

Abstract
Posttranslational modifications of histones in chromatin are emerging as an important mechanism in the regulation of gene expression. Changes in histone acetylation levels occur during many nuclear processes such as replication, transcriptional silencing, and activation. Histone acetylation levels represent the result of a dynamic equilibrium between competing histone deacetylase(s) and histone acetylase(s). We have used two new specific inhibitors of histone deacetylase, trichostatin A (TSA) and trapoxin (TPX), to probe the effect of histone hyperacetylation on gene expression. We confirm that both drugs block histone deacetylase activity and have no detectable effects on histone acetylation rates in human lymphoid cell lines. Treatment with either TSA or TPX results in the transcriptional activation of HIV-1 gene expression in latently infected cell lines. In contrast, TSA and TPX cause a rapid decrease in c-myc gene expression and no change in the expression of the gene for glycer aldehyde-3-phosphate dehydrogenase (GAPDH). Using differential display to compare the differences in gene expression between untreated cells and cells treated with TSA, we found that the expression of ∼2% of cellular genes (8 genes out of ∼340 examined) changes in response to TSA treatment. These results demonstrate that the transcriptional regulation of a restricted set of cellular genes is uniquely sensitive to the degree of histone acetylation in chromatin.
Keywords: Trichostatin, Trapoxin, HIV-1, Chromatin, Differential display, myc, Histone, Acetylation
CHROMATIN is emerging as an important participant in the transcriptional regulation of specific genes. The nucleosome core, the basic sub-unit of chromatin, is composed of 146 bp of DNA wrapped around a multiprotein particle, the histone octamer, which is itself composed of two copies of each of the four histones: H2A, H2B, H3, and H4. Histone proteins contain two domains: a globular carboxyl (C)-terminal domain, which forms the core of the nucleosome, and a flexible, highly basic amino (N)-terminal tail domain, located outside of the core particle and accessible to various modifying agents, such as proteases. The histone N-terminal tails are the subject of multiple posttranslational modifications including phosphorylation, methylation, acetylation, and poly-ADP-ribosylation. All four core histones are subject to reversible acetylation and this modification has been extensively studied [for recent reviews, see (7,29,41,42,46)]. Acetylation levels of histones reflect a dynamic equilibrium between competing histone acetylase(s) and histone deacetylase(s). Reversible acetylation of histones takes place on the ε-amino group of lysine residues and each additional acetyl group results in the neutralization of one positive charge on histone proteins. The functional consequences of histone acetylation are still unclear. Two broadly defined roles have been proposed for the N-terminal tails of histones (29,42), and the potential role of histone acetylation can be examined in the context of these two models. First, the histone N-terminal tails could play a structural role in which positively charges lysine residues neutralize negative charges in the DNA phosphodiester backbone and thereby stabilize the DNA/histone complex. The wrapping of the tails at the exposed surface of DNA could also impair the binding of transcription factors to DNA packaged into a nucleosome (25). An increase in acetylation in this model would result in a loosening of the DNA–histone interaction in the nucleosome, which might make it more amenable to transcription or to an unmasking of DNA regions necessary for transcription factor binding or for access for RNA polymerase (25). According to a second model, the N-terminal tails of histone interact with nuclear factors involved in nuclear processes such as DNA replication and transcriptional regulation (29,42). Modification of lysine residues would result in changes, positive or negative, in the ability of histone proteins to interact with other proteins (42). Elegant studies in yeast, using mutants in the N-terminal tails of histones, have demonstrated the validity of the main assumptions of this model (16,35).
Acetyl groups in histones demonstrate two distinct turnover rates: in the embryonic chicken erythrocyte for example, 30% of histones are stably acetylated whereas about 2% turn over rapidly (46). Evidence has been presented showing a positive correlation between the level of histone acetylation and the level of transcriptional activity in several systems [(12), reviewed in Davie (7)]. However, such correlation has not been observed in all systems (31).
All evidence accumulated thus far linking histone acetylation and nuclear processes has been correlative and, until recently, no experimental approach had permitted the study of the direct effect of histone acetylation on gene expression. Two newly isolated drugs, trichostatin A (TSA) and trapoxin (TPX), specifically inhibit histone deacetylase both in vitro and in vivo (17,47). Whereas n-butyrate, which also inhibits histone deacetylase, is used in the millimolar range concentration and therefore exhibits pleiotropic effects (20,22), both TSA and TPX exert their biological effects in the nanomolar range (17,47). Purification of histone deacetylase from a cell line selected for its resistance to TSA demonstrated that the purified enzyme was resistant to TSA in vitro (47). This cell line was found to also be resistant to TPX, indicating that both drugs exert their biological effects through a common effector, histone deacetylase. TSA also binds to the yeast enzyme, and this interaction has been used to purify the yeast histone deacetylase to homogeneity (M. Grunstein, personal communication).
Here we have used TPX and TSA to study the effect of histone hyperacetylation on gene expression. We report that histone hyperacetylation can modulate gene expression in either a positive (HIV-1 promoter), negative (c-myc), or neutral fashion (GAPDH). Using differential display to screen a large number of genes, we show that the expression of less than 5% of cellular genes is changed in response to hyperacetylation.
MATERIAL AND METHODS
Reagents
TSA was obtained from Dr. K. Sujita (Shionogi & Co., Osaka, Japan) and TPX was obtained from Dr. M. Yoshida (University of Tokyo, Tokyo, Japan).
