

Abstract
U1 small nuclear RNA plays an important role in early stages of intron recognition and spliceosome assembly. The 5′ splice site of the premessenger RNA base-pairs with the 5′ end of Ul; however, that interaction appears to be replaced by U5 and U6 at later stages of the splicing process. It has not been established when this transition occurs nor what factors are required for the transition. The PRP2 gene of Saccharomyces cerevisiae encodes an RNA-dependent ATPase that belongs to the DEAH putative RNA helicase family. A spliceosome can be assembled in the absence of PRP2 but the ATPase activity of PRP2 is required for the onset of the catalytic reactions in the spliceosome. By probing the precatalytic spliceosome formed in temperature-sensitive prp2 mutant extracts with oligonucleotides complementary to snRNAs, we found that the 5′ end of Ul was sensitive to RNase H digestion whereas the 5′ splice site-interacting region of U6 became resistant. Furthermore, by treating with heparin, a spliceosome depleted of Ul snRNA was isolated that subsequently underwent splicing with additional protein factors and ATP. Thus, these results indicate that PRP2 is not responsible for the transition from Ul to U6 in the spliceosome and that the entire Ul snRNA is not involved in the catalytic phase of pre-mRNA splicing.
Keywords: Pre-mRNA splicing, Spliceosome, Small nuclear RNAs, PRP2 ATPase, DEAH proteins, Saccharomyces cerevisiae
INTRONS are removed and exons are spliced from RNA precursors by a biochemical reaction called RNA splicing (1,3,41). Splicing of nuclear pre-mRNAs requires ATP, proteins, and small nuclear ribonucleoprotein particles (U1, U2, U5, and U4/U6 snRNPs). The pre-mRNA is first assembled with the snRNPs and non-snRNP protein factors into a large RNA–protein complex, called the spliceosome. Two sequential transesterification reactions (Reaction 1 and Reaction 2) then occur in the spliceosome to produce the spliced mRNA and the excised intron. Spliceosome formation is a dynamic process and a number of RNA interactions and RNA rearrangements have to occur before the spliceosome is fully assembled. For instance, the interaction between U4 and U6 snRNAs is replaced by that of U2 and U6 during the maturation of the spliceosome (24,26).
There are several lines of evidence indicating that U1 snRNP is involved in intron recognition and the early stages of spliceosome assembly (31). Splicing is completely abolished in vitro if U1 snRNA is inactivated or depleted in the extracts; and in yeast, inactivation of the U1 gene is lethal. The 5′ end of the U1 snRNA has been shown genetically to base-pair with the 5′ splice site of the pre-mRNA. Additional base-pairing between the 5′ end of U1 and the 3′ splice site was shown to be involved in splicing in fission yeast (29), although such an interaction is not essential in budding yeast (40). Interestingly, the subsequent steps of spliceosome assembly appear to require disruption of the interaction between the 5′ splice site and U1 snRNA (19). Instead, the 5′ splice site appears to interact with U5 and U6 snRNAs in the fully assembled spliceosome (14,21,36,37,43, 46,48). However, it has not been established when this transition occurs nor what factors are required for the transition.
It is generally believed that certain splicing factors with RNA unwinding or annealing activity are responsible for the RNA arrangements during the spliceosome assembly. Several PRP (pre-mRNA processing) genes encoding putative RNA helicases, which might be involved in some of the RNA rearrangements in the spliceosome, have been identified in the budding yeast Saccharomyces cerevisiae (12,33). These PRP proteins contain conserved motifs found in the DEAD-box family (e.g., PRP5 and PRP28) or in the DEAH-box family (e.g., PRP2, PRP16, PRP22) (8,38,45). Although these proteins exhibit RNA-dependent ATPase activity, none of them, thus far, has been shown to unwind RNA duplex in vitro. In fact, it is not even clear what the real cognate RNA ligands of these proteins are. Nonetheless, because mutants carrying temperature-sensitive alleles of these PRP genes are defective in pre-mRNA splicing in vivo and yield extracts that can be inactivated in vitro, they provide a very attractive system to study the mechanism of RNA rearrangements in pre-mRNA splicing (33,35,47).
We have been studying the role of the PRP2 ATPase in pre-mRNA splicing. Inactivation of the PRP2 gene in vivo causes the accumulation of un-spliced pre-mRNA and prevents cell growth (30). Heat-inactivated extracts derived from temperature-sensitive prp2 cells assemble pre-mRNA into spliceosome without the occurrence of transesterification reactions (22). This so-called prp2Δ spliceosome is stalled right before the onset of Reaction 1; and the addition of the wild-type PRP2 protein and ATP allows splicing to resume in the gradient-isolated prp2Δ spliceosome (15,16,28). Previously, we analyzed the snRNA content of this precatalytic prp2Δ spliceosome by Northern hybridization and found that U2, U5, and U6 snRNAs are indeed integral components of the spliceosome whereas U4 snRNA was not detected in the purified prp2Δ spliceosome (49). These results indicate that the U4 snRNA is not required for the transesterification reactions and that PRP2 cannot be responsible for the unwinding of the U4/U6 base-pairing.
The purpose of this study is to further examine whether PRP2 is involved in the transition from the earlier U1–5′ splice site pairing to the later U6–5′ splice site interaction by analyzing U1, U6, and U2 in the prp2Δ spliceosome. By probing with complementary oligonucleotides, we found that the 5′ end of U1 remained sensitive to RNase H digestion, whereas the 5′ splice site-interacting region of U6 and the branch point recognition region of U2 became resistant after the assembly of the prp2Δ spliceosome. Thus, these results suggest that PRP2 is not responsible for the transition from U1 to U6 in the spliceosome. Interestingly, by treating with heparin, a prp2Δ spliceosome depleted of U1 snRNA was isolated; the purified spliceosome subsequently underwent splicing in the presence of ATP and additional protein factors. This direct biochemical evidence demonstrates that the entire U1 snRNA is not involved in the catalytic phase of pre-mRNA splicing.
MATERIALS AND METHODS
Pre-mRNA Substrates and Splicing Extracts
32P-Labeled or unlabeled pre-mRNA substrates were prepared in parallel under the same conditions by transcribing SP6-actin plasmid DNA digested with HpaII or EcoRI followed by gel purification (23,49). The HpaII transcripts were used in all experiments except those described in Figs. 4 and 6, where the EcoRI transcripts were used. Splicing extracts were isolated from a wild-type strain, EJ101, and from a temperature-sensitive prp2-1 strain, CRL2101 (49).
FIG. 4.
