

Abstract
We have generated a series of fusion proteins carrying portions of subunit lie, the second largest subunit of Drosophila RNA polymerase I, and have used them in a domain interference assay to identify a fragment of the lie subunit that carries the binding site for a basal transcription factor. Fusion proteins carrying a subunit lie fragment spanning residues Ala519-Gly992 strongly inhibit promoter-driven transcription in both unfractionated nuclear extracts and in reconstituted systems. The same fusion proteins similarly inhibit dTFIIF stimulation of Pol II elongation on dC-tailed templates, suggesting that the IIc(A519-G992) fragment, which carries conserved regions D-H, interferes with transcription by binding to dTFIIF. Finally, dTFIIF can be specifically cross-linked to a GST-IIc(A519-G992) fusion protein or to subunit lie in intact Pol II.
Keywords: RNA polymerase II, TFIIF, Transcription factors, Domain interference, Drosophila, Fusion proteins
RNA polymerase II is a complex, multisubunit enzyme that plays a central role in eukaryotic gene expression (Sawadogo and Sentenac, 1990; Young, 1991, for recent reviews). Like the other nuclear RNA polymerases I and III, RNA polymerase II consists of two large subunits and eight or more small subunits. The largest subunit (ca. 215 kDa) is usually referred to as IIa (RPB1 in yeast), and the second largest subunit (ca. 140 kDa) is usually referred to as IIc (RBP2 in yeast) (Greenleaf, 1992). Although a complete picture of how individual subunits function during the transcription cycle has not yet emerged, the importance of subunits IIa and IIc during catalysis and in the regulation of transcription is clear from several lines of evidence. For example, sequence analysis of the two largest subunits from several organisms has revealed extensive evolutionary conservation (Palenik, 1992, and above references). Eukaryotic subnunit IIa shares eight regions of significant homology with the β′ subunit of eubacteria as well as with the corresponding subunit of archaebacteria and the largest subunits of eukaryotic RNA polymerases I and III. The conserved regions tend to concentrate toward the N-terminus. Eukaryotic subunit IIc contains nine regions homologous to the bacterial β subunit (and the archaebacterial and polymerase I and III counterparts), with the regions of strongest similarity concentrated toward the C-terminus.
Functional similarity between the prokaryotic and eukaryotic enzymes, suggested by the striking evolutionary conservation, has been borne out by biochemical analysis. For example, both large subunits were shown by antibody inhibition or UV cross-linking to contact the DNA template and the nascent transcript (Bartholomew et al., 1986; Breant et al., 1983; Dissinger and Hanna, 1990; Gundelfinger, 1983). Further, immunoprecipitation of epitope-tagged yeast enzyme revealed that formation of “premature core” yeast RNA polymerase II, containing the yeast counterparts to E. coli β, β′, and α, proceeds in a manner analogous to bacterial RNA polymerase assembly (Kolodziej and Young, 1991). In addition, E. coli β subunit and the second largest subunits of wheat germ pol II and yeast polymerases I, II, and III were affinity cross-linked to nucleotide substrate analogues, suggesting that the second largest subunits of prokaryotic and eukaryotic enzymes participate in binding the nucleotide substrates and in phosphodiester bond formation (Grachev et al., 1987; Grachev et al., 1989; Mustaev et al., 1991; Riva et al., 1987). Taken together, these results suggest that numerous aspects of RNA polymerase molecular architecture and function have been conserved throughout evolution.
Largely because of facile genetic selection schemes, much is known about possible functions of the β subunit. For example, in vitro studies of reconstituted bacterial RNA polymerase containing mutant subunits have shown that defects in the C-terminal one-third of β subunit cause biochemical defects such as decreased promoter clearance (Lee et al., 1991), changes in promoter selectivity (Glass et al., 1986b; Nene and Glass, 1984), altered polymerase propagation (Sagitov et al., 1993), changes in enzyme pausing and termination (Landick et al., 1990a; Landick et al., 1990b), or the inability to bind σ factor (Glass et al., 1986a).
In view of the structural similarities between bacterial β subunit and eukaryotic subunit IIc, it is reasonable to speculate that the eukaryotic subunit carries out functions similar to those of its bacterial counterpart. In contrast to the bacterial case, however, it has not yet been possible to dissociate any eukaryotic RNA polymerase into individual subunits and reconstitute active enzyme in vitro. On the other hand, mutations generated in yeast and Drosophila have provided some clues as to the role of the second largest subunit. For instance, certain mutations in the C-terminus of yeast subunit IIc (RPB2) are responsible for gene-specific transcriptional defects in vivo (Scafe et al., 1990b); this may indicate altered interactions with a transcription factor or factors. Other RPB2 mutations were isolated as suppressors of a mutation in the RPB1 gene, which suggests that a physical interaction may occur between the affected regions of the two largest subunits (Martin et al., 1990). Similarly, suppressors of a mutation in the largest subunit of Drosophila RNA polymerase II mapped to the RpII140 gene that encodes subunit IIc (Mortin, 1990). Other single-base substitutions in the C-terminal one-third of RpII140 caused developmental defects or were homozygous lethal, implying that alterations to these domains are critical to the enzyme’s structure or function in vivo (Chen et al., 1993; Mortin et al., 1992).
RNA polymerase II requires several accessory factors for accurate initiation and effective elongation in vitro (Zawel and Reinberg, 1993, for review); several of these interact directly with Pol II. Notably, TFIIF (RAP30/74) was originally isolated from HeLa cells on the basis of its binding to immobilized RNA polymerase (Sopta et al., 1985). The 30-kDa RAP30 subunit of TFIIF binds to RNA polymerase II in the absence of the 74-kDa RAP74 subunit (Killeen and Greenblatt, 1992) and is sufficient to recruit the enzyme into preinitiation complexes (Flores et al., 1991). Sequences required for binding to RNA polymerase II have been localized to the central portion of RAP30, a stretch of which shows homology to the σ subunit of E. coli RNA polymerase (Sopta et al., 1989; Yonaha et al., 1993). Binding of TFIIF, RAP30, or the homologous rat liver factor βγ to RNA polymerase II prevents the enzyme from associating with nonspecific DNA sequences in gel mobility shift assays, a function also exhibited by σ factors (Conaway and Conaway, 1990; Killeen and Greenblatt, 1992). Further, TFIIF can bind to E. coli RNA polymerase, and this binding is competed by σ70 (McCracken and Greenblatt, 1991). These results suggest that TFIIF probably binds to a region of RNA polymerase II that is conserved between prokaryotes and eukaryotes and that aspects of the mechanism of RNA polymerase II recruitment to the promoter may also be conserved. However, the RNA polymerase II subunits that are the direct target of TFIIF action have not been investigated previously, and the subunit of RNA polymerase II to which RAP30 binds has not been identified.
The Drosophila counterpart to mammalian TFIIF was originally isolated from Drosophila Kc cell nuclear extracts and called factor 5 (Price et al., 1987). Factor 5 is composed of two subunits of 86 and 34 kDa; it is essential for transcription initiation in vitro, it stimulates the elongation rate of pure RNA polymerase II in vitro, and it binds to RNA polymerase II (Price et al., 1989). Based on these properties and the sequence of a cloned gene for the large factor 5 subunit, it is clear that factor 5 is the Drosophila homolog of TFIIF (Kephart et al., 1993). In this report, we will refer to factor 5 as dTFIIF.
Although dTFIIF has a high affinity for free RNA polymerase II, template competition experiments and gel filtration of elongating ternary complexes have shown that in vitro it interacts transiently with paused RNA polymerase II, then dissociates upon reentry of the enzyme into a productive elongation cycle (Price et al., 1989). The mechanistic details of dTFIIF’s role in initiation and elongation, including its interactions with subunits of RNA polymerase II, are not clear. As an initial step toward clarifying details of these events, we sought to identify the dTFIIF binding site on Pol II. We hoped to identify not only the subunit to which the factor binds, but also the specific portion of that subunit involved in the interaction.
Our approach was based on the following ideas and facts. First, we reasoned that a recombinant fusion protein carrying some or all of the dTFIIF binding site, if added in excess to a transcription reaction, might compete with intact Pol II for dTFIIF binding and thereby inhibit factor-dependent transcription; this “domain interference assay” thus could potentially identify the Pol II subunit, or fragment thereof, carrying the dTFIIF-interacting domain. Next, genetic and biochemical experiments in E. coli, briefly summarized above, suggested the IIc subunit (the β homologue) as a likely candidate for interacting with dTFIIF. Further, the cloned gene for the Drosophila IIc subunit was available. Finally, a precedent for this general approach was in the literature (Rappaport et al., 1988).
In the work we describe here, fusion proteins containing portions of Drosophila RNA polymerase II subunit IIc fused to β-galactosidase or glutathione-S-transferase (GST) were expressed in E. coli, purified, and used as probes to examine the interaction of dTFIIF with this subunit of Pol II. We demonstrate that fusion proteins containing a particular region of the IIc subunit interfere with the activity of Drosophila dTFIIF during initiation and elongation. We also show that dTFIIF can be cross-linked to a fusion protein containing this region of subunit IIc as well as to intact subunit IIc within native RNA polymerase II.
MATERIALS AND METHODS
Chemicals
Restriction enzymes and terminal deoxynucleotidyl transferase were from BRL or Boehringer Mannheim. [α 32P]CTP and [125I]Bolton-Hunter reagent [3-(4-hydroxyphenyl) propionic acid N-hydroxysuccinimide ester] were from ICN Bio-medical. Ribonucleoside triphosphates were from Pharmacia. HEPES [4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid, free acid], phenylmethylsulfonyl fluoride (PMSF), and dithiothreitol (DTT) were from Calbiochem. Nitrocellulose used for blotting of iodinated GST-IIc(A519-G992) was from Bio-Rad. Nitrocellulose used for chemiluminescent detection of horseradish peroxidase-conjugated antibodies was from Amersham. Ethylene glycol-bis (succinimidyl succinate), or EGS, was from Pierce. All other reagents were from Sigma.
Expression and Purification of β-Galactosidase-IIc Fusion Proteins
A 2.0-kb EcoRI fragment from Drosophila RpII140 genomic DNA clone pBHgt-31 (Hamilton et al., 1993) was used to probe a λgt11 library of 0–12 h Drosophila embryo cDNA (from Tao Hsieh, Duke University) at high stringency. A phage containing a 2.0-kb cDNA fragment, corresponding to the C-terminal two-thirds of the RpII140 coding region, was obtained from this screen. A 2.0-kb partial EcoRI fragment containing the cDNA was subcloned from this phage into pUC19 to give pAS-Dl. A 1.9-kb SalI-HindIII fragment from pAS-Dl was then subcloned into the expression vector pUR290 to give pGAL-IIc(A519-K1157). The construct pGAL-IIc(A519-G992) was generated by cloning a 1.5-kb BamHI subfragment from pGAL-IIc(A519-K1157) back into pUR290. The plasmid pAS-W1 consists of a 1.0-kb EcoRI cDNA fragment from the 3′ end of RpII140, cloned into pUC19. Digestion of this plasmid with BamHI generated a 540-bp fragment that was subcloned into pUR291, resulting in plasmid pGAL-IIc(G992-T1175).