Cell Culture
All cell lines were obtained from the AIDS Research and Reference Reagent Program (NIAID, NIH, Bethesda, MD) and grown in RPMI 1640 medium (Gibco-BRL) supplemented with 10% fetal bovine serum (HyClone), 50 U/ml of penicillin, 50 μg/ml of streptomycin, and 2 mM glutamine at 37°C in a humidified 95% air/5% CO2 atmosphere and were maintained at densities between 0.25 and 1 × 106 cells/ml (exponential growth phase).
RNase Protection Analysis
HIV-1-specific transcripts were detected by RNase protection analysis after lysis of cells in guanidine thiocyanate (11) (Lysate Ribonuclease Protection kit, USB) and hybridization with an HIV-1-specific 32P-labeled antisense riboprobe synthesized in vitro by transcription of pGEM23 (23) with SP6 polymerase. A glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specific anti-sense probe (provided with the Lysate Ribonuclease Protection kit, USB) was synthesized using the same protocol and used in the same reaction with the HIV-1 probe, c-myc-specific transcripts were detected using an antisense RNA probe obtained by in vitro transcription of pTRI-c-myc-Human (Ambion, Austin, TX).
Histone Purification and Analysis
Nuclei isolated after lysis with NP40 were acid extracted (26), histones were ethanol precipitated, resuspended in water containing 2.5% thiodiglycol, and stored at −70°C (26). Histones, quantified by measuring their absorbance at 280 nm, were separated on Triton-acetic acid-urea gels following the protocol of Dimitrov (8). Gels were fixed in 24% methanol/4.5% glacial acetic acid followed by silver staining (Rapid-Ag-Stain, ICN Radiochemicals).
In Vivo Acetylation
Jurkat cells (2 × 106 cells/ml) were incubated in the presence of 250 μCi/ml of [3H]acetate (5.31 μCi/mmol, Dupont/NEN, Boston, MA) for 30 min. Cells were centrifuged and washed with fresh medium. After centrifugation, cells were resuspended in complete medium supplemented with TNF-α (800 U/ml) or TSA (400 nM) and chased for different lengths of time under these conditions.
RNA Differential Display
Jurkat and SupT1 cells (4 × 107 cells/point) untreated or treated with TSA (400 nM) for 2, 4, and 8 h were pelleted and RNA extracted with Trizol reagent according to the recommended protocol (GIBCO, Gaithersburg, MD). Purified RNA was treated with DNAse I in the presence of RNasin (Promega, WI) for 30 min at 37°C. RNA was purified by phenol/chloroform extractions and ethanol precipitation and resuspended in RNAse-free water. For each RNA, four reactions (0.2 μg RNA/reaction) containing a different one-base anchored 3′ primer were used for reverse transcription of RNA into cDNA according to the RNAimage kit protocol (27,28) (GenHunter Corporation, MA). PCR reactions were performed using 0.25 μCi of [α-33P]dATP (2000 Ci/mmol, NE/Dupont, Boston, MA) and the HAP1 to H-AP5 primers. Amplification reactions were run in a Perkin-Elmer 4800 thermocycler (94°C 30 s, 40°C 2 min, 72°C 30 s for 35 cycles, followed by 5′ at 72°C). PCR products were separated under denaturing conditions on 6% sequencing polyacrylamide gels. After electrophoresis, gels were dried, and autoradiographic exposures were carried out with Kodak XAR-Omat films for 16 h.
RESULTS
TSA and TPX Inhibit Histone Deacetylase in Lymphoid Cell Lines
As previous reports have determined that TSA and TPX inhibit histone deacetylase in specific cell lines (17,47), we first examined the effect of both drugs on histone acetylation in human CD4+ lymphoid cell lines. Histones were purified by acid extraction following treatment of Jurkat, SupT1, ACH2, and J49 cell lines with TSA or TPX for different lengths of time. Purified histones were separated on Triton-acid-urea gels, which allow the separation of differently acetylated forms of histones on the basis of their different charges. This analysis showed a marked increase in the degree of acetylation of histone H4 as early as 2 h following addition of the drugs (Fig. 1A). By 16 h, no unacetylated H4 was detectable, as demonstrated by the disappearance of unacetylated H40 in response to both TSA and TPX (Fig. 1A). Histone lysine groups exist in a dynamic equilibrium in terms of their acetylation level. This equilibrium results from the competing influence of ace-tylases and deacetylase. Consequently, an increase in acetylation could be secondary to an increase in acetylation or to a decrease in deacetylation. To distinguish between these two possibilities, we pulsed Jurkat cells with [3H]acetate for 30 min and chased them for 0, 1, 2, or 4 h in the absence or presence of TSA or in the presence of TNF-α as an additional control. Acid-extracted nuclear proteins (Fig. 1B) and total nuclear proteins (Fig. 1C) from such treated cells were separated on denaturing polyacrylamide gels and processed for autoradiography. This analysis showed the rapid incorporation of [3H]acetate into histones (Fig. 1B,C, lanes 0). Few proteins besides histones were found to incorporate acetate during the pulse (Fig. 1C, lanes 0). During the 4-h chase, the level of acetylation of histones was found to gradually decrease in control and TNF-α-treated cells. In contrast, the level of acetylation in histones actually increased when the pulse was performed in the presence of TSA. This experiment indicates that the major effect of TSA is to inhibit the constitutive histone deacetylase. These results are similar to what has been reported in other systems and confirm the inhibition of histone deacetylase by these drugs (17,47).
