

Abstract
Advances in the field of interferon research have identified a signal transduction pathway that initiates at a cell surface receptor and culminates at target genes in the nucleus. The binding of interferon to a transmembrane receptor stimulates the concomitant activation of tyrosine kinases of the Janus kinase (JAK) family. Subsequently, latent cytoplasmic transcription factors are activated by tyrosine phosphorylation and function as signal transducers and activators of transcription (STATs). The STATs form homomeric or heteromeric protein complexes that translocate to the nucleus to bind to specific DNA sequences in the promoters of stimulated genes. The discovery of this regulated pathway in the interferon system served as a paradigm for receptor to nucleus signal transmission by a variety of cytokines.
Keywords: Interferons, Signal transduction, Gene transcription activation, Tyrosine kinases, Cytokines
INTERFERONS (IFNs) are a family of polypep-tide hormones that share the ability to confer cellular resistance to many viruses (reviewed in Len-gyel, 1982; Pestka et al., 1987; DeMaeyer and DeMaeyer-Guiginard, 1988; Staeheli, 1990). This ability of IFN to interfere with the replication of viruses led both to its discovery and name, and serves to define a biological unit of activity (Isaacs and Lindenmann, 1957; Wheelock, 1965; Stewart, 1979). Since its discovery, many additional biological effects of IFN have been recognized, including the inhibition of cellular proliferation and the activation of cells in the immune system (reviewed in Baron et al., 1991). The production of IFN is a regulated and transient process stimulated by a variety of inducers that activate transcription of the IFN genes. The human IFN family of cytokines is comprised of two main types (Diaz et al., 1993). Type I IFN includes IFN-α, IFN-β, and IFN-ω, and nearly all cells of the body are capable of producing and secreting type I IFN. IFN-α is synthesized predominantly by leukocytes and monocytes, whereas IFN-β is produced mainly by fibroblasts. The primary inducers of type I IFN production are viruses and double-stranded RNA; however, bacteria (lipopolysaccharide) and various cytokines can also stimulate synthesis. Type II IFN is also known as immune IFN or IFN-γ and is produced by T lymphocytes and natural killer cells when the cells encounter foreign antigens or various mitogens.
The family of IFNs provides a fundamental defense system that acts to protect healthy cells in localized infections and acts via the bloodstream to curb dissemination of infection to distant sites. Because of their physiological effects, the United States Food and Drug Administration has approved the use of IFNs for a variety of infections, malignancies, and immune disorders. For these reasons, much effort has been dedicated to understanding the molecular mechanism of IFN action. Induction of specific cellular enzymes, such as the double-stranded RNA (dsRNA)-dependent protein kinase (PKR) and the dsRNA-dependent 2′-5′ oligoadenylate synthetase, appears to be involved in the antiviral actions of IFN (reviewed in Pestka et al., 1987). Three decades ago it was known that the antiviral response to IFN required new mRNA synthesis (Taylor, 1964). More recently, specific genes that are inducible by IFN have been isolated, and their as-regulatory DNA elements have been identified. These regulatory DNA elements have led to the characterization of trans-acting factors and an understanding of the IFN signal pathway that initiates at the cell surface.
GENE ACTIVATION BY IFNs-α/β
DNA Element Responsive to IFNs- α/β
To identify and characterize genes induced by IFN- α/β stimulation, two basic approaches were taken. Specific IFN-induced proteins were targeted, and antibodies, hybrid-selected translation, or amino acid sequences were used to derive cDNA clones (Merlin et al., 1983; Chebath et al., 1983; Blomstrom et al., 1986; Cheng et al., 1986; Staeheli et al., 1986). Alternatively, cDNA libraries prepared from IFN-treated cells were screened with radiolabeled probes complementary to mRNA from either untreated cells or IFN-treated cells (Larner et al., 1984; Friedman et al., 1984). Differential screening identified several IFN-α-stimulated genes (αlSGs) that hybridized specifically to probes from IFN-treated cells. This approach provided the ability to characterize and compare the expression of a group of novel αISGs. Northern blot hybridization confirmed expression of the αlSG mRNAs after IFN treatment, and nuclear run-on transcription assays assured that the genes were regulated at the level of tran-scriptional initiation. In unstimulated cells the αlSGs were silent, but they were activated within minutes following IFN-α stimulation. Transcrip-tional induction of the αlSGs did not require new protein synthesis because it was unaffected by translation inhibitors. However, gene induction was transient and declined within hours.
Because the transcriptional response to IFN-α did not require new protein synthesis, activation of this set of genes was thought to involve the modification of a preexisting transcription factor. To determine the identity of this factor, DNA sequences essential for induction of gene expression by IFN were first pursued. Genomic clones of the αISGs were isolated and DNA regulatory sequences were examined. Transfection studies performed with αISG reporter gene hybrids revealed an IFN-α-responsive region in the promoter of these genes (Vogel et al., 1986; Israel et al., 1986; Levy et al., 1986; Sugita et al., 1987; Korber et al., 1987; Benech et al., 1987; Gribaudo et al., 1987; Reich et al., 1987). Further characterization determined that the responsive sequence was required for activation by IFN-α and could function in a heterologous promoter, at a distance from the transcriptional start site, in an orientation-independent manner (Gribaudo et al., 1987; Reich et al., 1987). This inducible enhancer was found to be present in the promoters of other αISGs and was termed the IFN-α-stimulated response element (ISRE) (Friedman and Stark, 1985; Reich et al., 1987; Porter et al., 1988; Levy et al., 1988; Hug et al., 1988). The ISRE is a consensus sequence of approximately 15 nucleotides that is sufficient to confer transcriptional responsiveness to IFN-a (Reich and Darnell, 1989; Reid et al., 1989). Table 1 displays homologous ISRE sequences found in several alSGs, usually located approximately 100 nucleotides upstream of the transcriptional start site. The most notable characteristic of the sequence is the presence of a GAAA direct repeat.
TABLE 1.
EXAMPLES OF INTERFERON-STIMULATED GENES (ISGs) AND THEIR CORRESPONDING RESPONSE ELEMENTS
| αISG | ISRE | |
|---|---|---|
| ISG15 | GGGAAACCGAAACTG | (−108/−94 Reich et al., 1987) |
| ISG54 6-162′-5′7ISG | GGGAAAGTGAAACTA GGGAAAATGAAACTG OASE AGGAAAC-GAAACCAGAS | (−87/−102, Levy et al., 1988) (−112/−98, Porter etal., 1988) (−100/−87, Rutherford et al., 1988) |
| FcγRI | TATTTCCCAGAAAGG | (−32/−18, Pearseetal., 1991) |
| IRF-1 Ly6E GBP | GATTTCCCCGAAATG ATATTCCTGTAAGTG TTTCATATTACTCTCAAA | (−124/−111, Sims etal., 1993) (−1222/−1208, Kahn et al., 1993) (−111/−94, Decker etal., 1991) |
The interferon-stimulated response element (ISRE) of genes activated by interferon-a (alSGs) are shown above the γ-activated site (GAS) of genes activated by interferon-γ (γISGs).
The IFN-α/β Activated Transcription Factor, ISGF3
Once the ISRE was identified, the natural direction of study was to pursue DNA binding factors that mediated the response to IFN-α. Proteins that specifically recognized the ISRE were identified by DNA footprint analyses or electrophoretic mobility shift assays (EMSA) (Levy et al., 1988; Porter et al., 1988; Korber et al., 1988; Cohen et al., 1988; Rutherford et al., 1988; Reich and Darnell, 1989). EMSA analyses revealed the presence of a constitutive ISRE binding factor in the nuclei of untreated cells, termed the IFN-stimulated gene factor 1 (ISGF1). ISGF1 may be involved in quiescence of the αISGs. Following stimulation of cells with IFN-α, two additional ISRE binding factors appeared with distinct kinetics, ISGF2 and ISGF3. ISGF2 was not detectable until 2 h after IFN-α treatment, and its appearance required new protein synthesis (Levy et al., 1988). Because the transcriptional response to IFN does not require ongoing protein synthesis, ISGF2 is not the primary mediator of IFN action. Purification and sequence analysis of ISGF2 (Pine et al., 1990) revealed it to be the same as a previously isolated factor, the IFN regulatory factor−1 (IRF-1) (Miyamoto et al., 1988). The gene encoding IRF-1 was isolated by its ability to recognize a DNA sequence in the IFN-β promoter that shares homology with the core region of the ISRE. Indeed, both ISGF1 and IRF-1 /ISGF2 bind to an inner core region of the ISRE whereas ISGF3 requires nucleotides flanking this minimum recognition site (Reich and Darnell, 1989). IRF-1/ISGF2 likely plays a role in the secondary response to IFN-a because transfec-tion of the IRF-1/ISGF2 gene increases αlSG transcription, and constitutive expression confers resistance to viral infection (Pine, 1992).
Several lines of evidence suggested ISGF3 to be the primary transcription factor activated by IFN-α. Activation of ISGF3 occurred within minutes of IFN-a treatment in the absence of protein synthesis, revealing it to preexist in the cell (Levy et al., 1988). The rapid and transient appearance of ISGF3 correlated with αISG transcription. Activated ISGF3 was detected first in the cytoplasm of the cell prior to its translocation to the nucleus, indicating that it served as a signal to link the receptor to the nucleus (Dale et al., 1989; Levy et al., 1989). Purified ISGF3 stimulated the in vitro transcription of an ISRE-containing promoter (Fu et al., 1990). More recently, complementation of cells defective in their response to IFN- α has been achieved by transfection with genes encoding ISGF3(Müller etal., 1993a).
Characterization of ISGF3 revealed that is was composed of several subunits. Evidence for the multimeric nature of ISGF3 was first supported by the establishment of an in vitro reconstitution assay (Levy et al., 1990). The DNA binding activity of ISGF3 was found to be inhibited by treatment with the protein alkylating agent N-ethyl malemide (NEM). However, active ISGF3 could be reconstituted by in vitro mixing of NEM-inactivated ISGF3 with extracts from IFN-7-treated cells. Although IFN-γ does not activate ISGF3, it stimulates the synthesis of an NEM-sensitive component of ISGF3, termed ISGF3γ. Reconstitution of ISGF3 is achieved by mixing ISGF37 in vitro with the NEM-insensitive component of ISGF3, termed ISGF3α.
The multisubunit structure of ISGF3 became more apparent during its partial purification (Fu et al., 1990; Kessler et al., 1990). Photoaffinity UV cross-linking and protein renaturation experiments revealed ISGF3γ to be the major DNA binding subunit of ISGF3. ISGF3γ consists of a 48 kDa protein (p48) that has low intrinsic affinity for the ISRE and preexists mainly in the nuclear compartment of the cell, although it is also detectable in the cytoplasm. ISGF3 also contains three additional proteins of 84 kDa (p84), 91 kDa (p91), and 113 kDa (pi 13). These proteins reside in the cytoplasm of the cell until signal activation by IFN-α, whereupon they become phosphorylated on tyrosine residues, associate to form ISGF3α and translocate the nucleus to complex with ISGF3γ (p48) and form ISGF3.