The β-galactosidase (β-gal) fusion proteins were prepared by the following procedure. All steps were at 4°C or on ice unless noted otherwise. Five milliters of an overnight bacterial culture containing the expression plasmid were inoculated into 500 ml LB + ampicillin (50 μg/ml) and grown at 37°C until mid-log phase (1.5–2 h), then induced with 1 mM IPTG for 2 h. Cells were pelleted at 4000 rpm in a GSA rotor for 10 min, then frozen at −80°C for 15 min to overnight. Buffer L [20 mM Tris-HCl-Cl, pH 7.2, 200 mM KC1, 10 mM EDTA, 0.1 mM DTT, 0.1% PMSF (of a saturated solution in isopropanol)] was added at 3 ml per gram of wet cells and incubated on ice until thawed. Cells were sonicated with 3−4 × 30-s bursts with a microtip at maximum setting. Nine more cell volumes of buffer L were added and mixed gently. The diluted lysate was spun at 12,000 rpm for 30 min in an SA600 rotor. The pellet fraction was solubilized in buffer S (8 M urea, 50 mM Tris-HCl-Cl, pH 7.9, 1 mM EDTA, 50 mM KC1, 0.1% PMSF) and homogenized in a glass homogenizer (Dounce or Wheaton). Solid DTT was added to 50 mM. The solution was gently stirred at 4°C for 1–2 h. The protein was then dialyzed vs. 25 mM HEPES, pH 7.6, 15% glycerol, 0.1 mM EDTA, 0.1 mM DTT, 0.1% PMSF (HGEDP) overnight, with at least one change of buffer. After dialysis, the fusion protein solution was spun at 10,000 rpm for 10 min to remove any insoluble protein.
Further purification of β-gal fusion proteins was carried out on DEAE cellulose (DE52, Whatman). The salt of the fusion protein solution was increased to 100 mM KCl by the addition of HGKE (1 M KCl in buffer HGE). This solution was loaded onto a DE52 column equilibrated with 100 mM HGKEDP, washed with 5 column volumes of the same buffer, and step-eluted with 350 mM HGKEDP. The peak of protein was pooled, 25% w/v ammonium sulfate was added, and the solution stirred for 30 min. After centrifugation for 13 min at 45,000 rpm in a T865 rotor at 0°C, the pellets were resuspended in and dialyzed overnight versus HGEDP. The KCl concentration was then adjusted to 0.1 M with 1 M HGKEDP, and the fusion protein fractions were loaded onto a Pharmacia Superose 6 FPLC column equilibrated with 0.1 M HGKEDP. The extent of proteolysis of eluted fractions was assessed by SDS-PAGE. Selected fractions were pooled and concentrated in Centricon microconcentrators (Amicon). The protein concentration was determined using the Bio-Rad protein assay reagent. Commerically made β-gal used in the transcription assays was from Sigma. Lyophilized protein was denatured by suspension in buffer S and treated the same way as GAL-IIc(A519-K1157), except that β-gal was not subjected to chromatography. β-Galactosidase activity was measured by the method of Miller (1972).
Expression and Purification of GST-IIc Fusion Proteins
Plasmids based on the pGEX vectors (AMRAD Corp., Pharmacia) were generated, employing standard procedures analogous to those used for the β-gal fusions, to encode fragments of the IIc subunit linked C-terminally to GST (details in Skantar, 1993). The constructs analyzed are summarized in Fig. 1C.
FIG. 1.
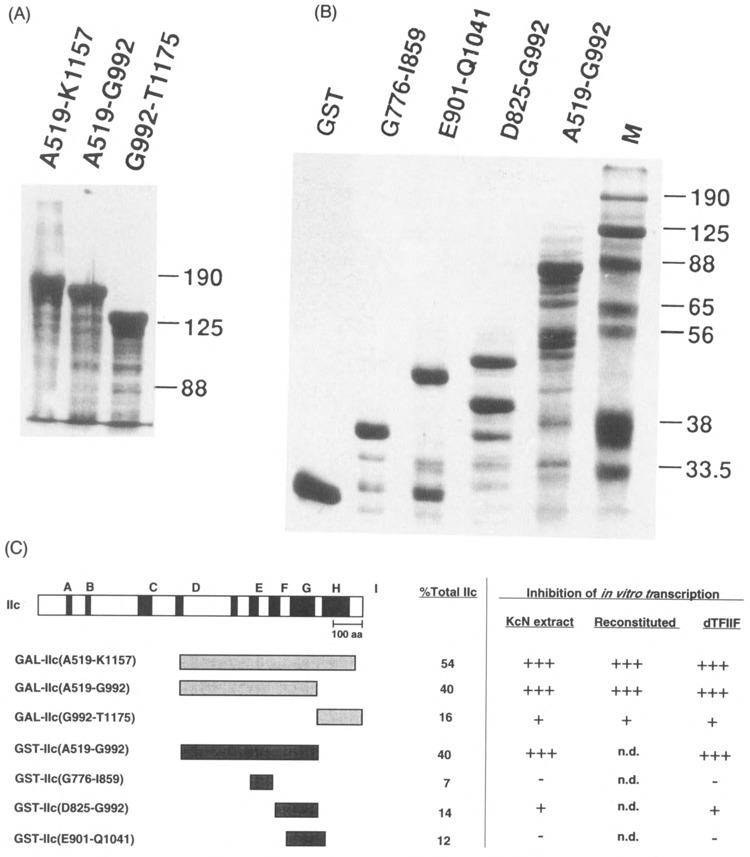
Fusion proteins used for inhibitions. Coomassie blue stained SDS-polyacrylamide gels of (A) the β-galactosidase-IIc fusion proteins (10 μg each lane) and (B) glutathione-S-transferase-IIc fusion proteins (2 μg unproteolyzed) used for inhibition studies. Purification of the fusion proteins is described in Materials and Methods. (C) Schematic diagram of the RNA polymerase IIc subunit and fusion proteins in this study. Black boxes denote regions conserved between eukaryotic subunits IIc and eubacterial β subunit. Gray boxes denote the extent of GAL-IIc and GST-IIc fusion proteins, compared to the full-length IIc sequence. The percent of the whole subunit in each fusion protein is indicated. To the right of the vertical line, the degree of inhibition by the listed fusion proteins is summarized (data from Results section). Inhibition of “KcN extract” and of “reconstituted” transcription refers to promoter-directed transcription of the A2 template; inhibition of “dTFIIF” refers to stimulation of elongation in the dC-tailed template assay, n.d. = not done.
Bacterial strains expressing GST fusion proteins were grown and induced generally as described by Smith and Johnson (1988). GST-IIc fusion proteins were found almost exclusively in inclusion bodies; thus, it was necessary to solubilize them from this insoluble fraction. This was accomplished basically as for the β-gal fusions, above. GST-IIc(E901-Q1041) and GST-IIc(D825-G992) were further subjected to GSH-agarose chromatography (Smith and Johnson, 1988). Solubilized GST-IIc(G776-I859) was isolated from a preparative SDS-polyacrylamide gel as described (Hager and Burgess, 1980), except that the acetone-precipitated protein eluted from the gel slice was resuspended in buffer S, then dialyzed against 50 mM Tris-HCl, pH 8, 1 mM EDTA, 1 mM DTT, 0.1% PMSF, to renature the protein. GST-IIc(G776-I859) was then subjected to GSH-agarose affinity chromatography. The GST used for in vitro transcription assays was purified from cell supernatant (Smith and Johnson, 1988). Each of these proteins was concentrated in Centricon-10 microconcentrators (Amicon) in order to remove the glutathione as well as to concentrate the fractions, so that a broad molar excess of fusion proteins could be examined in the transcription assays.
The binding of GST-IIc(A519-G992) to glutathione-agarose was poor under all conditions examined, most likely due to the size of the RNA polymerase IIc fragment relative to GST. Therefore, the inclusion bodies containing most of this fusion protein were prepared essentially as described (Frankel et al., 1991; Lin and Cheng, 1991). These protocols both involve hypotonic lysis of the outer membranes, believed to be the greatest source interference with proper fusion protein folding. The resulting inclusion body fraction was suspended in 10 ml buffer S and incubated at 4°C for 30 min, centrifuged at 60,000 × g for 30 min, then dialyzed against several exchanges of HGEDP and frozen at − 80°C.
Templates for In Vitro Transcription
The preparation of templates A2 (carrying the Act5C promoter) and pBalI-E for transcription, was described previously (Coulter and Greenleaf, 1985; Price et al., 1987). The template pPCP consists of a BamHI/EcoRI fragment of the Drosophila actin 5C gene subcloned into the vector pSP73, and was the kind gift of Dr. David Price (University of Iowa). PstI-digested template plasmids were dC-tailed using terminal deoxynucleotidyl transferase as described previously (Price and Parker, 1984).
Inhibition of KcN-Mediated and Reconstituted Transcription
A complete description of in vitro transcription assays and the procedures for preparing Drosophila Kc cell nuclear extract (KcN) and transcription factors can be found in Price et al. (1987). Reconstituted transcription assays typically contained factor 3 (the Drosophila homolog of TFIIE) and a fraction eluting from phosphocellulose betwen 0.3 and 0.4 M KCl (referred to as the P11-0.4 M step). This fraction contained transcription factors 5 (dTFIIF), DmS-II, and an RNaseH activity, in addition to other unidentified factors required for accurate initiation, such as TBP. Although not a transcription factor, addition of a DNAse inhibitor purified from Drosophila extracts was also necessary to prevent digestion of template by nucleases present in less pure transcription factor fractions (Sluder et al., 1987). For some inhibition reactions, the P11-0.4 M step fraction was preincubated with GAL-IIc(A519-K1157) or β-gal for 10 min at 25°C before the other required factors were added. In other reactions, all of the transcription factors were added together (see figure legends). A cocktail containing template, nucleotides, and other cofactors was added to start the reactions. After 20 min at 25°C, the reactions were stopped, extracted, and electrophoresed.
Elongation Assay Using dC-Tailed DNA Templates
Elongation assays were carried out essentially as described (Price et al., 1989; Sluder et al., 1988) except that the concentration of cold CTP was increased to 100 mM. dTFIIF used for elongation assays was purified essentially as described (Price et al., 1989) with some minor modifications. Reactions contained 10 mg/ml dC-tailed pPCP and 1–4 μCi [α 32P]CTP per reaction. A master reaction was prepared with 5–10 units of purified RNA polymerase II, template, GTP, CTP, and ATP, and buffer. This mixture was incubated under UTP-less conditions on ice for 5 min to allow the polymerase to bind to dC-tailed template. At the same time, dTFIIF was preincubated with fusion proteins or control proteins at 25°C for 5 min, in tubes that contained UTP and enough KCl and HGE to give 50–100 mM KCl in the final 12.5-μ1 reaction volume. After the preincubations, the master polymerase mixture was aliquotted into the dTFIIF fusion protein reactions, and elongation from the dC-tailed templates was allowed to proceed for 3–10 min. Reactions were stopped and samples prepared as described for the specific transcription assay.
