and encountered a single long open reading frame (Fig. 4). Because this coding sequence was contained entirely within the HincII fragment sufficient for suppression, and because the predicted gene product would be inactivated by deletion of the Bg/II fragment, we concluded that this open reading frame corresponds to the CES1 gene. (The CES1 DNA sequence has been deposited in Genbank; U32580.) CES1 has the capacity to encode a 103-kDa polypeptide (Fig. 4). The 915-amino acid CES1 protein is rich in serine, asparagine, and glutamic acid residues and is predicted to be strongly hydrophilic throughout its length. CES1 contains numerous clusters of repeating amino acids (e.g., a polyglutamine run at the C-terminus) and tandem repeats of short motifs (e.g., SSSP or SAPVQ, as underlined in Fig. 4).
FIG. 4.

Physical map of the CES1 locus and the amino acid sequence of the CES1 protein. Shown schematically is the 5-kbp insert of a 2μ-CES1 clone. HincII and Bg/II restriction sites used in mapping CES1 are indicated. The CES1 gene is located within the 3.8-kbp Hindi fragment, which was sequenced in its entirety. The location of a continuous open reading frame corresponding to the CES1 gene is shown, with the orientation (N-terminus to C-terminus) indicated by an arrow. The sequence of the 915-amino acid CES1 polypeptide is shown in single-letter amino acid code. Short tandemly repeated sequence elements are underlined.
FIG. 5.

Identification of the CES1 gene, ceg1-25 was transformed with the original 2μ-CES1 isolate containing a ∼9-kbp insert (CESl-9kb), with a genomic 2μ clone containing a 5-kbp insert that was isolated by colony hybridization (CESl-5kb), with a derivative of CESl-5kb in which the 1-kbp Bg/II fragment was deleted (CESl-ΔBg1), and with a YEP24-based plasmid containing a 3.8-kbp HincII fragment derived from CES1-5kb (CES1-Hinc2) (see Fig. 4). Ura+ transformants were selected and streaked on plates lacking uracil. Wild-type CEG1 and YEP24 vector transformants were streaked on the same plates as controls. The plates were photographed after incubation for 4 days at either 25°C or 37°C.
The CES1 gene was replaced by insertion of a hisG-URA3-hisG cassette (1). The disruption was performed in a diploid strain such that marker insertion eliminated the CES1 coding sequence from amino acid positions 13-712. Correct insertion into one CES1 locus was confirmed by Southern blotting of Ura+ transformants. After sporulation and tetrad dissection, we recovered viable Ura+ haploids in a 2:2 segregation pattern. CES1 was also disrupted by insertion of the hisG-URA3-hisG cassette into the CES1 locus of a haploid strain. The size of colonies formed by the Acesl null strains was indistinguishable from that of the parental CES1 strain at either 16°C, 25°C, or 37°C. We conclude that CES1 is nonessential.
Multicopy Suppression of ceg1-ts by a Yeast Homologue of CES1
The CES1 protein is homologous to a 942-amino acid polypeptide encoded by open reading frame 8339.10 on yeast chromosome XIII that was identified during genomic sequencing (Genbank Z49210; blastp score = 419). CES1 and 8339.10 display high similarity to each other (50-70% identity) within a series of conserved sequence blocks (Fig. 6). 8339.10 lacks the C-terminal polyglutamate tail and the tandemly repeated motifs found in CES1. CES1 bears little resemblance to other proteins in the data base.
FIG. 6.

Amino acid sequence similarity between CES1 and CES4. Segments of the CES1 protein were aligned with a homologous polypeptide encoded by an open reading frame on chromosome XIII (designated CES4, because this gene could function as a multicopy capping enzyme suppressor). The amino acid coordinates demarcating each segment are indicated. Amino acid identity is denoted by two dots (:); similarity is denoted by a single dot (.).