Partial purification of ISGF3 by affinity chro-matography provided sufficient protein to obtain amino acid sequence (Schindler et al., 1992a; Fu et al., 1992; Veals et al., 1992). The four ISGF3 components were separated by gel electrophoresis, transferred to nitrocellulose, excised, digested with trypsin, and subjected to peptide analysis. This information was used to create degenerate sense and antisense oligonucleotide primers for amplification of cDNA fragments in a polymerase chain reaction that were used to screen a cDNA library. Recombinant cloning of the genes encoding ISGF3 subunits by J. E. Darnell, Jr. and colleagues has been critical to our understanding of receptor to nucleus signaling by IFNs.
The p48/ISGF3γ gene was isolated from a cDNA library produced with mRNA from IFN-7-treated cells (Veals et al., 1992). To demonstrate that the correct cDNA was isolated, p48 was synthesized in vitro from a recombinant clone and mixed with a source of ISGF3a to successfully reconstitute ISGF3. Sequence analysis of the gene encoding p48 revealed the amino-terminus to possess significant homology to a family of DNA binding factors that includes IRF-1/ISGF2, and to contain a tryptophan repeat found in the DNA binding domain of the oncogene c-myb (Veals et al., 1993). Investigation of p48 functional domains by deletion analysis demonstrated that the amino-terminal region (a.a. 1–117) is responsible for DNA recognition. In addition, constitutive serine/threonine phosphorylation of p48 appears to be necessary for DNA binding. The carboxyl-terminal region of p48 (a.a. 217–377) was identified by deletion analysis to be responsible for interaction with ISGF3α (pll3/p91/p84). This association results in formation of the high-affinity ISGF3.
Isolation and sequence analysis of the p113, p91, and p84 cDNAs revealed the existence of a unique gene family of transcription factors. cDNAs corresponding to p91 and p84 clearly demonstrated that these two proteins were encoded by differentially spliced mRNAs from the same gene (Schindler et al., 1992a). p91 contained an additional 38 amino acids at the carboxy-terminus that were absent in p84. In addition, the cDNA of pi 13 encoded a protein with approximately 40% overall identity to p91, establishing it as a homologous family member (Fu et al., 1992; Fu, 1992). The sequences of p91 and pi 13 genes also provided a compelling clue to the signal activation pathway of ISGF3. Each protein contained a region of homology to the Src oncoprotein referred to as the Src homology 2 domain (SH2) (Fu, 1992). SH2 domains have been demonstrated to be binding sites for phosphotyrosine-containing peptides and thereby play a critical role in protein-protein interactions (reviewed in Pawson and Schlessinger, 1993). The presence of this domain suggested tyro-sine phosphorylation was involved in the regulation of ISGF3. Indeed, the p84, p91, and pi 13 subunits were found to be phosphorylated on tyrosine residues following IFN stimulation. These proteins were also found to possess two additional conserved structural motifs that serve to promote protein-protein interactions: a Src homology 3 (SH3) domain located adjacent to the SH2 domain, and a heptad leucine repeat within the amino-terminal portion of the proteins. SH3 domains appear to associate with components of the cytoskeleton that may direct cellular localization (reviewed in Pawson and Schlessinger, 1993). Heptad leucine repeats, known as leucine zippers, were first identified as protein dimerization domains in transcription factors (reviewed in Land-schulz et al., 1988). Because the p84, p91, and pi 13 components of ISGF3 remain latent in the cytoplasm and associate and translocate to the nucleus only after tyrosine phosphorylation, proteins with this structure and function have been termed signal transducers and activators of transcription (STATs) (reviewed in Darnell et al., 1994; Zhong et al., 1994a). p91 and p84 have been designated STATla and STAT1β, respectively, because they are encoded by the same gene, whereas pi 13 has been designated STAT2. The structure of p91/ STAT1α is depicted in Fig. 1 and represents the general structure of members of the STAT family.
FIG. 1.
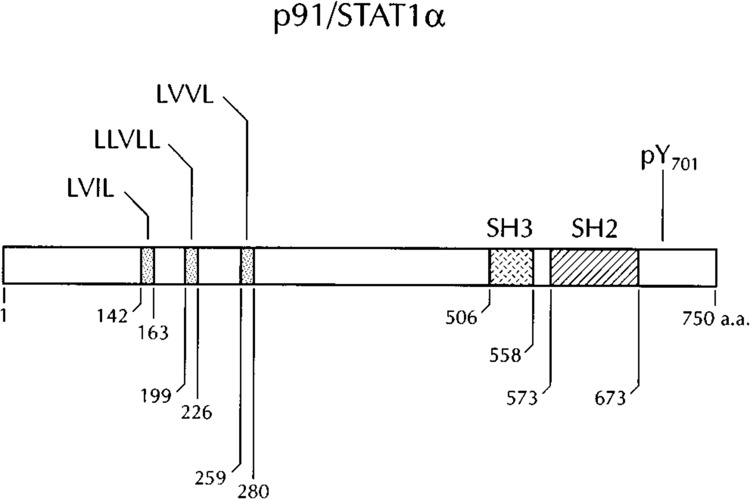
Diagrammatic representation of the p91 signal transducer and activator of transcription (STATlα). The leucine heptad repeats (containing leucine, valine, or isoleucine), the SH3 domain, and the SH2 domains are shown. The single tyrosine (Y701) that is phosphorylated following interferon stiumlation is indicated. Numbers correspond to amino acid residues.
Signal Activation of ISGF3
Many cellular responses are induced by rapid phosphorylation/dephosphorylation events triggered by extracellular stimuli. Earlier studies with the protein kinase inhibitor staurosporine, a broad spectrum inhibitor with potent effects on tyrosine kinases, demonstrated a requirement of phosphorylation for activation of latent ISGF3 by IFN-α (Reich and Pfeffer, 1990). In addition, treatment of cells with genistein, a specific tyrosine kinase inhibitor, was found to block signal activation of ISGF3 (Fu, 1992; Gutch et al., 1992). The inhibitory effects of genistein were found to be specific to the ISGF3α subunit of ISGF3 because in vitro reconstitution of ISGF3 could be achieved with ISGF3γ (p48), but not with ISGF3α, from gen-istein-treated cells (Gutch et al., 1992). Evidence that the ISGF3α proteins were themselves substrates of an IFN-induced tyrosine kinase was provided with the use of antibodies that recognize phosphotyrosine. Phosphotyrosine-specific antibodies reacted with ISGF3-ISRE complexes in EMSAs to produce a “supershift” complex and recognized p91/STATlα, p84/STATlβ, and pi 13/STAT2 in immunoblot assays only after signal activation by IFN (Fu, 1992; Gutch et al., 1992; Schindler et al., 1992b). Confirmations of phosphotyrosine content was provided with phos-phoamino acid analysis of STAT1 and STAT2 proteins metabolically labeled in vivo in the presence of [32P]orthophosphate and IFN. Both proteins contained phosphotyrosine only after IFN-a stimulation (Fu, 1992; Schindler et al., 1992b). It was determined subsequently that one specific tyrosine residue in each protein was phosphorylated in response to IFN: amino acid 701 of STATI, and amino acid 690 of STAT2 (Shuai et al., 1993a, 1993b; Improta et al., 1994) (Fig. 1).
Specific antisera prepared to the recombinant STAT1 and STAT2 proteins were used in coimmunoprecipitation analyses to demonstrate physical association of the STAT factors only following IFN-a stimulation (Schindler et al., 1992b). In addition, immunofluorescence studies confirmed the cytoplasmic to nuclear translocation of STAT1 and STAT2 within minutes of IFN-α binding to its receptor (Fig. 2).
FIG. 2.
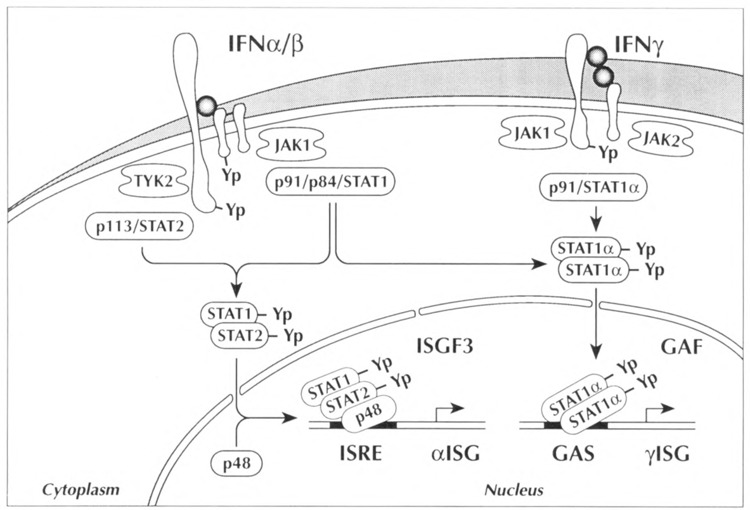
Illustrative model of the IFN signal transduction pathway. Binding of interferons (IFNs) to their respective receptors induces the activity of JAK kinases and tyrosine phosphorylation of STAT molecules as described in the text. The activated STAT-containing transcription factors, IFN stimulated gene factor 3 (ISGF3), and the IFN-γ activated factor (GAF) translocate to the nucleus and bind to the IFN stimulated response element (ISRE) or the γ activated site (GAS) in the promoters of inducible genes.
Activation of αlSGs in the Absence of IFN-α/β
In addition to transcriptional activation in response to IFNs-α/β, it has been found that the transcription of αISGs is induced by infection of cells with virus or transfection of cells with ds-RNA in the absence of IFN-α action (Wathelet et al., 1992; Bazzigher et al., 1992). This induction is mediated through the same DNA target that is responsive to IFN-α, the ISRE; however, αISG expression is not stimulated by ISGF3, but by unique dsRNA-activated factors (DRAFs) (Daly and Reich, 1993). Studies performed with dsRNA-transfected cells identified two ISRE binding factors in nuclear extracts, DRAF1 and DRAF2, that could be distinguished by different electrophoretic mobilities during EMSA analysis. The DRAFs can be activated independently of new protein synthesis in cells unresponsive to IFN-α or in cells deficient in the IFN gene locus and therefore independent of the IFN- α signal pathway. In addition, the effects of tyrosine kinase inhibitors demonstrated the requirement of tyrosine phosphorylation for both DRAF activation and stimulation of αISG transcription by dsRNA. Therefore, the primary defense response of a cell to virus infection appears to be the activation of latent DRAFs. The DRAFs in turn activate transcription of the antiviral alSGs before IFN synthesis, thus protecting the cell at an early stage of infection.
