

Abstract
The following review summarizes the state of the art in representative aspects of gene therapy/translational medicine and evolves from a symposium held at the School of Veterinary Medicine, University of Pennsylvania on November 16, 2017 honoring Dr. Gustavo Aguirre, recipient of ARVO's 2017 Proctor Medal. Focusing on the retina, speakers highlighted current work on moving therapies for inherited retinal degenerative diseases from the laboratory bench to the clinic.
Keywords: retinitis pigmentosa, X-linked Retinoschisis, Leber congenital amaurosis, animal models
Defining the Inherited Retinal Dystrophy Genome
Professor Alison J. Hardcastle
Abstract: Inherited retinal dystrophies display remarkable genetic heterogeneity, reflecting the complexity of retinal structure and function. Next-generation sequencing technologies have transformed our ability to identify mutation-causing diseases but also present additional challenges of interpreting exome and genome variants as potentially pathological or benign. Nevertheless, next-generation sequencing offers the opportunity to reveal the unusual and unexpected, such as expansion of phenotypes associated with specific genes, unexpected inheritance patterns and aberrant splicing, which is emerging as a relatively common mechanism of disease resulting from introduction of cryptic exons, and rare combinations of single-nucleotide polymorphism haplotypes. As a complementary approach to developing animal models to study disease mechanisms and test potential mutation-specific therapies, we can differentiate pluripotent stem cells to retinal pigment epithelium and optic cup organoids with mutations in human genomic and cellular context, another very useful tool in the study of inherited retinal degenerations.
The first gene mutation causing an inherited retinal degeneration was reported in the rhodopsin gene in 1990.1 Linkage studies and Sanger sequencing of candidate genes were instrumental in these early analyses, but the completion of the human genome sequence and next-generation sequencing (NGS) technologies has accelerated the rate of discovery.
Today, over 280 retinal disease genes have been mapped and/or identified (Fig. 1). These retinal gene mutations lead to many different types of retinal dystrophy, including Leber congenital amaurosis (LCA), which affects rod and cone photoreceptors, cone-specific diseases, such as achromatopsia, macular degeneration, such as Stargardt disease, and rare syndromic diseases, such as Usher Syndrome and Bardet-Biedl Syndrome. Mutations in a diverse number of genes can lead to a similar disease phenotype; for example, at least 14 genes have been identified whose phenotypic characteristics fall within the LCA classification. NGS has accelerated this gene discovery process, but this also presents challenges in interpreting potentially pathogenic variants compared with benign variants. Specifically, whole-exome sequencing, which will sequence all protein coding parts of genes (∼20,000 genes), can identify genetic causes in approximately 60% of cases. However, all 3 billion nucleotides can now be sequenced using whole-genome sequencing (WGS), but it can be very challenging to identify a causative mutation as WGS uncovers approximately 5 million differences in an individual compared with the reference human genome. Nevertheless, NGS has provided an unbiased opportunity to reveal unusual and unexpected phenotypic associations, inheritance patterns, and genetic mechanisms of disease (see examples below). Finally, animal models of retinal degeneration, particularly in the dog, have been instrumental in studying orthologous human retinal degenerations. For example, in XLPRA,2 physical mapping of the canine genome, linkage mapping and cloning with characterization of the RPGR gene, led to the identification of the causative RPGR variants in different dog breeds and finally to gene therapy.3 Several canine disease models have been developed by the Aguirre group, including models of photoreceptor dysplasia (1998), RPE65-based childhood blindness (2001), and achromatopsia (2002); stationary night blindness has even been studied in the Appaloosa horse (1978).
Figure 1.
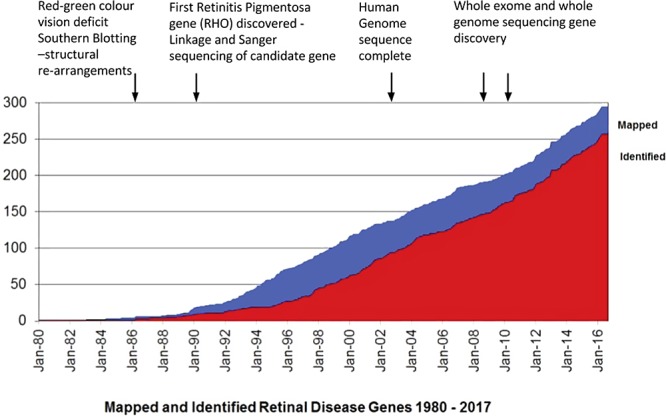
Mapped and identified retinal disease genes. Figure modified from RetNet (in the public domain, https://sph.uth.edu/retnet) to highlight landmark time points in path to disease gene discovery.
As mentioned above, NGS is a very valuable tool in studying retinal degenerations because it is unbiased and allows for all possible inheritance modes to be considered for association of gene variants with novel phenotypes. A good example of this is the study of missense variants in the X-linked gene PRPS1, which can cause retinal degeneration in females.4 These patients show a type of retinitis pigmentosa (RP) with tapetal-like reflexes and patchy retinal atrophy. Missense mutations in PRPS1 can have multiple effects, causing Arts Syndrome, X-linked Charcot-Marie Tooth 5, and X-linked nonsyndromic deafness. The absence of affected males in the RP families suggests some PRPS1 variants may be embryonic male lethal when inherited in the hemizygous state. In another family consistent with X-linked inheritance, no causative variants on the X-chromosome were found; however, two potentially deleterious compound heterozygous variants were detected in two autosomal recessive genes, indicating two recessive conditions in different branches of the family. Similarly, linkage analyses and NGS can uncover new loci and genes for genetically heterogeneous diseases. For example, the identification of the mutation responsible for the RP23 locus for X-linked RP5 was possible through targeted genomic NGS that revealed a deep intronic mutation in OFD1.6 Point mutations in the coding region and splice sites of OFD1 cause ciliopathies, including male-lethal orofaciodigital syndrome 1, Simpson-Golabi-Behmel syndrome, type 2, and Joubert syndrome. Interestingly, RP23 is not male lethal; lower levels of transcript are present, perhaps producing enough normal protein for other ciliated tissues, but not the retina.
Approximately 8% of human males have a color vision deficit, estimated at 250 million world wide. The long wavelength (LW) and medium wavelength (MW) cone opsins are highly homologous and share 98% identity at the nucleotide level. Recombination between LW and MW genes creates a variety of mutations and can also result in combinations of polymorphic variants in exon 3. Mutations in the OPN1LW (LW) and OPN1MW (MW) cone opsin genes can lead to a wide array of cone photoreceptor abnormalities, including progressive retinal degeneration. Genotypes can be clustered into the following 3 types: (1) deletions of the locus control region, (2) inactivating mutations, and (3) exon 3 single-nucleotide polymorphism (SNP) interchange haplotypes. Some mutations can cause blue cone monochromacy with no, or very slow progression, or are associated with a diagnosis of progressive cone dystrophy. Interestingly, in vitro splicing assays have shown that interchange haplotypes, which in isolation would not be considered to be detrimental to cone opsin function, affect splicing, causing skipping of exon 3.7 All in all, it is clear that mutations in the LW and MW cone genes can lead to a wide variety of dystrophies ranging from simple color blindness to severe cone cell dystrophies.
Along with mutant gene discovery and characterization, drugs and small molecules targeting specific gene mutations, such as translational read-through (TR-T) drugs, can be used in therapy. An example is the case of mutations in the RP2 gene, which leads to vision loss in a form of X-linked RP, and is often affected by nonsense mutations. Patient fibroblasts and iPSC-derived retinal pigment epithelial (RPE) cells from a patient with the R120X mutation lack RP2 protein, but, using translational read-through-inducing drugs, up to 20% of normal RP2 protein can be restored8 with reversal of cellular phenotype defects. This suggests that the TR-T drugs could be tested clinically for the restoration of vision in some RP2 patients with nonsense mutations. In a similar manner, specific gene mutations can be studied in patient iPSC-derived RPE and optic cups in genomic and cellular context, providing a model system to test disease mechanisms and potential therapies. In optic cups, not only can the underlying defects be identified, but corrective measures can also be designed and tested. Three-dimensional optic cups from iPSCs carrying a CEP290 mutation that causes LCA revealed that the optic cups have higher levels of aberrant splicing caused by the intronic mutation leading to fewer and shorter photoreceptor cilia. When the optic cups were treated with an antisense morpholino, aberrant splicing is blocked and full-length CEP290 was generated,9 leading to a 5-fold increase in the correctly spliced mRNA in the CEP290-LCA optic cups. Thus, personalized models, such as these optic cups as well as advanced molecular biology techniques, can lead to a better understanding of disease mechanisms as well as delivering therapeutic insights.
Conclusions and Future Directions
NGS helps us understand disease mechanisms resulting from factors, such as introduction of cryptic exons and rare combinations of SNP haplotypes. It is a complementary approach to developing animal models to study disease.
We also can differentiate pluripotent stem cells into RPE cells and optic cup organoids with mutations in human genomic and cellular context. This is another useful tool in the study of the mechanisms of inherited retinal degenerations.
