

Abstract
Key points
Augmented inositol 1,4,5‐trisphosphate (IP3) receptor (IP3R2) expression has been linked to a variety of cardiac pathologies.
Although cardiac IP3R2 function has been in the focus of research for some time, a detailed understanding of its potential role in ventricular myocyte excitation–contraction coupling under pathophysiological conditions remains elusive.
The present study focuses on mechanisms of IP3R2‐mediated sarcoplasmic reticulum (SR)‐Ca2+ release in ventricular excitation–contraction coupling under IP3R2‐overexpressing conditions by studying intracellular Ca2+ events.
We report that, upon IP3R2 overexpression in ventricular myocytes, IP3‐induced Ca2+ release (IP3ICR) modulates the SR‐Ca2+ content via “eventless” SR‐Ca2+ release, affecting the global SR‐Ca2+ leak. Thus, IP3R2 activation could act as a SR‐Ca2+ gateway mechanism to escape ominous SR‐Ca2+ overload.
Our approach unmasks a so far unrecognized mechanism by which “eventless” IP3ICR plays a protective role against ventricular Ca2+‐dependent arrhythmogenicity.
Abstract
Augmented inositol 1,4,5‐trisphosphate (IP3) receptor (IP3R2) function has been linked to a variety of cardiac pathologies including cardiac arrhythmias. The functional role of IP3‐induced Ca2+ release (IP3ICR) within ventricular excitation–contraction coupling (ECC) remains elusive. As part of pathophysiological cellular remodelling, IP3R2s are overexpressed and have been repeatedly linked to enhanced Ca2+‐dependent arrhythmogenicity. In this study we test the hypothesis that an opposite scenario might be plausible in which IP3ICR is part of an ECC protecting mechanism, resulting in a Ca2+‐dependent anti‐arrhythmogenic response on the cellular scale. IP3R2 activation was triggered via endothelin‐1 or IP3‐salt application in single ventricular myocytes from a cardiac‐specific IP3R type 2 overexpressing mouse model. Upon IP3R2 overexpression, IP3R activation reduced Ca2+‐wave occurrence (46 vs. 21.72%; P < 0.001) while its block increased SR‐Ca2+ content (∼29.4% 2‐aminoethoxydiphenyl borate, ∼16.4% xestospongin C; P < 0.001), suggesting an active role of IP3ICR in SR‐Ca2+ content regulation and anti‐arrhythmogenic function. Pharmacological separation of ryanodine receptor RyR2 and IP3R2 functions and two‐dimensional Ca2+ event analysis failed to identify local IP3ICR events (Ca2+ puffs). SR‐Ca2+ leak measurements revealed that under pathophysiological conditions, “eventless” SR‐Ca2+ efflux via enhanced IP3ICR maintains the SR‐Ca2+ content below Ca2+ spark threshold, preventing aberrant SR‐Ca2+ release and resulting in a protective mechanism against SR‐Ca2+ overload and arrhythmias. Our results support a so far unrecognized modulatory mechanism in ventricular myocytes working in an anti‐arrhythmogenic fashion.
Keywords: excitation‐contraction coupling, calcium cycling, cardiac arrhythmias, heart failure
Key points
Augmented inositol 1,4,5‐trisphosphate (IP3) receptor (IP3R2) expression has been linked to a variety of cardiac pathologies.
Although cardiac IP3R2 function has been in the focus of research for some time, a detailed understanding of its potential role in ventricular myocyte excitation–contraction coupling under pathophysiological conditions remains elusive.
The present study focuses on mechanisms of IP3R2‐mediated sarcoplasmic reticulum (SR)‐Ca2+ release in ventricular excitation–contraction coupling under IP3R2‐overexpressing conditions by studying intracellular Ca2+ events.
We report that, upon IP3R2 overexpression in ventricular myocytes, IP3‐induced Ca2+ release (IP3ICR) modulates the SR‐Ca2+ content via “eventless” SR‐Ca2+ release, affecting the global SR‐Ca2+ leak. Thus, IP3R2 activation could act as a SR‐Ca2+ gateway mechanism to escape ominous SR‐Ca2+ overload.
Our approach unmasks a so far unrecognized mechanism by which “eventless” IP3ICR plays a protective role against ventricular Ca2+‐dependent arrhythmogenicity.