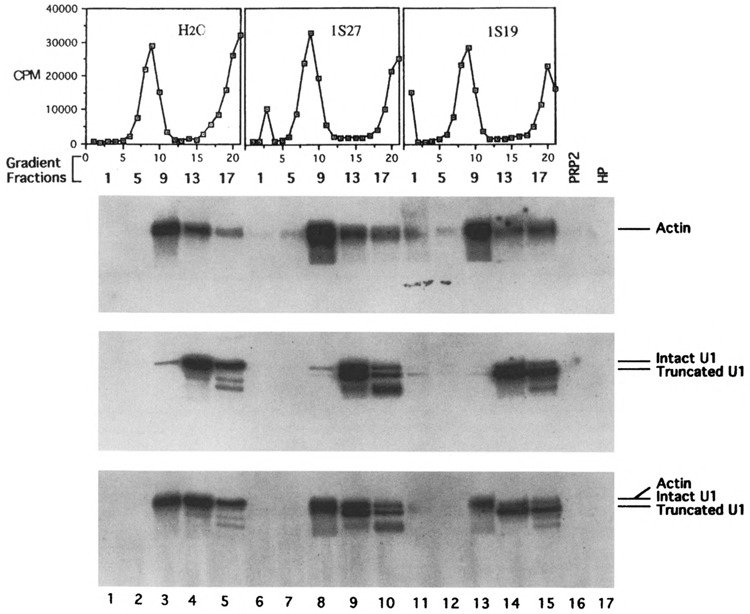
Gradient and Northern analyses of U1 snRNA after oligonucleotide-directed RNase H digestion. Oligonucleotide 1s27 or 1s19 was added to initiate RNase H digestion after the assembly of the spliceosome in prp2 Ts extracts. The reaction mixtures were treated with heparin at 0.5 μg/μl (water control and 1s27-treated samples) or 1.25 μg/μl (H19-treated sample) prior to centrifugation. The “hot” gradient profiles are displayed above the Northern autoradiograms: the top was probed with actin DNA (exposed to an X-ray film for 49 h with two LP screens), the middle probed with U1 DNA (78.5 h with two LP screens), and the bottom was probed with U1 plus actin sequences (25.5 h with one LP screen). Lanes 1–5: water control; lanes 6–10: 1s27-treated sample; lanes 11–15: 1s19-treated sample containing high concentration of heparin; lane 16: purified PRP2 protein; lane 17: purified HP factor. See Fig. 2 for additional description.
FIG. 6.
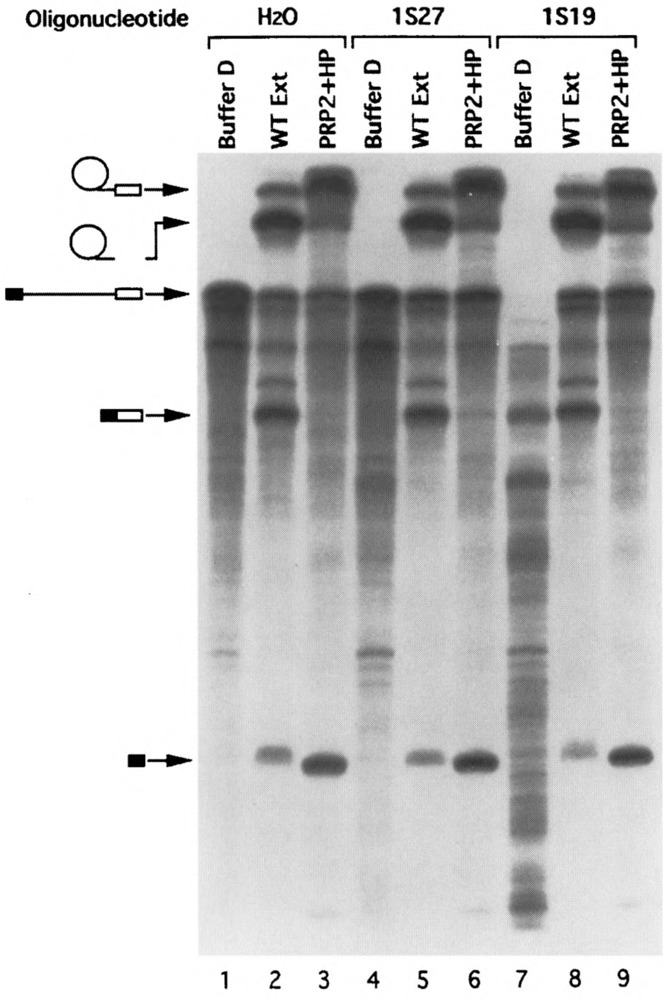
Splicing activity assays of the gradient-purified spliceosomes treated with U1 oligonucleotides as shown in Fig. 4. The “hot” spliceosome-containing fractions were assayed as described in Fig. 5. Lanes 1–3: water control spliceosomes; lanes 4–6: H27-digested spliceosomes; lanes 7–9: 1s19-digested spliceosomes treated with a higher concentration of heparin. Note that the longer EcoRI transcript was used in this experiment.
Primer Extension and Sequencing Reactions
To map the cleavage sites on U1 by primer extension, RNA recovered from the reaction mixture was resuspended in 8 μl of 10 mM methylmercury and denatured at room temperature for 10 min. The solution was adjusted to 10 mM MgCl2, 100 mM KCl, 50 mM Tris-HCl (pH 8.3), 10 mM DTT, 28 mM β-mercaptoethanol, 1.25 mM of all four dNTPs, 40 units of RNase inhibitor (Ambion), and 32P-labeled U1 primer (Table 1) in a total volume of 49 μl. After an incubation of 5 min at room temperature, 25 units of reverse transcriptase (New England Biolabs) in 1 μl was added and the mixture was incubated at 37°C for 60 min. The reaction was stopped by adding EDTA to 20 mM and the RNA in the mixture was digested with 1 μg of RNase A and 20 units of RNase T1 (Ambion) for 15 min at 37°C. The cDNA was recovered by phenol extraction and ethanol precipitation and was separated on a 6% polyacrylamide denaturing gel. The 32P-labeled U1 primer was prepared in a 25-μl reaction containing 250 mM potassium phosphate (pH 9.5), 250 mM magnesium acetate, 5 mM DTT, 60 μCi of [γ-32P]ATP (3000 Ci/mmol), 4 μg of the oligonucleotide, and 10 units of T4 polynucleotide kinase (New England BioLabs). After incubated at 37°C for 2 h, the reaction mixture was extracted with phenol followed by ethanol precipitation. Dideoxy sequencing reactions using U1 plasmid as template was performed according to the manufacturer’s protocol (Sequenase kit, USB).
TABLE 1.
OLIGODEOXYRIBONUCLEOTIDES USED IN THIS STUDY
| Name | Sequence (5′ to 3′) | Complementary to snRNA | Important Feature | Conc. Used |
|---|---|---|---|---|
| U1 primer | CAATGATAAATGCTTAACG | U1 (74th–92nd) | NA* | NA* |
| 1s19 | GATATCTTAAGGTAAGTAT | U1 (1st–19th) | 5′ splice site | 14 μM |
| 1s27 | TCTCCTCTGATATCTTAAGGTAAGTAT | U1 (1st–27th) | 5′ splice site | 20 μM |
| 2b19 | GAACAGATACTACACTTGA | U2 (28th–46th) | branch site | 0.31 μM |
| 6c27 | ATCTCTGTATTGTTCAAATTGACCAA | U6 (28th–54th) | ACAGAG sequence | 0.45 μM |
The names and the sequences of the oligodeoxyribonucleotides used are listed. The position of the snRNA to which the oligonucleotide base-pairs, the important feature of that segment of the snRNA, and the concentration of the oligonucleotide used for RNase H digestion are also described.
NA, not applicable.