We found that a 2μ clone containing the 8339.10 gene (a gift of Desmond Clark and Dan Burke, University of Virginia) suppressed the ts growth phenotype of cegl-25 to the same extent as CES1 (+ + growth at 37°C). Because CES1 and 8339.10 were functionally homologous with respect to capping enzyme suppression, we named the latter gene CES4. We deleted CES4 in a hap-loid strain by insertion of a hisG-URA-hisG cassette. Correct insertion was confirmed by Southern blotting. The Δces4 null strain grew at either 16°C, 25°C, or 37°C. Thus, CES4 is nonessential, like its homologue CES1.
We examined whether these genes might carry out essential, albeit redundant, functions by constructing a double-knockout of CES1 and CES4. This was done by disruption of the CES4 gene in a Δces1 haploid strain. We recovered viable haploids in which the CES1 and CES4 genes were both deleted. Thus, CES1 and CES4 are together dispensable for yeast cell growth.
CES2 Is Identical to ESP1
The 2μ-CES2 clone contained a 9.5-kbp genomic insert. Limited sequencing of the insert revealed that the clone included ESP1, a previously identified essential gene involved in nuclear division (16). A 2/x subclone of the CES2 insert containing only the ESP1 gene was just as effective as the original CES2 clone in suppressing the growth defect of cegl-25 (Fig. 7). We conclude, therefore, that CES2 is identical to ESP1.
FIG. 7.
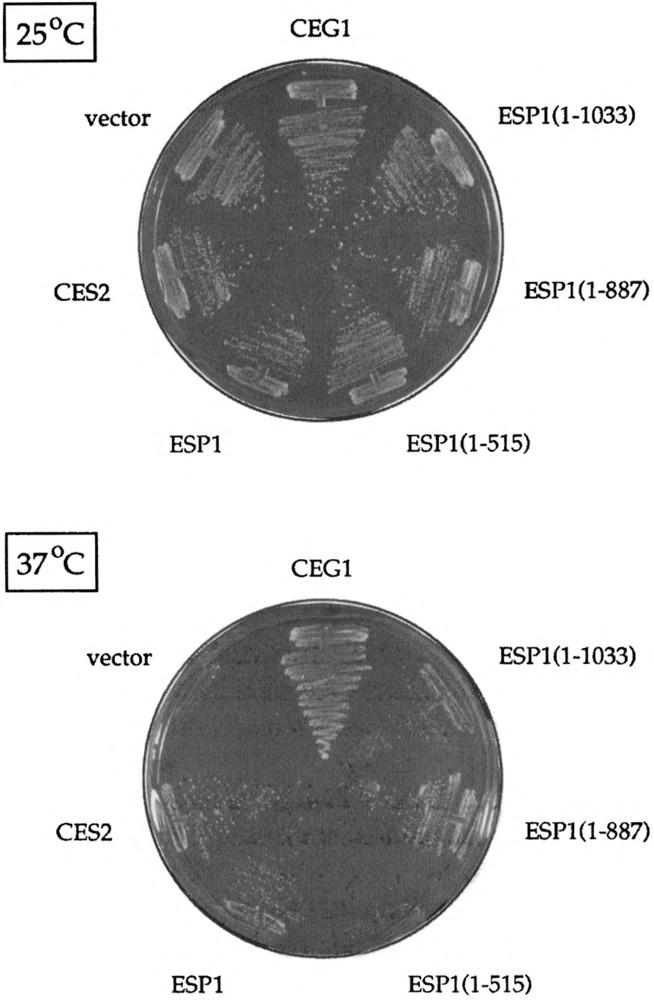
CES2 is identical to ESP1. cegl-5 was transformed with the original 2p-CES2 isolate (CES2), with a 2μ clone containing just the ESP1 gene (ESP1), and with three deletion clones-ESP1(1-1033), ESPl(l-887), and ESP1(1-515)-numbered according to the amino acid coordinates of the truncated ESP1 proteins they encode (see Fig. 8). Ura+ transform-ants were selected and streaked on plates lacking uracil. Wild-type CEG1 and YEP24 vector transformants were streaked on the same plates as controls. The plates were photographed after incubation for 4 days at either 25°C or 37°C.