FIG. 1.
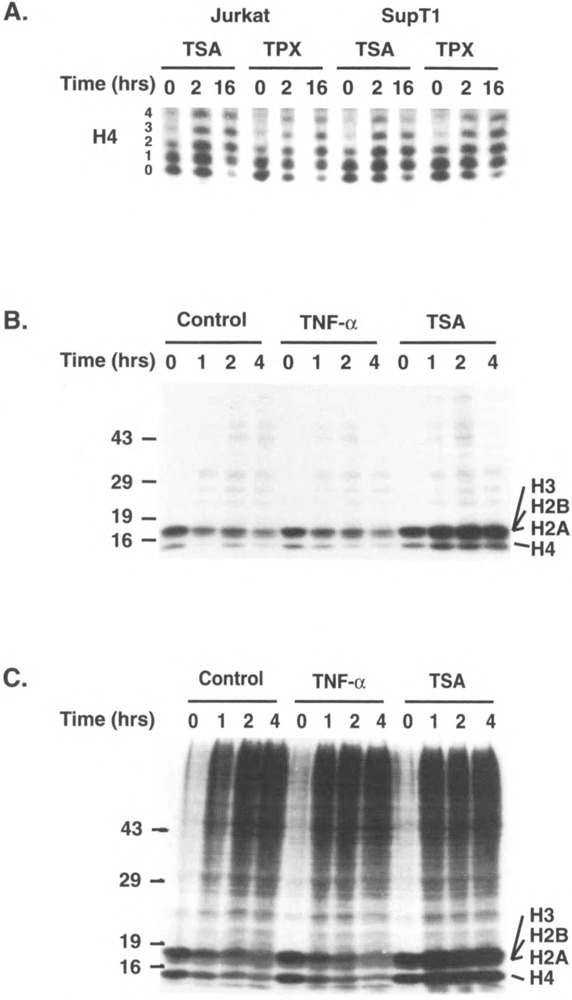
TSA and TPX inhibit histone deacetylation in lymphoid cell lines. (A) Histones were acid extracted from Jurkat or SupT1 cells treated with TSA (400 nM) or TPX (10 nM) for 0, 2, or 16 h and separated on a 15% Triton-acid-urea gel. Histones were visualized after silver staining and the portion of the gel corresponding to different acetylated isoforms of histone H4 is shown. (B, C) Jurkat cells were pulsed for 30 min with [3H]acetate (250 μCi/ml NEN), washed, and chased for 0–4 h in the presence of TNF-α (80 U/ml) or TSA (400 nM). Nuclei were either acid extracted and the purified histones loaded on a 12% polyacrylamide denaturing gel (B) or directly resuspended in Laemmli buffer and loaded on a 12% polyacrylamide denaturing gel to examine all nuclear proteins (C). Molecular weight markers in kDa are indicated on the left of the gel. The position of different histones is indicated on the right.
Modulation of HIV-1 Gene Expression by TPX and TSA
We have previously reported the presence of a chromatin transition in the HIV-1 promoter during transcriptional activation of latently infected cell lines (43–45). Latent cell lines, such as ACH2 or J49, contain a single integrated copy of the HIV-1 genome whose expression is blocked at the transcriptional level (44,45). Following treatment of these cells with phorbol esters or TNF-α, a nucleosome positioned immediately at the transcription start site, called nuc-1, is disrupted or displaced (45). The disruption of nuc-1 is independent of transcription as it occurs when the induction is performed in the presence of α-aminitin and is also independent of DNA replication as it occurs in less than an hour after stimulation in unsynchronized cell populations (45). These results are consistent with a model in which nuc-1 is responsible for the transcriptional block observed in latent cell lines either by impeding the progression of a poorly processive polymerase or by preventing the binding of a transcription factors critical for transcription initiation. To test this hypothesis, we have examined the effect of TPX and TSA on HIV-1 transcription in latent cell lines. The J49 cell line was incubated with different concentrations of TPX, TSA, or TNF-α (Fig. 2A). Eight hours following addition of the different agents, cellular RNA was prepared and virus-specific transcripts were detected using RNase protection using an HIV-1 specific antisense RNA probe (23). This probe protects two RNA fragments of 83 and 200 nt, which correspond to the 5′ and 3′ LTR, respectively (23). An antisense probe corresponding to the GAPDH gene was included as an internal standard in the hybridization reaction. This experiment showed that both TSA and TPX cause an increase in viral-specific transcripts at doses of TPX of 10 nM and of TSA of 100 nM (Fig. 2A), the same doses necessary to affect a change in histone acetylation levels (data not shown). In separate experiments, we showed that this increase in gene expression takes place at the transcriptional level as it was completely suppressed when the induction was conducted in the presence of α-amanitin used at concentrations known to inhibit RNA polymerase II elongation (data not shown). We also examined the time course of induction of HIV-1 expression in response to these drugs. An increase in virus-specific transcripts was observed as early as 2 h following addition of the drugs and progressively increased over time to reach a peak at 16 h (Fig. 2B). Similar results were obtained using another latently infected cell line, ACH2 (shown as part of Fig. 3). In a separate report (43), we further studied the mechanism of action of TSA and TPX on the HIV-1 promoter. We found that treatment with TSA and TPX is associated with the same disruption of nuc-1 that has been described following activation with TNF-α (43). In addition, the disruption of nuc-1 in response to TPX or TSA is also independent of transcription as it occurred in the presence of doses of α-amanitin sufficient to suppress transcription (43).