Splicing Reactions and Oligonucleotide-Directed RNase H Digestion
In vitro splicing was performed as described previously (49), except the concentration of PEG used was 1%. To cleave snRNAs by endogenous RNase H before the assembly of spliceosomes, a 2-μl aqueous solution containing the specified amount of oligonucleotide was added to 4 μl of heat-inactivated mutant extracts and incubated at 32°C for 30 min. Incubation was continued for 30 min at 23°C by the addition of 4 μl of a 2.5 × splicing buffer containing ATP and pre-mRNA substrate. To cleave snRNAs after the assembly of spliceosomes, the reaction was allowed to occur at 23°C for 30 min by mixing 4 μl of heat-inactivated mutant extracts and 6 μl of a 1.7 × splicing buffer containing ATP and pre-mRNA substrate. RNase H digestion was then initiated by the addition of 2 μ1 of the oligonucleotide solution and the incubation was continued at 32°C for 30 min. In some cases, an extra amount of ATP was added to a final concentration of 2 mM to facilitate cleavage (7,25). After the oligonucleotide treatment, the reaction mixture was incubated with purified PRP2 protein at 23°C for another 30 min to complete the splicing process. RNA extraction and gel electrophoresis were carried out as described (49).
Heparin Treatment and Gradient Sedimentation
To analyze and to isolate spliceosomes by glycerol gradient sedimentation, larger splicing reactions (50 μl) were carried out. After the RNase H digestion in the presence of oligonucleotides, the mixture was incubated with 10 μl (or 25 μl if a higher concentration of heparin is needed) of 10 μg/μl heparin for 10 min on ice. The volume of the mixture was increased to 200 μl by adding 1 × gradient buffer and the mixture was sedimented through a glycerol gradient by ultracentrifugation as described previously (49).
Northern Hybridization and Spliceosome Conversion Assay
To analyze the snRNAs by Northern hybridization, “cold” splicing reaction mixtures (10 μl) or gradient fractions (200 μl) were mixed with 250 μl of a stop buffer containing 25 μg/ml glycogen (49). RNA was recovered from the mixture by phenol/chloroform extraction and ethanol precipitation and separated on 2% agarose formaldehyde gels or 5% polyacrylamide urea gels (49). After gel electrophoresis, RNA was transferred onto a Biotrans nylon membrane (ICN) by using PosiBlot pressure blotter (Stratagene). The preparation of 32P-labeled probes and the hybridization procedure have been described previously (49). A plasmid, pBluescript-1A, containing both U1 and actin sequences was used to prepare U1 plus pre-mRNA probe. The pBluescript-1A plasmid was constructed by inserting a 0.5 kb BamHI-SacII fragment containing the actin intron and flanking exon sequences from plasmid SP6-actin (23) into the U1 plasmid, pBluescriptIISK-U1 (42). The splicing activity of the gradient-isolated spliceosome was assayed by the spliceosome conversion assay as described previously (49). The purification of PRP2 protein has been described (16), whereas the purification of the HP factor from wild-type yeast extracts will be described elsewhere (Kim and Lin, unpublished). Quantitation of autoradiograms was done by using the Speedlight Gel Documentation System (Lightools Research, Carlsbad, CA) and IPLab Gel Program (Signal Analytics Corporation, Vienna, VA).
RESULTS
Probing snRNAs With Oligonucleotide-Directed RNase H Digestion
Oligonucleotide-directed RNase H digestion has been used to degrade endogenous U1, U2, and U6 snRNAs from wild-type yeast extracts; the oligonucleotide-treated extracts cannot support the assembly of a fully active spliceosome and are therefore splicing incompetent (7,20,25,32). To test the susceptibility of U1, U6, and U2 snRNAs in prp2 temperature-sensitive (Ts) extracts before and after spliceosome formation, we chose three oligonucleotides similar or identical to those used previously: oligo 1s19 (complementary to the 5′ end of U1 that base-pairs with the 5′ splice site), oligo 6c27 (complementary to the central domain of U6 that interacts with the 5′ splice site), and oligo 2b l9 (complementary to the internal region of U2 that base-pairs with the branch point) (Table 1). We hypothesized that, after the assembly of the prp2Δ spliceosome, the 5′ end of U1 will still be susceptible to RNase H digestion, whereas the 5′ splice site-interacting region of U6 will become resistant to RNase H if the 5′ splice site of the pre-mRNA in the prp2Δ spliceosome interacts with U6 instead of U1 snRNA.
First, each of the three oligonucleotides with the amount needed for inactivation (Table 1) was incubated with the prp2 Ts extracts to allow the cleavage of the corresponding snRNA by the endogenous RNase H. The splicing activity of the digested extract was assayed by adding pre-mRNA, ATP, magnesium, and the wild-type PRP2 protein (Fig. 1A, lanes 2–4). As expected, splicing was inactive in all three cases. Preincubation of prp2 Ts extracts with water or a noncomplementary oligonucleotide did not prevent complementation by the PRP2 protein (Fig. 1A, lane 1; data not shown). To verify cleavage, total RNA was extracted from reaction mixtures containing unlabeled pre-mRNA and was subjected to Northern analysis. The RNA samples were separated on polyacrylamide gels (for probing U1 or U6) or agarose gel (for probing U2), blotted, and hybridized with U1, U2, or U6 probe. As shown in Fig. 1B, U1 snRNA was cleaved in the presence of oligo 1s19 (Fig. 1B, lane 2). The truncated U1 is very stable because its amount is comparable to the uncut control (cf. lanes 2 and 1 in Fig. 1B). The cleavage sites were mapped by primer extension to the 3′ end of the seventh and eighth bases of U1 snRNA (Fig. 1C, lane 7). It appeared that the 9th to the 19th nucleotides of U1 were inaccessible to RNase H cleavage, even though oligo 1s19 could potentially form duplex with the first 19 bases of U1 snRNA. The mapping data are important in demonstrating that virtually all base-pairing capacities of U1 to the 5′ end of the intron were destroyed by the oligonucleotide. U2 snRNA was cleaved in the presence of oligo 2b19 (Fig. 1B, lane 6) and the truncated U2 was less stable than the truncated U1 snRNA. The cleavage sites on U2 were not mapped. Unlike the case of U1 or U2, U6 snRNA was degraded in the presence of oligo 6c27 and no sizable fragments could be detected (Fig. 1B, lane 10; also see Fig. 3) (7). Note that a small amount of intact U1, U2, or U6 snRNA could be detected after RNase H digestion (Fig. 1B, lanes 2, 6, and 10), but evidently these low levels of snRNAs were not sufficient for the splicing process (Fig. 1A, lanes 2-4). Thus, the region of interest in U1, U2, or U6 snRNA is susceptible to oligonucleotide-directed RNase H digestion before the spliceosome assembly.
FIG. 1.