Thoughts on Human Ocular Gene Therapy: The Retinoschisis Gene Therapy Clinical Trial
Dr. Paul A. Sieving
Abstract: Successful retinal pigment epithelial (RPE) gene therapy for Leber congenital amaurosis (LCA) in 2008 demonstrated therapeutic proof-of-concept for treatment of human retinal degenerations. For the RPE65-LCA trial, the vector is delivered by a subretinal surgical approach, which treats a delimited area. Intravitreal injection would be preferred for ease of administration and to reach a wide expanse of retina. Unfortunately, current vectors do not readily penetrate into the retina. We are exploring the use of intravitreal application of vector for a gene therapy trial of X-linked retinoschisis (XLRS; Clinical trials.Gov #NCT02317887). The photoreceptor electroretinogram (ERG) a-wave is characteristically normal in XLRS but the postsynaptic bipolar cell B-wave is depressed. We have evidence that the retinoschisin (RS1) extracellular matrix protein is essential to maintain synaptic integrity of rod photoreceptor signaling onto bipolar cells. Hence, XLRS can be considered a synaptic disease. Adeno-associated virus (AAV)-RS1 gene therapy in XLRS retinoschisis knockout (RSI-KO) mice closes the schisis cavities seen in XLRS patients, improves synaptic function, and increases the ERG b-wave. Our clinical trial addresses whether this benefit extends to human vision. Gene replacement therapy for monogenic disease is simple in concept but implementation is not necessarily straight forward. Critical factors in AAV vector capsid biology that warrant attention are as follows: (1) vectors that can penetrate into the retina after intravitreal delivery, (2) increased transduction targeting and efficiency, and (3) lower immunogenicity. The field is moving forward at a deliberate pace through this and other ocular gene therapy trials that are underway.
Why gene therapy of the eye? To start with, inherited retinal disorders affect 1 in 3000 individuals around the world. They can cause progressive vision loss and blindness, which can even be apparent from early age as in Leber congenital amaurosis (LCA). Inherited retinal disorders are the most common cause of visual impairment registration in the United Kingdom among working age individuals and second in childhood. There are both personal and societal reasons for addressing this problem, not least of which involve the medical costs and the loss of productivity due to loss of vision. The eye is well suited for gene therapy, as it is a small, closed compartment that allows for targeted delivery to the retina. Thus, only small quantities of vector are needed for treatment. Further, the eye is relatively immune privileged so there is limited risk of systemic toxicity. Animal models exist for many of the inherited retinal diseases, and the cell types in the eye are evolutionarily conserved from mouse to human. Optical transparency of the eye allows for safe, noninvasive retinal imaging in vivo. Finally, the contralateral untreated eye can serve as a convenient and effective control.
Following the lead of the LCA clinical trials for mutations of the RPE65 gene, there are currently a number of gene therapy clinical trials in progress on different forms of retinal degeneration (Table). These range from types of rod-based diseases, such as retinitis pigmentosa (RP) to diseases mainly affecting the macula and cone cells (Stargardt disease), as well as conditions, such as XLRS, which involve progressive impairment of retinal synaptic function. Mostly, AAV viral vectors have been used (AAV2, AAV8) to deliver the normal gene replacement although lentivirus also appears to be an effective vehicle. The route of administration is also under study (i.e., subretinal versus intravitreal delivery).
Table.
Representative Gene Therapy Clinical Trials

Mutation of the RS gene is causative for XLRS and results in reduction or elimination of the RS protein. Immunolabelling reveals that the retinoschisin protein is found in two prominent locations in the retina, around the rod inner segments and around the rod-bipolar synapses. More specifically, it is an extracellular matrix protein with high lipid affinity and coats the rod inner segments, although it is not an integral membrane protein. Electron microscopy (EM) immunogold-histochemistry shows RS protein within the synaptic cleft between rod and depolarizing bipolar cells. Loss of RS protein possibly explains why the XLRS problem involves deficient activity postsynaptic to the photoreceptor. At the molecular level, cryo-EM three-dimensional imaging shows back-to-back RS protein rings with eight RS proteins aggregated into a planar disc and two octomers forming a bilayer complex.10 Octomer-octomer contacts may couple neighboring cell membranes, promoting cellular adhesion; loss of RS1 would lead to the disease-characteristic schisis cavities. All in all, a “Velcro Zipper Model” may explain one action of the RS protein.
The prevalence of XLRS is approximately 1 in 5000 to 1 in 25,000 depending on the particular population examined. Vision loss can be accompanied by strabismus, nystagmus, and amblyopia. Diagnosis is both clinical examination and electrophysiology, using techniques, such as optical coherence tomography (OCT) and ERG. OCT reveals typical cystic spaces within the retinal layers while dark-adapted ERG analysis demonstrates a typical decrease or loss of the b-wave amplitude relative to the a-wave. The intraretinal splitting results from mutations in the RS gene that cause decreased RS protein but the exact mechanism is not yet understood. Good genetic studies start with good genetic pedigrees, and we were fortunate at the University of Michigan to have an extensive pedigree of XLRS and to collect blood samples from 119 members of the family. In addition, preclinical studies on gene therapy safety and efficacy were facilitated by the construction of a RS1-KO in our lab.11 In this model, the transgene construct involved replacement of exon 1 by a neomycin cassette and resulted in retinal structural disorganization, no RS protein detectable and functional loss of the ERG b-wave implicating a synaptic deficit in the absence of the RS protein. Problems in delayed rod photoreceptor cell maturity as well as impaired light-driven transducin translocalization have been reported more recently.12 In this RS-KO model, we initially performed gene therapy using an AAV(2/2)-cytomegalovirus (CMV)-Rs1h vector containing Rs1h cDNA with a CMV promoter. Subsequent work expanded on these studies with an AAV8-RS1 vector restoring function and causing synaptic signaling proteins to re-localize properly to the dendritic tips of bipolar cells. This construct was injected into the vitreous cavity of the XLRS mouse at 4 weeks of age, and OCT and ERG studies were performed 12 weeks later. At that time, we found that the schisis cavities were closed in the treated eyes, and the b-wave had recovered (Fig. 2). Thus, such treatment “restored the normal ERG configuration,” demonstrated safety and efficacy of the procedure and allowing for movement toward a human clinical trial.
Figure 2.

AAV-RS1 rescues structure and function in the XLRS mouse. scAAV-hRS1/IRBP- hRS1 injected intravitreally at 4 weeks of age and OCT and ERG evaluated 12 weeks later. Left panel demonstrates that schisis cavities close. Right panel demonstrates restoration of ERG b-wave (green arrow).
For the clinical trial, we developed the scAAV8-hRS/interphotoreceptor retinoid-binding protein gene (IRBP)-RS1 as the clinical vector (produced by Dr. J. Fraser Wright at Children's Hospital in Philadelphia, PA). It is a nonpathogenic, replication-deficient AAV sero-type 8 capsid containing the complete human retinoschisis cDNA and a tissue-specific retinoschisis promoter augmented by an IRBP enhancer element. As demonstrated in the XLRS mouse, intravitreal delivery of AAV8 RS reached the entire retina,13 and distributed across all of the retina as seen histologically by 17 weeks of age. Importantly, cell-type specific gene expression was seen with no “off-target expression.” A preclinical dose-escalation study of intravitreal AAV8-RS1 gene therapy was performed in the RS1-KO mouse for the Food and Drug Administration (FDA) investigational new drug (IND) application.14 With vector applied in mice at 4 weeks of age and analysis 12 weeks thereafter, a dose-dependent expression was observed with improved retinal structure and function at 1 × 108 vector genome copies per eye. Significantly, the study indicated that a “fully normal level of RS1 expression” was not necessary for a therapeutic effect.
With this background, an application for an XLRS gene therapy trial was made with FDA authorization of the IND application in November of 2014, registration with ClinicalTrials.Gov in December of 2014 and the first participant dosed with vector in February 2015. Prior to this, however, important matters needed to be considered in preparing for the trial. What XLRS patient population should be used? What are appropriate outcome measures, and what is the reliability of those measures? In human XLRS, there is a broad ‘treatment window of opportunity' because the disease progresses to macular thinning and atrophy during middle age. After age 45 to 50, the schisis collapses and macular photoreceptors begin to atrophy. The therapeutic outcome can be estimated using visual acuity, full-field ERGs, microperimetric macular visual sensitivity, and retinal thickness measured by OCT. A test-retest intervisit variability study was performed on seven participants in four visits over a 6-month interval. We determined the 95% confidence intervals for visual acuity, ERG measurements, retinal sensitivity, and central retinal thickness relative to baseline in the XLRS patients studied, and thus these outcome measures are suitable for use in the trial.15
At present, we are pursuing a human Phase I/IIa trial of RS1 retinal gene therapy transfer for XLRS. The study objective is to determine the safety, tolerability, and biological activity of intravitreal administration of gene transfer vector scAAV8-hRS/IRBP-hRS to the retina of XLRS affected men. It is a prospective, single-center (National Institutes of Health), dose-escalation study using the AAV-RS1 vector administered in one eye by intravitreal injection. The primary and secondary outcomes are as follows: (1) adverse events that affect ocular structure or function, (2) retinal structure changes as determined by OCT, (3) retinal function changes as determined by ERG response, (4) visual field sensitivity as determined by MP-1 microperimetry, (5) change of systemic blood chemistry parameters, and (6) determination of circulating systemic anti-AAV8 antibodies. Study eyes received a dose of 1e11 vector genomes per eye. In one participant, extensive parafoveal schisis cavities were observed prior to treatment with complete closure by 14 days after vector administration.
Conclusions and Future Directions
With this good news, we can plan the next steps in our XLRS trial.
The dose range and number of participants can be extended.
We can plan to better understand the inflammatory immune response.
We seek to design measures to manage the acute ocular inflammation observed. For the field generally, there are areas that need further development.
Better vectors/synthetic capsids should be developed. In particular, we should develop intravitreal vectors to enhance retinal delivery.
Learn how to modulate the inevitable immune response.
Extend the durability of treatment benefit.
Beyond these factors, it should be remembered that each disease has unique features to understand and address. Outcome metrics must be tailored specifically for each condition. Finally, we must remember that, although patients are eager to participate in clinical trials, it is critical to manage their expectations.