The ESP1 protein is a 1573-amino acid poly-peptide; it contains a 371-amino acid region at its C-terminus that is homologous to the corresponding regions of S. pombe cut1 and Aspergillus nidu-lans bimB (16). The latter genes, like ESP1, are involved in nuclear division, espl mutations cause abnormal segregation of the nucleus during mitosis, such that the DNA and spindle poles are partitioned asymmetrically to the daughter cell (16). To examine the possible connection between the essential function of ESP1 in cell division and its action as a multicopy suppressor of capping enzyme mutants, we tested a series of 2μ-URA3 clones encoding C-terminally truncated versions of ESP1 for both genetic functions (Fig. 8). CES activity was tested in the cegl-25 strain. We found that the ESP1(1–1033) and ESPl(1–887) clones were just as effective as the full-sized ESP1 clone in supressing the cegl-ts mutation (Fig. 7). However, further truncation of the gene in the case of ESP1(1–515) abolished CES2 function (Fig. 7).
FIG. 8.
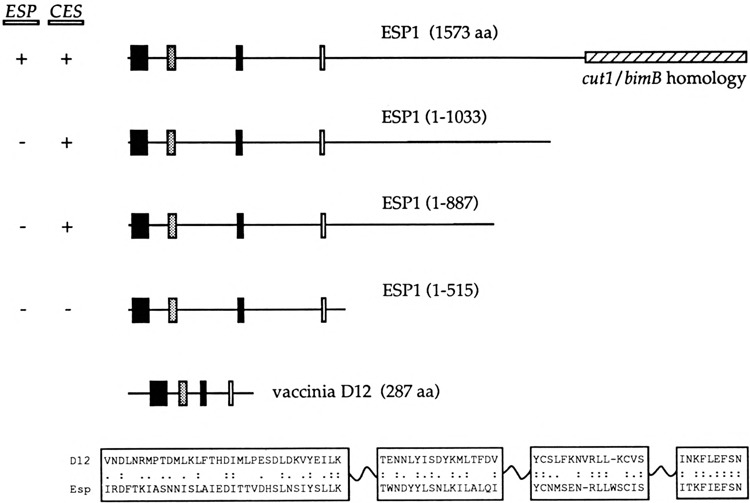
Deletion analysis of CES2/ESP1 and sequence similarity to a subunit of the vaccinia virus capping enzyme. 2μ plasmid clones containing the complete ESP1 gene and deleted versions of ESP1 were constructed as described in Materials and Methods. The polypeptides encoded by these constructs are shown schematically in the figure. The clones are numbered according to the amino acid coordinates of the truncated ESP1 proteins they encode. The C-terminal 371-amino acid region homologous to nuclear division proteins cutl and bimB is shown as a hatched box. Regions of sequence similarity between the N-terminus of ESP1 and the D12 subunit of the vaccinia virus capping enzyme are depicted as bars. The sequences of the D12 and ESP1 proteins in these regions are aligned at the bottom. Amino acid identity is indicated by two dots (:); similarity is denoted by a single dot (.). The results of functional analyses of the clones are summarized on the left. ESP function refers to the ability of the plasmid-encoded gene to complement an espl::LEU2 mutation. CES activity refers to the experiment of Fig. 7, in which the clones were tested for multicopy suppression of ceg1−25.