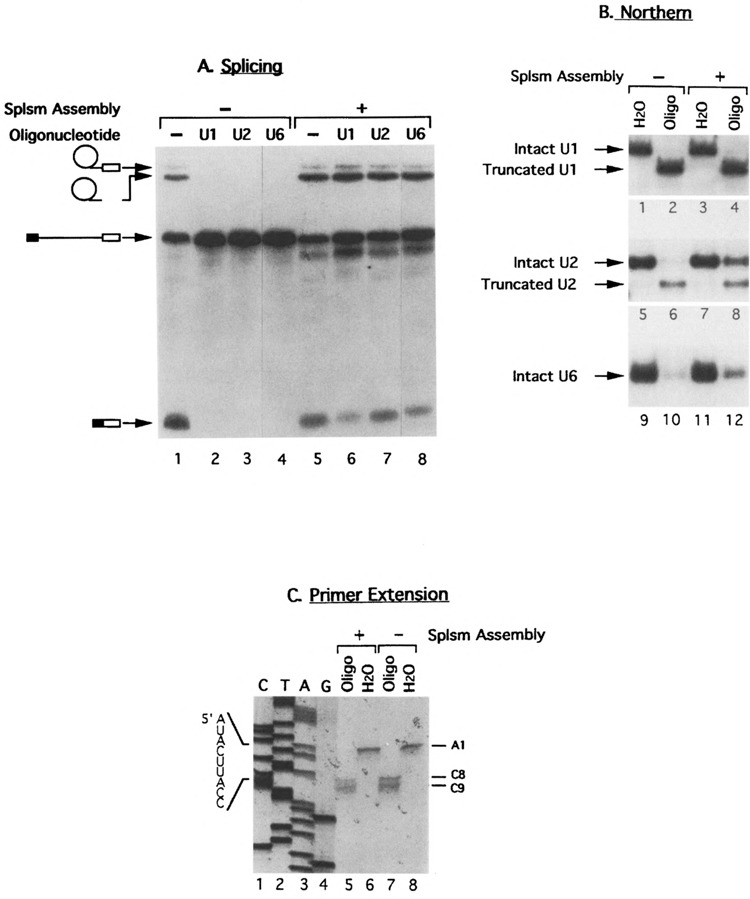
Cleaving snRNAs in extracts before or after spliceosome assembly by oligonucleotide-directed RNase H digestion. Three oligonucleotides, 1s19, 2b19, and 6c27, complementary to U1, U2, and U6 snRNA, respectively, were used to target specific snRNA cleavage by the endogenous RNase H. For cleaving before spliceosome formation, oligonucleotides were incubated with the prp2 Ts extracts prior to the addition of pre-mRNA substrate; for cutting afterwards, spliceosomes containing pre-mRNA were allowed to be assembled in the Ts extracts followed by the addition of oligonucleotides. Purified wild-type PRP2 protein was added last to complete the splicing reactions. In all figures, lanes of gels are numbered at the bottom of the autoradiograms. (A) Splicing activity assays of reactions containing 32P-labeled pre-mRNA substrates on denaturing polyacrylamide gels. Lanes 1–4: oligonucleotides were added prior to the assembly of the spliceosome; lanes 5–8: oligonucleotides were added after spliceosome assembly. Water, instead of oligonucleotide, was used in samples in lanes 1 and 5 as control. (B) Northern analysis of the cleavage events. RNA was recovered from reaction mixtures containing unlabeled pre-mRNA, separated on three gels (polyacrylamide gels for U1 and U6, agarose gel for U2), and probed with U1 (lanes 1–4), U2 (lanes 5–8), and U6 (lanes 9–12). No oligonucleotides were used in samples in odd-number lanes; samples in even-number lanes were treated with 1s19 (lanes 2 and 4), 2b19 (lanes 6 and 8), or 6c27 (lanes 10 and 12). (C) Mapping the cleavage sites on U1 snRNA by primer extension. RNA was recovered from samples treated with water (lanes 6 and 8) or with oligonucleotide 1s19 (lanes 5 and 7) and was reverse-transcribed with a 32P-labeled oligonucleotide complementary to the 74th to 92nd bases of U1 (Table 1). Dideoxy sequencing reactions on U1 DNA were used as size markers (lanes 1–4).
FIG. 3.
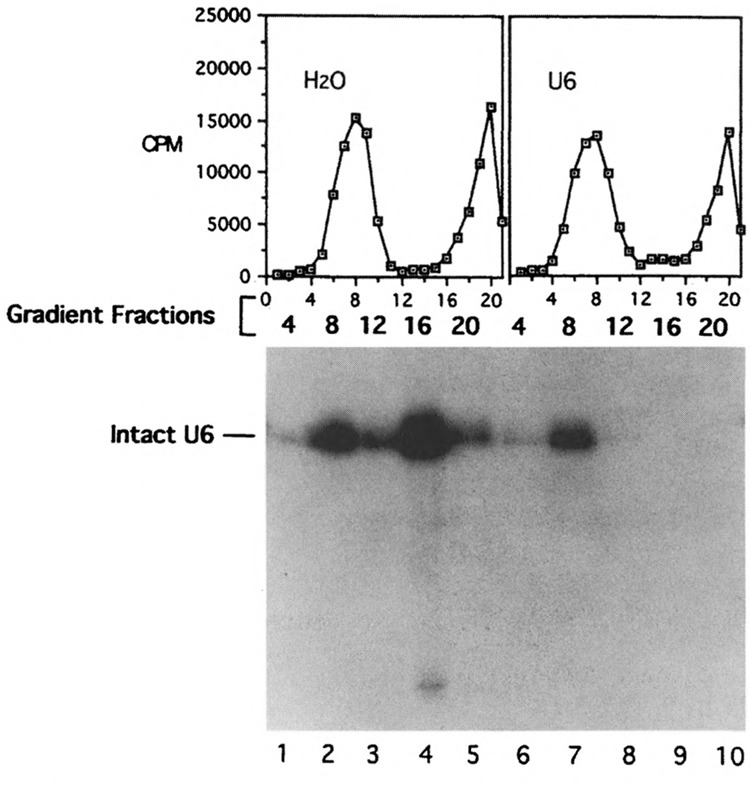
Gradient and Northern analyses of U6 snRNA after oligonucleotide-directed RNase H digestion. Oligonucleotide 6c27 was added to initiate RNase H digestion after the assembly of the spliceosome in prp2 Ts extracts. The “hot” gradient profiles and the Northern hybridization using “cold” fractions probed with U6 were displayed as in Fig. 2.
We then did a different experiment by adding each of the three oligonucleotides to the reaction mixture after the prp2Δ spliceosome was allowed to be assembled in prp2 Ts extracts. Interestingly, in all three cases splicing was able to resume when reaction mixtures were supplemented with PRP2 (Fig. 1A, lanes 6–8). These results indicated that either the snRNAs were protected from RNase H cleavage or they were no longer required after the assembly of the spliceosome. To monitor the cleavage events, total RNA was extracted from the reaction mixture and subjected to Northern hybridization. As shown in Fig. 1B, adding oligo 1s19 after the assembly of the prp2Δ spliceosome induced the truncation of U1 snRNA (lane 4) almost to the same extent as adding the oligonucleotide prior to spliceosome assembly (lane 2). The cleavage sites were, again, mapped by primer extension to the same positions (i.e., the 3′ end of the seventh and eighth bases of U1 snRNA) (Fig. 1C, lane 5). The fact that virtually all U1 molecules were cleaved to lose intron base-pairing ability favored the notion that U1 snRNA was no longer interacting with the 5′ splice site after the assembly of the prp2Δ spliceosome. The cleavage patterns for U2 and U6 were quite different from that of U1: substantial amounts of U2 and U6 snRNAs remained intact when RNase H digestion was initiated after spliceosome assembly (Fig. 1B, lanes 8 and 12). These results strongly suggest that, when U2 and U6 snRNAs were incorporated into the spliceosome, the U2 sequence involved in base-pairing with the branch point and the U6 sequence involved in interacting with the 5′ splice site were protected from oligonucleotide or RNase H.
Association of U2 and U6, Not U1, With the Spliceosome
To demonstrate that intact U2 and U6 snRNAs were indeed associated with the prp2Δ spliceosome as well as to test whether truncated U1 remained associated with the spliceosome, it is necessary to separate the spliceosome from free snRNPs. Several protocols including affinity chromatography (11,34), native gel electrophoresis (17,27), and gradient sedimentation (2,10) have been used for the separation of splicing complexes. Here we chose to use gradient sedimentation over the other methods because the spliceosome isolated from a glycerol gradient consistently retained its splicing potential, which could be subsequently activated in a spliceosome conversion reaction (15,22,49). Thus, the gradient isolation method can be used to simultaneously analyze the snRNA content and the splicing activity of the spliceosome.