Shooting in the Dark: Studies in Cone-Targeted Therapies
Dr. José-Alain Sahel
Abstract: We have started on a “fishing expedition” aiming at identifying the underlying mechanisms in cone degeneration as potential clues for therapies that preserve/restore light-adapted and central vision in patients with retinal dystrophies. The discovery of rod-derived cone viability factor (RdCVF), the identification of its receptor and mechanism of action, and the demonstration of its potential therapeutic benefit in several animal models paved the way for upcoming clinical trials. Emerging optogenetic strategies are now under evaluation to restore cone or inner retinal function. Selection of target populations of patients that might benefit from these strategies will rely on in depth phenotyping, while demonstration of the therapeutic value will require the development of novel, real life, functional outcome measurements.
Pathologies with photoreceptor degeneration are varied in type and number. Thirty million are affected, including age-related macular degeneration, with 10% to 20% severely impaired. Fewer are affected with retinitis pigmentosa (RP; 1/4000 individuals) and allied diseases, but profound vision loss and blindness is a common occurrence in these pathologies, often from birth. Our goal is to provide personalized therapies to all of these retinal dystrophies. Several of the following factors are involved in this process:
identification and correction of the causative gene defect
protecting the remaining photoreceptor cells
restoring light responses in the degenerated photoreceptor or other retinal cells
restoring retinal encoding
determining the status of the remaining tissue
demonstrating the restoration of useful vision
The process starts with identification, characterization, and ultimate correction of causative gene defects in the inherited retinal dystrophies. Once identified and characterized, correction can take several approaches, such as gene therapy or gene editing. Recent achievements in gene therapy, include progress in clinical trials for Leber congenital amaurosis (LCA), choroideremia, retinoschisis, achromatopsia, Stargardt disease, and Usher syndrome, with preclinical work on many disease-causing genes, including PDE6 and RPGR. Nowhere is this more important than in therapy for cones of the macula, because these photoreceptors serve central, sharp, and color vision. Gene replacement therapy must be safe and functional but practical challenges remain. There is, for example, a complexity and large number of mutations responsible for the various disease phenotypes—at least 63 genes identified for RP alone, while many disease entities are yet caused by unknown mutations. Planning for the clinical trial can be a challenge not least of which is deciding on the subject population and the stage of intervention.
Another challenge is to protect the remaining photoreceptor cells. This is difficult because of the intense remodeling of retinal tissue, including cell loss, loss of conductivity, and glial proliferation,16 that follow the sequential loss of rods and cones. In RP, most mutations are rod-specific with initial loss of rods followed by cones, giving a variable window of time for cone-directed strategies. As has been stated by Dr. Paul Sieving “50% cone loss is yet compatible with an acuity of 20/20” and “95% cone loss is compatible with a correct orientation and discrimination performance.”17,18 Similarly, Dr. Alan Wright has stated that “preserving cones would prevent 1.5 million people worldwide from becoming blind because, in the age of artificial lighting, we function very well without rods.”19 One possible strategy for cone rescue is through transplanting rods into the degenerating retina. Many studies from our group have demonstrated that cone survival is dependent on the presence of rods and selective transplantation of rods could slow the loss of cones, maintaining useful vision in RP.
Such selective transplantation of rods that was demonstrated to slow cone loss in RP models20 can possibly maintain useful vision in humans through a trophic activity exerted by a diffusible factor that stimulates cone survival.21 This trophic factor was identified as the rod- derived cone viability factor (RdCVF)22 a protein encoded by the nucleoredoxin-like-1 (Nxnl1) gene that also encodes the thioredoxin enzyme rod-derived cone viability factor long (RdCVFL). The cone-protective effect of RdCVF has been found to be independent of the causative gene, working both on autosomal recessive mutations, such as in the rd1 mouse, and autosomal-dominant diseases, such as in the P23H rat.
The mode of action of the NXNL1 gene product is complex, involving stimulation of glucose metabolism by RdDVF and repair of oxidative damage by RdCVFL.23,24 RdCVF helps to preserve the morphology of the cone outer segment by accelerated glucose entry into photoreceptors and enhanced aerobic glycolysis. In parallel, administration of the enzymatically active RdCVFL reduces the damage produced by photo-oxidative stress in the Nxnl1−/− mouse, restoring cone function and viability toward normal. These combined actions of RdCVF and RdCVFL have therapeutic implications as, even with significant reduction in visual acuity, theoretically, they will allow for years of preservation of central vision as outlined by Léveillard and Sahel.25
Two other challenges are to restore light responses of degenerated photoreceptor cells as well as restoration of retinal encoding. Although optogenetics can be used to insert photoswitches into retinal secondary neurons after loss of photoreceptors, it can also be used to reactivate “dormant” cones for restoration of visual responses. In mouse models of RP, the expression of archaebacterial halorhodopsin in light-insensitive cones can substitute for the native phototransduction cascade and “restore light sensitivity.”26 In human ex vivo retinas, halorhodopsin can also “reactivate light-insensitive human photoreceptors” as demonstrated by the groups of Roska, Picaud, and Sahel.26 Besides this application in cone photoreceptors, optogenetics can restore vision by using photoswitches in secondary retinal neurons, which can survive years after degeneration of photoreceptors in RP patients. Targeting channelrhodopsin-2 to ON-bipolar cells restores ON and OFF visual responses.27 This was done in rd1 blind mice through intravitreal injection using adeno-associated virus (AAV) gene therapy. A problem with optogenetics though is that cells expressing the photoswitch proteins are less sensitive to light, so vision under normal lighting conditions would not be achieved. To address this challenge, the company Gensight Biologics (Paris, France) has a clever solution in their developmental pipeline. In their technique, a red-shifted channelrhodopsin protein is first introduced into remaining retinal neurons via a modified AAV vector (AAV2-7m8). Biomimetic camera goggles then stimulate the bioengineered retinal cells with images projected onto the retina by a light source using a specific wavelength (light-emitting diode light amplification). Functional visual perception thus can be restored.
A fifth challenge involves determination of the status of the remaining tissue. This is most important in assessing foveal cone cells in late-stage RP and reanimating them. Complete photoreceptor maps are useful in these studies through fusion of images with different entry points.
Finally, there must be a demonstration of the restoration of useful vision. For this, we must get out of our comfort zone of reliable, highly standardized, precisely measured clinical tests into the messy world of patient-reported outcomes questionnaires and performance-based tests. Questions such as “Is a 1-line acuity gain clinically meaningful and does it make a difference to the patient?” should be asked. To better answer these questions, we have designed the Streetlab platforms at the Institut de la Vision that provides “training, consulting and evaluation services to companies that are developing products and services improving autonomy, mobility, and quality of life of visually impaired people”. Three-dimensional simulators are used to simulate and evaluate low vision. Also, an Artificial Street is simulated on an indoor platform that has the appearance of an urban area. There are behavioral recordings using a motion-capture system (Vicon, Oxford, UK) with specific markers, internal sensors, an eye tracker and surveillance cameras. A control room is used for monitoring and recording and there is postprocessing. Objective behavioral measurements can be made: gate analysis, for example, allows for measurement and analysis of movement patterns, kinematics, and kinetics, as well as forces produced by movements. Walking cycles can be assessed as well as cycles affected by aging (e.g., shorter stride length) and low vision (e.g., longer stance duration). Similarly, trajectory changes in tunnel vision–RP patient head direction as well as gaze behavior (head and eye orientation) can be assessed.28
Conclusions and Future Directions
The following factors need to be addressed to adequately provide effective, personalized therapies for the diverse population of patients with inherited retinal degenerations:
identification and correction of the causative gene defect
protection of the remaining photoreceptor cells
restoring light responses to the affected retina
restoration of coding ability to the affected retina
assessment of the status of the affected remaining tissue
demonstrating the restoration of functional vision
From Classifying Patterns of Visual Loss to Planning Therapies: Autosomal Dominant Retinitis Pigmentosa Caused by Rhodopsin Mutations
Dr. Samuel G. Jacobson and Dr. Artur V. Cideciyan
Abstract: We are advancing from an era of description of disease patterns in autosomal-dominant retinitis pigmentosa (adRP) to a time of potential therapies. To answer specific questions about how to administer therapy, we reinvestigated the phenotype. We studied adRP patients with rhodopsin gene mutations at different disease stages to try to understand more details of the disease expression in preparation for local and retina-wide therapies for the regionalized retinopathy known as Class B phenotype. At least three components of the phenotype were found in these cross-sectional studies.
Although originally thought of as a single entity, retinitis pigmentosa (RP) is now known to be a great number of disease conditions caused by different genetic mutations that lead to degeneration of retinal photoreceptors and loss of vision. Treatment strategies are beginning to be designed for these individual conditions. This progress requires careful classification and characterization of the disease entities both genetically and phenotypically to ensure thorough evaluation of any proposed therapy.
The first step in this process usually occurs in the clinic with a history, pedigree, and examination. For autosomal-dominant retinitis pigmentosa (adRP), premolecular phenotyping led to the understanding that there were two major forms.29,30 In Type 1 (Type D), there is early-onset retina-wide diffuse and severe loss of rod function with later loss of cone function. Type 2 (Type R or sector RP) exhibits a milder phenotype. Rod and cone function can be lost in one retinal region but retained in the remaining regions. A mutation in the rhodopsin (RHO) gene was the earliest report of a cause for RP, specifically, adRP. Over the years though, this complexity has increased from one mutation to more than 100 mutations in the RHO gene. The question then became “Was there any way to make sense of the phenotypes of this large number of RHO genotypes?” In the 1990s, many investigators cataloged the subjective onset of night blindness and the degree of rod dysfunction and patterns of field loss in subjects carrying RHO mutations. In 1998, we divided the RHO diseases into two main phenotypic classes with accompanying genotyping.31 Class A RHO mutants have severe, early rod-cell loss with residual cone function following normal cone density. Genotypes include, for example, R135G, R135L, R135W, E181K, V345L, and P347L mutations. Class B RHO mutants exhibit slow loss of rod function in certain retinal regions; cone function can remain normal until over 75% of rod outer segments are lost. Genotypes include, for example, T17M, T58R, V87D, G106R, D190G, and P23H mutations.