Inclusion of μg quantities of the IIc fusion proteins sometimes resulted in inconsistent recovery of the RNA after transcription. Thus, a radioactively labeled, 186 bp size and recovery standard was made by PCR amplification of RpII140 cDNA pAS-D1 with the primers [5′-CCGTACCGCGTACACATCTGCAAC and [5′-AAGCTTATCGATCGGCGCAATGTT-3′]. The PCR products were radioactively labeled during amplification with approximately 1 [α 32P]dAMP incorporated per DNA strand. The labeled standard was phenol extracted, ethanol precipitated, washed with 70% ethanol, dried, and resuspended in TE. An aliquot of standard was added to the Sarkosyl solution used to stop the transcription reactions. This provided an internal standard for monitoring nucleic acid recovery during the subsequent extraction steps.
Iodination of GST-IIc(A519-G992) With Bolton-Hunter Reagent
The iodination of GST-IIc(A519-G992) was carried out as described (Bolton and Hunter, 1973). After passage through a Sephadex G50 column (buffer 0.1 M HGEDK), BSA (50 μg/ml) was added to the peak fractions as a stabilizer. A portion of each peak fraction fraction was denatured by adding an equal volume of 10 M urea, 15 mM DTT, and 0.1% of PMSF. The protein was allowed to denature on ice for 15 min, then was dialyzed versus HGEDP, and stored at 4°C.
Chemical Cross-Linking of dTFIIF to GST-IIc(A519-G992) and RNA Polymerase II
dTFIIF was incubated with iodinated GST-Hc(A519-G992) or with purified RNA polymerase II for 15–60 min at 25°C, under conditions similar to in vitro transcription reactions, including 25 mM HEPES, pH 7.6, 12% glycerol, 0.1 mM EDTA, and 50–150 mM KCl. A solution of 50 mM EGS was prepared fresh in DMSO; working dilutions were prepared in HGE such that 2 ml added to the 15-ml reactions would give the desired final concentration of EGS. After cross-linking for 15 min, the reactions were quenched with 75 mM lysine; 6 × SDS gel sample buffer was added immediately. The samples were heated to 100°C for 5 min, spun briefly, then loaded onto an SDS-polyacrylamide gel (Laemmli, 1970). The gels were electroblotted for 2 h as described (Towbin et al., 1979; Weeks et al., 1982). When iodinated GST-IIc(A519-G992) was used, the blots were air dried, wrapped in Saran Wrap, and auto-radiographed. Before immunodetection of cross-linked products, the nitrocellulose membranes were dried under a heat lamp for 10 min per side to ensure protein adsorption to the membrane.
Affinity-Purified Antibodies
The preparation of Pol II subunit IIa antibodies was described previously (Lee et al., 1991). Antibodies to domains of RNA polymerase IIc were obtained by subcutaneous injection of 0.5 mg denatured IIc(A519-G992) into a rabbit, 0.5 mg IIc(G992-T1175) into another rabbit, and 1 mg each protein into a goat as described (Weeks et al., 1982). After a second injection, serum was obtained from the animals and stored at −20°C. The titer of antibodies was checked by Western blotting (Towbin et al., 1979). Fusion protein columns were constructed by binding 9.1 mg GAL-IIc(A519-G992) or 4.7 mg GAL-IIc(G992-T1175) to 1-ml aliquots of Reacti-gel (Pierce) according to the manufacturer’s instructions. Affinity purification of the antibodies was performed as described (Robbinsetal., 1984).
Western Blotting and Chemiluminescent Detection
Western blotting was as previously described (Weeks et al., 1993). Identical blots of dTFIIF cross-linked to RNA polymerase II were probed with antibodies affinity purified against the 215-kDa (anti-exon 2) or 140-kDa subunits. The blots were incubated with a 1:5000 dilution of primary antibody overnight at 4°C. Bound antibody was detected by incubation for 1 h with 1:10,000 dilution of horseradish peroxidase-conjugated swine anti-goat IgG (Tago). The blots were developed using ECL chemiluminescent antibody detection system according to the manufacturer’s instructions (Amersham).
RESULTS
Fusion Protein GAL-IIc(A519-K1157) Inhibits Promoter-Dependent Basal Transcription In Vitro
Drosophila Kc cell nuclear extracts (KcN) support specific, promoter-driven transcription in vitro. To identify critical transcription factor-RNA polymerase II interactions, we first examined the ability of a fusion protein carrying a large fragment of the RNA polymerase IIc subunit to inhibit this process. The nuclear extract was incubated with a molar excess of GAL-IIc(A519-K1157) (Fig. 1) or β-gal for 5 min, before the addition of DNA template, nucleotides, and other cofactors (see Materials and Methods). A 50–75% reduction in the 450-base run-off transcript from the actin A2 promoter was observed when fusion protein GAL-IIc(A519-K1157) was present in a 500–1000-fold molar excess over the endogenous Pol II (Fig. 2, lanes 7–9). A similar excess of β-gal caused slight if any reduction in the level of transcript (Fig. 2, lanes 5–6). Because neither fusion protein nor β-gal exhibited RNase activies (Fig. 2, lanes 2-3), this experiment clearly shows that the RNA polymerase II domain present in the GAL-IIc(A519-K1157) fusion protein interferes with some component of the KcN extract that is required for transcription in vitro.
FIG. 2.
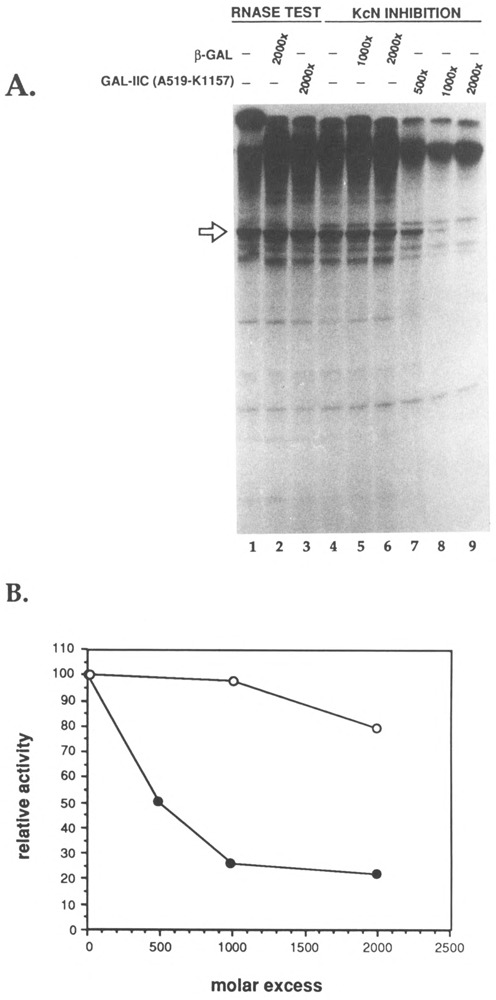
Inhibition of specific initiation by GAL-IIc(A519-K1157) or β-galactosidase. The amount of RNA polymerase II (mass ≈ 600,000 Da) in Kc cell nuclear extract (KcN) was estimated to be 10 ng/μ1 (A. Sluder, personal communication). Thus, reactions containing 3 μ1 KcN had an estimated 50 fmol of RNA polymerase II. The molar excess of fusion protein added to transcription reactions was calculated using the concentration of unproteolyzed GAL-IIc(A519-K1157) determined by comparison to a known amount of pure 0-galactosidase on a Coomassie blue-stained SDS-polyacrylamide gel (not shown). (A) Run-off transcription assay. Lanes 1-3: test for RNase activity of the fusion proteins. Identical transcription reactions were incubated for 20 min, stopped by the addition of a-amanitin (2 μg/ml final), then incubated for an additional 5 min with the indicated proteins. In lanes 4-9, HGE buffer, 0-galactosidase, or fusion protein GAL-IIc(A519-Kl157) as indicated were incubated with 3 μ1. KcN extract for 5 min before addition of the mixtures to DNA, nucleotides, and cofactors as described in Materials and Methods. Transcriptions were allowed to proceed for 20 min. The 450-base run-off transcript (linearized A2 template) is indicated by the arrow. In (B), the amount of run-off transcript was quantitated by densitometer scanning of the autoradiograph on an ImageQuant computing densitometer (Molecular Dynamics). The entire autoradiograph was scanned, and the desired bands were marked for quantitation. An equal-sized area next to the RNA bands was chosen as the background value and subtracted from each sample. The values from the uninhibited transcription reactions were set at 100%. The relative activity of inhibition reactions was calculated by dividing by this value.
We attribute the high molar excess of fusion protein required for significant inhibition in this and subsequent experiments to the necessity to denature and then renature insoluble fusion proteins (Skantar, 1993, and Materials and Methods). Presumably only a fraction of the polymerase portion of the fusion protein adopts its correct conformation after this treatment. We base this statement on measurements of the specific activity of the β-gal moiety of the fusion protein, which we monitored frequently and often found to be ≤ 25% of that of commercial β-gal treated similarly (Lee and Greenleaf, 1991; Skantar, 1993). Note that we always subjected the control proteins (β-gal, GST) to the same denaturation/renaturation treatment as the fusion proteins.
To determine which factor within the Drosophila extract was inhibited by the fusion protein, the effect of GAL-IIc(A519-K1157) on reconstituted in vitro transcription was investigated. Initial experiments were done using partially purified transcription factors obtained from fractionation of Drosophila Kc cell nuclear extracts (Price et al., 1989). Figure 3 shows that including up to a 300-fold molar excess of GAL-IIc(A519-K1157) over the polymerase in the reaction reduced the level of reconstituted transcription by as much as 75% (Fig. 3, lanes 2–8). This inhibition was enhanced 30–50% by preincubation of the fusion protein with the P11-0.4 M fraction before addition of the other factors (Fig. 3, lanes 10–12). Inclusion of β-gal did not significantly inhibit transcription, even at 3000-fold excess over RNA polymerase II (Fig. 3, lanes 8–9). Taken together, these results show that the subunit IIc segment carried by GAL-IIc(A519-K1157) interacts with one or more factors in the P11-0.4 M fraction that are required for in vitro transcription.
FIG. 3.