Because it was shown previously that, in the presence of heparin, U1 snRNA was not associated with spliceosomes during native gel electrophoresis (4,18,27), heparin was added to the reaction mixture right before the gradient sedimentation, which was not used in the previous protocol (15,22,49). The heparin treatment did not affect the splicing potential of the isolated spliceosome except that additional protein factors were needed for the subsequent spliceosome activity assay (see below). The prp2Δ spliceosome was assembled with the same amount of 32P-labeled or unlabeled pre-mRNA in prp2 Ts extracts followed by the addition of an snRNA-specific oligonucleotide to initiate RNase H cleavage. The mixture was further incubated in the presence of heparin and then sedimented through a glycerol gradient. The reaction using 32P-labeled pre-mRNA provided “hot” spliceosomes as marker for sedimentation as well as the source of radioactive spliceosomes for the transesterification assay. The glycerol gradient fractions derived from reactions containing unlabeled pre-mRNA (the “cold” samples) would be used for Northern analysis. Because the two sets of reactions were carried out in parallel under the same conditions, the results of these analyses were directly comparable.
The Northern analysis of the gradient fractions derived from oligo 2b19-digested “cold” sample showed that the intact U2 snRNA sedimented with the spliceosome (Fig. 2, fraction 8 of the right gradient, lane 7), whereas the truncated U2 snRNAs were found in the less dense fractions (Fig. 2, fraction 16 of the right gradient, lane 9). Similar results were seen in the case of 6c27-digested samples: intact U6 snRNA was cosedimented with the spliceosome (Fig. 3, fraction 8 of the right gradient, lane 7), whereas no U6 snRNA was found in the less dense fraction (Fig. 3, fraction 16 of the right gradient, lane 9). These results indicated that U2 and U6 snRNAs in nonspliceosomal forms are accessible to oligonucleotides and RNase H; however, they become inaccessible after being incorporated into the spliceosome. Because the prp2Δ spliceosome contains pre-mRNA without the splicing intermediates or products, the protection of U2 and U6 snRNAs must occur prior to the ATP hydrolysis by PRP2 and the transesterification reactions.
FIG. 2.
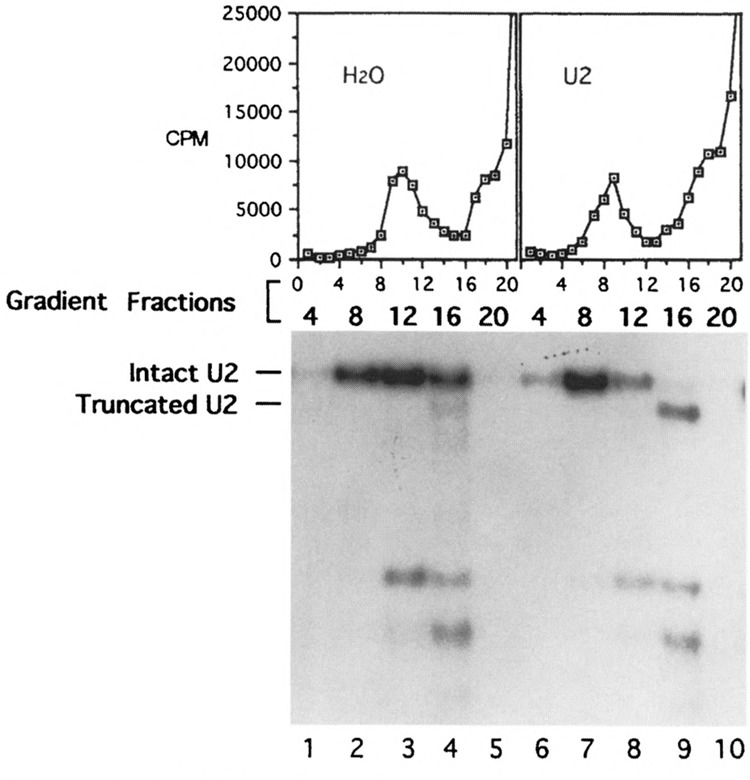
Gradient and Northern analyses of U2 snRNA after oligonucleotide-directed RNase H digestion. Oligonucleotide 2b19 was added to initiate RNase H digestion after the assembly of the spliceosome in prp2 Ts extracts. The reaction mixtures were treated with heparin at 0.5 μg/μl, then sedimented through glycerol gradients. The graphs show the gradient profiles (fractions with smaller numbers were closer to the bottom of the gradient) of the control reaction containing 32P-labeled pre-mRNA without oligonucleotide (H2O) and the 2b19-treated, radioactive sample (U2). Total RNA was isolated from “cold” gradient fractions 4, 8, 12, 16, and 20 (water control, lanes 1–5; 2b19-treated, lanes 6–10), separated on an agarose gel, and probed with U2. Fraction numbers are given on the top of the autoradiogram and lanes are numbered at the bottom.
Three sets of reactions were carried out in the U1 experiments and their gradient profiles were shown in Fig. 4: no oligonucleotides added (Fig. 4, left), oligo 1s27 (Fig. 4, middle; Table 1), and oligo 1s19 (Fig. 4, right). The heparin concentration used in the third reaction was 2.5-fold higher that the other samples. The Northern analysis of the gradient fractions using the U1 probe (Fig. 4, middle autoradiogram) showed that the majority of the U1 snRNA was detected in fractions 13 and 17 (lanes 4, 5, 9, 10, 14, and 15) and that only a small amount of U1 snRNA was detected in the main spliceosome fraction, #9 (lanes 3, 8, and 13), regardless of the presence of the oligonucleotide. In the presence of a higher concentration of heparin (2.5-fold), only a trace amount of U1 snRNA was detected in the main spliceosome fraction, #9 (Fig. 4, middle autoradiogram, lane 13). It appeared that the higher the concentration of heparin the lower the amount of U1 snRNA cosedimented with the spliceosome. In any event, these results indicate that the 5′ end of the U1 snRNA is still sensitive to RNase H digestion after the assembly of the spliceosome in prp2 Ts extracts; furthermore, the U1 snRNA is not an integral component of the prp2Δ spliceosome.
Taken together, the Northern analysis of the gradient samples showed that the entire U1 snRNA was no longer associated with the prp2Δ spliceosome, especially in the presence of heparin, whereas U2 and U6 snRNAs remained as integral components. Furthermore, the U1 to U6 transition must have occurred prior to the PRP2-dependent step.