FIG. 2.
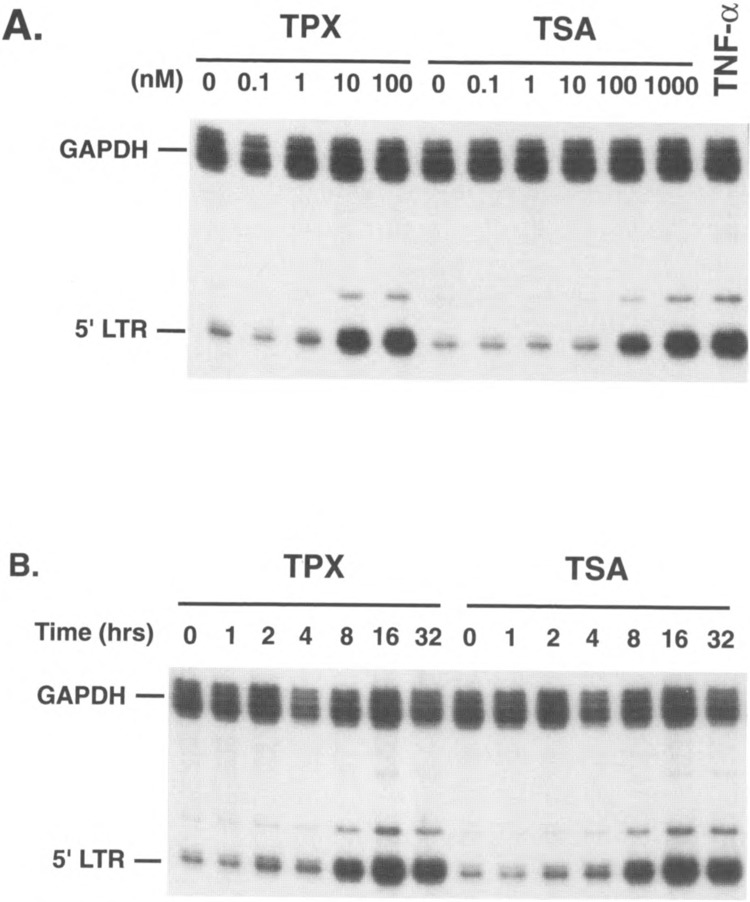
Induction of HIV-1 expression in J49 cells by TSA and TPX. (A) J49 cells were incubated in the presence of different concentrations of both TPX and TSA as indicated and RNA was harvested at 8 h posttreatment. TNF-α was used at 800 U/ml. (B) TPX (100 nM) or TSA (400 nM) was added to J49 cells, for 0–32 h. Harvested RNA were analyzed by RNase protection analysis using an HIV-1 LTR-specific antisense probe (23) and a GAPDH antisense probe. The HIV-1 probe protects two fragments corresponding to the 5′ and 3′ LTR and only the 5′ LTR protected band is shown.
FIG. 3.
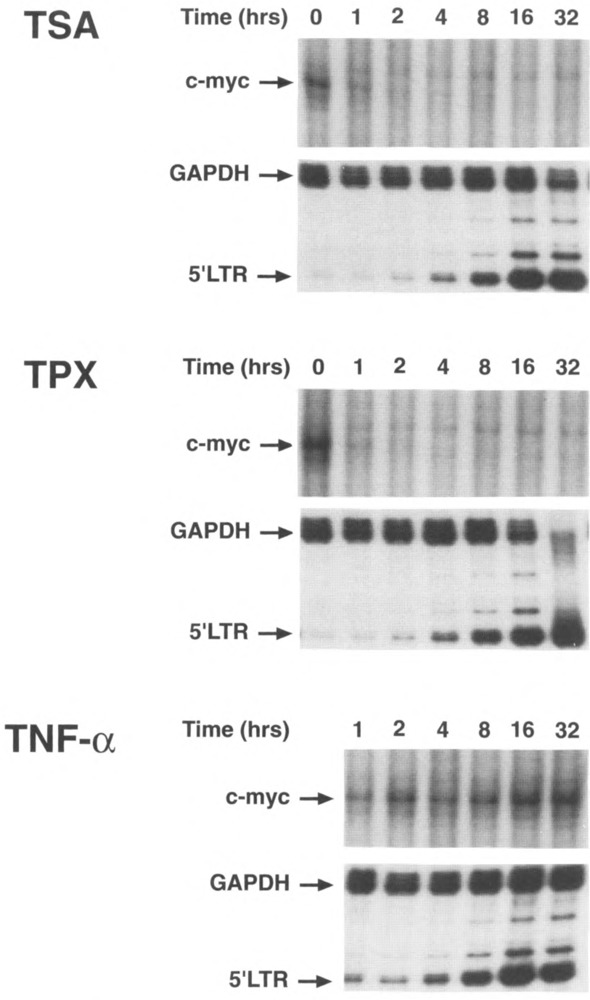
TPX and TSA suppresses c-myc gene expression. ACH2 cells were treated for different periods of time (0–32 h) with TSA (400 nM), TPX (100 nM), or TNF-α (800 U/ml). RNase protection analysis was performed using specific anti-sense probes for c-myc, GAPDH, and the HIV-1 LTR (23).