More recently, we examined a large cohort of RHO Class B patients clinically, and with psychophysics and imaging (cross sectional and en face) at different disease stages. As a consequence, we were able to better describe the complexity of the Class B phenotype.32 Patients could have hemifield dysfunction, pericentral loss of function, or a diffuse rod sensitivity loss across the visual field. There could be combinations of the different patterns. Photoreceptor layer thickness measurements, co-localized with the psychophysical data, were in agreement with these patterns.
With this updated knowledge of genotyping and phenotyping, attention can now turn to some very practical questions about treatment of RHO mutations, specifically therapy of Class B RHO mutants with remaining rod vision and structure. For a local (subretinal) therapy, is it best at a certain eccentricity? Maybe just determine a transition zone from healthier to unhealthier retina and inject there? This could be at the in-out boundary at the edge of the ellipsoid zone line of OCT? For a systemic therapy (e.g., oral) what retinal regions should be monitored for efficacy? Is there improvement? Lack of progression? Toxicity? What disease stage should be monitored?
Conclusions and Future Directions
There is a three-component pattern of visual loss in Class B RHO patients.
Whether focal or systemic treatment, we must know which and how many of these patterns are present in each patient and design the treatment or monitoring accordingly.
A “one-injection site fits all” protocol in a clinical trial would seem to be the easiest treatment delivery approach but, with what we now know, this would be ill advised for Class B RHO patients.
For example, a macular, subretinal injection into the cone-rich central retina is potentially risking the 20/20 vision in this region and not serving the purpose of trying to positively alter the natural history of rod disease. Equally, an injection in an inferior retinal area devoid of photoreceptors is obviously ill conceived. Pretreatment mapping of function and structure in this disease group is needed to make enlightened decisions about planning any therapeutic intervention.
Optogenetic Vision Restoration
Dr. John G. Flannery
Abstract: Electronic retinal prostheses can restore useful vision in patients affected by retinal degenerations. Optogenetics is an alternative therapeutic approach. To date, however, a major limitation of the microbial opsins used for restoration of retinal light sensitivity in optogenetics is the high light intensity required for activating channelrhodopsins. One solution to this caveat is the use of mammalian opsins with higher light sensitivity but sufficiently fast kinetics for useful motion vision. We are developing a novel approach to restore vision to patients by expressing light sensitive G-protein coupled receptors (GPCR) proteins in specific, second-order retinal neurons to make them light sensitive. Our approach uses a common neuronal receptor, modified to add a light receptive function to the remaining light-insensitive retinal neurons that survive after photoreceptor degeneration. The receptor uses either retinal, which is available in the eye or a synthetic chemical photoswitch delivered by intravitreal injection. In this way, the cells in which the receptor is located respond to light with a change in neural firing. This compensates for their loss of input from photoreceptors, restoring light responsiveness to the retina, and sending information to the brain to restore vision. In most cases, this approach is independent of the mutation that causes the photoreceptor degeneration. Exceptions to this approach may be diseases that cause RPE dysfunction, such as Leber congenital amaurosis (LCA) 2, or retinal pigment epithelial (RPE) cell death as in choroideremia. To date, versions of this approach, developed by our group and others in the field, have employed receptors that are rather insensitive to light or very slow in response so could not support normal motion vision. We are developing a new strategy that uses the natural amplification properties of GPCR signaling to increase sensitivity by 1000 times and response speed. GPCR signaling cascades are intrinsic to rods and cones as well as to bipolar, ganglion, and other retinal cells. We are also pursuing a new discovery, a combinatorial approach that uses more than one optical sensor molecule at a time to recreate the diversity of natural signaling in the retina that earlier had been missing. We have employed sophisticated behavioral analyses to test not only the restoration of the ability to tell light from dark or flashing from steady light but to determine if the animal is able to see images.
Gene therapy for early-stage retinal diseases primarily targets photoreceptors and RPE cells, mainly through gene replacement therapy. Numerous clinical trials are in progress, and one product (voretigene neparvovec; Spark Therapeutics, Philadelphia, PA) has been approved by the Food and Drug Administration for clinical use. In contrast, gene therapy for late-stage retinal diseases (where photoreceptors have been lost) targets bipolar and retinal ganglion cells (RGCs), mainly using optogenetic techniques. Several companies are planning clinical trials in this area. Acucela (Seattle, WA) is investigating adeno-associated virus (AAV) vectors to deliver human rhodopsin33,34 to ON-bipolar cells. Allergan (Dublin, Republic of Ireland) has an approach using channelrhodopsin-2 targeted to retinal ganglion cells. Gensight (Paris, France) proposes to use a red-shifted channelrhodopsin (ChrimsonR) expressed in RGCs.
Delivery
The design of the viral vectors and their surgical application varies between early- and late-stage gene therapies. A major consideration in evaluating a potential therapy in animal models is that the tropism of a specific virus changes significantly when applied in a different species. For example, gene expression is restricted to macular RGCs in the nonhuman primate (NHP) retina after intravitreal injection of AAV serotype 2 while there is panretinal expression in rodent RGCs with this vector. This is largely due to the thicker inner limiting membrane (ILM), posing a greater barrier to transduction from the vitreous in NHP retina compared with rodents.35 AAV vectors efficiently transduce NHP RGCs in the region of the macular pit, where the ILM is thinner, but poorly transduce RGCs in the equatorial and peripheral retina of NHP, due to the penetration barrier of the ILM. This is not observed in rodents, where the ILM is thinner throughout and much less of a physical barrier to viral particles.36 In contrast, intravitreal injection of AAV serotype 2 does not efficiently transduce Müller glial cells in rodent or NHP.37,38 AAV can access Müller cells from the vitreous but glia lack the cell surface receptors required for binding and internalizing most natural AAV serotypes.37 Subretinal injection of AAV2 efficiently transduces photoreceptors and RPE cells but not Müller glia. Neither route efficiently transduces inner retinal neurons such as bipolar, horizontal, and amacrine cells. We are developing new AAV vectors with improved characteristics for use in ophthalmology. We have applied a directed evolution technique to identify AAV variants with improved gene delivery to specific retinal cells following intravitreal delivery.36 The intravitreal route has significant advantages over subretinal injection, particularly for the central retina, as it avoids detachment of the delicate macular and foveal photoreceptors from the RPE.
Discovery and characterization of novel AAV vectors was performed first in mouse retina,39 and more recently in the retina of a dog and NHP. To begin a “directed evolution” study, libraries of AAV viruses are created by modifying the ‘cap' gene that encodes the three structural proteins that form the AAV capsid. One type of library is made by using error-prone polymerase chain reaction to introduce random point mutations into the cap open reading frame. Additional libraries are created by an in vivo recombination method or cap gene shuffling, generating random chimeras of AAV cap genes, yielding AAV libraries with millions of novel serotypes. Testing in the dog (with Dr. William Beltran) has been productive and has generated improved AAV variants for transduction of canine retina. We combined directed evolution with DNA-barcoded vectors and deep sequencing to track the best performing variants through successive rounds of selection. We found that the AAV pool becomes enriched for an increasingly small number of winning AAV variants over successive rounds of selection. Specifically, the application of barcoded AAV libraries and deep sequencing identifies the best performing variants in the outer nuclear layer and in RPE cells. We are currently screening AAV libraries In NHP retina through six rounds of screening following intravitreal injections. In this way, new AAV capsid variants specifically optimized for primates are identified.
Application of New AAV Vectors to Optogenetics
Current optogenetic approaches have been limited by the fact that, although the naturally occurring optogenetic molecules are extremely fast in their light response, they are not very light sensitive. In addition, unlike the retinal rod and cone photoreceptors, these molecules cannot adapt their range or adjust their sensitivity. As a result, their function is limited to 1 or 2 log units of illumination, and do not cover the many (∼11–12) log units of light intensities that encompass natural scotopic and photopic vision (Fig. 3). One design consideration is “How fast and how sensitive does optogenetics for vision restoration need to be?” Our current goal is to restore useful motion vision at normal lighting levels to blind patients. When selecting a light-sensing molecule, physics imposes an unavoidable tradeoff of light sensitivity versus response speed. The “persistence of vision” phenomenon is exploited by cathode ray tube monitors to make single pixels appear continuous when flickering at 1/20 of a second. Light-emitting diode monitors appear stable when illuminating a narrow strip at 60 Hz.
Figure 3.