To assess ESP1 -dependent cell growth, we constructed a diploid strain in which one copy of ESP1 was deleted and replaced by a LEU2 marker. This diploid was transformed with the full-length and truncated ESP1 clones shown in Fig. 8. The Ura+ diploids were then sporulated. If the 2μ plasmid encoded a form of ESP1 that was competent to carry out its essential function, then we expected the following: i) to recover Leu+ haploids in a 2:2 segregation pattern, and ii) that all Leu+ progeny would be 2μ plasmid dependent for growth and therefore unable to grow on 5-FOA. This is what we observed when diploids containing the full-length ESP1 clone were sporulated and markers were subsequently tested (five tetrads analyzed). However, if the plasmid-encoded ESP1 protein was not able to perform its essential function, we predicted that no viable Leu+ haploids would be recovered. This is just what we observed for the ESP1(1−1033), ESPl(1−887), and ESP1(1−515) clones; that is, all tetrads yielded two viable spores and all haploids were Leu— (20 tetrads analyzed). Thirty-three of the 40 haploid Leu— progeny were Ura+, indicating that there was no problem with plasmid maintenace during meiosis and sporulation.
We conclude from this analysis that there are distinct functional domains of the ESP1 protein. ESP1 function during cell growth clearly requires the C-terminal protein segment that includes the 371-amino acid homology to cutl and bimB. In contrast, CES2 function is unaffected by removal of 686 residues from the carboxyl end.
The CES2 Domain of ESP1: Similarity to the Small Subunit of Vaccinia Virus Capping Enzyme
A search of the Genbank data base highlighted a resemblance between the ESP1 protein and the D12 subunit of the vaccinia virus mRNA capping enzyme. Four short segments of sequence similarity are arrayed in collinear fashion in the ESP1 and D12 polypeptides (Fig. 8). Remarkably, these motifs are located in the N-terminal portion of ESP1 that constitutes the CES2 domain. The vaccinia virus capping enzyme is a heterodimer of D1 (95 kDa) and D12 (33 kDa) subunits that catalyzes all three steps in cap synthesis (23). The three catalytic functions are organized in a modular fashion within the native enzyme. The RNA triphosphatase and RNA guanylyltranferase activities reside within a 60-kDa N-terminal region of the D1 sub-unit. The 287-amino acid D12 subunit, together with the C-terminal 305-amino acid portion of the D1 subunit, catalyzes the cap methylation reaction. The D12 protein itself has no catalytic activity; rather, it interacts tightly with the D1 subunit and stimulates its RNA (guanine-7)-methyl-transferase activity by 50- to 100-fold (3,9,15). In light of our earlier findings regarding the structural and functional homology between the vaccinia virus and yeast capping enzyme systems (14,21,24), we regard it as significant that a genetic screen in yeast for suppression of ts capping enzyme function yielded a protein resembling a known subunit of the vaccinia capping enzyme. We consider this further in the Discussion.
CES3 Corresponds to BUD5
The four independent CES3 isolates contained genomic DNA inserts of 6 to 9 kbp. We localized the CES3 inserts by limited DNA sequencing to the vicinity of the MAT locus on yeast chromosome III. The four clones overlapped within an —4-kbp region that included the entire BUD5 gene. BUDS is a nonessential gene encoding a 537-amino acid polypeptide with sequence similarity to guanine nucleotide exchange factors (2,18). A 2.8-kbp Pstl-Scal subclone containing sequence from nucleotides −568 to +2242 relative to the translation start site of the BUD5 gene (which includes the entire coding sequence and 631 bp of DNA 3′ of the translation stop codon) was capable of suppressing cegl-5. However, a 2.9-kbp Pvull subclone, extending from +681 within the BUD5 open reading frame to position +3642 located 2031 bp downstream of the BUD5 stop codon, did not suppress cegl−5. We conclude that CES3 corresponds to BUD5.