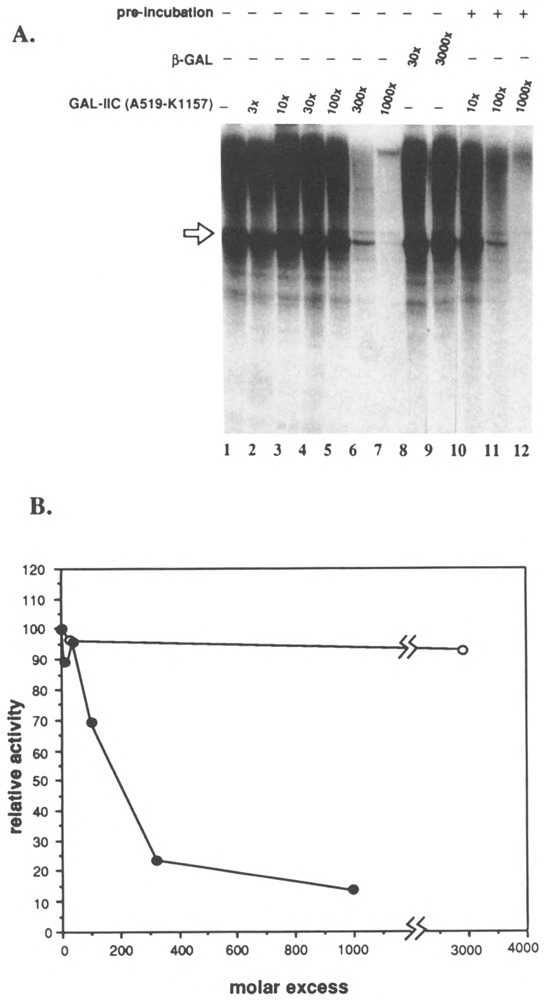
Inhibition of reconstituted transcription by GAL-IIc(A519-Kl157). Each reaction contained 5/μg/ml linear A2 template, 10 units (4.8 fmol) embryo RNA polymerase II, 0.0625 μl DNAse inhibitor (Sluder et al., 1987, MonoQ fraction), 0.5 pi factor 3 (MonoQ fraction), 2 μl P11-0.4 M step, and the indicated molar excess of GAL-IIc(A519-Kl 157) or -galactosidase over RNA polymerase II. The 450-base actin run-off transcript is indicated by the arrow. (A) Run-off transcription assay. In lanes 1-9, GAL-IIc(A519-K1157) or β-galactosidase was added to reactions just before the addition of the transcription factors. In lanes 10-12, GAL-IIc(A519-K1157) was preincubated with the P11-0.4 M fraction for 10 min as described in Materials and Methods. (B) Quantitation of transcripts by scanning laser densitometry.
Localization of the Transcription Factor Interacting Domain of RNA Polymerase II Subunit IIc
To determine whether fusion proteins containing subfragments of GAL-IIc(A519-K1157) were capable of interfering with transcription factors during promoter-dependent transcription, we studied the effect of GAL-IIc(A519-G992) and GAL-IIc(G992-Tl 175) on the P11-0.4 M step fraction during reconstituted in vitro transcription assays. Increasing amounts of these fusion proteins were preincubated with the P11-0.4 M fraction as described above. Figure 4 shows that a 28-fold molar excess of GAL-IIc(A519-G992) over the RNA polymerase II present in the reaction is sufficient to achieve a 50% reduction in the amount of run-off transcript. In the presence of a 138-fold or greater excess GAL-IIc(A519-G992), less than 20% of the control activity remains. In contrast, GAL-IIc(G992-T1175) inhibited transcription weakly, requiring more than seven times as much fusion protein to diminish the amount of transcript by 50%. Thus, we have identified an RNA polymerase subunit IIc segment required for a functional interaction between a basal transcription factor and RNA polymerase during promoter-dependent transcription.
FIG. 4.
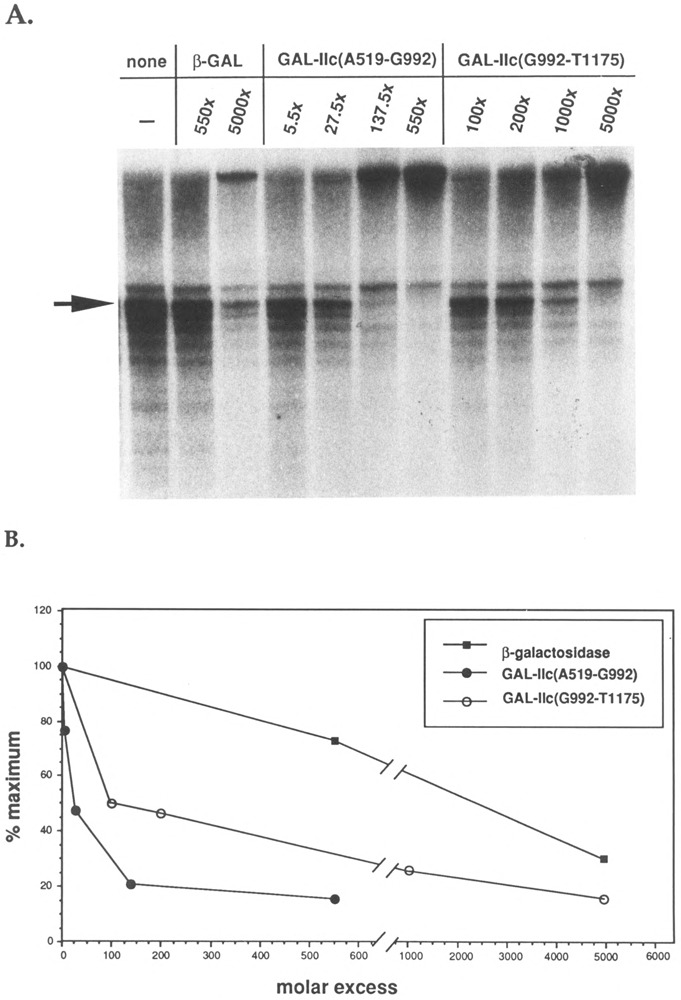
Inhibition of reconstituted transcription by GAL-IIc(A519-G992) and GAL-IIc(G992-T1175). (A) Run-off transcription assays were conducted as described in Fig. 3. Transcriptions contained 10 units (4.8 fmol) RNA polymer-ase II, 0.1 μ1 RNase inhibitor, 0.5 μ1 factor 3, and 3 Ml PI 1-0.4 M step fraction. In these reactions, the P11-0.4 M fraction was preincubated for 5 min at 25°C with lane 1, no addition; lanes 2–3, /3-galactosidase; lanes 4–7, GAL-IIc(A519-G992); lanes 8–11, GAL-IIc(G992-T1175). The actin run-off transcript is indicated by the arrow. (B) Quantitation of run-off transcript produced in (A).
GAL-IIc(A519-K1157) and GAL-IIc(A519-G992) Inhibit dTFIIF (Factor 5) Stimulation of RNA Polymerase II Elongation
Having localized the inhibitory region of subunit IIc to within the fragment defined by amino acids 519–992, we next sought to determine the basal transcription factor that was the target of the inhibition. Price et al. had purified Drosophila transcription dTFIIF (factor 5) from the P11-0.4 M step and showed that this factor was required for initiation of transcription (Price et al., 1987). Further characterization of highly purified dTFIIF revealed that it could also dramatically stimulate the elongation rate of RNA polymerase II during a promoterless transcription assay, and that it could bind stably to free RNA polymerase II in glycerol gradients (Price et al., 1989). Given the cosedimentation of dTFIIF and RNA polymerase II on glycerol gradients, its requirement for initiation, and its stimulatory effects during elongation, we decided to investigate whether interactions between GAL-IIc fusion protein and dTFIIF could lead to inhibition of dTFIIF activity during in vitro transcription. The dC-tailed template assay provides a way to evaluate the effect of individual transcription factors on the Pol II elongation rate, in the absence of initiation from a promoter. To establish that the fusion proteins did not interfere with the elongation rate of RNA polymerase II itself, GAL-IIc(A519-K1157), GAL-IIc(A519-G992), GAL-IIc(G992-T1175), or β-gal were added to dC assays in the absence of dTFIIF. RNA polymerase II was allowed to bind to the dC-tailed template and then fusion proteins or 0-gal were incubated with the complex, and then elongation was initiated with UTP (see Materials and Methods). None of the proteins reduced the elongation rate of RNA polymerase II alone; rather, they all slightly stimulated the number of transcripts seen. In addition, the transcripts were not degraded by fusion protein fractions either in the presence or absence of dTFIIF, indicating an absence of RNase activities (not shown).
GAL-IIc(A519-Kl 157), GAL-IIc(A519-G992), and GAL-IIc(G992-T1175) were tested individually over a wide range of molar excess relative to RNA polymerase II for reduction of dTFIIF stimulation. Increasing amounts of GAL-IIc(A519-K1157) caused a decrease in dTFIIF stimulation of the Pol II elongation rate. Although transcription was slightly stimulated at the lowest levels of GAL-IIc(A519-K1157) tested, at higher amounts the production of dTFIIF-dependent RNAs in the 1000-2000 base size range was dramatically reduced. In contrast, production of short, largely dTFIIF-independent RNAs was not affected. Fusion protein-dependent reduction in transcription leveled off between 80-fold or greater GAL-IIc(A519-K1157) (data not shown). Similarly, when excess GAL-IIc(A519-G992) was incubated with dTFIIF, between 1- and 20-fold GAL-IIc(A519-G992) slightly stimulated elongation (Fig. 5, lanes 5-8). However, at greater than 20-fold excess GAL-IIc(A519-G992), dTFIIF stimulation was dramatically reduced (Fig. 5, lanes 9-12). Similar to the results with GAL-IIc(A519-K1157), greater than 500-fold GAL-Hc(A519-G992) caused no further inhibition of dTFIIF activity (not shown). GAL-IIc(A519-G992) primarily diminished the production of long RNA, but a slight increase in some shorter transcripts was seen with 500-fold excess GAL-IIc(A519-G992) (Fig. 5, lane 12). Note that the level of transcription in the presence of 500-fold GAL-IIc(A519-G992) was only slightly higher than in the presence of /3-gal but no dTFIIF, indicating that the fusion protein almost completely abolished the dTFIIF stimulatory activity (Fig. 5, compare lanes 12 and 13). In contrast to these results, GAL-IIc(G992-T1175) did not efficiently inhibit dTFIIF stimulation of RNA polymerase II. Greater than 500-fold excess of this fusion protein was required to see appreciable reduction in the level of transcription (data not shown).
FIG. 5.