GENE ACTIVATION BY IFN-γ
IFN-γ elicits many of the physiological effects induced by IFNs-α/β, including the generation of a cellular antiviral response and the inhibition of cellular proliferation. However, IFN-γ also has unique stimulatory effects in cells of the immune system (reviewed in Farrer and Schreiber, 1993). It is a major stimulator of macrophages, activating both nonspecific cytocidal activity against pathogens and antibody-dependent cytotoxicity via induced expression of cell surface immuno-globulin receptors (FcR). It induces major histo-compatibility class (MHC) II protein expression in many cells, thereby promoting antigen presentation to T lymphocytes, and it regulates immuno-globulin class switching in B lymphocytes. Many of these regulatory effects are generated by the transcriptional activation of a subset of genes, in some cases restricted to a specific cell type.
DNA Element Responsive to IFN-γ
The experimental approach that identified cis-regulatory DNA sequences and trans-acting factors in the IFN-α/β signal pathway likewise was employed to investigate the IFN-7 pathway. Genes were isolated that encoded known IFN-γ-induced proteins (Boss and Strominger, 1986; Cheng et al., 1986; LeClair et al., 1986; Newburger et al., 1988; Pearse et al., 1991), or IFN- γ -induced genes were isolated by differential screening of cDNA libraries prepared from IFN- γ -treated cells (Luster et al., 1985; Caplen and Gupta, 1988; Fan et al., 1988; Wright and Farber, 1991). Studies of transcriptional expression revealed that induction of a subset of these genes by IFN-γ, including MHC II, required new protein synthesis (Blanar et al., 1988). Activation of this subset therefore appears to be a secondary response to IFN-γ and dependent on the synthesis of an intermediary transcription factor. The genes that respond in a primary manner to IFN-γ are induced within minutes, independent of protein synthesis. Characterization of IFN-γ-stimulated genes (γISGS) that respond immediately to IFN-γ has revealed the requirement of a conserved DNA response element (Table 1).
Identification of the DNA regulatory sequences involved in activation of γISGs was determined with transfection studies. Hybrid constructs containing the promoter region of a γISG linked to a reporter gene were introduced into cells and induced by IFN-γ treatment. Deletion analyses of the transfected hybrids identified DNA promoter sequences necessary for gene activation. The promoter of a gene encoding a guanylate binding protein (GBP) was found to contain an IFN-a-responsive ISRE that overlapped with a novel IFN-γ-responsive sequence, termed the IFN-γ activation site (GAS) (Lew et al., 1991). The GBP gene was known to be induced by either IFN-α or IFN-γ, and the presence of two distinct DNA response elements accounted for the response to both stimuli. Introduction of mutations within the ISRE impaired IFN-a induction of the GBP gene, but had little effect on transcriptional induction by IFN-γ.
The gene encoding the IRF-1/ISGF2 transcription factor that binds to the inner core of the ISRE is induced preferentially by IFN-γ but also is induced by IFN-α. Transfection analyses of genes containing the IRF-1 promoter identified a single palindromic sequence at 123 nucleotides upstream of the transcriptional start site that was required for the response to both types of IFN. The DNA palindrome contains an inverted repeat of the sequence GAAA (Table 1). This result suggested that both IFN-a and IFN-γ could mediate a primary response via the same DNA target site.
IFN-γ induces expression of the high-affinity immunoglobulin receptor FCγRI, but expression is restricted to cells of the myeloid lineage (Pearse et al., 1994). A region of the FCγRI promoter was identified by transfection analyses to confer IFN-γ inducibility to a heterologous promoter. This DNA site, originally called the IFN-γ response region (GRR), contains a palindromic GAAA sequence similar to the inverted repeat found in the IRF-1 gene and can confer a transcriptional response to IFN-γ (Pearse et al., 1991, 1993; Perez et al., 1993, 1994). Recent studies have demonstrated the restricted myeloid expression of the Fc-γRI gene is due to the presence of a DNA binding site for the PU.l/Spi-1 ets family transcription factor adjacent to the IFN-7 response sequence. PU.l/Spi-1 is expressed in myeloid and B cells and appears to be required for expression of the FC7RI promoter (Perez et al., 1994).
DNA response elements have been characterized in the promoters of other γISGS, including the IFN consensus sequence binding protein (ICSBP) gene (Kanno et al., 1993), the monokine-induced gene (mig) (Wright and Farber, 1991), and the Ly6A/E gene (Khan et al., 1993). The ICSBP gene contains a conserved palindromic sequence homologous to the GAAA inverted repeat identified in IRF-1 and FC7RI, and the responsive sites in GBP, Ly6A/E, and mig contain imperfect palindromes. Although the sites are not identical in these genes, the response sequence has been termed the IFN-γ activated site (GAS). The GAS sequence in IRF-1, ICSBP, and FC7RI contains two half-sites of GAAA in an inverted palindrome, whereas the IFN-a activated ISRE contains two half-sites of GAAA in a tandem orientation (Table 1).
The IFN-γ Activated Transcription Factor, GAF
Because the γISGS are induced immediately following IFN-γ stimulation, independent of new protein synthesis, transcription must depend on activation of a preexisting transcription factor. An IFN-γ-induced DNA binding factor was identified by exonuclease protection experiments of the GAS in the GBP gene (Decker et al., 1991a). This DNA binding factor was called the IFN-γ activated factor (GAF), and was activated in the cytoplasm of the cell because it appeared in enucleated cells (cytoplasts) within minutes of IFN-γ treatment. The kinetics of activation of GAF paralleled the induction of GBP gene transcription. A similar DNA binding factor rapidly induced by IFN-γ was detected by EMSA analysis to bind to the GAS of the FcγRI gene (Wilson and Finbloom, 1992).This DNA binding activity originally was called the FcRFγ, but it appears to be similar to GAF. Activation of an IFN-γ-induced factor that recognizes a GAS site in the IRF-1 gene also was identified and originally termed IRFi, but it appears to be the same as GAF (Sims et al., 1993). Subsequent experiments demonstrated IFN-α induced the same GAF activity, although at reduced levels, in addition to the IFN-α-induced ISGF3 activity (Decker et al., 1991b; Sims et al., 1993).
To determine the molecular size of the protein in the GAF that contacts the DNA, UV cross-linking experiments were performed (Shuai et al, 1992). GAF was partially purified, bound to the GAS site of the Ly6E gene in an EMSA, and exposed to UV radiation. SDS polyacrylamide gel electrophoresis (PAGE) revealed a protein of approximately 90 kDa cross-linked to the radiola-beled DNA. Because the protein was similar in size to the p91/STATla subunit of ISGF3, antisera that recognized the STAT1α and STATβ proteins of ISGF3 were tested for reactivity with GAF. Indeed, the STAT1 antisera specifically bound to GAF-GAS complex in a gel shift assay, but the STAT2 antisera did not. These surprising results demonstrated that the signal transduction pathways of IFN-α/β and IFN-γ shared activation of a similar factor, STATlα.
Signal Activation of GAF
Similar to the IFN-α/β activation signal, the IFN-γ signal transduction pathway is dependent on the tyrosine phosphorylation of STATla. Phosphoamino acid analysis of STAT1α from [32P]orthophosphate-labeled cells detected phosphorylated tyrosine within minutes following IFN-γ stimulation (Schindler et al., 1992b). Sequence analysis of tryptic phosphopeptides identified the same tyrosine residue 701 to be phosphor-ylated on STATlα in response to IFN-γ or to IFN-a (Shuai et al., 1993a, 1993b). Protein kinase inhibitors such as staurosporine blocked the induced tyrosine phosphorylation of STATlα/GAF (Shuai et al., 1992). In addition, immunofluores-cence studies demonstrated cytoplasmic to nuclear translocation of STAT1, but not STAT2, following IFN-γ stimulation (Shuai et al., 1992).
IFN-α stimulation leads to the activation of both STAT1 and STAT2, which associate with p48 to form ISGF3, whereas IFN-γ induces the activation of only STAT1 to form GAF. However, STATla is phosphorylated on the same tyrosine residue 701 following stimulation with either IFN-α or IFN-γ (Shuai et al., 1993a, 1993b). Analysis of the sedimentation coefficient of GAF indicates that it is a homodimer of STATlα (Shuai et al., 1994). STATlα thereby appears to serve as a combinatorial partner in signal transduction. It can associate with STAT2 and p48 to generate the IFN-α-induced heteromeric complex ISGF3, or associate with itself to form homomeric STATla complexes as GAF (Fig. 2). The ability of STAT la to associate with STAT2 or other STAT molecules creates a diverse repertoire of transcription factor combinations activated by distinct extracellular ligands.
FUNCTIONAL ANALYSES OF STAT MOLECULES
To affirm the role of STAT1, STAT2, and p48 in the IFN signal transduction pathway, transfection analyses were performed with the recombinant cDNAs. These studies were enabled by the isolation of cell lines deficient in their response to IFNs (reviewed in Darnell et al., 1994). The laboratories of George Stark and Ian Kerr used a genetic approach to obtain mutant cell lines that do not respond to IFN (Pellegrini et al., 1989; John et al., 1991; McKendry et al., 1991). A cell line was established that expresses a transfected recombinant plasmid containing an IFN responsive promoter linked to the E. coli guanine phos-phoribosyltransferase (gpt) gene. These cells lack the hypoxanthine-guanine phosphoribosyltrans-ferase genes and cannot grow in media with hypo-xanthine-aminopterin-thymidine (HAT) unless they express the gpt gene. If the cells are induced by IFN to express the gpt gene they can grow in HAT media but will die in the presence of 6-thioguanine (6-TG). The stable transfected cells were treated with a frameshift mutagen, and subsequently placed in both 6-TG and IFN to select for the growth of mutant cells that do not respond to IFN. Several mutant cell lines unresponsive to IFN-α and/or IFN-γ were isolated in this manner. Somatic cell fusions with parent cells and with different mutant clones demonstrated defects that were recessive and belonged to discrete complementation groups.
The mutant cells were transfected with the STATI, STAT2, or p48 cDNA expression vectors in an attempt to rescue the IFN-responsive pheno-type and thereby identify the genetic defect. In addition, this technique corroborated the role of the cloned factors in the signal response to IFN. Complementation of one mutant line unresponsive to IFN-a was achieved by transfection with the p48 cDNA (John et al., 1991; Darnell et al., 1994). Complementation of a different line unresponsive to IFN-a was achieved by transfection with the STAT2 cDNA (Darnell et al., 1994). A third cell line unresponsive to either IFN-α or IFN-γ was complemented by transfection with STAT1 cDNA. This latter result attested to the role of STAT1 in the signal pathways of both IFNs (formation of ISGF3 and GAF) (Fig. 2).