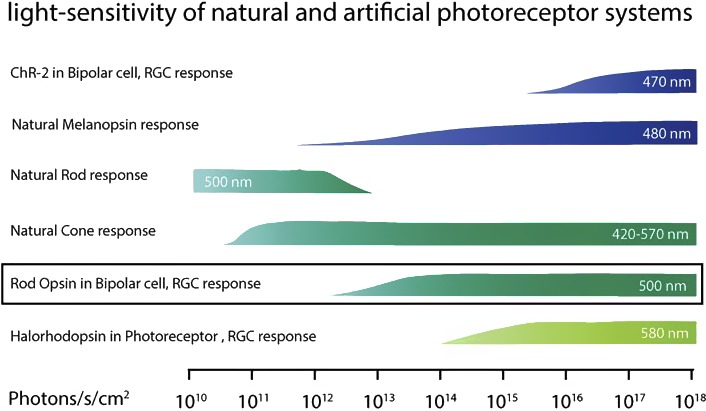
Light sensitivity of natural and artificial photoreceptor systems. Reprinted with permission from Gaub BM, Berry MH, Visel M, Holt A, Isacoff EY, Flannery JG. Optogenetic retinal gene therapy with the light-gated gpcr vertebrate rhodopsin. In: Boon C, Wijnholds J, eds. Retinal Gene Therapy. Methods in Molecular Biology. Humana Press: New York; 2018:177–189.
Optogenetic sensor proteins can be modified to increase their sensitivity, but a system using a single protein as sensor and effector will have a limited range of adaptation. Optogenetic proteins, such as channelrhodopsin (Chr2) and halorhodopsin (NphR), are a “1-component” system—they function as both the light sensor and the ion channel effector. A light sensitive ion channel, such as Chr2, can have exceptionally fast responses in terms of channel opening and closing in response to light, well above 50 Hz, substantially faster than rod or cone photoreceptors. One tradeoff for the high response speeds is a small adaptation range. When used in the retina, this will require the use of electronic goggles to intensify the projected image and to provide adaptation. It is useful to remember that natural microbial switches, such as channelrhodopsin and halorhodopsin function in algae for phototaxis, not vision. Melanopsin primarily plays a nonimage-forming role in setting the circadian rhythm. Other systems have been evaluated, such as synthetic photoswitches (e.g., the ionotropic glutamate receptor; LiGluR).40–42 This light-gated ion channel is also “1-component” and similarly lacks the amplification and adaptation found in the phototransduction cascade native to rod and cone photoreceptors.43–47
We asked, “What are the advantages and disadvantages of targeting different cell types in the degenerating retina?” ON-bipolar cells, for example, are one potential target for optogenetic activation to treat blindness. They are upstream of the RGCs, allowing the possibility to recapture some of the intrinsic retinal image processing functions of the retinal circuity. Retinal ganglion cells are a more ‘down-stream' target for late-stage retinal disease.
In a 6-month-old photoreceptor-less rd1 mouse retina that is insensitive to light, LiGluR gene expression in RGCs elicits a strong light response.38 This restoration of light sensitivity was also seen with LiGluR expression in ON-bipolar cells42; however, there is more diversity in RGC responses driven by ON-bipolar expression than by making all the RGCs intrinsically light sensitive. In an alternative approach, rhodopsin can also be expressed ectopically in ON-bipolar cells34 where it drives light responses in blind rd-1 mouse retinas.
With rhodopsin, we employ a system more similar to the native photoreceptor phototransduction cascade, by separating the light sensor (rhodopsin) from the ion channel effector with an interconnecting G-protein coupled receptors (GPCR) cascade. This approach gives 1000× higher sensitivity and adds several log units of adaptation above the 1-component approaches. However, we found that rhodopsin, ectopically expressed in bipolar cells, responds to light much more slowly than it does in rods. This response is likely too slow for motion vision and may be due to different G-protein cascades with slower kinetics in bipolars than in photoreceptors. Searching for a response speed more useable for vision in motion for patients, we are currently testing cone opsin responses in –ON bipolars.
We also studied whether combinations of genetically and chemically engineered LiGluRs could be useful in vision restoration. We previously found that, in the rd1 mouse model, LiGluR “restores light sensitivity to RGCs, reinstates light responsiveness to the primary visual cortex and restores both the pupillary reflex and a natural light-avoidance behavior.”40 We extended this work to investigate the possibility of combining LiGLuR with the light-agonized and light-antagonized metabotropic glutamate receptors (LimGluRs).48 In this work, Levitz, et al., found that light-agonized LimGluR2 was “fast, bistable and supported multiple rounds of on/off switching.” We found that AAV-SNAP-mGLUR2 expression in rd1 mouse RGCs endows a light-sensitive inhibitory OFF-response. The treated animals could learn to recognize an illuminated pattern on an organic light-emitting diode display to avoid a mild foot shock.
We then asked, “Can we express two optogenetic switches with opposite response properties to restore more complex vision?” For this, we co-expressed an −ON agonist and an −OFF agonist in RGCs. Functionally, we co-injected two AAV viral DNA expression vectors, LiGluR and SNAP-mGluR2. A random distribution of expression of two photoswitches in RGCs was generated by the two AAV vectors. We found that mice expressing either the −ON and the −OFF sensor proteins could perform a learned behavioral response requiring visual discrimination of ‘+' versus ‘−’ sign to avoid a mild foot shock. Combined expression of the −ON and −OFF response yields improved line discrimination behavior over either sensor acting alone.
Conclusions and Future Directions
Optogenetics can provide useful vision restoration for late-stage retinal degenerations:
We can target proteins to specific retinal cell classes using engineered AAV variants. The light-sensitive proteins drive light responses in the retina and enable innate and learned visually guided behavior in mice.
Expression of a light-activated On–transient ion channel in –ON bipolar cells generates diverse responses in ganglion cells, whereby expression of a light-activated On–transient ion channel in all functional subtypes of ganglion cells generates a uniform retinal output with little diversity.
“One-component” light-sensitive ion channels like channelrhodopsin and LiGluR are very fast but not very light sensitive and provide very limited adaptation range.
Light-sensitive GPCRs like rhodopsin have increased range of adaptation and increased sensitivity but have slower responses than single-component sensors like channelrhodopsin2 and LiGluR when expressed ectopically in ON-bipolar cells. This is likely due to the kinetics of the G-protein cascade and ion channel “hijacked” in the bipolar cell.
Combinations of optogenetic sensors may be able to restore pattern vision.
Preclinical Development of a Single Vector Strategy for Rhodopsin Autosomal Dominant Retinitis Pigmentosa Gene Therapy
Dr. William A. Beltran
Abstract: Mutations in the rhodopsin (RHO) gene are the most common cause of autosomal dominant retinitis pigmentosa (adRP) and no treatment is currently available. We have developed an allele-independent single adeno-associated virus (AAV) vector-mediated gene therapy strategy that combines two important treatment aspects. First, knocking down via shRNA technology the expression of endogenous RHO, both wild-type (WT) and mutant copies. Secondly, replacing endogenous RHO expression with that from a normal human RHO cDNA made resistant to the shRNA via silent mutations. This combined gene therapy approach was tested in dogs that carry a mutation in RHO and show similar features of disease as reported in Class B RHO-adRP patients. Our results show successful and stable prevention of disease onset in areas of the retina that were treated.
Mutations in the rhodopsin (RHO) gene are a common cause of autosomal-dominant retinitis pigmentosa (adRP) and constitute 30% to 40% of this type of retinal degeneration (1/40,000). Over 150 different mutations in the RHO gene are known to be responsible for adRP. Many of these result in a toxic gain of function mechanism so are not amenable to treatment by simple gene augmentation therapy. One approach to this problem is to eliminate (knockdown) endogenous production of rhodopsin, both the wild-type (WT) and mutant alleles, and then replace them with a WT RHO cDNA that has been modified to be resistant to the knockdown tool (e.g., shRNA) and results in the production of a normal RHO protein. Certain dog breeds like the English mastiff in which a form of retinal degeneration caused by a point mutation, Thr4Arg (T4R), in the RHO gene are the only naturally occurring animal models of RHO adRP identified to this date.49 The phenotype, characterized by a topographic pattern of disease that affects primarily the central retina, closely mimics the altitudinal distribution of disease reported in some RHO-adRP patients. Besides its large (human-sized) eye and the presence of a central bouquet of cone photoreceptors similar to the fovea of the primate that has a predilection for naturally occurring inherited macular degenerations,50 a significant value of the heterozygote RHO-adRP dog model is that it expresses natural levels of both mutant and WT RHO protein. This is extremely valuable for identifying the optimal levels of RHO knockdown and replacement that are necessary to correct, in particular, the deleterious effects of toxic gain of function mutations. Cideciyan et al.51 studied this canine model and found that relatively modest levels of light “dramatically accelerated the neurodegeneration.” It is well known that visible light can cause photoreceptor cell death in many animal species52 with light sensitivity especially prominent in RHO mutants, experimentally produced or naturally occurring, and ranging from Xenopus laevis to mouse, rat, and dog. The possible deleterious effect of environmental light exposure has also been suggested in some RHO-adRP patients with an altitudinal pattern of disease.53
The extreme light sensitivity of the canine T4R RHO retina has been recently further characterized, and a new light-damage paradigm developed to better understand the degenerative process of rods, and to enable rapid assessment of novel therapeutic strategies in this large animal model. Following acute (1-minute) exposure to white light (1 mW/cm2 corneal irradiance, ∼1590 lux), ultrastructural alterations of rod outer segment discs were seen within 15 minutes, followed by severe disruption of rod inner segment integrity within 6 hours.54 TUNEL assay, which was used to examine cell death, showed the first events of rod loss to begin at 6 hours and to peak by 24 hours postlight exposure. The effect of a 1-minute exposure to light (0.1–1.0 mW/cm2) on photoreceptor loss was recently found to be dose dependent, and the threshold for causing light-induced rod death to be between 0.1 and 0.2 mW/cm2 (170–320 lux).55 Two weeks following exposure to a “middose” of light (0.5 mW/cm2), there was approximately a 50% loss of the outer nuclear layer (ONL) in the tapetal region of heterozygous mutant dogs, which was more severe at 6 and 36 weeks postexposure, suggesting a slower yet ongoing process.55,56 Thus, exposure to this “middose” light intensity may be used to assess whether therapeutic intervention, delivered after the degeneration has been triggered, can stall the progression of degeneration. With the highest dose of light (1 mW/cm2), there was at 2 weeks postexposure extensive rod photoreceptor cell death in the central to midperipheral retina with an ONL limited to a single row of cones, and activation of Müller cell gliosis.56 This “high-dose” light damage setting could therefore provide a way of rapidly (within a couple of weeks) assessing the beneficial effect of therapeutic intervention preventing degeneration. In summary, a light-damage paradigm that uses short exposure to a range of white light intensities that are not themselves toxic to retinas of WT animals, causes substantial acute and progressive loss of rod cells in the outer nuclear layer of the central to midperipheral zones of the T4R RHO retina. Outcome measures of structural integrity using both in vivo retinal imaging and histomorphometric methods have been established. Functional assessment may include electroretinography, but novel methods for assessing visual behavior need to be developed. Indeed, the use of an obstacle avoidance course to test both rod- and cone-mediated vision failed to show any impairment in T4R RHO mutant dogs exposed to the highest dose of light damage. Retention of far peripheral rods, and central cones (in particular in the fovea-like area) likely supports sufficient vision for mutant dogs to successfully complete this psychophysical test.54
With this clinically relevant large animal model in hand, an National Institutes of Health–funded consortium of investigators from the Schools of Veterinary Medicine (Aguirre, Beltran) and Medicine (Cideciyan, Jacobson) at the University of Pennsylvania, and at the University of Florida (Hauswirth, Lewin) was set up to develop and test a gene therapy for RHO-adRP. As a strategy that could provide a treatment for all RHO mutations irrespective of their mechanism of action, the investigators opted to develop an allele-independent knockdown and replacement approach. This gene therapy strategy requires the following: (1) inhibition of the expression of both the normal (WT) and the mutant alleles of RHO, and (2) replacement of endogenous RHO expression with a copy of the normal RHO gene, which is made resistant to siRNA or ribosome degradation through silent mutations. After screening various knockdown reagents (including ribozymes and shRNAs) both in vitro and in vivo in WT dogs, an optimal shRNA (shRNA820) that substantially reduced expression of endogenous RHO levels (both at the RNA and protein levels) in WT and T4R RHO mutant dogs was identified. Following validation of this lead shRNA820, a resistant human RHO cDNA was developed (RHO820). An in vitro assay confirmed that shRNA820 did not reduce levels of RHO820 protein expression.