DISCUSSION
The RNA capping activity of the yeast CEG1 protein is required for cell growth (21). Yet the essential role played by the RNA cap in vivo remains unclear, and is likely to be complex, given that in vitro studies have implicated the cap in so many aspects of cellular RNA metabolism. As a first step toward analyzing cap function genetically, we isolated a collection of temperature-sensitive cegl alleles from a pool of mutagenized CEG1 clones. Conditional mutations constitute valuable tools for two levels of inquiry: i) what happens to RNA metabolism and gene expression when capping enzyme is inactivated? and ii) what other genes impact on CEG1 function in vivo? Both areas are under investigation in our laboratories. Initial phenotypic analysis confirms the prediction that many aspects of gene expression are affected when cegl-ts mutants are shifted to the nonpermissive temperature (e.g., we find that protein synthesis is shut off, pre-mRNA splicing is inhibited, and the steady-state level of mRNA declines sharply) (Schwer, Mao, and Shuman, unpublished).
The present report describes the use of the ceg1-ts mutants to identify interactions between CEG1 and other yeast genes. We isolated and characterized three yeast CES genes that, when present in high copy, can suppress the conditional growth phenotype of specific mutations in the capping enzyme. CES1 encodes a novel 103-kDa polypeptide. CES2 corresponds to ESP1, a gene required for proper nuclear division during mitosis. CES3 corresponds to BUD5, which encodes a putative guanine nucleotide exchange factor involved in bud site selection. An additional capping enzyme suppressor, CES4, was identified by homology to CES1.
Suppression of defects in mRNA capping could occur by increasing the dosage of a protein that interacts with CEG1 Alternatively, a high copy suppressor might act downstream of cap synthesis to enhance some essential cap-dependent RNA transaction that had become limiting for growth at the nonpermissive temperature. We consider below how these ideas might apply to multicopy suppression by the three CES genes identified in our screen.
ESP1
The suppressor gene identified initially as CES2 is identical to ESP1, a gene required for proper nuclear division (16). ESP1 encodes a fairly large polypeptide (181 kDa). No specific role of the ESP1 protein in nuclear division has been described. The C-terminal homology between the ESP1, cut1, and bimB gene products has been suggested to confer a common function in nuclear division (16). Our findings support this model, insofar as deletion of the C-terminal segment of ESP1 that includes the cutl/bimB domain inactivates the essential ESP1 function. [This extends an earlier finding that ESP1 was inactivated by a transposon insertion near the C-terminus of the open reading frame (16).] The capping enzyme suppressor activity of ESP1 is clearly distinct from its function in nuclear division, because nearly half of the protein can be deleted from the C-terminus without affecting CES2 activity. It remains to be determined if the capping enzyme suppressor and nuclear division functions are carried out by autonomous or overlapping protein domains (i.e., whether the N-terminal half of the protein that is sufficient for CES2 function is necessary for ESP1 activity in nuclear division).
The N-terminal CES2 domain of ESP1 includes segments of homology with the D12 subunit of the vaccinia virus capping enzyme. The extent of the homology is modest, but is likely to be functionally significant. Indeed, the extent of sequence similarity in this case is not much different than what has been noted for other vaccinia and yeast capping proteins that perform the same biochemical function [e.g., the CEG1 protein versus the guanylyltransferase domain of the vaccinia capping enzyme D1 subunit (24,26), or the yeast ABD1 cap methyltransferase versus the methyl-transferase domain of the vaccinia D1 protein (14)]. The D12 subunit of the vaccinia capping enzyme binds tightly to the D1 subunit and stimulates the intrinsic D1 methyltransf erase activity (9,15). The stimulation of catalysis by D12 depends on the ability of the two subunits to hetero-dimerize (3,15). The D12 subunit is also essential for the action of the vaccinia capping enzyme as a transcription termination factor (13). A potentially relevant property of the D12 subunit is its ability to protect the D1 subunit from proteolysis in vivo (8,25). Nothing is known about the domain structure of the D12 subunit or about specific structural requirements for subunit association. Hence, it is not obvious what function might be conferred by the D12 motifs found in ESP1. We speculate, by analogy to the vaccinia system, that increased dosage of CES2/ESP1 drives its physical interaction with CEG1, either directly or through a third protein (such as ABD1), the effect of which is to either stabilize CEG1 against dena-turation or proteolysis at the nonpermissive temperature or else to enhance its catalytic activity above a threshold level required for viability. Attempts to directly test the effects of ESP1 on CEG1 guanylyltransferase activity in vitro have been hampered at the stage of expressing the ESP1 protein in bacteria.