Because the STAT1 gene encodes two proteins produced from differentially spliced mRNAs, p91/STATlα and p84/STATlβ, the role of each individual protein in IFN signaling was examined. The STATlα or the STAT1β cDNAs were transfected into a cell line lacking expression of ST AT1 and were analyzed for their ability to complement the IFN-α or IFN-γ response (Müller et al., 1993a). Both STAT1 cDNAs were found to complement the IFN-α response. Transfected cells displayed IFN-α-dependent growth in HAT media and death in 6-TG. Expression of either STATlα or STAT10 conferred inducibility of the ISGF3 transcription factor following IFN-a stimulation. Furthermore, expression of IFN-a-induced genes was activated in cells complemented with either STATlα or STAT1β cDNAs. Therefore, the 38 amino acids that are located at the carboxy-terminus of STATlα, but are absent in STAT1β, are not required for factor activation or association with STAT2 and p48 in the formation of ISGF3.
Complementation of the cell line lacking STAT1 also demonstrated a specific role of STAT1α, but not STAT1β, in the response to IFN-γ (Müller et al., 1993a). Only constructs expressing STAT1α restored the transcriptional response of genes to IFN-γ stimulation. Following IFN-γ treatment, both STAT1α and STATlβ became phosphorylated on tyrosine 701 and translocated to the nucleus (Shuai et al., 1993a). In addition, both STATlα and STAT1β formed a specific DNA binding complex in response to IFN-γ that recognized the GAS target sequence. However, transcriptional activation by IFN-γ of endogenous genes or transfected genes containing a GAS element was dependent on expression of STATlα, not STAT1β. The carboxy-terminus of STATlα may therefore be required for interaction with basal transcription factors of the cell.
To determine the functional importance of various protein domains in STATlα, a mutational analysis was undertaken. The TAT codon for tyrosine at position 701 was altered to a TTT codon for phenylalanine (F) (Shuai et al., 1993a). The F701 mutant STATlα was not tyrosine phosphorylated in response to IFN-γ after transfection into the STAT1-deficient cells. Immunofluorescence studies demonstrated that the F701 mutant protein did not translocate to the nucleus in response to IFN-γ, nor did it form a DNA binding GAF-GAS complex. Not surprisingly, the F701 mutant STATla did not activate expression of IFN-γ-stimulated genes. Because the STATI protein contains a region of similarity to SH2 domains (sites of binding to phosphotyrosine-containing proteins), the requirement of this region was tested (Shuai et al., 1993b). The function of an arginine residue within an SH2 domain appears to be critical for binding to phosphotyrosine residues. For this reason, the CGG codon for arginine at position 602 in STATlα was changed to a CTG codon for leucine (L). Transfection analyses of the L602 mutant protein revealed that it did not become tyrosine phosphorylated in response to IFN-γ. Therefore, the SH2 domain of STATlα is required for it to be a substrate for tyrosine phos-phorylation. Recent experiments have demonstrated STATlα can bind to a phosphotyrosine site on the intracellular domain of the receptor, an association that appears to be required for activation (Greenlund et al., 1994).
Glycerol gradient sedimentation analyses and native gel electrophoresis studies of the physical properties of STATlα suggested that the factor exists as a monomer in the cytoplasm prior to IFN-γ stimulation and as a dimer in the nucleus following IFN-γ stimulation (Shuai et al., 1994). These studies were further supported with the demonstration of heterodimerization between STATlα and STAT1β in vivo by coexpression of the two corresponding cDNAs in the STAT1-deficient cell line. The coexpression produced a novel DNA binding complex following IFN-γ treatment that appeared as a band of intermediate migration in a gel shift assay, faster than the STATlα homodimer and slower than the STAT1β homodimer, representing the STATI heterodimer. An in vitro association of the ST ATI factors also can be demonstrated after denaturation and rena-turation of the dimers, indicating association is noncovalent. For this reason, the role of the SH2 domain and phosphotyrosine residues in the formation of dimers was examined. A specific tyrosyl phosphorylated peptide corresponding to STATlα was shown to dissociate dimers at high concentration (160 μM) and to promote subunit interchange at lower concentrations (4 μM). A phosphotyrosyl peptide corresponding to STAT2 of ISGF3 could also displace the STAT1 subunits of GAF, but a phosphotyrosyl peptide of the Src protein had no effect. In addition, the SH2 domain of STATlα could promote STATI subunit interchange. These results suggest that the STATI dimerization required for GAF formation is dependent on interaction of a phosphotyrosine residue on one sub-unit with the SH2 domain of the other subunit. The peptide of ST ATI that is tyrosine phosphorylated (pYIKTELI) and the homologous peptide of STAT2 that is tyrosine phosphorylated (pYLKHRLI) may interact with other unidentified SH2-containing molecules. This phosphopeptide motif appears to represent a novel consensus domain that has not been described to interact with other known SH2-containing proteins (Songyang etal., 1993).
A mutational analysis of protein domains in STATlα was also performed to determine their role in signal activation of ISGF3 by IFN-α (Improta et al., 1994). The tyrosine mutant, F701, and the SH2 region mutant, L602, both were defective in their ability to complement STATl-deficient cells in the formation of ISGF3. The mutant proteins were not tyrosine phosphorylated, did not associate with STAT2, and did not translocate to the nucleus in response to IFN-a stimulation. A deletion of the amino-terminal 216 amino acids of STATlα or a deletion of amino acids 199-220 in the central leucine repeat (LLVL) also abrogated the tyrosine phosphorylation of STATlα and the formation of ISGF3 in response to IFN-α. The tyrosine phosphorylation of STATlα may therefore require a variety of distinct protein domains that are necessary for protein-protein interactions. In addition, expression of these STATla mutants did not interfere with tyrosine phosphorylation of STAT2 on amino acid 690 or translocation of STAT2 to the nucleus following IFN-α stimulation.
JANUS AT THE GATES
Tyrosine phosphorylation of the STAT sub-units is critical for formation of the multimeric transcription factors ISGF3 and GAF in response to IFN-α/β or IFN-γ. Signal transduction must therefore depend on the activation of latent tyrosine kinases by IFN binding. However, the trans-membrane receptors that bind IFN do not possess intrinsic tyrosine kinase activity. The identity of the targeted kinase(s) remained unknown until a breakthrough experiment revealed the ability of a gene encoding a specific tyrosine kinase could complement a cell line unresponsive to IFN (Velazquez et al., 1992). This discovery provided a missing link in the IFN pathway that transduced a signal from the IFN receptor to latent transcription factors in the cytoplasm (Fig. 2).
A genetic system employing a panel of mutant cell lines unresponsive to IFN-α was used in a similar manner as that described to analyze the function of the STAT proteins (Velazquez et al., 1992). In seeking to identify genes that could complement the mutant cells, transfections were performed with genomic DNA and a gene encoding neomycin resistance. Neomycin-resistant clones were isolated and tested for reversion to an IFN-α-responsive phenotype (death in HAT or death in IFN plus 6-GT). DNA from a complemented cell line was used to construct a cosmid library for subsequent transfections, cDNA isolation, and gene identification. Nucleotide sequence analysis revealed the complementing gene to be tyk2, previously isolated by homology to the tyrosine ki-nase domain of c-fms (Krolewski et al., 1990). The function of Tyk2 remained unknown until its role in IFN-a signaling became apparent with these complementation experiments. Tyk2 is found to be phosphorylated on tyrosine within minutes following IFN-α/β stimulation (Müller et al., 1993b; Barbierietal., 1994).
Tyk2 is now known to be a member of the Janus kinase (JAK) family of nonreceptor tyrosine kinases. This family consists of three additional members, JAK1, JAK2, and JAK3, that were isolated by the polymerase chain reaction with oligo-nucleotide primers homologous to consensus tyrosine kinase domains (Wilks, 1989; Wilks et al., 1991; Cance et al., 1993; Sanchez et al., 1994; Kawamura et al., 1994; Witthuhn et al., 1994). Janus is a mythological Roman god and protector of the gates of Rome and he is portrayed with two faces looking in opposite directions. The Janus nomenclature appears appropriate because these enzymes contain two tandem kinase domains and assist in the transduction of extracellular signals.
Because Tyk2 restored the IFN-α response of one mutant cell line, the natural course was to determine if transfection of genes encoding the other JAK members could complement the deficiency of other mutant cells. A cell line defective in its response to both IFN-α and IFN-γ was found to be restored by transfection with JAK1 cDNA (Müller et al., 1993b). In addition, JAK1 is tyrosine phosphorylated within minutes following IFN-α or IFN-γ stimulation (Müller et al., 1993b; Shuai et al., 1993b; Silvennoinen et al., 1993a). The signal transduction pathway of IFN-α therefore activates two homologous kinases, Tyk2 and JAK1. This kinase duo acts in concert to phos-phorylate the STAT1 and STAT2 factors leading to the formation of ISGF3. It appears that there is a reciprocal interdependence for activation of these kinases by IFN-α. Tyrosine phosphorylation of Tyk2 is dependent on the presence of JAK1, and, in turn, tyrosine phosphorylation of JAK1 is dependent on the presence of Tyk2 (Müller et al., 1993b). The lack of a demonstrable hierarchy in Tyk2 and JAK1 activation suggests that the kinases are juxtaposed following receptor oligomerization (Fig. 2).
The IFN-γ signal pathway also stimulates the activation of two JAK kinases, JAK1 and JAK2. Genetic complementation of a cell line unresponsive to IFN-γ, but normally responsive to IFN-α, was achieved by transfection of cDNA encoding JAK2 (Watling et al., 1993). This result, in conjunction with the ability of JAK1 to complement the cell line unresponsive to both IFN-α and IFN-γ, provided evidence that activation of both JAK1 and JAK2 is required to respond to IFN-γ. Similar to the IFN-α pathway, there is an interdependence of each kinase for tyrosine phosphorylation, phosphorylation of STATlα, and activation of gene expression (Fig. 2).
The only evidence for the involvement of a protein tyrosine phosphatase (PTPase) in the IFN-γ signal pathway has relied on studies with vana-date, a specific PTPase inhibitor. Vanadate treatment of cells appears to stimulate the GAS binding activity of STATlα in the absence of added IFN-γ (Igarashi et al., 1993). In addition, vanadate prolongs the transcriptional activation induced by IFN-α (David et al., 1993).
INTERFERON RECEPTORS
The biological effects of IFN are initiated by their binding to specific high-affinity transmem-brane receptors. IFNs-α and -β bind to a common type I receptor, whereas IFN-γ binds to a distinct type II receptor. Both type I and type II receptors are multimeric and are composed of at least two nonidentical subunit proteins.