The efficacy of a combination virus that contained both shRNA820 and RHO820 (scAAV2/5-HOP-RHO820-H1-shRNA820) was tested in T4R RHO mutant dogs. Subretinal injection (titer: 5 × 1011 vector genomes/mL) under infrared illumination was shown to prevent damage from single or repeated light exposures (1 mW/cm2) that, in the untreated animal, cause end-stage retinal degeneration.55,56 Expression of human RHO820 was associated with both retention of normal ONL thickness, and rod outer segment structure after light exposure. Animals that were repeatedly light exposed had preservation of photoreceptors in the treated area and sustained rod-mediated ERG function in the AAV-injected eyes while balance sat solution–injected eyes showed severely reduced ONL thickness and electroretinogram amplitudes (see Note added in proof before Reference section).
Conclusions and Future Directions
The RHO dog is a large animal model that recapitulates features of Class B RHO mutations seen in the human.
The model is extremely light sensitive allowing for a rapid assessment of a therapeutic intervention.
A single vector with dual function (knockdown and replacement) can prevent onset of rod loss and preserve rod and cone function.
The question now remains—can a similar strategy rescue rods in degenerating retinas of the human?
Preclinical Gene Therapy Development for Leber Congenital Amaurosis Caused by NPHP5 Mutations
Dr. Gustavo D. Aguirre
Abstract: Leber congenital amaurosis (LCA) poses a therapeutic challenge given that there is early, severe visual deficit or blindness, and there is an overlap between normal development and early degeneration of photoreceptor cells. To develop therapies for eventual translation to the clinic, we have used the canine NPHP5 (IQCB1) model where a C-terminal truncation causes a photoreceptor ciliopathy with early onset and rapid degeneration. By 6 weeks of age, electroretinogram (ERG) rod responses are markedly reduced in amplitude or absent and cone responses are not recordable. The absence of cone function correlates with the lack of cone outer segments even though there is preservation of cone inner segments. To examine therapeutic approaches, we used adeno-associated virus (AAV) vectors for gene augmentation. We injected subretinally in one eye AAV vectors with hIRBP or hGRK1 promoters and the therapeutic transgene. The fellow eye served as the untreated control. Photoreceptor structure and function were quantified by spectral-domain optical coherence tomography and full-field ERGs. Visual behavior was assessed at 1 year or later after treatment using an obstacle-avoidance course to test separately the treated and contralateral control eyes. Treatment at 5 to 6 weeks of age resulted in remarkable recovery of cone function and preservation of rod ERG and vision for the 1+ year observation time period. Treatment blebs were associated with a significantly retained photoreceptor layer. Treatment at 13 weeks of age, an age at which all cones have lost their inner and outer segments and rod outer segments are degenerated in the affected animals, showed rapid recovery of rod and cone function by 8 weeks after treatment as well as preservation of function and vision for at least 1 year. Our results show that, in spite of the very severe and rapidly progressive photoreceptor degeneration in mutant dogs, AAV-mediated gene augmentation restores rod and cone function and preserves retinal structure and vision long term. These positive preclinical studies suggest a path forward for translating this treatment approach to the clinic.
Much progress has been made in the last few years in moving from gene discovery to gene therapy. For example, proof of concept studies for Leber congenital amaurosis (LCA), achromatopsia and X-linked retinitis pigmentosa were first done in canine models, and subsequently in mouse disease models. Best disease and now, LCA due to NPHP5 mutations, have been described in the dog. These pathologies have the same general features as in the human. It is particularly fortunate to have canine models of these diseases since they are viewed favorably by the Food and Drug Administration as models for assessing efficacy and safety in preclinical testing.
Defects in the NPHP5 (IQCB1) gene are known to cause an early onset, severe form of retinal degeneration within the LCA group of dystrophies (NPHP5-LCA). The disease is a ciliopathy in which defects are present in the photoreceptor connecting cilium, a critical transition zone for passing soluble and membrane components from inner segments to outer segments. Mutations in NPHP5 are also found in Senior Loken Syndrome (SLSN) in which there is cystic kidney disease (nephronophthisis, NPHP) as well as LCA. Ciliopathies, such as those caused by CEP290 and RPGR mutations, have been extensively reviewed and a framework has been proposed to investigate aspects of cilial development and pathology.57 In 2013,58 we described an aggressive, early-onset canine retinal ciliopathy in the Pit Bull Terrier caused by a single nucleotide insertion in exon 10 of the NPHP5 gene. This causes a premature stop that truncates the C-terminus of the native protein. Clinically, this results in several signs including early-onset retinal degeneration, nystagmus and widely dilated pupils. The condition is characterized by abnormal photoreceptor cell development with concurrent, early, and progressive degeneration.59 Disease pathology in the dog was found to parallel and recapitulate that seen in clinical testing of human patients. This includes an NPHP5 mutation with early loss of rods but relative retention of central retinal cone photoreceptors cells although lack of function in these cells was documented (Figs. 4A, 4B). In the canine model by 6 weeks of age, rod responses as measured by electroretinogram (ERG) are markedly reduced in amplitude or fully absent; cone responses are not recordable. The absence of cone responses is due to a lack of cone outer segments; cone cell bodies and inner segments are present but, simply, outer segments fail to form.
Figure 4.

NPHP5 disease phenotypes and response to gene therapy. (A1) ONL thickness topography of NPHP5 patient shows the distribution of the detectable photoreceptors to be limited to a central ellipse corresponding to the cone-dominant region, and more peripherally corresponding to the rod-dominant region. Wide annular region shows no detectable photoreceptors (black). (A2) Cross-sectional OCT scans along the vertical meridian through the fovea, extending 15° into superior (S) and inferior (I) retina in a healthy subject (age 30 years; top) and NPHP5-LCA patient P4 (age 13 years). Magnified views of the central retina (right; white rectangles) with overlapping longitudinal reflectivity profiles (LRP). Photoreceptor nuclear layer (ONL) is highlighted in blue; ellipsoid zone is highlighted in yellow. OLM, outer limiting membrane; COS, cone outer segments; RPE/BrM, retinal pigment epithelium/Bruch's membrane. In the patients, there are wide hyperscattering bands (S+) and more narrow hyposcattering bands (S−) distal to the ONL (far right along image). Icon (upper left) is location of the scans on a retinal schematic. (B) Pseudocolor maps of ONL thickness topography (upper) in a 35-week-old control dog (left) and an NPHP5-mutant (right) at 33 weeks of age. Insets, near-infrared reflectance images. Arrows on the maps localize the reconstituted OCT scans (lower) along a superior–inferior meridian crossing the central visual streak at the gaps of the lines. ONL on reconstituted scans is highlighted in blue and location of the scan relative to the vertical arrows is shown with a white wedge. All eyes shown as equivalent right eyes and optic nerve, major blood vessels (black) and tapetum boundary (yellow) are overlaid for ease of orientation. T, temporal; N, nasal retina. (C1) Right eye of a 5.7-week-old vector-treated NPHP5 mutant with large subretinal bleb that encompasses the fovea-like central region. (C2) Six-week-old NPHP5 mutant dog retina showing that most cones lack an outer segment (red = hCAR labeling; arrows indicate present cone outer segments); rod outer segments (green = rod opsin labeling) form, but are abnormal and there is extensive rod opsin mislocalization to the ONL. (C3) Treatment at 5.7 weeks with AAV-cNPHP5 vector (red traces = treated eye; black traces = untreated fellow eye) results in robust recovery rod, mixed rod-cone and cone ERG responses by 13 weeks. Fig. A1 modified and reprinted with permission from Cideciyan AV, Rachel RA, Aleman TS, et al. Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy. Hum Mol Genet 2011; 20:1411–1423. Figs. B, C2, and C3 modified and reprinted with permission from Downs LM, Scott EM, Cideciyan AV, et al. Overlap of abnormal photoreceptor development and progressive degeneration in Leber congenital amaurosis caused by NPHP5 mutation. Hum Mol Genet 2016;25:4211–4226.