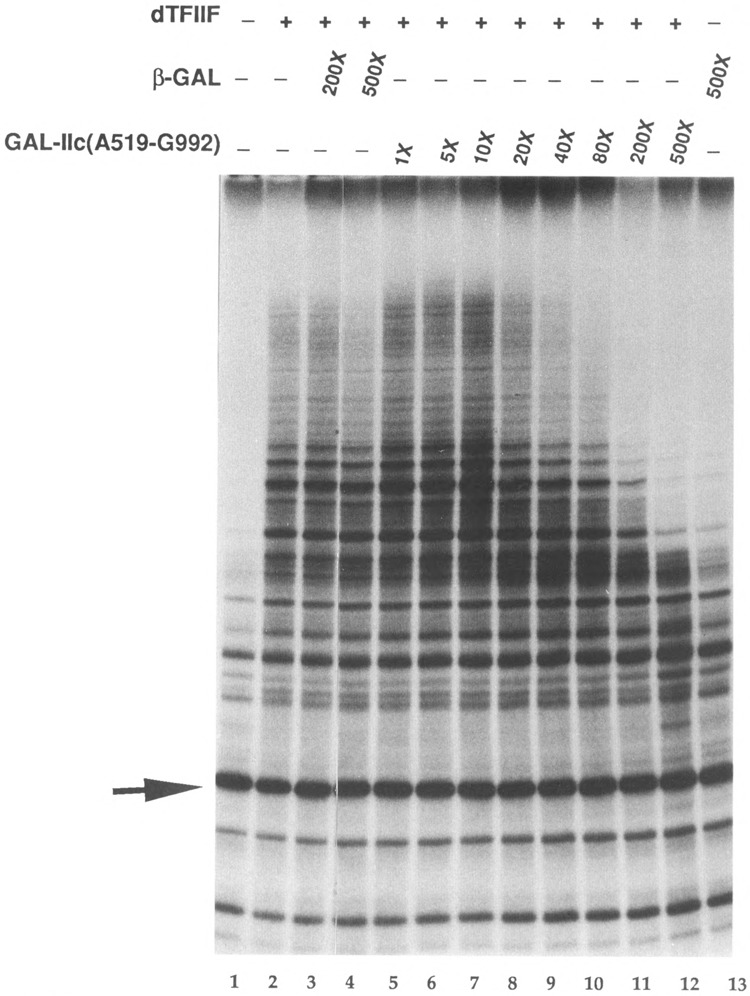
Inhibition of dTFIIF elongation stimulation during transcription of dC-tailed template. The final KCl concentration in each reaction was 80 mM. Each reaction contained 4.8 fmol Pol II (29 ng), 10 /xg/ml template dC-PCP, 140 ng MonoQ fraction DNAse inhibitor, 5 units RNase inhibitor (Boehringer Mannheim), and 1.1 μg β-galactosidase. The indicated reactions contained 100 ng of MonoQ-purified dTFIIF. GAL-IIc(A529-G992) was preincubated with dTFIIF for 5 min at 25°C. Following preincubations of Pol II with template and dTFIIF with fusion protein, the master polymerase mixture was aliquotted into the individual reaction tubes, and transcripts were elongated for 3 min. Lane 1: Pol II only; lane 2: Pol II + dTFIIF; lane 3-4: same as lane 2 with indicated amount of 0-galactosidase in molar excess over Pol II; lanes 5-12: same as lane 2 with indicated amount of GAL-IIc(A519-G992); lane 13: Pol II + β-galactosidase. The arrow indicates the position of a 186 bp size and recovery standard added with the transcription stop solution. Note that the length of the longest transcripts in lanes 5–8isca. 1.5 kb.
Inhibition of Promoter-Dependent Basal Transcription and dTFIIF-Mediated Stimulation of Elongation by GST-IIc Fusion Proteins
An investigation of the physical association between transcription factors and RNA polymerase II is essential to a clear understanding of how gene expression is controlled. In particular, localization of Drosophila transcription dTFIIF binding to a particular RNA polymerase II subunit or subunit domain would provide some insight into the factor’s mode of action and the role played by that subunit during the transcription cycle. We have demonstrated that the fragment of RNA polymerase subunit He consisting of amino acids 519-992 can inhibit dTFIIF elongation-stimulatory activity, thus providing strong, albeit indirect, evidence for a specific interaction between dTFIIF and this subunit domain. The following experiments were designed to obtain direct physical evidence for the binding of dTFIIF to the IIc(A519-G992) domain. The large, tetrameric structure of β-gal-IIc fusion proteins precluded their use in most types of binding experiments and, in part, motivated the construction of the GST-IIc fusion proteins.
The GST fusion proteins are summarized schematically in Fig. 1 (and see Materials and Methods). GST-IIc(A519-G992) contained the lie fragment from GAL-IIc(A519-G992) cloned into the GST vector. In addition, smaller portions of He were constructed as GST fusions, to see if the interacting domain could be localized further. The conserved domains F, G, H, and I were targeted for these fusion proteins, due to the genetic and biochemical evidence, which suggests that these regions are critical for the enzyme’s function (see Discussion).
The demonstration of equivalent dTFIIF inhibition by GST-IIc(A519-G992) and GAL-IIc(A519-G992) would provide persuasive evidence for the specificity of this effect. Indeed, we found that GST-IIc(A519-G992) and GAL-IIc(A519-G992) interfere similarly with Kc cell nuclear extract-mediated transcription from the actin promoter (not shown). Parallel inhibition by the two fusion proteins was also observed during dTFIIF-dependent stimulation of RNA polymerase II elongation. Figure 6 shows that comparable levels of GST-IIc(A519-G992) and GAL-IIc(A519-G992) caused similar reductions in the size and number of transcripts elongated. GST-IIc(A519-G992) did not interfere with elongation by RNA polymerase II itself. These data strongly suggest that the IIc(A519-G992) domain plays an important role in the association of transcription dTFIIF with RNA polymerase II during initiation and elongation.
FIG. 6.
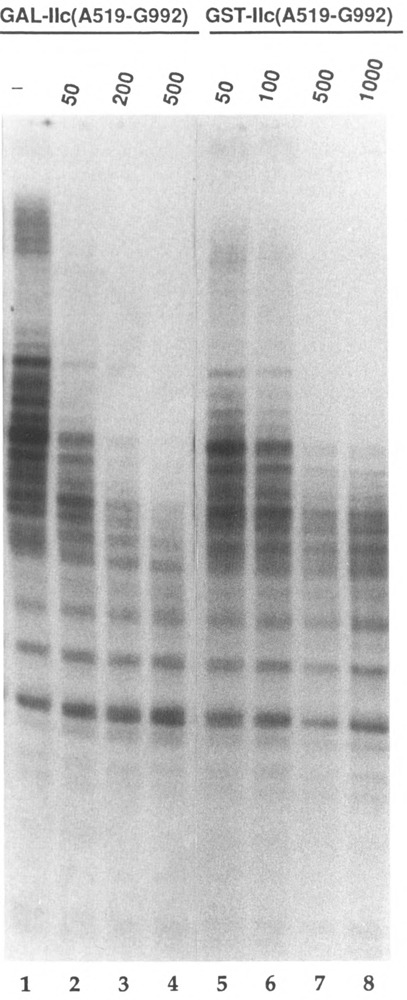
Comparison of GAL-IIc(A519-G992) and GST-IIc(A519-G992) inhibition of dTFIIF-mediated elongation stimulation. The final KC1 concentration was 80 mM; transcripts were elongated for 3 min. Each reaction contained 4.8 fmol Pol II (29 ng), 140 ng MonoQ fraction DNAse inhibitor, 5 units RNase inhibitor (Boehringer Mannheim), and 20 μg/ ml BSA. All reactions contained 100 ng of MonoQ-purified dTFIIF. Lane 1: Pol II + dTFIIF; lanes 2-4: same as lane 1 plus the indicated excess of GAL-IIc(A519-G992); lanes 5-8: same as lane 1 plus the indicated excess of GST-IIc(A519-G992).
To determine whether the inhibitory lie domain could be localized any further, we conducted preliminary tests for inhibition by small GST-IIc fusion proteins in KcN transcription and elongation assays. The results of these studies are summarized in Fig. 1C. None of the smaller fusion proteins significantly inhibited either KcN-mediated transcription from a promoter or dTFIIF-specific stimulation of RNA polymerase II elongation (data not shown). Overall, the inability of GST-IIc(G776-I859), GST-IIc(E901-Q1041), and GST-IIc(D825-G992) to interfere clearly with either KcN-dependent transcription or dTFIIF-mediated elongation stimulation provides evidence that a fairly large portion of the second largest subunit is necessary for the association with dTFIIF, either via a direct interaction or by indirectly stabilizing the interacting domain.
Chemical Cross-Linking of dTFIIF to Iodinated GST-IIc(A519-G992)
Protein-protein interactions involved in many processes have been studied by an increasingly diverse range of methods, including gel mobility shift assays (Williams et al., 1991), blot-overlay techniques (Homann et al., 1991; Wood et al., 1992), and affinity chromatography (Ing et al., 1992; Koleske et al., 1992). Of course, the intrinsic affinity of the associating proteins ultimately determines whether a complex can be detected by a particular assay method. In addition, the lifetime of association of a complex is affected by differences in reaction pH, salt concentration, and concentrations of the interacting proteins. Binding of mammalian dTFIIF analogue, RAP30/74, to immobilized RNA polymerase II is modest, with a reported dissociation constant of 2 x 10−8(T8 M (Formosa et al., 1991). We had hoped that the association of dTFIIF with the GST-IIc fusion protein might be similarly detected by using immobilized fusion proteins; however, such efforts were unsuccessful (data not shown). We then sought an alternative method to study the association of dTFIIF with RNA polymerase II, namely chemical cross-linking with ethylene glycol bis-succinimidyl succinate (EGS).
Chemical cross-linking has been used to study the spatial relationships between components of complex structures such as virions (Mariani et al., 1990), cytochromes (Font et al., 1991), heat shock factor (Clos et al., 1990), or ribosomes (Peters and Richards, 1977). In general, the spacing and geometry of the target moieties and the linear dimensions and geometry of the reagent used are critically important for the establishment of a successful cross-link. Naturally, when intermolecular protein-protein interactions are being studied, the concentrations of the reacting species must be sufficiently high for the complexes to be detected. Ideally, the amount of reagent used should be enough to cross-link the proteins of interest without modifying every available reactive group. Although a single modification is unlikely to cause drastic changes, it is possible that modification of several or key hyperreactive groups may markedly affect the protein’s physical properties or biological activity. The contribution of most small cross-linking agents to molecular weight differences observed between species is negligible; however, possible changes in hydrodynamic properties of the cross-linked species must not be overlooked.
Radiolabeled GST-IIc(A519-G992) was incubated either alone or with an excess of Mono Q-purified dTFIIF for 1 h before cross-linking of the proteins with EGS. Figure 7 shows that three protein bands, with apparent molecular weights of approximately 190, 170, and 120, had cross-linked to labeled GST-IIc(A519-G992) in the presence of added dTFIIF, but not in its absence. This effect was reproduced in three separate experiments (unpublished results). Factor dTFIIF (factor 5) has subunits of 86 kDa (dTFIIFa or factor 5a) and 34 kDa (dTFIIFb or factor 5b), and a total mass of 120 kDa, whereas GST-IIc(A519-G992) has an estimated mass of 86 kDa also. Although this experiment does not show directly that dTFIIF subunits are present in these cross-linked species, the band sizes are consistent with fusion protein cross-linking to dTFIIF(a + b), dTFIIFa (factor 5a) only, or dTFIIFb (factor 5b) only, respectively. In a separate experiment using unlabeled fusion protein, antibodies specific for the IIc(A519-G992) domain detected the 190-kDa band, confirming the presence of GST-IIc(A519-G992) in the cross-linked complex with dTFIIF (data not shown). The smaller cross-linked bands were not detected by chromogenic detection of the antibodies, probably due to the limited sensitivity of this method.