CES1
CES1 encodes a novel hydrophilic 103-kDa polypeptide. The sequence of the CES1 protein is uninformative, except for its extensive similarity to the product of CES4. CES4 is a functional homolog of CES1 in that it too acts as a multicopy suppressor of ts mutations in the capping enzyme. We found that neither gene is essential and that a double-knockout is viable.
CES4 has been isolated as a multicopy suppressor of mutations in cdc20 and sin4 (D. Burke, personal communication; Yu et al., Genbank U32938), two genes that have no apparent connection to CEG1. The fact that CES1 and/or CES4 have been identified in diverse high copy suppressor screens suggests that they may affect some aspect of gene expression. For example, multiple defects might be suppressed by genes that enhance protein stability or upregulate gene expression at the transcriptional or posttranscriptional level. Given the unusual amino acid composition of CES1, it is conceivable that it interacts physically with a variety of other proteins and that this is relevant to its suppressor activity. Yet, we favor the idea that CES1 affects gene expression. Of the various proteins that are suppressed by CES1 or CES4, the capping enzyme seems most closely tied to gene expression. Our preliminary analysis of the cegl-ts phenotype reveals, among other effects, a significant reduction in the steady-state level of mRNA after shift of cegl-ts cells to the nonpermissive temperature (unpublished). We speculate that CES1 may suppress the capping enzyme defect either by stimulating the utilization of residual mRNAs that are capped or by enhancing the use of uncapped RNAs. This effect could occur at the level of RNA stability or translation or both. A possible role for CES1 in translation is suggested by the independent isolation of CES1 as a multicopy supressor of a ts mutation in the translation initiation factor eIF4A (P. Linder, personal communication).
BUD5
BUDS is not essential for cell growth; however, a Δbud5 deletion elicits a random budding pattern in lieu of the axial or bipolar patterns seen in wild-type cells (2). BUD5 encodes a putative guanine nucleotide exchange factor, which is believed to regulate the activity of the BUD1 and CDC42 proteins; the latter are Ras-like GTPases involved in bud site selection and bud formation, respectively (2). BUDS, when overexpressed from a high copy plasmid, can also interact with RAS2 and suppress the growth defect of a dominant-negative RAS2 mutation (18).
How might BUD5, a component of a GTPase signaling pathway, act in high dosage to suppress mutations in the capping enzyme? Although capping enzyme binds GTP, it is an unlikely target for a guanine nucleotide exchange factor, because GDP is neither an intermediate nor a product in the guanylyltransferase reaction (23). We speculate that BUD5 overexpression may impact on a downstream phase of gene expression. Yeast RNA processing and RNA transport are affected by mutations in genes that encode guanine nucleotide signaling proteins (10,19,20). In addition, GTPases and guanine nucleotide exchange factors play key roles in translation. The cap structure is thought to facilitate each of these RNA events.
Capping and Budding
A strange nexus between cap synthesis and cell polarity is emerging that involves at least four yeast genes: CEG1, ABD1, CES1, and BUDS. For example: i) ABD1, which encodes RNA (guanine-7)-methyltransferase, the enzyme that catalyzes RNA cap methylation (14), has been isolated in a genetic screen for mutational synergy with BEM1, a gene involved in bud emergence (Corrado and Pringle, Genbank L12000); ii) bem1 mutations display synergy with budS mutations (2); iii) BUD5 in high copy partially suppresses ts mutations in mRNA guanylyltransferase (this study); iv) ceg1-ts mutations are suppressed by CES1 in high copy (this study); v) CES1 has been isolated in an indepedent high copy screen as a negative regulator of cell polarity (NRC1) (Bi and Pringle, Genbank L42821). What does 5′ end modification of RNA have to do with cell polarity? Budding entails the recruitment of numerous structural components and regulatory factors to a site on the cell cortex. It is possible that synthesis of some of these components or factors might be especially cap dependent in vivo. It is even conceivable that mRNA encoding one of the proteins involved in budding is itself localized within the cell (e.g., at the bud site) and that this process is cap dependent.