With this knowledge, the dog NPHP5-LCA model appeared to be suitable for preclinical studies on gene replacement therapy. To begin this process, we modified a subretinal injection device originally developed for human applications, and now used in dogs.60 This was the RetinaJet Subretinal Cannula from SurModics, Inc. (product no longer manufactured). This device has the advantage of not needing surgical dissection, leading to a “shorter procedure time and milder postoperative conjunctival swelling.” In the LCA-NPHP5 dog, we injected the NPHP5 transgene, using AAV5, scAAV8Y733F vectors with interphotoreceptor retinoid-binding protein (IRBP) and hGRK1 promoters. We thought it appropriate to target treatment to the fovea-like region of the dog retina (as described in this review by Dr. Beltran). Cideciyan et al.61 have previously demonstrated from studies in human patients that NPHP5-LCA is an excellent candidate for “cone-directed gene augmentation therapy” (Fig. 4A). We also found that not all gene therapy approaches are effective in that choice of vector was critical in achieving positive results.
Treatment using the vector AAV2/5-IRBP-cNPHP5 in 7.5-week-old dogs at a titer of 1.5 × 1011 vector genomes/mL did not rescue retinal function when assessed up to 33 weeks after treatment. A 10-fold increase in titer and treatment at an earlier age resulted in improved retinal rod and cone function. In contrast, treatment at 6 weeks with scAAV2/8(Y733F)-hGRK1-cNPHP5 vector at a comparable titer resulted in functional recovery comparable to the higher dose of AAV2/5-hIRBP-cNPHP5 vector (Fig. 4C). Furthermore, raising the dose 10-fold led to excellent recovery of cone function, preservation of cone and rod ERGs as well as functional vision measured up to 1.3 years after treatment (Aguirre GD, et al. IOVS 2016;57:ARVO E-Abstract 2293). This therapy allowed for retention of a significant number of photoreceptor nuclei but there was thinning long term. It was also apparent that early treatment resulted in longer lasting structural and functional rescue. Thus, further vector and promoter optimization will be needed to ensure a permanent treatment modality.
Conclusions and Future Directions
An NPHP5 exon 10 insertion in dogs results in an early-onset, aggressive LCA-ciliopathy, similar to that seen in the human.
Most cone outer segments fail to form during development; thus, cone function is absent. Rod outer segments develop but they are abnormal. There is early loss of rod function indicating dissociation of structure and function.
The mutant NPHP5 protein permits rod outer segment formation, albeit abnormal. Failure of cone outer segments to form suggests that the functional domains and role(s) of NPHP5 in the rod and cone sensory cilium are not identical in each photoreceptor class.
Following gene therapy, cone outer segments form and function is restored. Rod structural abnormalities are reversed and structure and function are preserved.
The following three pending questions remain: (1) identification of optimal vector promoter for translational therapies; (2) achieving long-term stability and efficacy of the treatment, and (3) can we treat more advanced disease stages and what are the structural and functional outcomes?
Acknowledgments
The authors thank Oliver A. Garden, Professor and Chair, Department of Clinical Sciences & Advanced Medicine, School of Veterinary Medicine, University of Pennsylvania for his unwavering support in planning and chairing the original research symposium and for support of this publication. They also thank Gerald J. Chader for help in translating the meeting presentations into the present written publication format.
Supported by grants from Merck, Sanofi, agtc, and Nightstar Therapeutics.
Disclosure: A.J. Hardcastle, None; P.A. Sieving, None; J.-A. Sahel, Pixium Vision (C), GenSight Biologics (C, I), Genesignal (C), Chronocam (I), Chronolife (I), Pixium Vision (I), Tilak Healthcare (I), Sparing Vision (I); S.G. Jacobson, Provisional patent on rhodopsin adRP gene therapy (P); A.V. Cideciyan, Provisional patent on rhodopsin adRP gene therapy (P); J.G. Flannery, None; W.A. Beltran, Provisional patent on rhodopsin adRP gene therapy (P); G.D. Aguirre, Provisional patent on NPHP5 gene therapy (P)
Note added in proof: Cideciyan AV, et al. (2018) Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector [published online August 20, 2018]. Proc. Natl. Acad. Sci. U. S. A. https://doi.org/10.1073/pnas.1805055115
References
- 1.Dryja TP, McGee TL, Hahn LB, et al. Mutation within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Eng J Med. 1990;323:1302–1307. doi: 10.1056/NEJM199011083231903. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Q, Acland GM, Wu WX, et al. Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum Mol Genet. 2002;11:993–1003. doi: 10.1093/hmg/11.9.993. [DOI] [PubMed] [Google Scholar]
- 3.Beltran WA, Cideciyan AV, Lewin AS, et al. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc Natl Acad Sci U S A. 2012;109(6):2132–2137. doi: 10.1073/pnas.1118847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fiorentino A, Fujinami K, Arno G, et al. Missense variants in the X-linked gene PRPS1 cause retinal degeneration in females. Hum Mutat. 2018;39:80–91. doi: 10.1002/humu.23349. [DOI] [PubMed] [Google Scholar]
- 5.Hardcastle AJ, Thiselton DL, Zito I, et al. Evidence for a new locus for X-linked retinitis pigmentosa (RP23) Invest Ophthalmol Vis Sci. 2000;41:2080–2086. [PubMed] [Google Scholar]
- 6.Webb TR, Parfitt DA, Gardner JC, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23) Hum Mol Genet. 2012;21:3647–3654. doi: 10.1093/hmg/dds194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gardner JC, Liew G, Quan YH, et al. Three different cone opsin gene array mutational mechanisms with genotype-phenotype correlation and functional investigation of cone opsin variants. Hum Mutat. 2014;35:1354–1362. doi: 10.1002/humu.22679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwarz N, Carr AJ, Lane A, et al. Translational read-through of the RP2 Arg120stop mutation in patient iPSC-derived retinal pigment epithelium cells. Hum Mol Genet. 2015;15:972–986. doi: 10.1093/hmg/ddu509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parfitt DA, Lane A, Ramsden CM, et al. Identification and correction of mechanisms underlying inherited blindness in human iPSC-derived optic cups. Cell Stem Cell. 2016;18:769–781. doi: 10.1016/j.stem.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tolun G, Vijayasarathy C, Huang R, Zang Y. Paired octamer rings of retinoschisis suggest a junctional model for cell-cell adhesion in the retina. Proc Natl Acad Sci U S A. 2016;113:5287–5292. doi: 10.1073/pnas.1519048113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeng Y, Takada Y, Kjellstrom S, et al. RS-1 gene delivery to an adult Rs1h knockout mouse model restores ERG b-wave with reversal of the electronegative waveform of X-linked retinoschisis. Invest Ophthalmol Vis Sci. 2004;45:3279–3285. doi: 10.1167/iovs.04-0576. [DOI] [PubMed] [Google Scholar]
- 12.Ziccardi L, Vijayasarathy C, Bush RA, Sieving PA. Photoreceptor pathology in the X-linked retinoschisis (XLRS) mouse results in delayed rod maturity and impaired light-driven transducin translocation. Adv Exp Med Biol. 2014;801:559–566. doi: 10.1007/978-1-4614-3209-8_71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park TK, Wu Z, Kjellstrom S, et al. Intravitreal delivery of AAV8 retinoschisis results in cell type-specific gene expression and retinal rescue in the Rs-1 KO mouse. Gene Ther. 2009;16:916–926. doi: 10.1038/gt.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bush RA, Zeng Y, Colosi P, et al. Preclinical dose-escalation study of intravitreal AAV-RS1 gene therapy in a mouse model of X-linked retinoschisis: dose-dependent expression and improved retinal structure and function. Hum Gene Ther. 2016;27:376–389. doi: 10.1089/hum.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeffreys BJ, Cukras CA, Vitale S, et al. Test-retest intervisit variability of functional and structural parameters in x-linked retinoschisis. Transl Vis Sci Technol. 2014;3(5):5. doi: 10.1167/tvst.3.5.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones BW, Marc RE. Retinal remodeling during retinal degeneration. Exp Eye Res. 2005;81:123–137. doi: 10.1016/j.exer.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Geller AM, Sieving PA. Assessment of foveal cone photoreceptors in Stargatdt's macular dystrophy using a small dot detection task. Vision Res. 1993;33:1509–1524. doi: 10.1016/0042-6989(93)90144-l. [DOI] [PubMed] [Google Scholar]
- 18.Geller AM, Sieving PA, Green DG. Effect on grating identification of sampling with degenerative arrays. J Opt Soc Am A. 1992;9:474–477. doi: 10.1364/josaa.9.000472. [DOI] [PubMed] [Google Scholar]
- 19.Wright AF. A searchlight through the fog. Nat Genet. 1997;17:132–134. doi: 10.1038/ng1097-132. [DOI] [PubMed] [Google Scholar]
- 20.Mohand-Said S, Hicks D, Dreyfus H, Sahel JA. Selective transplantation of rods delays cone loss in a retinitis pigmentosa model. Arch Ophthalmol. 2000;118:807–811. doi: 10.1001/archopht.118.6.807. [DOI] [PubMed] [Google Scholar]