FIG. 7.
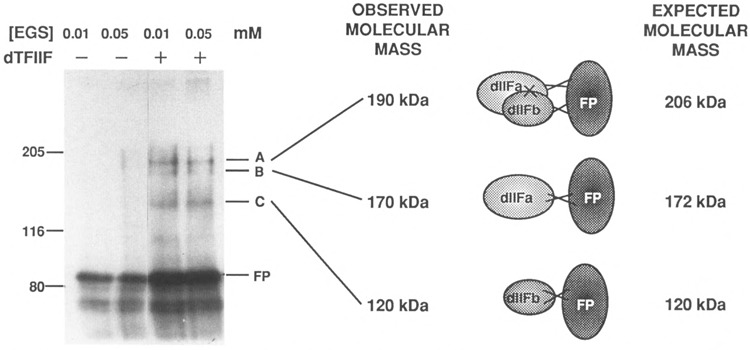
Chemical cross-linking of iodinated GST-IIc(A519-G992) to dTFIIF. Each 15-μ1 reaction contained 5 μl [l25I]GST-IIc(9A519-G992) [5.6 × 105 cpm] and either no factor (lanes 1 and 2) or 1.5 μg dTFIIF fraction Q15 (lanes 3 and 4) at a final salt concentration of 100 mM KC1. The reactions were incubated for 1 h at 25°C; cross-linking with EGS proceeded for 15 min and was then quenched by the addition of 30 mM lysine. Electrophoresis sample buffer was added (from 6x stock), the samples were heated to 100°C for 5 min, and then loaded on a 7.5% polyacrylamide-SDS gel. The gel was blotted to nitrocellulose for 2 h at 0.3 amps, air dried, then exposed to X-ray film. Reactions contained 0.01 mM EGS (lanes 1 and 3) or 0.05 mM EGS (lanes 2 and 4). The positions of molecular weight markers are indicated at the left. The observed and expected molecular masses of GST-IIc(A519-G992) cross-linked to one or both subunits of dTFIIF are indicated at the right.
Chemical Cross-Linking of dTFIIF to RNA Polymerase II
dTFIIF bound stably to Drosophila RNA polymerase II during glycerol gradient sedimentation (Price et al., 1989). Therefore, a complex of dTFIIF and intact RNA polymerase II should be detectable by chemical cross-linking. RNA polymerase II and an excess of TFIIF (two different preparations) were incubated together for 15 min, cross-linked with EGS, and subjected to SDS-PAGE. Identical Western blots of these gels were then incubated with antibodies to exon 2 of the D rosophila RNA polymerase II 215-kDa subunit Ha (Fig. 8A) or with antibodies to the IIc(A519-G992) domain of the 140-kDa subunit (Fig. 8B). Two major and several minor bands were detected only by antibodies to the 140-kDa subunit and not by antibodies to the 215-kDa subunit. As was shown for dTFIIF cross-linking to the fusion protein, the estimated sizes of some of the bands detected by anti-IIc antibodies are consistent with the sizes predicted for cross-linking of one or both dTFIIF subunits to RNA polymerase lk [dTFIIF + IIc = 260 kDa (bands “A”); dTFIIFa + IIc = 226 kDa (band “Bl”); dTFIIFb + IIc = 174 kDa (bands “C”)]. Note that the intensity of bands C is > > that of Bl, an observation that is consistent with dTFIIFb (the RAP 30 homologue) directly contacting RNA polymerase II (see the Introduction).
FIG. 8.
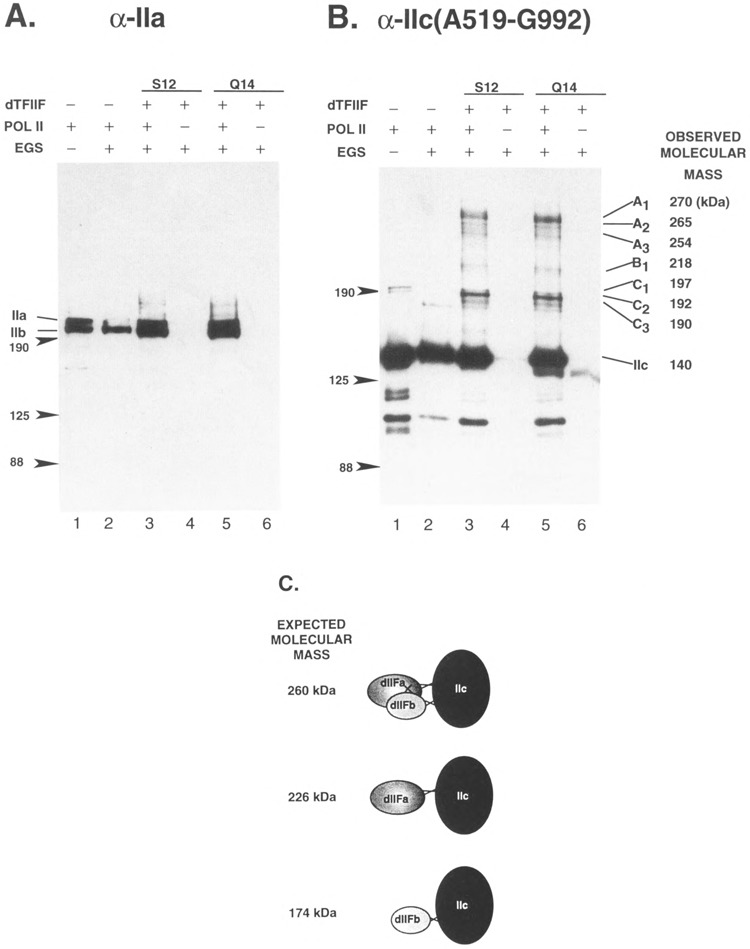
Cross-linking of RNA polymerase II to dTFIIF. Identical reactions containing 23.5 ng RNA polymerase II were incubated alone or with one of two different preparations of dTFIIF, fraction S12 (500 ng protein of a MonoS fraction) or fraction Q14 (1 mg protein of a MonoQ fraction) (Skantar, 1993, Fig. A-4), and cross-linked with 0.02 mM EGS as described in the legend to Fig. 7. Identical nitrocellulose blots were blocked and probed with goat antibodies affinity purified to the second exon of RNA polymerase subunit Ha (anti-exon 2) (Lee and Greenleaf, 1991; Weeks et al., 1993) or to IIc(A519-G992). The blots were incubated with a 1:5000 dilution of primary antibody overnight at 4°C, then for 1 h with 1:10,000 horseradish peroxidase-conjugated swine anti-goat IgG. The blots were developed using ECL chemiluminescent antibody detection system (Amersham). (A) Blot probed with goat anti-exon 2 (directed against exon 2 of the 215-kDa subunit Ha). (B) Blot probed with anti-IIc(A519-G992). To the left of each figure, the positions of molecular size markers are indicated. In (A), the positions of 215-kDa subunit including the carboxy-terminal domain (Ila) or missing this feature (lib) are also indicated. In (B), the cross-linked species that appear only in the presence of the dTFIIF fractions and Pol II are grouped A, B, and C, along with their observed molecular masses. The molecular composition and sizes expected for cross-linking of dTFIIF subunits to He are illustrated in (C). Note that the 260-kDa species may result from individual cross-linking of the dTFIIF subunits to He, or by cross-linking of dTFIIF subunits to each other with only one being cross-linked directly to IIc.
Although some of the cross-linking may have been due to specific or nonspecific binding to other proteins in the dTFIIF fractions, some, if not all, of the bands are due to specific cross-linking of the 140-kDa subunit to dTFIIF. First, two different dTFIIF preparations gave virtually identical results. Also, when an equivalent amount of BSA was incubated with RNA polymerase II in the presence of EGS, no BSA-dependent cross-linking was detected by either large subunit antibody (data not shown). Furthermore, that anti-IIc(A519-G992) antibodies affinity purified from two different goats, one injected with purified native RNA polymerase II and one injected with bac-terially expressed IIc(A519-G992), detected the same pattern of cross-linked bands shows that the effect was not due to nonspecific cross-reaction of antibodies to proteins in the dTFIIF fraction (data not shown). Taken together, all of these results point to the specific cross-linking of dTFIIF to subunit He.
DISCUSSION
Domain Interference and the Role of dTFIIF During Transcription
The elongation efficiency of both prokaryotic and eukaryotic RNA polymerases is limited significantly by the amount of time they spend at pause sites in the DNA. Previous work from this lab and others has shown that RNA polymerase II pauses frequently during transcription on dC-tailed templates, with a very strong, apparently sequence-independent pause occurring at base 13 (formerly called the 14-mer) and many sequence-dependent pauses occurring downstream (Kadesh and Cham-berlin, 1982; Coulter and Greenleaf, 1985; Gao and Price, 1993; Reines, 1992; Sluder et al., 1989). Unlike factor DmS-II, which can suppress pausing, dTFIIF does not alter the ability of RNA polymerase II to recognize a particular site as a pause; it simply reduces the amount of time Pol II spends at the pause. Sluder et al. (1989) also found that dTFIIF increased the elongation rate of RNA polymerase II through the 13-mer region, and demonstrated the ability of dTFIIF to increase the total number of polymerase molecules productively engaged in transcription on dC-tailed templates (reproduced here in, for example, Fig. 5A, lanes 1 and 2). They also discovered that dTFIIF reduced the residence time of Pol II at multiple sites downstream of the 13-mer, causing a three-to fivefold stimulation of the overall elongation rate (Price et al., 1989; Sluder, 1988). However, dilution experiments and size fractionation of actively transcribing complexes have shown that dTFIIF is not stably bound to RNA polymerase II during elongation on dC-tailed templates (Price et al., 1989). Therefore, under these conditions dTFIIF apparently interacts independently with polymerase each time it pauses, and an excess of dTFIIF over RNA polymerase II is necessary to ensure that this encounter is not rate limiting.
In the studies reported here, we observed inhibition of dTFIIF-stimulated transcription of dC-tailed templates by fusion proteins carrying portions of RNA polymerase subunit lie. This observation is consistent with the proposals that the described effects of dTFIIF on elongation occur by virtue of its binding to a site on RNA polymerase that includes portions of the lie subunit, and that excess fusion protein present in the reaction titrates dTFIIF, diminishing its productive interactions with the transcribing polymerase. We note that the effective fusion proteins, GAL-IIc(A519-Kl157), GAL-IIc(A519-G992), and GST-IIc(A519-G992), did not significantly change the relative strength or pattern of pause sites observed with RNA polymerase II alone or with added dTFIIF during elongation from either the dC-Ball-E or dC-PCP templates (Figs. 5 and 6B, and data not shown). Thus, the fusion proteins apparently exerted no additional effects on the elongation reaction other than to prevent dTFIIF from productively engaging the polymerase. In addition, that GAL-IIc(A519-K1157), GAL-IIc(A519-G992), and GST-IIc(A519-G992) most dramatically affected the production of long transcripts in these dC assays illustrates that the inhibition by these three fusion proteins is neither instantaneous nor irreversible.