IFN-α/β Receptor
Human IFNs-α generally have a low affinity for murine IFN-α receptors. This species specificity formed the basis of the molecular cloning of a subunit of the IFN-α receptor (IFNAR1) (Uzét al., 1990). Murine cells were transfected with human DNA and selected for expression of a functional human IFN-α receptor. The positive selection was based on the ability of human IFN-a to confer resistance to infection of murine cells with vesicular stomatitis virus. DNA from a secondary transfectant was used to construct a genomic library in bacteriophage λ that was screened with a human Alu repeat to isolate the transfected human DNA. This genetic transfer technique identified a cDNA encoding a protein of 557 a.a. with a single transmembrane spanning segment. This protein is now defined as a class II cytokine receptor and contains two extracellular homologous domains with repeats of four cysteine residues (Ba-zan, 1990).
Following IFN-α binding, the IFNAR1 receptor subunit is tyrosine phosphorylated in a dose-and time-dependent manner (Platanias and Cola-monici, 1992). In addition, IFNAR1 has been demonstrated to be physically associated with the Tyk2 tyrosine kinase (Colamonici et al., 1994). Antibodies that recognize IFNAR1 can coimmu-noprecipitate Tyk2 as assayed by immunoblotting, and antibodies that recognize Tyk2 can coimmu-noprecipitate IFNAR1 bound to radiolabeled [125I]IFN-α2. The physical association of IFNAR1 and Tyk2 is found both in the absence and presence of IFN, suggesting a stable complex exists prior to ligand binding, and that Tyk2 phosphory-lates IFNAR1 after ligand binding.
Recently, a second component of the IFN-α receptor (IFNAR2) was identified (Novick et al., 1994). This subunit was detected initially as a soluble factor able to block the activity of IFN-a. The protein was purified and used to prepare specific antibodies. These antibodies blocked the biological response to IFN-α, and immunoprecipitated a disulfide-linked dimer of 51 kDa subunits associated with the membrane. Degenerate oligonucleo-tides corresponding to the protein sequence were used in a polymerase chain reaction to generate a DNA fragment for subsequent isolation of the gene from a bacteriophage λ library. The soluble form of the receptor (41 kDa) was found to be encoded by the amino-terminal region and did not contain the transmembrane domain. The extracellular sequence of IFNAR2 is approximately 23% identical with the extracellular ligand binding domain of IFNAR1, and binds to [125I]IFN-α2 or IFN-β. Of particular significance to our understanding of the IFN-α signal transduction pathway was the demonstration of a physical association of IFNAR2 with JAK1 both before and after IFN binding. Antibodies that recognize IFNAR2 coimmunoprecipitate JAK1, but not Tyk2 or JAK2. It therefore appears that IFNAR1 is associated with Tyk2, and that IFNAR2 is associated with JAK1. IFN binding likely causes the oligo-merization of these receptor components, leading to the codependent activation of Tyk2 and JAK1 and subsequent tyrosine phosphorylation of STATI and STAT2.
IFN-γ Receptor
The molecular cloning of the human IFN-γ receptor gene was achieved with the use of a poly-clonal antiserum prepared against the purified receptor to screen a bacteriophage γgtll cDNA expression library (Aguet et al., 1988). The isolated IFN-γ receptor cDNA encoded a protein of 489 a.a. with a single putative transmembrane domain. To ensure that the isolated gene was functional, the human IFN-γ receptor gene was transfected and expressed in murine cells because human IFN-γ does not bind to murine IFN- γ receptors. Murine cells transfected with the human IFN-γ receptor gene were found to express high-affinity binding to human IFN- γ. However, human IFN- γ binding to the transfected receptor did not stimulate a biological response. Therefore, the IFN- γ receptor is required for binding to IFN- γ, but it is not sufficient to activate signal transduction.
Recently, a species-specific accessory factor (AF-1) required for signal transduction via the human IFN- γ receptor was identified (Soh et al., 1994). The strategy for isolation of this factor was based on construction of a yeast artificial chromosome (YAC) with genomic human DNA that could complement the function of the IFN- γ receptor. Probes derived from the YAC clone were used to isolate the cDNA encoding AF-1, a protein of 337 a.a. with a single putative transmembrane domain. Expression of both the AF-1 cDNA and the IFN- γ receptor gene reconstituted IFN- γ -induced gene expression. An analogous receptor system has been demonstrated in murine cells with the isolation of the murine IFN- γ receptor gene (Hemmi et al., 1989; Gray et al., 1989) and a murine accessory component designated the IFN-γ receptor β chain (Hemmi et al., 1994).
An enhanced understanding of the mechanism whereby the IFN-γ receptor transmits a signal into the cell was provided with the demonstration of a physical association between the IFN-γ receptor and STATlα following ligand binding (Greenlund et al., 1994). IFN-γ induced the rapid and transient tyrosine phosphorylation of a specific intra-cellular site (Y440) of the IFN-γ receptor, a site previously shown to be required for signal transduction. The presence of the AF-1 (or β chain) of the IFN-γ receptor was required for this ligand-induced tyrosine phosphorylation. The specific peptide corresponding to phosphorylated Y440 of the receptor was shown to associate physically with STATlα; the phosphorylated receptor peptide disrupted the STATlα homodimer of GAF when added to the DNA binding reaction of a gel mobility shift assay, and STATlα coimmunopre-cipitated with the tyrosine phosphorylated receptor peptide. In addition, the phosphorylated receptor peptide blocked STATlα tyrosine phosphorylation induced in a ligand-dependent cell-free system. These results have provided a conceptual model for STATlα activation via the IFN-γ receptor. A membrane proximal intracellular region of the IFN-γ receptor chains may be associated with the JAK kinases that are activated following IFN-γ binding. Activated JAK kinase phosphorylates the Y440 region of the receptor chain, and this tyrosine phosphorylated peptide recruits STATlα via its SH2 domain. Once STATlα is recruited to the receptor, it becomes tyrosine phosphorylated by the JAK kinase and subsequently dissociates from the receptor to homodi-merize with another tyrosine phosphorylated STATlα molecule and translocate to the nucleus.
JAK-STAT ACTIVATION BY DIVERSE CYTOKINES AND GROWTH FACTORS
The discovery of the JAK-STAT pathways in the interferon system has served as a model for receptor to nucleus signal transduction by a variety of cytokines and growth factors (Table 2). Growth factors such as epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) bind to transmembrane receptors that possess intracellular domains with tyrosine kinase activity. The binding of the growth factor induces homodi-merization of the receptors, subsequent activation of the intrinsic tyrosine kinase, and receptor auto- phosphorylation (reviewed in Ulrich and Schlessinger, 1990; Fantl et al., 1993). Following EGF or PDGF binding, specific expression of the c-fos gene is induced. This transcriptional induction appears to be a consequence of the rapid activation of a specific c-sxs-inducible factor (SIF) that binds to a DNA target site in the c-fos promoter called the c-sis-inducible element (SIE) (Hayes et al., 1987; Wagner et al., 1990). The c-fos SIE is an imperfect palindromic sequence similar to the GAS found in IFN-y-inducible genes. Electropho-retic mobility shift analyses with the SIE (usually performed with a mutated SIE possessing improved dyad symmetry) revealed three distinct SIF complexes (Sadowski et al., 1993; Ruff-Jamison et al., 1993; Silvennoinen et al., 1993b; Fu and Zhang, 1993). With the use of specific antibodies, STATlα was shown to be present in two of the SIF complexes; as a STATlα homodimer and as a heterodimer with a novel STAT molecule, STAT3. The third SIF complex contained a homodimer of STAT3 (Zhong et al., 1994a). This novel STAT3 family member was isolated initially by amplifying the STATla SH2 domain with degenerate primers in a polymerase chain reaction and screening a cDNA library (Zhong et al., 1994b). STAT3 possesses domain homology to STAT1 and approximately 50% amino acid identity. This cloning approach also generated a fourth member of the family, STAT4, for which expression appears to be restricted to specific cell lineages (Zhong et al., 1994b; Yamamoto et al., 1994).
TABLE 2.
A LIST OF CYTOKINES AND THEIR IDENTIFIED JAK AND STAT SIGNALING COMPONENTS AS DESCRIBED IN THE TEXT
| Cytokine | JAK | STAT | DNA Target |
|---|---|---|---|
| IFN-α | Tyk2, JAK1 | STAT1,STAT2 | ISRE/GAS |
| IFN-γ | JAK1, JAK2 | STATlα | GAS |
| EGF | JAK1 | STATlα, STAT3 | GAS/SIE |
| PRL | JAK2 | STATlα, STAT5 | GAS |
| G-CSF | JAK1, JAK2 | STAT3 | GAS |
| IL-4 | JAK1, JAK3 | STAT6 | GAS |
| IL-6 | Tyk2, JAK1, JAK2 | STATlα, STAT3 | GAS/APRE |
| IL-2 | JAK1, JAK3 | IL-2-STAT | GAS |
| IL-3 | JAK2 | IL-3-STAT | GAS |
The activation of STATlα by EGF results in the tyrosine phosphorylation of STATlα on the same tyrosine 701 residue that is phosphorylated following IFN-α or IFN-y stimulation (Shuai et al., 1993a). In addition, STATlα was shown to physically associate with the EGF receptor after ligand binding (Fu and Zhang, 1993). Association with the activated EGF receptor requires the presence of the SH2 domain of the STATlα protein, suggesting that a tyrosine phosphorylated residue on the EGF receptor may initiate association. However, it remains to be determined whether the EGF receptor phosphorylates STATla, because it has been found that JAK1 kinase is also activated following EGF binding to its receptor and STATlα may be a substrate for this kinase (Shuai et al., 1993b). The ability of STATlα to hetero-dimerize with the coactivated STAT3 represents another example of the combinatorial power of STATla not only in the IFN system but also in response to diverse extracellular stimuli like EGF.
Cytokines that function in immune and hema-topoietic systems bind to cell surface receptors that do not possess intrinsic tyrosine kinase activity (reviewed in Kishimoto et al., 1994). However, signal transduction and gene expression stimulated by many of these cytokines requires tyrosine phosphorylation. Cytokines, including interleu-kin-6 (IL-6), ciliary neurotrophic factor (CNTF), leukemia inhibitory factor, and oncostatin M, bind to receptors that share a common gpl30 sub-unit. In the liver, IL-6 is the major inducer of acute phase response genes. The promoter region of these genes contains two different IL-6 response elements that bind either nuclear factor IL-6 (NF IL-6, a member of the C/EBP family) or the acute phase response factor (APRF). APRF binds to an acute phase response element (APRE) that resembles a GAS sequence. Purification and subsequent cloning of APRF revealed it to be STAT3 (Akira et al., 1994). As with stimulation by EGF, ligand binding to gpl30 stimulates activation of both STATlα and STAT3 (Sadowski et al., 1993; Bonni et al., 1993; Wegenka et al., 1994; Zhong et al., 1994a; Akira et al., 1994). Examination of kinase activation revealed induction of Tyk2, JAK1, and JAK2 following ligand binding to gpl30 (Narazaki et al., 1994; Lutticken et al., 1994; Stahl et al., 1994). In addition, STAT3 and JAK1 were demonstrated to physically associate with the gpl30 receptor subunit (Lutticken et al., 1994).