Transesterification Occurs in Spliceosome Lacking U1
To test if the spliceosomes isolated from the oligonucleotide-digested, heparin-treated samples are bona fide spliceosomes, the main spliceosomal fractions from the 32P-labeled pre-mRNA-containing samples shown in Figs. 2, 3, and 4 were assayed for splicing activity by supplementing with ATP and extrinsic splicing factors. Previously we have shown that the prp2Δ spliceosome isolated in the absence of heparin needed only PRP2 for activity (15); however, heparin-treated spliceosomes required additional protein factors (Kim and Lin, unpublished). These protein factors, called HP or bn (22), were apparently removed from spliceosomal fractions by the heparin treatment and needed to be reconstituted (Kim and Lin, unpublished). As shown in Figs. 5 and 6, splicing was restored in both RNase H- and heparin-treated spliceosomal fractions upon addition of ATP and wild-type extracts (Fig. 5, lanes 2, 5, 8, and 11; Fig. 6, lanes 2, 5, and 8). Note that the amount of extract used (1 μl in 20 μ1 of reaction) was not sufficient to initiate splicing without the preassembly of the spliceosome (22,49). Thus, the gradient-isolated spliceosomes were fully functional because both Reaction 1 and Reaction 2 could efficiently occur in the assembled spliceosome. When the spliceosomal fractions were incubated with ATP and purified extrinsic splicing factors PRP2 and HP, the pre-mRNA in the fractions was converted to the splicing intermediates (i.e., the lariat intron-exon 2 and linear exon 1) (Fig. 5, lanes 3, 6, 9, and 12; Fig. 6, lanes 3,6, and 9). It is important to note that the preparations containing the extrinsic splicing factors were free of U1 contamination as indicated by Northern hybridization (Fig. 4, lanes 16 and 17).
FIG. 5.
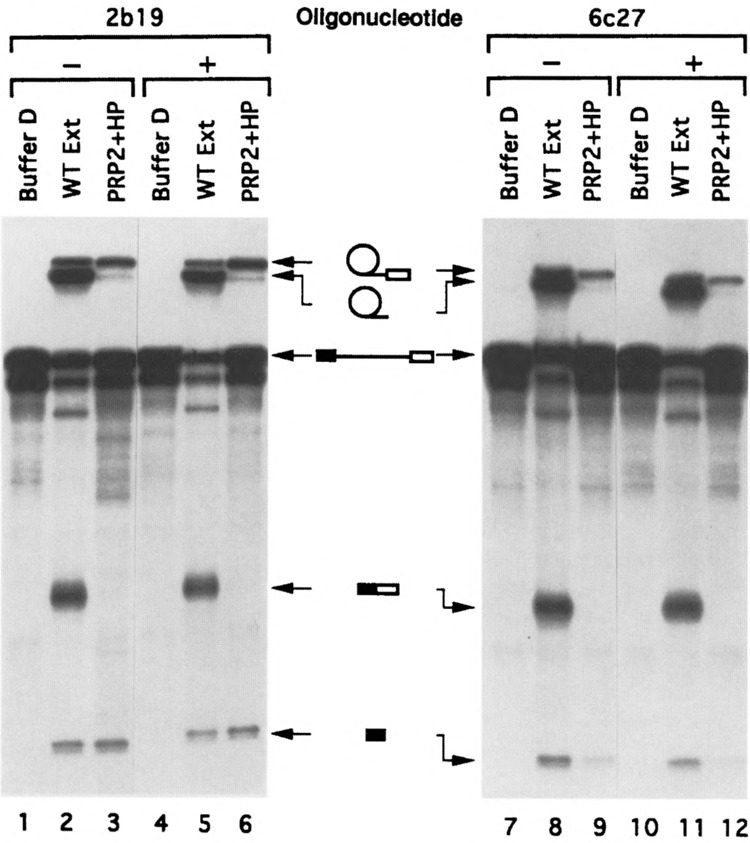
Splicing activity assays of the gradient-purified spliceosomes treated with U2 (2b19) or U6 (6c27) oligonucleotide as shown in Figs. 2 and 3. The “hot” spliceosome-containing fractions were incubated with Buffer D (lanes 1, 4, 7, 10), wild-type extracts (lanes 2, 5, 8, 11), or two extrinsic splicing factors, PRP2 and HP (lanes 3, 6, 9, 12) under splicing conditions. After the incubation RNA was analyzed on sequencing gels followed by autoradiography. Lanes 1–3 and 7–9: water control spliceosomes; lanes 4–6: 2b19-digested spliceosomes; lanes 10–12: 6c27-digested spliceosomes.
To demonstrate that U1 snRNA was not involved in the catalytic reactions that occurred in the gradient-isolated spliceosome, we estimated the “convertible” portion of the pre-mRNA in the spliceosome fraction (Fig. 6) and the ratio of U1 to the pre-mRNA in the same fraction (Fig. 4). First, quantitation of the autoradiogram shown in Fig. 6 indicated that more than 65% of the pre-mRNA in the heparin-treated spliceosomal fractions underwent Reaction 1 to produce the splicing intermediates when ATP and extrinsic protein factors were supplemented. Second, the blot containing the U1 oligonucleotide-treated samples was stripped and reprobed (Fig. 4). When probed with actin DNA, the pre-mRNA was detected in spliceosomal fractions (Fig. 4, lanes 3, 8, and 13 of the top autoradiogram). More importantly, the same blot was reprobed with 32P-labeled plasmid pBluescript-1A, which contains both U1 and actin DNA to directly compare the amount of U1 snRNA and actin pre-mRNA substrate in the gradient fractions (Fig. 4, bottom autoradiogram). Using labeled pBluescript-1A plasmid would ensure that the amount and the specific activity of U1 and actin pre-mRNA probes were as close as possible during hybridization. Note that the lengths of the U1 snRNA and the pre-mRNA substrate, 569 bases and 566 bases, respectively, are quite close and cannot be separated on the gel. By quantitating and comparing the auto-radiograms derived from the actin-probed, U1-probed, and the U1/actin double-probed Northern blots (Fig. 4, top, middle, and bottom autoradiograms, respectively), we found that less than 4% of the spliceosome (represented by the pre-mRNA) in the 1s19-treated sample (fraction 9, lane 13) could possibly contain U1 snRNA (Fig. 4). Because the ratio of U1 snRNA to the pre-mRNA in the 1s19-digested spliceosome fraction (Fig. 4, fraction 9) was less than 1:25 and because U1 was absent from the complementing protein preparations, the U1 snRNA (either full-length or truncated) could not be responsible for the conversion rate of more than 65% observed in that spliceosome preparation (Fig. 6, lane 9). Moreover, heparin-treated prp2A spliceosomes could undergo both transesterification reactions to generate spliced exons and excised intron when PRP16, the ATPase required for Reaction 2 (39), was also added (Kim and Lin, unpublished). Thus, it was very unlikely that the spliceosome underwent aberrant splicing to form dead-end products under these “U1-less” conditions. In sum, we conclude that these experiments provide direct biochemical evidence that U1 snRNA does not participate in the catalytic phase of pre-mRNA splicing in yeast.
DISCUSSION
Cleaving U1, U2, or U6 snRNAs by RNase H with specific oligonucleotides prior to the assembly of the spliceosome abolishes the splicing activity of yeast extracts, whereas adding the same oligonucleotides after the spliceosome has been assembled has little effect on splicing. Northern hybridization and gradient sedimentation analyses demonstrated that U2 and U6 are protected in the spliceosome, whereas U1 is not. Treating the reaction mixtures with heparin effectively removed almost all U1 snRNAs from the spliceosome-containing gradient fractions. The gradient-purified spliceosome can efficiently carry out the first transesterification reaction upon the addition of ATP and extrinsic splicing protein factors free of detectable U1 snRNA. Thus, the study presented here indicates that the U1 to U6 transition occurs prior to the binding of PRP2 to the spliceosome and that U1 snRNA is not required for the catalytic phase of pre-mRNA splicing.