That large molar excesses of these fusion proteins were required to produce a clear reduction in elongation rate is consistent with the notion that dTFIIF acts via a transient interaction with this Pol II domain. Similarly, the inability to show association between dTFIIF and the lie fusion proteins in the absence of cross-linker may also reflect the dynamic nature of this interaction. It may also be that part of the necessity for excess fusion proteins can be attributed to only a fraction of the fusion protein being folded correctly. All of the fusion proteins we generated were insoluble as expressed in E. coli, and obtaining soluble protein necessitated denaturation, renaturation, and dialysis into appropriate buffers. As one approximation of correct refolding, we monitored the specific activity of 0-gal in the GAL-IIc constructs. However, it is not possible to know how accurately this reflects the refolding of the lie portion of the fusions.
The domain interference results are consistent with the findings of Price and coworkers (Kephart et al., 1992), who showed that three agents known to disrupt protein-protein interactions inhibited the activity of dTFIIF during transcription from dC-tailed templates. In the presence of 250 mM KC1, 0.3% Sarkosyl, or 1 mg/ml heparin, dTFIIF stimulation of RNA polymerase II elongation rate was nearly or completely eliminated. That disruptive agents or an excess of dTFIIF binding sites can inhibit dTFIIF activity to a similar degree confirms that this factor is not tightly bound to RNA polymerase II during elongation as measured here in vitro.
The transcription inhibition data strongly suggest that dTFIIF makes direct contact with the second largest subunit of RNA polymerase II. On the other hand, our attempts to demonstrate binding between fusion protein GST-IIc(A519-G992) and dTFIIF in the absence of cross-linking agents were not successful (data not shown). The dissociation constant measured for RAP30/74 binding to immobilized RNA polymerase II is 2 × 10−8 M (Formosa et al., 1991), which is probably a reasonable approximation of dTFIIF binding as well. However, one might predict that the absence of certain lie domains or additional RNA polymerase subunits that stabilize dTFIIF binding could increase the dissociation constant, as could altered folding or stability of the fusion proteins. Therefore, we propose that the binding of dTFIIF to the fusion proteins is likely much weaker than to native polymerase, rendering it undetectable in the absence of cross-linking agents.
The cross-linking of dTFIIF to [125I]GST-IIc(A519-G992) (Fig. 7) or to subunit IIc of native Drosophila RNA polymerase II (Fig. 8) adds some physical evidence to support a direct involvement of this subunit in the modulation of RNA polymerase II activity by dTFIIF. As shown in the Results, some of the shifted bands containing fusion protein or subunit lie had apparent molecular masses in SDS-PAGE gels consistent with cross-linking to one or both dTFIIF subunits. Due to the complexity of cross-linking reactions, we cannot say whether both dTFIIF subunits bound directly to lie domains or whether only one dTFIIF subunit was directly attached. Both possibilities are illustrated in the schematic representations of expected cross-linking products (Figs. 7 and 9C). However, others have shown that mammalian RAP30 (the dTFIIFb analogue) can bind to RNA polymerase II in the absence of RAP74 (the dTFIIFa analogue) (Killeen and Greenblatt, 1992). Therefore, it is possible that dTFIIFb bound directly to and was cross-linked to subunit He, and that dTFIIFa cross-linked to dTFIIFb. dTFIIFa may also cross-link directly to IIc if its attachment to dTFIIFb brings it into proximity with He, such that cross-linking can take place. Interestingly, σ factor and the β subunit of E. coli RNA polymerase cross-link to the lac UV5 promoter only five nucleotides apart (Paddon and Hartley, 1987). Given the apparent sequence and functional similarity between RAP30 and a70 (McCracken and Greenblatt, 1991), and between Drosophila Pol lie and β (Falkenburg et al., 1987), it is perhaps not surprising that dTFIIF was cross-linked to IIc or that the IIc fusion proteins interfered with dTFIIF function.
Defining Functionally Significant Regions of RNA Polymerase II
Table 1 summarizes critically important amino acid residues or domains defined by genetic manipulations or functional analyses of second largest subunits from several different organisms, focusing on mutations that overlap the Drosophila IIc sequences that inhibited dTFIIF in this study. Interestingly, mutations in many of these sites affected the propagation of RNA polymerase (i.e., promoter clearance and pausing, Sagitov et al., 1993), or interfered with interactions of transcription factors GreB (Borukhov et al., 1993) or a (Glass et al., 1988). Other mutations have been localized near the enzyme’s catalytic center (Scafe et al., 1990a; Chen et al., 1993; Kashlev et al., 1990; Lee et al., 1991). Additionally, alterations in Drosophila RpII140 or yeast RPB2 suppress mutations in the large subunit of RNA polymerase II, thus implicating domains E and I in subunit-subunit contacts (Table 1).
TABLE 1.
FUNCTIONALLY DEFINED RESIDUES OF SUBUNIT IIC HOMOLOGUES IN DOMAINS THAT OVERLAP THE dTFIIF-INTERACTING FUSION PROTEINS
| Species | Amino Acids(s) | Domain | Defect or Function | Reference |
|---|---|---|---|---|
| E. coli-RPOB | 813(G→K) | F | decreased promoter clearance; abortive initiation & pausing | Lee etal., 1991 |
| Several | F&I | altered termination and pausing | Landick et al., 1990a | |
| 1065 (K→R) | H | near priming nucleotide; fails to elongate properly | Kashlev etal., 1990 | |
| D1064-K1073 | H | polymerase propagation | Sagitov et al., 1993 | |
| Δ 889-1342 | I | fails to bind σ | Glass etal., 1988 | |
| S. cerevisiae-RPB2 | 913 (G→D) | G | near proposed purine binding site | Scafe etal., 1990b |
| K936 | H | active site labelling (priming nucleotide) | Treich et al., 1992 | |
| 1142(G→D) | I | gene-specific transcription defects | Scafe etal., 1990a | |
| 1145 (S→L) | I | suppress rpbl-1 mutation | Martin et al., 1990 | |
| 1136(D→N) | I | suppress rpbl-1 mutation | Martin et al., 1990 | |
| Drosophila-RplUM | SI, 728 (S→C) | E | suppress RpII215 WKJ1 | Mortin, 1990, and personal communication |
| 57, 735 (M→I) | E | suppress RpII215WKJ1 | Mortin, 1990, and personal communication | |
| Z19, 1028 (E→K) | I | recessive lethal | Chen etal., 1993 | |
| A5,Δ1047-1051 | I | recessive lethal | Chen etal., 1993 | |
| Z43, 940 (R→H) | H | enhance Ubx phenotype | Chen etal., 1993 | |
| Z45, 1098 (S→F) | I | recessive lethal | Chen etal., 1993 | |
| M39, 992 (G→E) | H/I | recessive lethal | Chen et al., 1993 |
The inability of the smaller fusion proteins GST-IIc(G776-I859), GST-IIc(D825-G992), or GST-IIc(E901-1041) to interfere with dTFIIF activity in the elongation assay as effectively as GST-IIc(A519-G992) suggests that much of the region encompassed by residues 519-992 is important for the interaction of dTFIIF with lie (Fig. 7). However, at this time, some combinations of domains D-I have not been investigated, and we therefore do not know whether certain other fusion proteins would inhibit dTFIIF as effectively as GST-HC(A519-G992). Because conserved domain I has been implicated in functions such as a factor binding (Glass et al., 1988), it remains possible that this C-terminal domain plays a role, possibly an indirect one, in the dTFIIF interaction with IIc.
Potential Future Applications of the Domain Interference Assay
Given that mammalian TFIIF (RAP30/74) can be dissociated by salt from immobilized RNA polymerase II columns (Sopta et al., 1985), and that dTFIIF stimulation of elongation on dC-tailed templates is inhibited by 0.3% Sarkosyl, 1 mg/ml heparin, or 250 mM salt (Kephart et al., 1993), it seems likely that the einding gf TFIIF to RNA polymerase II involves electrostatic interactions. Intriguingly, long stretches of charged residues have been located in RAP74 (Finkelstein et al., 1992; Kephart et al., 1993), and this TFIIF subunit is also believed to be phosphorylated in vivo (Sopta et al., 1985). It will be important to know what role if any these properties play in the association of the factor with the enzyme, and preliminary functional dissections of this TFIIF sub-unit have been carried out (Kephart et al., 1994, and refs, therein). When both dTFIIF subunit genes are cloned, it should be possible to exploit their availability in combination with the lie constructs and approaches we have described in this paper to carry out more detailed investigations of interactions between dTFIIF and Pol II.
Several groups have examined the entry of basal transcription factors and RNA polymerase II into initiation complexes (Buratowski et al., 1989; Killeen et al., 1992; Lu et al., 1992). As pointed out recently, in order to understand the architecture of the initiation complex it will be important to map the factor-polymerase interactions to specific Pol II subunits (Buratowski, 1994). .he eomain nnterference eechnique eould be quite useful in helping to determine critical protein contacts within the initiation complex. In addition, Pol II subunit domain fusion proteins might be used as probes to determine whether transcriptional regulatory proteins act via interactions with specific polymerase II subunit domains.
Finally, application of the domain interference assay to the study of existing mutant Drosophila RNA polymerase II subunits might complement genetic studies of these altered enzymes. Due to the homozygous lethality of most known RpII140 mutations, these mutant enzymes are not available for direct biochemical analysis (Chen et al., 1993, and refs, therein). However, it would be possible to express as fusion proteins subunit domains containing characterized mutations; these could then be analyzed by the techniques described in this work. For example, interactions between mutant domains and transcription factors could be examined, potentially pinpointing defective protein-protein interactions responsible for lethality in vivo. Additional point mutations, deletions, or sequence substitutions could be introduced into critical regions by in vitro mutagenesis, providing an array of altered subunit domains whose properties could then be examined. Ultimately such a combination of in vivo and in vitro approaches will contribute to an increased understanding of factor-polymerase interactions and of mechanisms involved in transcribing genetic information.
ACKNOWLEDGEMENTS
We express our gratitude to Deborah Danaher, Muh-Lin Lee, and Meei Liu for expert technical assistance; and to John Weeks, David Price, and Steven Hardin for helpful discussions. This work was supported by National Institutes of Health Grant GM 28078.