A common receptor β subunit is also shared by the cytokines interleukin-3 (IL-3), interleukin-5 (IL-5), and granulocyte/macrophage colony stimulating factor (GM-CSF) (reviewed in Kishimoto et al., 1994). Stimulation of these cytokine receptors induces the tyrosine phosphorylation of a latent STAT factor that can bind to a GAS sequence (Larner et al., 1993). The activated STAT molecule does not react with antibodies that recognize STATla, and the identification of this STAT family member remains to be determined. However, with the use of specific JAK antibodies, IL-3 was shown to induce the rapid tyrosine phosphorylation of JAK2 (Silvennoinen et al., 1993c).
Cytokines such as erythropoietin (EPO), granu-locyte colony stimulatory factor (G-CSF), growth hormone (GH), and prolactin (PRL) generate ho-modimerization of their respective receptors and subsequent signal transduction. EPO and GH both stimulate the tyrosine phosphorylation of JAK2, but activate distinct STAT molecules (Witthuhn et al., 1993; Argetsinger et al., 1993; Finbloom et al., 1994). G-CSF stimulates the tyrosine phosphorylation of JAK1 and JAK2, leading to the activation of latent STAT3 (Nicholson et al., 1994; Tian et al., 1994). Studies of PRL signal activation in mammary epithelial cells led to the purification and molecular cloning of a fifth member of the STAT family, called mammary gland factor (MGF) or STAT5, that binds to a GAS-like element in the 0-casein gene promoter (Wakao et al., 1994). T lymphocytes also respond to PRL with the activation of a latent PRL-induced factor (PRLIF) that binds to a GAS site in the IRF-1 gene, and PRLIF may be identical to STAT5 (Gil-mour and Reich, 1994). In addition to PRLIF, PRL also activates STATlα in T lymphocytes (Gilmour and Reich, 1994; David et al., 1994). STAT5 and STATlα activation by PRL appears to be the result of tyrosine phosphorylation by JAK2 kinase.
IFN-γ and interleukin-4 (IL-4) have been known to generate antagonistic as well as analogous effects on specific gene expression. Indeed, IL-4 was found to activate a specific IL-4 nuclear activated factor (IL-4 NAF) that recognized a GAS site in regulated genes (Kotanides and Reich, 1993; Kohler and Rieber, 1993). IL-4 NAF was subsequently purified and molecularly cloned and is known as IL-4 STAT or STAT6 (Hou et al., 1994). Two JAK family members appear to be activated following IL-4 stimulation, JAK1 and JAK3 (Johnston et al., 1994; Witthuhn et al., 1994). JAK3 is a new member of the JAK family that was isolated by amplification of tyrosine kinase domains with the polymerase chain reaction (Cance et al., 1993; Sanchez et al., 1994; Kawa-mura et al., 1994). JAK1 and JAK3 are also activated in response to interleukin-2 (IL-2) (Johnston et al., 1994; Witthuhn et al., 1994). This finding is in accord with the fact that IL-4 binds to a heteromeric receptor that contains the γ chain of the IL-2 receptor (Kondo et al., 1993; Russell et al., 1993). IL-2 has also been demonstrated to induce a specific DNA binding protein, IL-2 nuclear activated factor (IL-2 NAF), which recognizes the GAS DNA element of the IRF-1 gene (Gilmour and Reich, 1994). IL-2 NAF has a similar mobility to PRLIF in gel mobility shift assays and may be related to STAT5 (Gilmour and Reich, 1994).
WHEREIN LIES THE SPECIFICITY?
The signal transduction pathway of IFN culminates in the transcriptional induction of IFN-responsive genes via the activation of specific JAK and STAT molecules. The IFN pathway has served as a model for signal transmission by other cytokines that stimulate specific gene expression. Because a number of cytokines share JAK and STAT components in their signal transduction pathway, and because many genes contain GAS elements in their promoters, it is important to consider the manner in which specificity of transcriptional activation is achieved.
A critical determinant of physiological response to a cytokine is the expression of specific cell surface receptors. Receptor expression in many cases is restricted to a particular cell lineage. In addition, many receptors, such as the IL-2 receptor, consist of heteromeric protein subunits encoded by separate genes. Furthermore, a specific receptor component can be shared with alternate sub-units to create distinct receptors recognized by different cytokines. For example, the IL-2 γ chain is a shared component of the IL-4 receptor, as the IL-3 β common chain is shared by the IL-5 receptor. Regulating expression of particular receptors limits biological response to the ligand.
Activation of individual members of the JAK family appears to be regulated both by tissue-specific expression and by physical association with a particular receptor. For instance, the molecular cloning of JAK3 revealed its expression to be restricted to lymphocytes and myeloid cells. With respect to JAK recognition of specific receptors, association of Tyk2 kinase is seen with the IFNAR1 chain of the IFN-α receptor whereas JAK1 kinase is associated with the IFNAR2 chain of the IFN-α receptor. It appears that a membrane proximal intracellular region of the receptor chain is associated with a specific JAK kinase prior to ligand binding. Subsequent to receptor oligomerization induced by cytokine binding, the JAK kinases may interact, resulting in their autophos-phorylation or cross-phosphorylation. Different combinations of JAKs will be activated dependent upon receptor subunit composition. The receptor subunit itself is thought to be a substrate for an activated JAK and the resultant tyrosine phos-phorylated site on the receptor creates a new docking site for proteins with specific SH2 domains.
The STAT transcription factors possess SH2 domains that can recognize particular tyrosine phosphorylated sites in the receptors. As with individual members of the JAK family, expression of some STAT family members, such as STAT4, is restricted to specific cell lineages. In addition, different STAT molecules appear to recognize distinct tyrosine phosphorylated receptors. A conceptual model has been proposed wherein a specific STAT molecule docks to a phosphorylated receptor, thereby bringing it to the vicinity of the activated JAK kinase. The JAK kinase phospho-rylates the STAT molecule, promoting its association with other STAT molecules via SH2 and phosphotyrosine domains. The tyrosine phosphorylated STATs can function as homodimers (STATla in GAF), heterodimers (STATlα and STAT3 in SIFc), or oligomers (STAT1 and STAT2 with p48 in ISGF3). The variety of STAT molecules and theoretical STAT combinations contribute to signal diversity and specificity. The STAT-containing transcription factors translocate from the cytoplasm to the nucleus, bind to specific DNA response elements, and thereby regulate transcription.
The specificity of gene activation induced by distinct cytokines must in part lie in differential affinity/recognition of the STAT transcription factors for variant DNA sequences. The IFN-α-stimulated response element possesses a tandem GAA repeat whereas the response elements of IFN-γ and the growth factors and cytokines described here possess an inverted GAA repeat or palindrome (Table 1). Variations in the palindrome can be achieved either by specific nucleo-tide substitution or by differential nucleotide spacing between the inverted repeats. The importance of spacing previously has been demonstrated for the retinoic acid family of receptors. The spacing between a direct repeat sequence determines which hormone receptors can bind (reviewed in Mangels-dorf and Evans, 1992). Different STAT dimers may require specific spacing between the inverted repeat structure or specific nucleotide sequences and therefore different homomeric or heteromeric STATs may bind distinct GAS elements. In addition, the promoter context of the STAT binding site has also been found to influence gene expression (Perez et al., 1993).
The physiological response to cytokines is dependent on the transcriptional induction of specific genes. Specificity can be conferred by the stimulation of distinct receptors, JAKs, STATs, and DNA targets. Achieving specificity by using various combinations of signaling components is an evolutionarily sound approach. Cells can elicit specific biological responses to various cytokines while limiting the number of proteins they express. The future will surely lead to the discovery of new members of the JAK and STAT families, and to an understanding of STAT interaction with the basal transcriptional machinery of the cell.
ACKNOWLEDGEMENTS
We would like to thank the members of the laboratory for their support, the National Institutes of Health (CA50773), the American Cancer Society (NP-882), and the Council for Tobacco Research (3317M).