Because PRP2 is unlikely to be responsible for replacing U1 with U6 at the 5′ splice site, other RNA helicases might be involved. There are two other DEAH proteins required for splicing: PRP16 (39) and PRP22 (5); however, they are unlikely candidates because both act after the PRP2 step. Other PRP proteins of the DEAD family could potentially be responsible for the transition of base-pairing from U1 5′ splice site to U6 5′ splice site. It is also possible that the transition of U1 to U6 is induced by factors other than RNA helicases, such as RNA binding proteins or RNA annealing proteins.
The analysis of the snRNAs in the prp2Δ spliceosome in this article and a previous article (49) demonstrates that U2 interaction with the branch point, U6 interaction with the 5′ splice site, the loss of U4, and the dissociation of U1 all occur prior to the ATP-dependent activation of the spliceosome by PRP2. What would then be the role of PRP2 in splicing? Perhaps PRP2 does not participate in the gross rearrangement of RNA base-pairing but instead promotes specific RNA-RNA or RNA-protein interactions that are critical to the catalytic activity of the spliceosome. In fact, recently we have obtained evidence indicating that the ATP hydrolysis by PRP2 causes the protection of a single nucleotide in U6 snRNA against chemical modification (Yean and Lin, unpublished). Therefore, we suspect that PRP2 may not be a classical RNA helicase (16).
Although this study provides the first direct biochemical evidence that U1 is not involved in the catalytic phase of yeast splicing, several lines of evidence have accumulated from other studies suggesting this possibility. By using oligonucleotides containing the 5′ splice site sequence in HeLa extracts, Konforti et al. showed that later stages of spliceosome assembly require disruption of the interaction between the 5′ splice site and U1 snRNA (19). Moreover, several studies indicate that the 5′ splice site interacts with U5 and U6 snRNAs in the fully assembled spliceosome (14,21,36,37,43,46,48). More importantly, two recent reports showed that, in the presence of high levels of SR proteins (a group of splicing factors containing arginine-serine dipeptide repeats), pre-mRNA splicing can occur in HeLa extracts that were depleted or inactivated of U1 snRNA (6,44). In those studies, U1 is either removed from the extract by binding to anti-U1 oligonucleotides immobilized on a solid support or is inactivated by sequestering the 5′ splice site interacting region of U1 with an anti-U1 oligonucleotide. Addition of SR proteins complements these U1-inactivated extracts. Interestingly, certain cryptic 5′ splice sites were also used for splicing in the SR protein-complemented reactions, whereas these cryptic sites are mostly silent in reactions with intact U1. Although in the chemical sense U1 snRNA is not essential for the transesterification reactions occurring under these in vitro conditions, U1 must play an important role in discriminating potential 5’ splice sites within a pre-mRNA molecule. Together with our results, these in vitro elimination experiments demonstrated that U1 snRNA cannot be an essential part of the catalytic center in yeast or mammalian spliceosome.
Among the five essential snRNAs in pre-mRNA splicing, U1 and U4 apparently dissociate from or become weakly associated with the spliceosome prior to the first transesterification reaction. Although it is still unclear as to whether any of the U1 or U4 snRNP proteins remains on the spliceosome during splicing, these observations impose an interesting problem for the regeneration of these snRNPs where multiple splicing cycles are likely to occur in vivo. It is of interest to note that some yeast PRP proteins may play a role in the regeneration of snRNPs at the end of a splicing event (9,13).
ACKNOWLEDGEMENTS
We thank Dr. John Abelson for providing oligonucleotide 6c27 as well as the U2 and U6 plasmids, and Dr. Paul Siliciano for the U1 plasmid. We also thank Drs. Douglas Black, Timothy Nilsen, and Joan Steitz for suggestions and comments, and Dr. John Termini for critical reading of the manuscript. S.-L.Y. thanks Sahn-Ho Kim for providing PRP2 and HP. This work was supported by grant GM40639 from the National Institutes of Health to R.-J.L.
REFERENCES
- 1. Abelson J. RNA processing and the intervening sequence problem. Annu Rev. Biochem. 48:1035–1069; 1979. [DOI] [PubMed] [Google Scholar]
- 2. Brody E.; Abelson J. The spliceosome: Yeast pre-messenger RNA associates with a 40S complex in a splicing-dependent reaction. Science 228:963–967; 1985. [DOI] [PubMed] [Google Scholar]
- 3. Cech T. R. The generality of self-splicing RNA: Relationship to nuclear mRNA splicing. Cell 44:207–210; 1986. [DOI] [PubMed] [Google Scholar]
- 4. Cheng S. C.; Abelson J. Spliceosome assembly in yeast. Genes Dev. 1:1014–1027; 1987. [DOI] [PubMed] [Google Scholar]
- 5. Company M.; Arenas J.; Abelson J. Requirement of the RNA helicase-like protein PRP22 for release of messenger RNA from spliceosomes. Nature 349:487–493; 1991. [DOI] [PubMed] [Google Scholar]
- 6. Crispino J. D.; Blencowe B. J.; Sharp P. A. Complementation by SR proteins of pre-mRNA splicing reactions depleted of U1 snRNP. Science 265:1866–1869; 1994. [DOI] [PubMed] [Google Scholar]
- 7. Fabrizio P.; McPheeters D. S.; Abelson J. In vitro assembly of yeast U6 snRNP: A functional assay. Genes Dev. 3:2137–2150; 1989. [DOI] [PubMed] [Google Scholar]
- 8. Fuller-Pace F. V. RNA helicases: Modulators of RNA structure. Trends Cell Biol. 4:271–274; 1994. [DOI] [PubMed] [Google Scholar]
- 9. Ghetti A.; Company M.; Abelson J. Specificity of Prp24 binding to RNA: A role for Prp24 in the dynamic interaction of U4 and U6 snRNAs. RNA 1:132–145; 1995. [PMC free article] [PubMed] [Google Scholar]
- 10. Grabowski P. J.; Seiler S. R.; Sharp P. A. A multi-component complex is involved in the splicing of messenger RNA precursors. Cell 42:345–353; 1985. [DOI] [PubMed] [Google Scholar]
- 11. Grabowski P. J.; Sharp P. A. Affinity chromatography of splicing complexes: U2, U5, and U4 + U6 small nuclear ribonucleoprotein particles in the spliceosome. Science 233:1294–1299; 1986. [DOI] [PubMed] [Google Scholar]
- 12. Guthrie C. Messenger RNA splicing in yeast: Clues to why the spliceosome is a ribonucleoprotein. Science 253:157–163; 1991. [DOI] [PubMed] [Google Scholar]
- 13. Jandrositz A.; Guthrie C. Evidence for a Prp24 binding site in U6 snRNA and in a putative intermediate in the annealing of U6 and U4 snRNAs. EMBO J. 14:820–832; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kandels-Lewis S.; Seraphin B. Involvement of U6 snRNA in 5′ splice site selection. Science 262:2035–269; 1993. [DOI] [PubMed] [Google Scholar]