Conclusions
We have identified a genetic connection between capping enzyme and four other yeast proteins. CES2/ESP1 displays sequence similarity to the small subunit of the vaccinia capping enzyme and is a candidate to interact physically or functionally with yeast RNA guanylyltransferase. CES1 and CES4, which are novel proteins, and CES3/BUD5, a putative guanine-nucleotide exchange factor, have no obvious connection to capping, but may impact on downstream transactions that are cap dependent in vivo.
ACKNOWLEDGEMENTS
Yizhi Liu provided valuable technical assistance. This work was supported by NIH Grant GM52470 (B.S. and S.S), and Grants FRA-432 (S.S.) and JFRA-571 (B.S.) from the American Cancer Society.
REFERENCES
- 1. Alani E.; Cao L.; Kleckner N. A method for gene disruption that allows repeated use of URA3 selection in the construction of mutiply dirupted yeast strains. Genetics 116:541–545; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chant J.; Corrado K.; Pringle J. R.; Herskowitz I. Yeast BUD5, encoding a putative GDP-GTP exchange factor, is necessary for bud site selection and interacts with bud formation gene BEM1. Cell 65:1213–1224; 1991. [DOI] [PubMed] [Google Scholar]
- 3. Cong P.; Shuman S. Methyltransferase and subunit association domains of vaccinia virus mRNA capping enzyme. J. Biol. Chem. 267:16424–16429; 1992. [PubMed] [Google Scholar]
- 4. Cong P.; Shuman S. Covalent catalysis in nucleotidyl transfer: A KTDG motif essential for enzyme-GMP complex formation by mRNA capping enzyme is conserved at the active sites of RNA and DNA ligases. J. Biol. Chem. 268:7256–7260; 1993. [PubMed] [Google Scholar]
- 5. Edery I.; Sonenberg N. Cap-dependent RNA splicing in a HeLa nuclear extract. Proc. Natl. Acad. Sci. USA 82:7590–7594; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fresco L. D.; Buratowski S. Active site of the mRNA capping enzyme guanylyltransferase from Saccharomyces cerevisiaet Similarity to the nucleotidyl attachment motif of DNA and RNA ligases. Proc. Natl. Acad. Sci. USA 91:6624–6628; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Furuichi Y.; LaFiandra A.; Shatkin A. J. 5′-Terminal structure and mRNA stability. Nature 266:235–239; 1977. [DOI] [PubMed] [Google Scholar]
- 8. Guo P.; Moss B. Interaction and mutual stabilization of the two subunits of vaccinia virus mRNA capping enzyme coexpressed in Escherichia coli . Proc. Natl. Acad. Sci. USA 87:4023–4027; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Higman M. A.; Christen L. A.; Niles E. G. The mRNA (guanine-7-) methyltranferase domain of the vaccinia virus mRNA capping enzyme: Expression in Escherichia coli and structural and kinetic comparison to the intact capping enzyme. J. Biol. Chem. 269:14974–14981; 1994. [PubMed] [Google Scholar]
- 10. Kadowaki T.; Goldfarb D.; Spitz L. M.; Tartakoff A. M.; Ohno M. Regulation of RNA processing and transport by a nuclear guanine nucleotide release protein and members of the Ras superfamily. EMBO J. 12:2929–2937; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Konarska M. M.; Padgett R. A.; Sharp P. A. Recognition of cap structure in splicing in vitro of mRNA precursors. Cell 38:731–736; 1984. [DOI] [PubMed] [Google Scholar]