- 21.Mohand-Said S, Deudon-Combe A, Hicks D, et al. Normal retina releases a diffusible factor stimulating cone survival in the retinal degeneration mouse. Proc Natl Acad Sci U S A. 1998;95:8357–8362. doi: 10.1073/pnas.95.14.8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leveillard T, Mohand-Said S, Lorentz O, et al. identification and characterization of rod-derived cone viability factor. Nat Genet. 2004;36:755–759. doi: 10.1038/ng1386. [DOI] [PubMed] [Google Scholar]
- 23.Aït-Ali N, Fridlich R, Millet-Puel G, et al. Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell. 2015;161:817–832. doi: 10.1016/j.cell.2015.03.023. [DOI] [PubMed] [Google Scholar]
- 24.Leveillard T, Sahel JA. Metabolic and redox signaling in the retina. Cell Mol Life Sci. 2017;74:3649–3665. doi: 10.1007/s00018-016-2318-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leveillard T, Sahel JA. Rod-derived cone viability factor for treating blinding diseases: from clinic to redox signaling. Sci Transl Med. 2010;2:26ps16. doi: 10.1126/scitranslmed.3000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Busskamp V, Duebel J, Balya D, et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science. 2010;329:413–417. doi: 10.1126/science.1190897. [DOI] [PubMed] [Google Scholar]
- 27.Mace E, Caplette R, Marre O, et al. Targeting channelrhodopsin-2 to ON-bipolar cells with vitreally administered AAV restores ON and OFF visual responses in blind mice. Mol Ther. 2015;23:7–16. doi: 10.1038/mt.2014.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Authie CN, Berthoz A, Sahel JA, Safran AB. Adaptive gaze strategies for locomotion with constricted visual field. Front Hum Neurosci. 2017;11:387–391. doi: 10.3389/fnhum.2017.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Massof RW, Finkelstein D. Two forms of autosomal dominant primary retinitis pigmentosa. Doc Ophthalmol. 1981;51:289–346. doi: 10.1007/BF00143336. [DOI] [PubMed] [Google Scholar]
- 30.Lyness AL, Ernst W, Quinlan MP, et al. A clinical, psychophysical and electroretinographic survey of patients with autosomal dominant retinitis pigmentosa. Br J Ophthalmol. 1985;69:326–339. doi: 10.1136/bjo.69.5.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cideciyan AV, Hood DC, Huang Y, et al. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci U S A. 1998;95:7103–7118. doi: 10.1073/pnas.95.12.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacobson SG, McGuigan DB, III,, Sumaroka A, et al. Complexity of the Class B phenotype in autosomal dominant retinitis pigmentosa due to rhodopsin mutations. Invest Ophthalmol Vis Sci. 2016;57;:4847–4858. doi: 10.1167/iovs.16-19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cehijac-Kapetanovic J, Eleftheriou C, Allen AE, et al. Restoration of vision with ectopic expression of human rod opsin. Curr Biol. 2015;25:2111–2122. doi: 10.1016/j.cub.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaub BM, Berry MH, Holt AE, Iscoff EY, Flannery JG. Optogenetic vision restoration using rhodopsin for enhanced sensitivity. Mol Ther. 2015;23:1562–1571. doi: 10.1038/mt.2015.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolstad KD, Dalkara D, Guerin K, et al. Changes in adeno-associated virus-mediated gene delivery in retinal degeneration. Hum Gene Ther. 2010;21:571–578. doi: 10.1089/hum.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dalkara D, Byrns LC, Klimczak RR, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5:189ra76. doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 37.Dalkara D, Kolstad KD, Caporle N, et al. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol Ther. 2009;17:2096–2102. doi: 10.1038/mt.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klimczak RR, Koerber JT, Dalkara D, Flannery JG, Schaffer DV. A novel adeno-associated viral variant for efficient and selective transduction of rat Muller cells. PLoS One. 2009;4:e7467. doi: 10.1371/journal.pone.0007467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalkara D, Byrne LC, Klimczak RR, et al. In-vitro-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5:189ra76. doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 40.Caporale N, Kolstad KD, Lee T, et al. LiGluR restores visual responses in rodent models of inherited blindness. Mol Ther. 2011;12:1212–1219. doi: 10.1038/mt.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berry MH, Holt A, Levitz J, et al. Restoration of patterned vision with an engineered photoactivatable G protein-coupled receptor. Nat Commun. 2017;8:1862. doi: 10.1038/s41467-017-01990-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaub BM, Berry MH, Holt AE, et al. Restoration of visual function by expression of a light-gated mammalian ion channel in retinal ganglion cells or ON-bipolar cells. Proc Natl Acad Sci U S A. 2014;111:E5574–E5583. doi: 10.1073/pnas.1414162111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamb TD, Pugh EN., Jr. Phorotransduction, dark adaptation and rhodopsin regeneration. The Proctor Lecture. Invest Ophthalmol Vis Sci. 2006;47:5137–5152. doi: 10.1167/iovs.06-0849. [DOI] [PubMed] [Google Scholar]
- 44.Lamb TD, Pugh EN., Jr. Dark adaptation and the retinoid cycle of vision. Prog Retin Eye Res. 2004;23:307–380. doi: 10.1016/j.preteyeres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 45.Arshavsky VY, Lamb TD, Pugh EN., Jr. G proteins and phototransduction. Ann Rev Physiol. 2002;64:153–187. doi: 10.1146/annurev.physiol.64.082701.102229. [DOI] [PubMed] [Google Scholar]
- 46.Pugh EN, Jr,, Nikonov S, Lamb TD. Molecular mechanisms of vertebrate photoreceptor light adaptation. Curr Opin Neurobiol. 1999;9:410–418. doi: 10.1016/S0959-4388(99)80062-2. [DOI] [PubMed] [Google Scholar]
- 47.Lamb TD, Pugh EN., Jr. G-protein cascades: gain and kinetics. Trends Neurosci. 1992;15:291–298. doi: 10.1016/0166-2236(92)90079-n. [DOI] [PubMed] [Google Scholar]
- 48.Levitz J, Pantoja C, Gaub B, et al. Optical control of metabolic glutamate receptors. Nat Neurosci. 2013;16:507–516. doi: 10.1038/nn.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kijas JW, Cideciyan AV, Aleman TS, et al. Naturally occurring rhodopsin mutation in dog causes retinal dysfunction and degeneration mimicking human dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 2002;99:6328–6333. doi: 10.1073/pnas.082714499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beltran WA, Cideciyan AV, Guziewicz KA, et al. Canine retina has a primate fovea-like bouquet of cone photoreceptors which is affected by inherited macular degeneration. PLoS One. 2014;5:9e90390. doi: 10.1371/journal.pone.0090390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cideciyan AV, Jacobson SG, Aleman TS, et al. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc Natl Acad Sci U S A. 2005;102:5233–5238. doi: 10.1073/pnas.0408892102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wenzel A, Grimm C, Samarddzija M, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. 2005;24:275–306. doi: 10.1016/j.preteyeres.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 53.Paskowitz DM, LaVail MM, Duncan JL. Light and inherited retinal degeneration. Br J Ophthalmol. 2006;90:1060–1066. doi: 10.1136/bjo.2006.097436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marsili S, Genini S, Sudharsan R, et al. Exclusion of the unfolded protein response in light-induced retinal degeneration in the canine T4R RHO model of autosomal dominant retinitis pigmentosa. PLoS One. 2015;10:e0115723. doi: 10.1371/journal.pone.0115723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sudharsan R, Simone KM, Anderson NP, Aguirre GD, Beltran WA. Acute and protracted cell death in light-induced retinal degeneration in the canine model of rhodopsin autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2017;58;:270–281. doi: 10.1167/iovs.16-20749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwabe S, Ying GS, Aguirre GD, Beltran WA. Assessment of visual function and retinal structure following acute light exposure in the light sensitive T4R mutant dog. Exp Eye Res. 2016;146:341–353. doi: 10.1016/j.exer.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rachel RA, Swaroop A. Photoreceptor sensory cilia and ciliopathies: focus on CEP290, RPGR and their interacting proteins. Cilia. 2012;1:22. doi: 10.1186/2046-2530-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldstein O, Mezey JG, Schweitzer PA, et al. IQCB1 and PDE6B mutations cause similar early onset retinal degenerations in two closely related terrier dog breeds. Invest Ophthalmol Vis Sci. 2013;54:7005–7019. doi: 10.1167/iovs.13-12915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Downs LM, Scott EM, Cideciyan AV, et al. Overlap of abnormal photoreceptor development and progressive degeneration in Leber congenital amaurosis caused by NPHP5 mutation. Hum Mol Gen. 2016;25:4211–4226. doi: 10.1093/hmg/ddw254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Komaromy AM, Varner SE, deJuan E, Acland GM, Aguirre GD. Application of a new subretinal injection device in the dog. Cell Transplant. 2006;15:511–519. doi: 10.3727/000000006783981701. [DOI] [PubMed] [Google Scholar]
- 61.Cideciyan AV, Rachel RA, Aleman TS, et al. Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all cone NPHP6 homomorph mouse that mimics the human ciliopathy. Hum Mol Gen. 2011;20:1411–1423. doi: 10.1093/hmg/ddr022. [DOI] [PMC free article] [PubMed] [Google Scholar]