REFERENCES
- Aguet M., Dembic Z., and Merlin G. (1988), Cell 55, 273–280. [DOI] [PubMed] [Google Scholar]
- Akira S., Nishio Y., Inoue M., Wang X-j., Wei S., Matsusaka T., Yoshida K., Sudo T., Naruto M., and Kishimoto T. (1994), Cell 77, 63–71. [DOI] [PubMed] [Google Scholar]
- Argetsinger L. S., Campbell G. S., Yang X., Witthuhn B. A., Silvennoinen O., Ihle J. N., and Carter-Su C. (1993), Cell 74, 237–244. [DOI] [PubMed] [Google Scholar]
- Barbieri G., Velaquez L., Scrobogna M., Fellous M., and Pelligrini S. (1994), Eur J Biochem 223, 427–435. [DOI] [PubMed] [Google Scholar]
- Baron S., Tyring S. K., Fleischman W. R., Coppenhaver D. H., Niesel D. W., Klimpel G. R., Stanton J., and Hughes T. K. (1991), JAMA 266, 1375–1383. [DOI] [PubMed] [Google Scholar]
- Bazan J. F. (1990), Immunol Today 11, 350–354. [DOI] [PubMed] [Google Scholar]
- Bazzingher L., Pavlovic J., Haller O., and Staeheli P. (1992), Virology 186, 154–160. [DOI] [PubMed] [Google Scholar]
- Benach P., Vigneron M., Peretz D., Revel M., and Chebath J. (1987), Mol Cell Biol 7, 4498–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanar M. A., Boettger E. C., and Flavell R. A. (1988), Proc Natl Acad Sci USA 85, 4672–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomstrom D. C., Fahey D., Kutny R., Korant B., and Knight E. Jr. (1986), J Biol Chem 261, 8811–8816. [PubMed] [Google Scholar]
- Bonni A., Frank D. A., Schindler C., and Greenberg M. E. (1993), Science 262, 1575–1579. [DOI] [PubMed] [Google Scholar]
- Boss J. M. and Strominger J. L. (1988), Proc Natl Acad Sci USA 83, 9139–9143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cance W. G., Craven R. J., Weiner T. M., and Liu E. T. (1993), Int J Cancer 54, 571–577. [DOI] [PubMed] [Google Scholar]
- Caplen H. S. and Gupta S. (1988), J Biol Chem 263, 332–339. [PubMed] [Google Scholar]
- Chebath J., Merlin G., Metz R., Benech P., and Revel M. (1983), Nucleic Acids Res 11, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y-S., Becker-Manley M. F., Nguyen T., Degrado W., and Jonak G. (1986), J Interferon Res 6, 417–427. [DOI] [PubMed] [Google Scholar]
- Cohen B., Peretz D., Viaman D., Benech P., and Chebath J. (1988), EMBO J 7, 1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colamonici O. R., Uyttendaele H., Domonski P., Yan H., and Krolewski J. J. (1994), J Biol Chem 269, 3518–3522. [PubMed] [Google Scholar]
- Dale T. C., Ali Iman A. M., Kerr I. M., and Stark G. R. (1989), Proc Natl Acad Sci USA 86, 1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly C. and Reich N. C. (1993), Mol Cell Biol 13, 3756–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J. E. Jr., Kerr I. M., and Stark G. R. (1994), Science 264, 1415–1421. [DOI] [PubMed] [Google Scholar]
- David M., Grimley P. M., Finbloom D. S., and Larner A. C. (1993), Mol Cell Biol 13, 7515–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David M., Petricoin E. F. III, Igarashi K-I., Feldman G., Finbloom D. S., and Larner A. C. (1994), Proc Natl Acad Sci USA 91, 7174–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker T., Lew D., Mirkovitch J., and Darnell J. (1991a), EMBO J 10, 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker T., Lew D. J., and Darnell J. E. (1991b), Mol Cell Biol 11, 5147–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maeyer E. and De Maeyer-Guignard J. (1988), Interferons and Other Regulatory Cytokines, Wiley, New York. [Google Scholar]
- Diaz M. O., Bohlander S., and Allen G. (1993), J Interferon Res 13, 61–62. [DOI] [PubMed] [Google Scholar]
- Fan X-D., Goldberg M., and Bloom B. (1988), Proc Natl Acad Sci USA 85, 5122–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantl W. J., Johnson D. E., and Williams L. T. (1993), Annu Rev Biochem 62, 453–481. [DOI] [PubMed] [Google Scholar]
- Farrar M. A. and Schreiber R. D. (1993), Annu Rev Immunol 11, 571–611. [DOI] [PubMed] [Google Scholar]
- Finbloom D. S., Petricoin E. F. III, Hackett R. H., David M., Feldman G. M., Igarashi K-I., Fiebach E., Weber M. J., Thorner M. O., Silva C. M., and Larner A. C. (1994), Mol Cell Biol, 14, 2113–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freidman R. L., Manley S. P., McMahon M., Kerr I. M., and Stark G. R. (1984), Cell 38, 745–755. [DOI] [PubMed] [Google Scholar]
- Freidman R. L. and Stark G. F. (1985), Nature 314, 637–639. [DOI] [PubMed] [Google Scholar]
- Fu X-Y. (1992), Cell 70, 323–335. [DOI] [PubMed] [Google Scholar]
- Fu X-Y., Kessler D. S., Veals S. A., Levy D. E., and Darnell J. E. Jr. (1990), Proc Natl Acad Sci USA 87, 8555–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X-Y., Schindler C., Improta T., Aebersold R., and Darnell J. E. Jr. (1992), Proc Natl Acad Sci USA 89, 7840–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X-Y. and Zhang J-J. (1993), Cell 74, 1135–1145. [DOI] [PubMed] [Google Scholar]
- Gilmour K. C. and Reich N. C. (1994), Proc Natl Acad Sci USA 91, 68–50–6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray P. W., Leong S., Fennie E. H., Farrar M. A., Pingel J. T., Fernandez L. J., and Schreiber R. D. (1989), Proc Natl Acad Sci USA 86, 8497–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlund A. C., Farrar M. A., Viviano B. L., and Schreiber R. D. (1994), EMBO J 13, 1591–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribaudo G., Toniato E., Engel D. A., and Lengyel P. (1987), J Biol Chem 262, 11878–11883. [PubMed] [Google Scholar]
- Gutch M. J., Daly C., and Reich N. C. (1992), Proc Natl Acad Sci USA 89, 11411–11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes T. E., Kitchen A. M., and Cochran B. H. (1987), J Cell Physiol Suppl 5, 63–68. [DOI] [PubMed] [Google Scholar]
- Hemmi S., Peghini P., Metzler M., Merlin G., Dembic Z., and Aguet M. (1989), Proc Natl Acad Sci USA 86, 9901–9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmmi S., Bohni R., Stark G., Di Marco F., and Aguet M. (1994), Cell 76, 803–810. [DOI] [PubMed] [Google Scholar]
- Hou J., Schindler U., Henzel W. J., Ho T. C., Brasseur M., and McKnight S. L. (1994), Science 265, 1701–1706. [DOI] [PubMed] [Google Scholar]
- Hug H., Costas M., Staeheli P., Aebi M., and Weissmann C. (1988), Mol Cell Biol 8, 3065–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi K., David M., Finbloom D. S., and Larner A. (1993), Mol Cell Biol, 13, 1634–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Improta T., Schindler C., Horvath C. M., Kerr I. M., Stark G. R., and Darnell J. E. Jr. (1994), Proc Natl Acad Sci USA 91, 4776–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs A. and Lindemann J. (1957), Proc R Soc Ser B 147, 258.13465720 [Google Scholar]
- Isreal A., Kimura A., Fournier A., Fellous M., and Kourilsky P. (1986), Nature 322, 743–746. [DOI] [PubMed] [Google Scholar]
- John J., McKendry R., Pellegrini S., Flavell D., Kerr I. K., and Stark G. R. (1991), Mol Cell Biol 11, 4189–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. A., Kawamura M., Kirken R. A., Chen Y-Q., Blake T. B., Shibuya K., Ortaldo J. R., McVicar D. W., and O’Shea J. J. (1994), Nature 370, 151–153. [DOI] [PubMed] [Google Scholar]
- Kahn K. D., Shuai K., Lindwall G., Maher S. E., Darnell J. E. Jr., and Bothwell A. L. M. (1993), Proc Natl Acad Sci USA 90, 6806–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno Y., Kozak C. A., Schindler C., Driggers P. H., Ennist D. L., Gleason S. L., Darnell J. E. Jr., and Ozato K. (1993), Mol Cell Biol 13, 3951–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura M., McVicar D. W., Johnston J. A., Blake T. B., Chen Y-Q., Lai B. K., Lloyd A. R., Kelvin D. J., Stapes J. E., Ortaldo J. R., and O’Shea J. J. (1994), Proc Natl Acad Sci USA 91, 6374–6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D. S., Veals S. A., Fu X-Y., and Levy D. E. (1990), Genes Dev 4, 1753–1765. [DOI] [PubMed] [Google Scholar]
- Kishimoto T., Taga T., and Akira S. (1994), Cell 76, 253–262. [DOI] [PubMed] [Google Scholar]
- Köhler J. and Rieber E. P. (1993), Eur J Immunol 23, 3066–3071. [DOI] [PubMed] [Google Scholar]
- Kondo M., Takeshita T., Ishii N., Nakamura M., Wantanabe S., Arai K., and Sugamura K. (1993), Science 262, 1874–1877. [DOI] [PubMed] [Google Scholar]
- Korber B., Mermond N., Hood L., and Stroynowski I. (1987), Science 239, 1302–1306. [DOI] [PubMed] [Google Scholar]
- Kotanides H. and Reich N. C. (1993), Science 262, 1265–1267. [DOI] [PubMed] [Google Scholar]
- Krolweski J. J., Lee R., Eddy R., Shows T. B., and Dalla-Favera R. (1990), Oncogene 5, 277–282. [PubMed] [Google Scholar]
- Larner A. C., Jonak G., Cheng Y.-S. E., Korant B., Knight E., and Darnell J. E. Jr. (1984), Proc Natl Acad Sci USA 81, 6733–6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larner A. C., David M., Feldman G. M., Igarashi K-I., Hachett R. H., Webb D. S. A., Sweitzer S. M., Petricoin E. F. III, and Finbloom D. S. (1993), Science 261, 1730–1733. [DOI] [PubMed] [Google Scholar]
- Landschulz W. H., Johnson P. F., and McKnight S. L. (1988), Science 240, 1759–1764. [DOI] [PubMed] [Google Scholar]
- LeClair K. P., Palfree R. G. E., Flood P. M., Hammerling U., and Bothwell A. (1986), EMBO J 5, 3227–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel P. (1982), Annu Rev Biochem 51, 251–282. [DOI] [PubMed] [Google Scholar]
- Levy D. E., Larner A., Chauduri A., Babiss L. E., and Darnell J. E. Jr. (1986), Proc Natl Acad Sci USA 83, 8929–8933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D., Kessler D., Pine R., Reich N., and Darnell J. E. Jr. (1989), Genes Dev 2, 282–393. [DOI] [PubMed] [Google Scholar]
- Levy D. E., Kessler D. S., Pine R., and Darnell J. E. Jr. (1989), Genes Dev 3, 1362–1371. [DOI] [PubMed] [Google Scholar]
- Levy D. E., Lew D. J., Decker T., Kessler D. S., and Darnell J. E. Jr. (1990), EMBO J 9, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew D. J., Decker T., Strehlow I., and Darnell J. E. Jr. (1991), Mol Cell Biol 11, 182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luster A. D., Unkeless J. C., and Ravetch J. V. (1985), Nature 315, 671–676. [DOI] [PubMed] [Google Scholar]