- 12. Leung D. W.; Chen E.; Goeddel D. V. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique J. Methods Cell Mol. Biol. 1:11–15; 1989. [Google Scholar]
- 13. Luo Y.; Mao X.; Deng L.; Cong P.; Shuman S. The D1 and D12 subunits are both essential for the transcription termination factor activity of vaccinia capping enzyme. J. Virol. 69:3852–3856; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mao X.; Schwer B.; Shuman S. Yeast mRNA cap methyltransferase is a 50-kilodalton protein encoded by an essential gene. Mol. Cell. Biol. 15:4167–4174; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mao X.; Shuman S. Intrinsic RNA (guanine-7) methyltransferase activity of the vaccinia virus capping enzyme D1 subunit is stimulated by the D12 subunit: Identification of amino acid residues in the D1 protein required for subunit association and methyl group transfer. J. Biol. Chem. 269:24472–24479; 1994. [PubMed] [Google Scholar]
- 16. McGrew J. T.; Goetsch L.; Beyers B.; Baum P. Requirment for ESP1 in the nuclear division of Saccharomyces cerevisiae . Mol. Biol. Cell 3:1443–1454; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muthukrishnan S.; Both G. W.; Furuichi Y.; Shatkin A. J. 5′-Terminal 7-methylguanosine in eukaryotic mRNA is required for translation. Nature 255:33–37; 1975. [DOI] [PubMed] [Google Scholar]
- 18. Powers S.; Gonzales E.; Christensen T.; Cubert J.; Broek D. Functional cloning of BUD5, a CDC25-related gene from S. cerevisiae that can suppress a dominant-negative RAS2 mutant. Cell 65:1225–1231; 1991. [DOI] [PubMed] [Google Scholar]
- 19. Schlenstedt G.; Saavedra C.; Loeb J. D.; Cole C. N.; Silver P. A. The GTP-bound form of the yeast Ran/TC4 homologue blocks nuclear protein import and appearance of poly(A)+ RNA in the cytoplasm. Proc. Natl. Acad. Sci. USA 92:225–229; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schneiter R.; Kadowaki T.; Tartakoff A. M. mRNA transport in yeast: Time to reinvestigate the role of the nucleolus. Mol. Biol. Cell 6:357–370; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schwer B.; Shuman S. Mutational analaysis of yeast mRNA capping enzyme. Proc. Natl. Acad. Sci. USA 91:4328–4332; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shibagaki Y.; Itoh N.; Yamada H.; Hagata S.; Mizumoto K. mRNA capping enzyme: Isolation and characterization of the gene encoding mRNA guanylyltransferase subunit from Saccharomyces cerevisiae . J. Biol. Chem. 267:9521–9528; 1992. [PubMed] [Google Scholar]
- 23. Shuman S. Capping enzyme in eukaryotic mRNA synthesis. Prog. Nucleic Acid Res. Mol. Biol. 50:101–129; 1995. [DOI] [PubMed] [Google Scholar]
- 24. Shuman S.; Liu Y.; Schwer B. Covalent catalysis in nucleotidyl transfer reactions: Essential motifs in Saccharomyces cerevisiae RNA capping enzyme are conserved in Schizosaccharomyces pombe and viral capping enzymes and among polynucleotide ligases. Proc. Natl. Acad. Sci. USA 91:12046–12050; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shuman S.; Morham S. G. Domain structure of vaccinia virus mRNA capping enzyme: Activity of the Mr 95,000 subunit expressed in Escherichia coli . J. Biol. Chem. 265:11967–11972; 1990. [PubMed] [Google Scholar]
- 26. Shuman S.; Schwer B. RNA capping enzyme and DNA ligase—a superfamily of covalent nucleotidyl transferases. Mol. Microbiol. 17:405–410; 1995. [DOI] [PubMed] [Google Scholar]