REFERENCES
- Bartholomew B., Dahmus M. E., and Meares C. F. (1986), J Biol Chem 261, 14226–14231. [PubMed] [Google Scholar]
- Bolton A. E. and Hunter W. M. (1973), Biochem J 133, 529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borukhov S., Sagitov V., and Goldfarb A. (1993), Cell 72, 459–466. [DOI] [PubMed] [Google Scholar]
- Breant B., Huet J., Sentenac A., and Fromageot P. (1983), J Biol Chem 258, 11968–11973. [PubMed] [Google Scholar]
- Buratowski S. (1994), Cell 77, 1–3. [DOI] [PubMed] [Google Scholar]
- Buratowski S., Hahn S., Guarente L., and Sharp P. A. (1989), Cell 56, 549–561. [DOI] [PubMed] [Google Scholar]
- Chen Y., Weeks J., Mortin M. A., and Greenleaf A. L. (1993), Mol Cell Biol 13, 4214–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clos J., Westwood J. T., Becker P. B., Wilson S., Lambert K., and Wu C. (1990), Cell 63, 1085–1097. [DOI] [PubMed] [Google Scholar]
- Conaway J. W. and Conaway R. C. (1990), Science 248, 1550–1553. [DOI] [PubMed] [Google Scholar]
- Coulter D. E. and Greenleaf A. L. (1985), J Biol Chem 260, 13190–13198. [PubMed] [Google Scholar]
- Dissinger S. and Hanna M. M. (1990), J Biol Chem 265, 7662–7668. [PubMed] [Google Scholar]
- Falkenburg D., Dworniczak B., Faust D. M., and Bautz E. K. (1987), J Mol Biol 195, 929–937. [DOI] [PubMed] [Google Scholar]
- Finkelstein A., Kostrub C. F., Li J., Chavez D. P., Wang B. Q., Fang S. M., Greenblatt J., and Burton Z. F. (1992), Nature 355, 464–467. [DOI] [PubMed] [Google Scholar]
- Flores O., Lu H., Killeen M., Greenblatt J., Burton Z. F., and Reinberg D. (1991), Proc Natl Acad Sci USA 88, 9999–10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Font B., Goldschmidt D., Chich J. F., Thieffry M., Henry J. P., and Gautheron D. C. (1991), FEBS Lett 279, 105–109. [DOI] [PubMed] [Google Scholar]
- Formosa T., Barry J., Alberts B. M., and Greenblatt J. (1991), Methods Enzymol 208, 24–45. [DOI] [PubMed] [Google Scholar]
- Frankel S., Sohn R., and Leinwand L. (1991), Proc Natl Acad Sci USA 88, 1192–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H. and Price D. H. (1993), J Biol Chem 268, 18762–18770. [PubMed] [Google Scholar]
- Glass R. E., Honda A., and Ishihama A. (1986a), Mol Gen Genet 203, 492–495. [DOI] [PubMed] [Google Scholar]
- Glass R. E., Jones S. T., Nene V., Nomura T., Fujita N., and Ishihama A. (1986b), Mol Gen Genet 203, 487–491. [DOI] [PubMed] [Google Scholar]
- Glass R. E., Ralphs N. T., Fujita N., and Ishihama A. (1988), Eur J Biochem 176, 403–407. [DOI] [PubMed] [Google Scholar]
- Grachev M. A., Kolocheva T. I., Lukhtanov E. A., and Mustaev A. A. (1987), FEBS Lett 103, 113–121. [DOI] [PubMed] [Google Scholar]
- Grachev M. A., Lukhtanov E. A., Mustaev A. A., Zaychikov E. F., Abdukayumov M. N., Rabinov I. V., Richter V. I., Skoblov Y. S., and Chistyakov P. G. (1989), Eur J Biochem 180, 577–585. [DOI] [PubMed] [Google Scholar]
- Greenleaf A. L. (1992), in Transcriptional Regulation, vol. 1 (Yamomoto K., and McKnight S., eds.), Cold Spring Harbor Press, Cold Spring Harbor, NY, pp 55–80. [Google Scholar]
- Gundelfinger E. D. (1983), FEBS Lett 157, 133–138. [DOI] [PubMed]
- Hager D. A. and Burgess R. R. (1980), Anal Biochem 109, 76–86. [DOI] [PubMed] [Google Scholar]
- Hamilton B. J., Mortin M. A., and Greenleaf A. L. (1993), Genetics 134, 517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann H. E., Willenbrink W., Buchholz C. J., and Neubert W. J. (1991), J Virol 65, 1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ing N. H., Beekman J. M., Tsai S. Y., Tsai M.-J., and O’Malley B. W. (1992), J Biol Chem 267, 17617–17623. [PubMed] [Google Scholar]
- Kadesh T. R. and Chamberlin M. J. (1982), J Biol Chem 257, 5286–5295. [PubMed] [Google Scholar]
- Kashlev M., Lee J., Zalenskaya K., Nikiforov V., and Goldfarb A. (1990), Science 248, 1006–1009. [DOI] [PubMed] [Google Scholar]
- Kephart D. D., Marshall N. F., and Price D.H. (1992), Mol Cell Biol 12, 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kephart D. D., Price M. P., Burton Z. F., Finkelstein A., Greenblatt J., and Price D. H. (1993), Nucleic Acids Res 21, 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kephart D. D., Wang B. Q., Burton Z. F., and Price D. H. (1994), J Biol Chem 269, 13536–13543. [PubMed] [Google Scholar]
- Killeen M. T., Coulombe B., and Greenblatt J. (1992), J Biol Chem 267, 9463–9466. [PubMed] [Google Scholar]
- Killeen M. T. and Greenblatt J. F. (1992), Mol Cell Biol 12, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske A. J., Buratowski S., Nonet M., and Young R. A. (1992), Cell 69, 883–894. [DOI] [PubMed] [Google Scholar]
- Kolodziej P. A. and Young R. A. (1991), Mol Cell Biol 11, 4669–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U. K. (1970), Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Landick R., Colwell A., and Stewart J. (1990a), J Bacteriol 172, 2844–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landick R., Stewart J., and Lee D. N. (1990b), Genes Dev 4, 1623–1636. [DOI] [PubMed] [Google Scholar]
- Lee J., Kashlev M., Borukhov S., and Goldfarb A. (1991), Proc Natl Acad Sci USA 88, 6018–6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. M. and Greenleaf A. L. (1991), Gene Expr 1, 149–167. [PMC free article] [PubMed] [Google Scholar]
- Lin K.-H. and Cheng S. Y. (1991), Biotechniques 11, 748–751. [PubMed] [Google Scholar]
- Lu H., Zawel L., Fisher L., Egly J. M., and Reinberg D. (1992), Nature 358, 641–645. [DOI] [PubMed] [Google Scholar]
- Mariani C., De Beuckeleer M., Truettner J., Leemans J., and Goldberg R. B. (1990), Nature 347, 737–741. [Google Scholar]
- Martin C., Okamura S., and Young R. (1990), Mol Cell Biol 10, 1908–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken S. and Greenblatt J. (1991), Science 253, 900–902. [DOI] [PubMed] [Google Scholar]
- Miller J. H. (1972), in Experiments in Molecular Genetics, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, pp. 398–404. [Google Scholar]
- Mortin M. (1990), Proc Natl Acad Sci USA 87, 4864–4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortin M., Zuerner R., Berger S., and Hamilton B. J. (1992), Genetics 131, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustaev A., Kashlev M., Lee J., Polyakov A., Lebedev A., Zalenskaya K., Grachev M., Goldfarb A., and Nikiforov V. (1991), J Biol Chem 266, 23927–23931. [PubMed] [Google Scholar]
- Nene V. and Glass R. (1984), Mol Gen Genet 194, 166–172. [DOI] [PubMed] [Google Scholar]
- Paddon C. J. and Hartley R. W. (1987), Gene 53, 11–19. [DOI] [PubMed] [Google Scholar]
- Palenik B. (1992), Curr Opin Genet Dev 2, 931–936. [DOI] [PubMed] [Google Scholar]
- Peters K. and Richards F. M. (1977), Annu Rev Biochem 46, 523–551. [DOI] [PubMed] [Google Scholar]
- Price D. H. and Parker C. S. (1984), Cell 38, 423–429. [DOI] [PubMed] [Google Scholar]
- Price D. H., Sluder A. E., and Greenleaf A. L. (1987), J Biol Chem 262, 3244–3255. [PubMed] [Google Scholar]
- Price D. H., Sluder A. E., and Greenleaf A. L. (1989), Mol Cell Biol 9, 1465–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappaport J., Cho K., Saltzman A., Prenger J., Golomb M., and Weinmann R. (1988), Mol Cell Biol 8, 3136–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reines D. (1992), J Biol Chem 267, 3795–3800. [PMC free article] [PubMed] [Google Scholar]
- Riva M., Schaffner A. R., Sentenac A., Hartmann G. R., Mustaev A. A., Zaychikov E. F., and Grachev M. A. (1987), J Biol Chem 262, 14377–14380. [PubMed] [Google Scholar]
- Robbins A., Dynan W. S., Greenleaf A. L., and Tjian R. (1984), J Mol Appl Genet 2, 343–353. [PubMed] [Google Scholar]
- Sawadogo M. and Sentenac A. (1990), Annu Rev Biochem 59, 711–754. [DOI] [PubMed] [Google Scholar]
- Scafe C., Martin C., Nonet M., Podos S., Okamura S., and Young R. A. (1990a), Mol Cell Biol 10, 1270–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafe C., Nonet M., and Young R. A. (1990b), Mol Cell Biol 10, 1010–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skantar A. M. (1993), PhD Thesis, Duke University. [Google Scholar]
- Sluder A. E. (1988), PhD Thesis, Duke University. [Google Scholar]
- Sluder A. E., Price D. H. and Greenleaf A. L. (1987), Biochimie 69, 1199–1205. [DOI] [PubMed] [Google Scholar]
- Sluder A. E., Price D. H., and Greenleaf A. L. (1988), J Biol Chem 263, 9917–9925. [PubMed] [Google Scholar]
- Sluder A. E., Greenleaf A. L., and Price D. H. (1989), J Biol Chem 264, 8963–8969. [PubMed] [Google Scholar]
- Smith D. B. and Johnson K. S. (1988), Gene 67, 31–40. [DOI] [PubMed] [Google Scholar]
- Sopta M., Carthew R. W., and Greenblatt J. (1985), J Biol Chem 260, 10353–10360. [PubMed] [Google Scholar]
- Sopta M., Burton Z. F., and Greenblatt J. (1989), Nature 341, 410–414. [DOI] [PubMed] [Google Scholar]
- Towbin H., Staehelin T., and Gordon J. (1979), Proc Natl Acad Sci USA 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks J. R., Coulter D. E., and Greenleaf A. L. (1982), J Biol Chem 257, 5884–5892. [PubMed] [Google Scholar]
- Weeks J. R., Hardin S. E., Shen J., Lee J. M., and Greenleaf A. L. (1993), Genes Dev 7, 2329–2344. [DOI] [PubMed] [Google Scholar]
- Williams F. E., Varanasi U., and Trumbly R. J. (1991), Mol Cell Biol 11, 3307–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood E. R., McDonald O. B., and Sahyoun N. (1992), J Biol Chem 267, 14138–14144. [PubMed] [Google Scholar]
- Yonaha M., Aso T., Kobayashi Y., Vasavada H., Yasukochi Y., Weissman S. M., and Kitajima S. (1993), Nucleic Acids Res 21, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R. A. (1991), Annu Rev Biochem 60, 689–715. [DOI] [PubMed] [Google Scholar]
- Zawel L. and Reinberg D. (1993), Prog Nucleic Acid Res Mol Biol, 67–108. [DOI] [PubMed]