Suppression of c-myc Gene Expression in Response to Hyperacetylation
As described in the Introduction, histone hyperacetylation has been implicated in gene transcription but also in silencing of gene expression. We were intrigued by the possibility that treatment with histone deacetylase inhibitors might result in the silencing of specific genes. n-Butyrate, a non-competitive inhibitor of histone deacetylase, has been shown to block the expression of c-myc (3,6,13,14,18,19,32,34,39,40). This inhibition is secondary both to a decrease in the transcription rate (32) and to an increase in the block to transcriptional elongation present in the c-myc gene (14). However, butyrate also demonstrates many pleiotropic effects that are independent of histone acetylation (22). Consequently, the relationship between histone hyperacetylation and suppression of c-myc expression has not been clearly established.
To examine the effect of histone acetylation on c-myc expression, ACH2 cells were treated with TSA, TPX, or TNF-α as a control. The expression of c-myc, GAPDH, and HIV-1 was examined by RNase protection using specific antisense RNA probes. The c-myc probe spans 118 bases of the 3′ end of the first coding exon and 132 bp of the flanking intron and when hybridized with mRNA results in the protection of a 118 nt fragment. This region of c-myc has no significant homology to other myc genes (N-myc, L-myc). Using this probe, we showed that c-myc expression was rapidly down-regulated following addition of either TSA or TPX so that no detectable signal was measured 2 h following the addition of TSA or TPX (Fig. 3). In contrast, TNF-α had no effect on c-myc expression. In the same experiment, GAPDH was unaffected by any of the agents and HIV-1 expression was induced by all three. We have also examined c-myc expression in other cell lines, including Jurkat, SupT1, J49, and found the same downregulation of c-myc in all cell lines studied (data not shown).
The Expression of a Small Number of Cellular Genes Is Modulated by Histone Acetylation
As the experiments described above demonstrate, the degree of histone acetylation can have profound effects on the expression of specific genes. However, the generality of this phenomenon is unknown and in particular the fraction of genes in the genome whose expression is modulated in response to histone hyperacetylation has not been determined. To address this question, we treated Jurkat and SupT1 cells with TSA and extracted total RNA from these cells after different times. After reverse transcription of mRNAs using an anchored (A, C, G, or T) oligo dT primer, differential display (27,28) was used to compare the relative abundance of approximately 340 genes following treatment with TSA. Differential display on RNA samples not submitted to the reverse transcription reaction yielded no detectable signal, confirming the cDNA nature of the bands observed (data not shown). Several genes were found to be either induced (Fig. 4A–C) or suppressed (Fig. 4D–E) in response to TSA treatment. The time course of induction was similar to what had been observed with the HIV-1 LTR as the amount of mRNA slowly increased from 2 to 8 h postinduction and closely matched the progressive increase in histone acetylation levels (data not shown). In the case of genes whose expression was inhibited by TSA treatment, the decrease followed a similar time course with maximal inhibition at 8 h posttreatment. Out of approximately 340 cDNAs tested, we found 8 genes whose expression appeared modulated in response to TSA. The expression of some genes was modified in both cell lines in response to TSA; however, for some genes, cell-specific induction (or suppression) was observed. This experiment indicates that the expression of a small proportion of cellular genes (∼2%) can be modulated by changes in the degree of acetylation of histones.
FIG. 4.
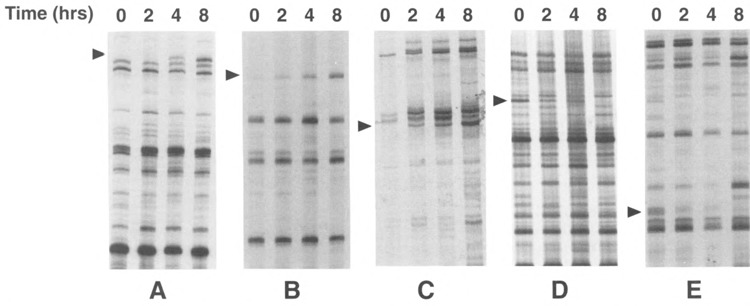
Differential display analysis of Jurkat, SupT1 mRNA in response to TSA. Total RNA from Jurkat and SupT1 cells was analyzed by differential display and the amplified products separated on sequencing gels. Regions from the gels containing mRNAs whose expression is modulated by TSA are shown. cDNAs whose expression is modulated in response to TSA are indicated by an arrow. (A) Jurkat RNA amplified with primers AP4 and C, (B) Jurkat with primers AP2 and A, (C) SupT1 RNA with primers API and G, (D) Jurkat RNA with primers AP5 and A, and (E) SupT1 RNA with primers AP5 and G.