- Lütticken C., Wegenka U. M., Yuan J., Bischmann J., Schindler C., Ziemiecki A., Harpur A. G., Wilks A. F., Yasukawa K., Tage T., Kishimoto T., Barbieri G., Pellegrini S., Sendtner M., Heinrich P. A., and Horn F. (1994), Science 263, 89–92. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf D. J. and Evans R. M. (1992), Transcriptional Regulation Book 2 (McKnight S. L. and Yamamoto K. R., eds.), Cold Spring Harbor Press, Cold Spring Harbor, NY, pp. 1137–1167. [Google Scholar]
- McKendry R., John J., Flavell D., Muller M., Kerr I. M., and Stark G. R. (1991), Proc Natl Acad Sci USA 88, 11455–11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin G., Chebath J., Benech P., Metz R., and Revel M. (1983), Biochemistry 80, 4904–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto M., Fujita T., Kimura Y., Maruyama M., Harada H., Sudo Y., Miyata T., and Taniguchi T. (1988), Cell 54, 903–913. [DOI] [PubMed] [Google Scholar]
- Müller M., Laxton C., Biscoe J., Schlinder C., Improta T., Darnell J. E. Jr., Stark G. R., and Kerr I. M. (1993a), EMBO J 12, 4221–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M., Briscoe J., Laxton C., Guschin D., Ziemiecki A., Silvennoinen O., Harpur A. G., Barbieri G., Witthuhn B. A., Schindler C., Pellegrini S., Wilks A. F., Ihle J. N., Stark G. R., and Kerr I. M. (1993b), Nature 366, 129–135. [DOI] [PubMed] [Google Scholar]
- Narazaki M., Witthuhn B. A., Yshida K., Silvennoinen O., Yasukawa K., Ihle J. N., Kishimoto T., and Tage T. (1994), Proc Natl Acad Sci USA 91, 2285–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburger P. E., Ezekowitz R. A. B., Whitney C., Wright J., and Orkin S. H. (1988), Proc Natl Acad Sci USA 85, 5215–5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson S. E., Oates A. C., Harpur A. G., Ziemiecki A., Wilks A. F., and Layton J. (1994), Proc Natl Acad Sci USA 91, 2985–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick D., Cohen B., and Rubinstein M. (1994), Cell 77, 391–400. [DOI] [PubMed] [Google Scholar]
- Pawson T. and Schlessinger J. (1993) Curr Biol 3, 434–442. [DOI] [PubMed] [Google Scholar]
- Pearse R. N., Feinman R., and Ravetch J. V. (1991), Proc Natl Acad Sci USA 88, 11305–11309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse R. N., Feinman R., Shuai K., Darnell J. E. Jr., and Ravetch J. V. (1993), Proc Natl Acad Sci USA 90, 4314–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini S., John J., Shearer M., Kerr I. M., and Stark G. R. (1989), Mol Cell Biol 9, 4605–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C., Wietzerbin J., and Benech P. D. (1993), Mol Cell Biol 13, 2182–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C., Coeffier E., Moreau-Gachelin F., Weitzerbin J., and Benech P. D. (1994), Mol Cell Biol 14, 5023–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S., Langer T. L., Zoon K. K., and Samuel C. E. (1987), Annu Rev Biochem 56, 727–778. [DOI] [PubMed] [Google Scholar]
- Pine R. (1992), J Virol 66, 4470–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pine R., Decker T., Kessler D. S., Levy D. E., and Darnell J. E. Jr. (1990), Mol Cell Biol 10, 2448–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias L. C. and Colamonici O. R. (1992), J Biol Chem 267, 24053–24057. [PubMed] [Google Scholar]
- Porter A. C. G., Chernajovsky Y., Dale T. C., Gilbert C. S., Stark G. R., and Kerr I. M. (1988), EMBO J 7, 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich N., Evans B., Levy D., Fahey D., Knight E., and Darnell J. (1987), Proc Natl Acad Sci USA 84, 6394–6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich N. C. and Darnell J. E. Jr. (1989), Nucleic Acids Res 17, 3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich N. C. and Pfeffer L. M. (1990), Proc Natl Acad Sci USA 87, 8761–8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid L. E., Brasnett A. H., Gilbert C. S., Porter A. C. G., Gewert D. R., Stark G. R., and Kerr I. M. (1989), Proc Natl Acad Sci USA 86, 840–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff-Jamison S., Chen K., and Cohen S. (1993), Science 261, 1733–1736. [DOI] [PubMed] [Google Scholar]
- Russell S. M., Keegan A. D., Harada N., Nakamura Y., Noguchi M., Leland P., Friedmann M. C., Miyajima A., Puri R. K., Paul W. E., and Leonard W. J. (1993), Science 262, 1880–1883. [DOI] [PubMed] [Google Scholar]
- Rutherford M. N., Hannigan G. E., and Williams B. R. G. (1988), EMBO J 7, 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski H. B., Shuai K., Darnell J. E. Jr., and Gilman M. Z. (1993), Science 261, 1739–1744. [DOI] [PubMed] [Google Scholar]
- Sánchez M. P., Tapley P., Saini S. S., He B., Pulido B., and Barbacid M. (1994), Proc Natl Acad Sci USA 91, 1819–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C., Fu X., Improta T., Aebersold R., and Darnell J. E. Jr. (1992a), Proc Natl Acad Sci USA 89, 7836–7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C., Shuai K., Prezioso V., and Darnell J. (1992b), Science 257, 809–813. [DOI] [PubMed] [Google Scholar]
- Shuai K., Schindler C., Prezioso V. R., and Darnell J. E. Jr. (1992), Science 258, 1808–1812. [DOI] [PubMed] [Google Scholar]
- Shuai K., Stark G. R., Kerr I. M., and Darnell J. E. Jr. (1993a), Science 261, 1744–1746. [DOI] [PubMed] [Google Scholar]
- Shuai K., Ziemiecki A., Wilks A. F., Harpur A. G., Sadowski H. B., Gilman M. Z., and Darnell J. E. (1993b), Nature 366, 580–583. [DOI] [PubMed] [Google Scholar]
- Shuai K., Horvath C. M., Tsai Huang L. H., Qureshi S. A., Cowburn D., and Darnell J. E. Jr. (1994), Cell 76, 821–828. [DOI] [PubMed] [Google Scholar]
- Silvennoinen O., Ihle J. N., Schlessinger J., and Levy D. E. (1993a), Nature 366, 583–585. [DOI] [PubMed] [Google Scholar]
- Silvennoinen O., Schindler C., Schlessinger J., and Levy D. E. (1993b), Science 261, 1736–1739. [DOI] [PubMed] [Google Scholar]
- Silvennoinen O., Witthuhn B. A., Quelle F. W., Cleveland J. L., Yi T., and Ihle J. N. (1993c), Proc Natl Acad Sci USA 90, 8429–8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims S. H., Cha Y., Romine M. F., Gao P., and Gottlieb K. (1993), Mol Cell Biol 13, 690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh J., Donnelly R. J., Kotenko S., Mariano T. M., Cook J. R., Wang N., Emmanuel S., Schwartz B., Miki T., and Pestka S. (1994), Cell 76, 793–802. [DOI] [PubMed] [Google Scholar]
- Songyang Z., Shoelson S. E., Chauduri M., Gish G., Pawson T., Haser W. G., King F., Roberts T., Ratnofsky S., Lechleider R. J., Neel B. G., Birge R. B., Fajardo J. E., Chou M. M., Hanafusa H., Schaffhausen B., and Cantley L. C. (1993), Cell 72, 767–778. [DOI] [PubMed] [Google Scholar]
- Staeheli P. (1990), Adv Virus Res 38, 147–200. [DOI] [PubMed] [Google Scholar]
- Staeheli P., Haller O., Boll W., Lindenmann J., and Weissman C. (1986), Cell 44, 147–158. [DOI] [PubMed] [Google Scholar]
- Stahl N., Boulton T. G., Farruggella T., Ip N. Y., Davis S., Witthuhn B. A., Quelle F. R., Silvennoinen O., Barbieri G., Pelligrini S., Ihle J. N., and Yancopoulos G. D. (1994), Science 263, 92–95. [DOI] [PubMed] [Google Scholar]
- Stewart W. E. II (1979), The Interferon System, Springer, New York, p. 421. [Google Scholar]
- Sugita K., Miyazaki J-I., Appella E., and Ozato K. (1987), Mol Cell Biol 7, 2625–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J. (1964), Biochem Biophys Res Commun 14, 447–451. [DOI] [PubMed] [Google Scholar]
- Tian S-S., Lamb P., Seidel H. M., Stein R. B., and Rosen J. (1994), Blood 84, 1760–1764. [PubMed] [Google Scholar]
- Ullrich A. and Schlessinger J. (1990), Cell 61, 203–212. [DOI] [PubMed] [Google Scholar]
- Uzé G., Lutfalla G., and Gresser I. (1990), Cell 60, 225–234. [DOI] [PubMed] [Google Scholar]
- Veals S. A., Schindler C., Leonard D., Fu X., Aebersold R., Darnell J. Jr., and Levy D. (1992), Mol Cell Biol 12, 3315–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veals S. A., Santa Maria T., and Levy D. E. (1993), Mol Cell Biol 13, 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez L., Fellows M., Stark G. R., and Pelligrini S. (1992), Cell 70, 313–322. [DOI] [PubMed] [Google Scholar]
- Vogel J., Kress M., Khoury G., and Jay G. (1986), Mol Cell Biol 6, 3550–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner B. J., Hayes T. E., Hoben C. J., and Cochran B. H. (1990), EMBO J 9, 4477–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakao H., Gouilleux F., and Groner B. (1994), EMBO J 13, 2182–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watling D., Guschin D., Muller M., Silvennoinen O., Witthuhn B. A., Quelle F. W., Rogers N. C., Schindler C., Stark G. F., and Kerr I. M. (1993), Nature 366, 166–170. [DOI] [PubMed] [Google Scholar]
- Wathelet M. G., Berr P. M., and Huez G. A. (1992), Eur J Biochem 206, 901–910. [DOI] [PubMed] [Google Scholar]
- Wegenka U. M., Lutticken C., Buschmann J., Yuan J., Lottspeich F., Muller-Esterl W., Schindler C., Roeb E., Heinrich P. C., and Horn F. (1994), Mol Cell Biol 14, 3186–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelock E. F. (1965), Science 149, 310–311. [PubMed] [Google Scholar]
- Wilks A. F. (1989), Proc Natl Acad Sci USA 86, 1603–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks A. F., Harpur A. G., Kurban R. R., Ralph S. J., Zurcher G., and Zlemleckl A. (1991), Mol Cell Biol 11, 2057–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K. C. and Finbloom D. S. (1992), Proc Natl Acad Sci USA 89, 11964–11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witthuhn B. A., Quelle F. W., Silvennoinen O., Yi T., Tang B., Miura O., and Ihle J. N. (1993), Cell 74, 227–236. [DOI] [PubMed] [Google Scholar]
- Witthuhn B. A., Silvennoinen O., Miura O., Lai K. S., Cwik C., Liu E. T., and Ihle J. N. (1994), Nature 370, 153–157. [DOI] [PubMed] [Google Scholar]
- Wright T. M. and Farber J. M. (1991), J Exp Med 173, 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K., Quelle F. W., Thierfelder W. E., Kreider B. L., Gilbert D. J., Jerkins N. A., Copeland N. G., Silvennoinen O., and Ihle J. N. (1994), Mol Cell Biol 14, 4342–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z., Wen Z., and Darnell J. E. Jr. (1994a), Science 264, 95–98. [DOI] [PubMed] [Google Scholar]
- Zhong Z., Wen Z., and Darnell J. E. Jr. (1994b), Proc Natl Acad Sci USA 91, 4806–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]