- 15. Kim S. H.; Lin R. J. Pre-mRNA splicing within an assembled yeast spliceosome requires an RNA-dependent ATPase and ATP hydrolysis. Proc. Natl. Acad. Sci. USA 90:888–892; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim S. H.; Smith J.; Claude A.; Lin R. J. The purified yeast pre-mRNA splicing factor PRP2 is an RNA-dependent NTPase. EMBO J. 11:2319–2326; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Konarska M. M. Analysis of splicing complexes and small nuclear ribonucleoprotein particles by native gel electrophoresis. Methods Enzymol. 180:442–453; 1989. [DOI] [PubMed] [Google Scholar]
- 18. Konarska M. M.; Sharp P. A. Electrophoretic separation of complexes involved in the splicing of precursors to mRNAs. Cell 46:845–855; 1986. [DOI] [PubMed] [Google Scholar]
- 19. Konforti B. B.; Koziolkiewicz M. J.; Konarska M. M. Disruption of base pairing between the 5′ splice site and the 5′ end of U1 snRNA is required for spliceosome assembly. Cell 75:863–873; 1993. [DOI] [PubMed] [Google Scholar]
- 20. Kretzner L.; Rymond B. C.; Rosbash M. S. cerevisiae U1 RNA is large and has limited primary sequence homology to metazoan U1 snRNA. Cell 50:593–602; 1987. [DOI] [PubMed] [Google Scholar]
- 21. Lesser C. F.; Guthrie C. Mutations in U6 snRNA that alter splice site specificity: Implications for the active site. Science 262:1982–1988; 1993. [DOI] [PubMed] [Google Scholar]
- 22. Lin R. J.; Lustig A. J.; Abelson J. Splicing of yeast nuclear pre mRNA in vitro requires a functional 40S spliceosome and several extrinsic factors. Genes Dev. 1:7–18; 1987. [DOI] [PubMed] [Google Scholar]
- 23. Lin R. J.; Newman A. J.; Cheng S. C.; Abelson J. Yeast mRNA splicing in vitro . J. Biol. Chem. 260:14780–14792; 1985. [PubMed] [Google Scholar]
- 24. Madhani H. D.; Guthrie C. Dynamic RNA-RNA interactions in the spliceosome. Annu. Rev. Genet. 28:1–26; 1994. [DOI] [PubMed] [Google Scholar]
- 25. McPheeters D. S.; Fabrizio P.; Abelson J. In vitro reconstitution of functional yeast U2 snRNPs. Genes Dev. 3:2124–2136; 1989. [DOI] [PubMed] [Google Scholar]
- 26. Moore M. J.; Query C. C.; Sharp P. A. Splicing of precursors to mRNAs by the spliceosome. In: Gesteland R. F.; Atkins J. F., eds RNA world. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1993:303–358. [Google Scholar]
- 27. Pikielny C. W.; Rosbash M. Specific small nuclear RNAs are associated with yeast spliceosomes. Cell 45:869–877; 1986. [DOI] [PubMed] [Google Scholar]
- 28. Plumpton M.; McGarvey M.; Beggs J. D. A dominant negative mutation in the conserved RNA helicase motif ‘SAT’ causes splicing factor PRP2 to stall in spliceosomes. EMBO J. 13:879–887; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reich C. I.; VanHoy R. W.; Porter G. L.; Wise J. A. Mutations at the 3′ splice site can be suppressed by compensatory base changes in U1 snRNA in fission yeast. Cell 69:1159–1169; 1992. [DOI] [PubMed] [Google Scholar]
- 30. Rosbash M.; Harris P. K. W.; Woolford J. L.; Teem J. L. The effect of temperature-sensitive RNA mutants on the transcription products from cloned ribosomal protein genes of yeast. Cell 24:679–686; 1981. [DOI] [PubMed] [Google Scholar]
- 31. Rosbash M.; Seraphin B. Who’s on first? The U1 snRNP-5′ splice site interaction and splicing. Trends Biochem. Sci. 16:187–190; 1991. [DOI] [PubMed] [Google Scholar]
- 32. Ruby S. W.; Abelson J. An early hierarchic role of U1 small nuclear ribonucleoprotein in spliceosome assembly. Science 242:1028–1035; 1988. [DOI] [PubMed] [Google Scholar]
- 33. Ruby S. W.; Abelson J. Pre-mRNA splicing in yeast. Trends Genet. 7:79–85; 1991. [DOI] [PubMed] [Google Scholar]
- 34. Ruby S. W.; Goelz S. E.; Hostomsky Z.; Abelson J. N. Affinity chromatography with biotinylated RNAs. Methods Enzymol. 181:97–121; 1990. [DOI] [PubMed] [Google Scholar]
- 35. Rymond B. C.; Rosbash M. Yeast pre-mRNA splicing. In: Broach J. R.; Pringle J. R.; Jones E. W., eds. The molecular and celkar biology of the yeast Saccharomyces: Gene expression. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992:143–192. [Google Scholar]
- 36. Sawa H.; Abelson J. Evidence for a base-pairing interaction between U6 small nuclear RNA and 5′ splice site during the splicing reaction in yeast. Proc. Natl. Acad. Sci. USA 89:11269–11273; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sawa H.; Shimura Y. Association of U6 snRNA with the 5′-splice site region of pre-mRNA in the spliceosome. Genes Dev. 6:244–254; 1992. [DOI] [PubMed] [Google Scholar]
- 38. Schmid S. R.; Linder P. D-E-A-D protein family of putative RNA helicases. Mol. Microbiol. 6:283–291; 1992. [DOI] [PubMed] [Google Scholar]
- 39. Schwer B.; Guthrie C. PRP16 is an RNA-dependent ATPase that interacts transiently with the spliceosome. Nature 349:494–499; 1991. [DOI] [PubMed] [Google Scholar]
- 40. Seraphin B.; Kandels-Lewis S. 3′ splice site recognition in S. cerevisiae does not require base pairing with U1 snRNA. Cell 73:803–812; 1993. [DOI] [PubMed] [Google Scholar]
- 41. Sharp P. A. Split genes and RNA splicing. Cell 77:805–815; 1994. [DOI] [PubMed] [Google Scholar]
- 42. Siliciano P. G.; Jones M. H.; Guthrie C. Saccharomyces cerevisiae has a U1-like small nuclear RNA with unexpected properties. Science 237:1484–1487; 1987. [DOI] [PubMed] [Google Scholar]
- 43. Sontheimer E. J.; Steitz J. A. The U5 and U6 small nuclear RNAs as active site components of the spliceosome. Science 262:1989–1996; 1993. [DOI] [PubMed] [Google Scholar]
- 44. Tarn W. Y.; Steitz J. A. SR proteins can compensate for the loss of U1 snRNP functions in vitro. Genes Dev. 8:2704–2717; 1994. [DOI] [PubMed] [Google Scholar]
- 45. Wassarman D. A.; Steitz J. A. RNA splicing. Alive with DEAD proteins. Nature 349:463–464; 1991. [DOI] [PubMed] [Google Scholar]
- 46. Wassarman D. A.; Steitz J. A. Interactions of small nuclear RNA’s with precursor messenger RNA during in vitro splicing. Science 257:1918–1925; 1992. [DOI] [PubMed] [Google Scholar]
- 47. Woolford J. L. Nuclear pre-mRNA splicing in yeast. Yeast 5:439–457; 1989. [DOI] [PubMed] [Google Scholar]
- 48. Wyatt J. R.; Sontheimer E. J.; Steitz J. A. Site- specific cross-linking of mammalian U5 snRNP to the 5′ splice site before the first step of pre-mRNA splicing. Genes Dev. 6:2542–2553; 1992. [DOI] [PubMed] [Google Scholar]
- 49. Yean S. L.; Lin R. J. U4 small nuclear RNA dissociates from a yeast spliceosome and does not participate in the subsequent splicing reaction. Mol Cell Biol. 11:5571–5577; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]