DISCUSSION
We have examined the expression of cellular genes in response to TSA and TPX. We confirm that both drugs inhibit histone deacetylase in human lymphoid cell lines in the nanomolar range as previously shown in other cellular systems (17,47). Both drugs cause a relative hyperacetylation, which is associated with the induction of HIV-1 expression in latently infected cell lines. In the same cell lines, the expression of GAPDH is unaffected and the expression of c-myc is completely suppressed. Comparison of the pattern of gene expression between cells treated with TSA and untreated cells using differential display demonstrates that the expression of a small subset of cellular genes (∼2%) is changed in response to histone hyperacetylation.
Similar changes in gene expression for the HIV-1 LTR and c-myc have been reported using n-butyrate (3,5,6,10,13,14,18,19,24,32,34,36,38–40). However, because of the pleitropic effects of butyrate (20–22), a direct correlation between hyperacetylation and the changes in gene expression could not be demonstrated.
In a separate report (43), we have demonstrated that both TPX and TSA treatment result in the disruption of a specific nucleosome (called nuc-1) (45) positioned at the transcriptional start in the HIV-1 promoter. This nucleosome is also disrupted during transcriptional activation of the HIV-1 promoter in response to other activators of HIV-1 transcription, such as phorbol esters and TNF-α (44,45). We have proposed that nuc-1 inhibits HIV-1 transcription in latently infected cell lines either by blocking the binding of a transcription factor crucial for transcription initiation or by inhibiting the progression of a poorly processive polymerase (43,45). The nature of the change occurring in nuc-1 in response to hyperacetylation has not been determined: nuc-1 could either be totally displaced from DNA or could undergo a conformational change in response to hyperacetylation. In the case of c-myc, previous studies have demonstrated that the inhibition of gene expression in response to butyrate occurs at the transcriptional level (33), possibly by causing an increase in a block to transcriptional elongation present in this gene (14). However, a detailed study of the chromatin organization of the gene in response to butyrate or TPX/TSA is not available and will be necessary for a full understanding of their mechanism of action in this context.
Several studies have examined the effect of TSA and TPX on the expression of specific genes (1,2,9,15,30,37,48). Using differential display, we have examined a large number of genes for changes in their pattern of expression in response to hyperacetylation. The small percentage of genes whose expression is modified in response to the drugs, less than 5%, is surprising in view of the fact that the hyperacetylation of histones appears global (see Fig. 1A). This discrepancy argues against a model in which these drugs activate transcription by “loosening” the interaction between DNA and histones through histone hyperacetylation, allowing the polymerase to transcribe the DNA more easily. Instead, these observations argue for an additional level of specifity in the mechanism underlying the changes observed in expression levels of specific genes. In this regard, it is interesting to note that the fraction of genes whose expression is modified by hyperacetylation (∼2%) approximates the fraction of histones whose acetyl groups turnover rapidly (2%) (46). From these two observations, one would predict that a small subset of nucleosomes contains histones that are highly sensitive to hyperacetylation, a prediction that was recently confirmed experimentally (4). Such hypersensitivity to hyperacetylation could be conferred to a nucleosome by its location in a nuclear compartment where acetylases or deacetylases are highly active or concentrated, or, alternatively, through the direct or indirect interaction of an acetylase or a deacetylase with a specific DNA binding factor. We cannot rule out at this point that the changes in expression levels for some genes are secondary (i.e., not immediately dependent on histone hyperacetylation per se but rather dependent on the primary activation of another gene). However, our time course analysis of gene expression indicates that the expression levels of all genes appear to change with the same kinetics (see Fig. 4), in close parallel with the changes in histone acetylation levels. Finally, the distinct responses of individual genes to hyperacetylation in terms of gene expression (i.e., stimulation, inhibition, or no change in gene expression) indicate that no general mechanism can explain the role of acetylation in gene expression and that the local chromatin environment (as shown for nuc-1 in the case of HIV-1) probably plays a critical role in the effect of the drugs. The crucial role of the local environment probably explains some of the apparently contradictory results that have been recently discussed in the litterature with respect to the role of acetylation in gene transcription (12,31).
ACKNOWLEDGEMENTS
We thank Anuj Dalai for excellent technical assistance. We thank Dr. Michael Laspia for his gift of pGEM23. We thank Dr. Sujita (Shionogi & Co., Osaka, Japan) and Dr. M. Yoshida (University of Tokyo, Tokyo, Japan) for their generous gift of TSA and TPX, respectively. We also thank Anthony Fauci, Tom Folks, Guido Poli, and Sal Butera for making their cell lines available to us for these studies through the the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. Carine Van Lint is “Charge de Recherches” of the “Fonds National de la Recherche Scientifique” (FNRS, Belgium). This work was supported in part by a grant from the National Institutes of Health of the United States Public Health Service (Grant GM51671-01A1) and by institutional funds from the Picower Institute for Medical Research.
REFERENCES
- 1. Almouzni G.; Khochbin S.; Dimitrov S.; Wolffe A. P. Histone acetylation influences both gene expression and development of Xenopus laevis . Dev. Biol. 165:654–669; 1994. [DOI] [PubMed] [Google Scholar]
- 2. Arts J.; Lansink M.; Grimbergen J.; Toet K. H.; Kooistra T. Stimulation of tissue-type plasminogen activator gene expression by sodium butyrate and trichostatin A in human endothelial cells involves histone acetylation. Biochem. J. 310:171–176; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barnard J. A.; Warwick G. Butyrate rapidly induces growth inhibition and differentiation in HT-29 cells. Cell Growth Differ. 4:495–501; 1993. [PubMed] [Google Scholar]
- 4. Barratt M. J.; Hazzalin C. A.; Cano E.; Mahadevan L. C. Mitogen-stimulated phosphorylation of histone H3 is targeted to a small hyperacetylation-sensitive fraction. Proc. Natl. Acad. Sci. USA 91:4781–4785; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bohan C. A.; Robinson R. A.; Luciw P. A.; Sjrinivasan A. Mutational analysis of sodium buty-rate inducible elements in the human immunodeficiency virus type 1 long terminal repeat. Virology 172:573–583; 1989. [DOI] [PubMed] [Google Scholar]
- 6. Collins J. F.; Herman P.; Schuch C.; Bagby G. C. Jr. c-myc antisense oligonucleotides inhibit the colony-forming capacity of Colo 320 colonic carcinoma cells. J. Clin. Invest. 89:1523–1527; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davie J. R.; Hendzel M. J. Multiple functions of dynamic histone acetylation. J. Cell Biochem. 55:98–105; 1994. [DOI] [PubMed] [Google Scholar]
- 8. Dimitrov S.; Dasso M. C.; Wolffe A. P. Remodeling sperm chromatin in Xenopus laevis egg extracts: The role of core histone phosphorylation and linker histone B4 in chromatin assembly. J. Cell Biol. 126:591–601; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Girardot V.; Rabilloud T.; Yoshida M.; Beppu T.; Lawrence J. J.; Khochbin S. Relationship between core histone acetylation and histone H1(0) gene activity. Eur. J. Biochem. 224:885–892; 1994. [DOI] [PubMed] [Google Scholar]
- 10. Golub E. I.; Li G. R.; Volsky D. J. Induction of dormant HIV-1 by sodium butyrate: Involvement of the TATA box in the activation of the HIV-1 promoter. AIDS 5:663–668; 1991. [PubMed] [Google Scholar]
- 11. Haines D. S.; Gillespie D. H. RNA abundance measured by a lysate RNase protection assay. Biotechniques 12:736–741; 1992. [PubMed] [Google Scholar]
- 12. Hebbes T. R.; Clayton A. L.; Thorne A. W.; Crane-Robinson C. Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken beta-globin chromosomal domain. EMBO J. 13:1823–1830; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herold K. M.; Rothberg P. G. Evidence for a labile intermediate in the butyrate induced reduction of the level of c-myc RNA in SW837 rectal carcinoma cells. Oncogene 3:423–428; 1988. [PubMed] [Google Scholar]
- 14. Heruth D. P.; Zirnstein G. W.; Bradley J. F.; Rothberg P. G. Sodium butyrate causes an increase in the block to transcriptional elongation in the c-myc gene in SW837 rectal carcinoma cells. J. Biol. Chem. 268:20466–20472; 1993. [PubMed] [Google Scholar]
- 15. Hoshikawa Y.; Kwon H. J.; Yoshida M.; Horinouchi S.; Beppu T. Trichostatin A induces morphological changes and gelsolin expression by inhibiting histone deacetylase in human carcinoma cell lines. Exp. Cell Res. 214:189–197; 1994. [DOI] [PubMed] [Google Scholar]
- 16. Johnson L. M.; Kayne P. S.; Kahn E. S.; Grunstein M. Genetic evidence for an interaction between SIR3 and histone H4 in the repression of the silent mating loci in Saccharomyces cerevisiae . Proc. Natl. Acad. Sci. USA 87:6286–6290; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kijima M.; Yoshida M.; Sujita K.; Horinouchi S.; Beppu T. Trapoxin, an antitumour cyclic tetra-peptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 268:22429–22435; 1993. [PubMed] [Google Scholar]
- 18. Kondoh N.; Oikawa T.; Satoh C.; Kuzumaki N. Effects of sodium butyrate on the rearranged c-myc expression in mouse plasmacytoma cells. Exp. Cell Res. 196:146–149; 1991. [DOI] [PubMed] [Google Scholar]
- 19. Kosaka M.; Nishina Y.; Takeda M.; Matsumoto K.; Nishimune Y. Reversible effects of sodium butyrate on the differentiation of F9 embryonal carcinoma cells. Exp. Cell Res. 192:46–51; 1991. [DOI] [PubMed] [Google Scholar]
- 20. Kruh J. Effect of sodium butyrate, a new pharmacological agent, on cells in culture. Mol. Cell. Biochem. 42:65–82; 1982. [DOI] [PubMed] [Google Scholar]
- 21. Kruh J.; Defer N.; Tichonicky L. Molecular and cellular action of butyrate. C R Seances Soc. Biol. Fil. 186:12–25; 1992. [PubMed] [Google Scholar]
- 22. Kruh J.; Defer N.; Tichonicky L. Action moleculaire et cellulaire du butyrate. C R Soc. Biol. 186:12–25; 1992. [PubMed] [Google Scholar]
- 23. Laspia M. F.; Wendel P.; Mathews M. B. HIV-1 Tat overcomes inefficient transcriptional elongation in vitro. J. Mol. Biol. 232:732–746; 1993. [DOI] [PubMed] [Google Scholar]
- 24. Laughlin M. A.; Zeichner S.; Kolson D.; Alwine J. C.; Seshamma T.; Pomerantz R. J.; Gonzalez-Scarano F. Sodium butyrate treatment of cells latently infected with HIV-1 results in the expression of unspliced viral RNA. Virology 196:496–505; 1993. [DOI] [PubMed] [Google Scholar]
- 25. Lee D.; Hayes J.; Pruss D.; Wolffe A. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 72:73–84; 1993. [DOI] [PubMed] [Google Scholar]
- 26. Lennox R. W.; Cohen L. H. In: Wassarman P. M.; Kornberg R. D., eds. Methods in enzymology—volume 170—nucleosomes. San Diego, CA: Academic Press, Inc.; 1989:532–549. [DOI] [PubMed] [Google Scholar]
- 27. Liang P.; Pardee A. B. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science 257:967–971; 1992. [DOI] [PubMed] [Google Scholar]
- 28. Liang P.; Averboukh L.; Pardee A. B. Distribution and cloning of eukaryotic mRNAs by means of differential display: Refinements and optimization. Nucleic Acids Res. 21:3269–3275; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loidl P. Histone acetylation: Facts and questions. Chromosoma 103:441–449; 1994. [DOI] [PubMed] [Google Scholar]
- 30. Miyashita T.; Yamamoto H.; Nishimune Y.; Nozaki M.; Morita T.; Matsushiro A. Activation of the mouse cytokeratin A (endo A) gene in teratocarcinoma F9 cells by the histone deacetylase inhibitor trichostatin A. FEBS Lett. 353:225–229; 1994. [DOI] [PubMed] [Google Scholar]
- 31. O’Neill L. P.; Turner B. M. Histone H4 acetylation distinguishes coding regions of the human genome from heterochromatin in a differentiation-dependent but transcription-independent manner. EMBO J. 16:3946–3957; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Polack A.; Eick D.; Koch E.; Bornkamm G. W. Truncation does not abrogate transcriptional down-regulation of the c-myc gene by sodium butyrate in Burkitt’s lymphoma cells. EMBO J. 6:2959–2964; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Polack A.; Feederle R.; Klobeck G.; Hortnagel K. Regulatory elements in the immunoglobulin kappa locus induce c-myc activation and the promoter shift in Burkitt’s lymphoma cells. EMBO J. 12:3913–3920; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rabizadeh E.; Shaklai M.; Nudelman A.; Eisenbach L.; Rephaeli A. Rapid alteration of c-myc and c-jun expression in leukemic cells induced to differentiate by a butyric acid prodrug. FEBS Lett. 328:225–229; 1993. [DOI] [PubMed] [Google Scholar]
- 35. Roth S. Y.; Shimizu M.; Johnson L.; Grunstein M.; Simpson R. T. Stable nucleosome positioning and complete repression by the yeast alpha 2 repressor are disrupted by amino-terminal mutations in histone H4. Genes Dev. 6:411–425; 1992. [DOI] [PubMed] [Google Scholar]
- 36. Sadaie M. R.; Hager G. L. Induction of developmentally programmed cell death and activation of HIV by sodium butyrate. Virology 202:513–518; 1994. [DOI] [PubMed] [Google Scholar]
- 37. Schlake T.; Klehr-Wirth D.; Yoshida M.; Beppu T.; Bode J. Gene expression within a chromatin domain: The role of core histone hyperacetylation. Biochemistry 33:4197–4206; 1994. [DOI] [PubMed] [Google Scholar]
- 38. Shahabuddin M.; Volsky B.; Kim H.; Sakai K.; Volsky D. J. Regulated expression of human immunodeficiency virus type 1 in human glial cells: Induction of dormant virus. Pathobiology 60:195–205; 1992. [DOI] [PubMed] [Google Scholar]
- 39. Souleimani A.; Asselin C. Regulation of c-myc expression by sodium butyrate in the colon carcinoma cell line Caco-2. FEBS Lett. 326:45–50; 1993. [DOI] [PubMed] [Google Scholar]
- 40. Taylor C. W.; Kim Y. S.; Childress-Fields K. E.; Yeoman L. C. Sensitivity of nuclear c-myc levels and induction to differentiation-inducing agents in human colon tumor cell lines. Cancer Lett. 62:95–105; 1992. [DOI] [PubMed] [Google Scholar]
- 41. Tsanev R.; Russev G.; Pashev I.; Zlatanova J. Replication and transcription of chromatin. Boca Raton, FL: CRC Press, Inc.; 1993. [Google Scholar]
- 42. Turner B. M. Decoding the nucleosome. Cell 75:5–8; 1993. [PubMed] [Google Scholar]
- 43. Van Lint C.; Emiliani S.; Ott M.; Verdin E. Transcriptional activation and chromatin remodelling of the HIV-1 promoter in response to histone acetylation. EMBO J. (in press). [PMC free article] [PubMed] [Google Scholar]
- 44. Verdin E. DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. J. Virol. 65:6790–6799; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Verdin E.; Paras P. Jr.; Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 12:3249–3259; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wolffe A. Chromatin—structure & function. San Diego, CA: Academic Press; 1992. [Google Scholar]
- 47. Yoshida M.; Kijima M.; Akita M.; Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 265:17174–17179; 1990. [PubMed] [Google Scholar]
- 48. Yoshida M.; Horinouchi S.; Beppu T. Trichostatin A and trapoxin: Novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays 17:423–430; 1995. [DOI] [PubMed] [Google Scholar]