Introduction
Sudden cardiac death due to arrhythmia is responsible for 10–30% of deaths among patients with hypertrophic cardiomyopathy (Ullal et al. 2016). In cardiac hypertrophy, alterations in Ca2+‐induced Ca2+ release (CICR) and excitation–contraction coupling (ECC) have been associated with cardiac instability (Gusev et al. 2009). Thus understanding the underlying mechanisms essentially contributes to the development of novel treatment strategies.
Ca2+ release from the sarcoplasmic reticulum (SR) in ventricular cardiomyocytes is mediated by two Ca2+ release channels: the ryanodine receptor type 2 (RyR2) and the inositol 1,4,5‐trisphosphate receptor type 2 (IP3R2; Bers, 2002; Hohendanner et al. 2014). While RyR2s play a central role in ventricular ECC, the contribution of functionally expressed IP3R2s under physiological and pathophysiological conditions is less well understood. In contrast to RyR2s, which are exclusively activated by Ca2+, IP3R2s are sensitive to Ca2+ and IP3. The latter SR‐Ca2+ release mechanism has been established as IP3‐induced Ca2+ release (IP3ICR; Zima & Blatter, 2004; Harzheim et al. 2009). Intracellular IP3 synthesis is triggered by a variety of vaso‐ and cardioactive hormones (e.g. endothelin‐1, angiotensin II) by stimulating G protein‐coupled receptors (GPCRs) that couple to phospholipase C, liberating IP3 from phosphatidylinositol 4,5‐bisphosphate (Proven et al. 2006; Berridge, 2009).
Although the ratio of functionally expressed RyR2s versus IP3R2s is approximately 100:1, a modulatory function of IP3ICR in ventricular myocyte ECC has been suggested and is the subject of an ongoing controversial discussion (Lipp et al. 2000; Kockskämper et al. 2008). In contrast to the physiological condition, a more pronounced role of IP3ICR in the framework of cardiac pathophysiology seems to be reasonable. It has been found that remodelling of ECC under various cardiac pathologies (e.g. cardiac hypertrophy, ischaemic dilated cardiomyopathy, atrial fibrillation, failing myocardium and hypertension) is associated with IP3R2s overexpression and, in this context, enhanced IP3ICR has been repeatedly linked to arrhythmogenicity (Zhang et al. 2002; Harzheim et al. 2009, 2010; Nakayama et al. 2010; Signore et al. 2013). Supporting evidence comes from the observation that endothelin‐1 and angiotensin II – both GPCR agonists – have similar positive inotropic effects in ventricular myocytes, suggesting a role of IP3ICR in the regulation of cytosolic Ca2+ homeostasis (Zima & Blatter, 2004). In addition, RyR2–IP3R2 local bi‐directional crosstalk mechanisms in cardiomyocytes have been recently described and may be involved in Ca2+‐dependent arrhythmogenicity at the cellular level (Wullschleger et al. 2017). Moreover, these studies support the concept of distinct RyR2‐IP3R2 Ca2+ signalling compartments within cardiomyocytes – either in the cytosol and/or in the nucleus – during pathophysiological signalling driven by IP3 and IP3R2 (Wu & Bers, 2006; Drawnel et al. 2010). In contrast, it has also been suggested that increased synthesis of IP3 as well as IP3R2 expression, may be a common feature of cardiac pathologies but without having functional significance for the ECC mechanism (Cooley et al. 2013).
At the subcellular level, synchronized and functionally clustered RyR2 openings result in “Ca2+ sparks”, the building blocks of global Ca2+ transients in ventricular myocytes; analogously coordinated openings of clustered IP3R2s give rise to “Ca2+ puffs” (Cheng et al. 1993; Yao et al. 1995). Although similar, the distinct spatio‐temporal characteristics of Ca2+ puffs and Ca2+ sparks and their pharmacological profile can be used for classification and separation of SR‐Ca2+ release events (Hohendanner et al. 2015). Openings of singular and/or “misplaced” RyR2s or IP3R2s that do not belong to channel clusters may not generate detectable SR‐Ca2+ release events but can still contribute to the intracellular Ca2+ homeostasis (Lipp & Niggli, 1996; Sobie et al. 2006; Brochet et al. 2011). Analogously to recently described “rogue” RyR2s, “rogue” IP3R2s may generate an “eventless” SR‐Ca2+ release flux which might have a significant impact on the total SR‐Ca2+ leak in ventricular myocytes without giving rise to localized Ca2+ events (Sobie et al. 2006).
Among other factors, the open probability (P o) of both IP3R2s and RyR2s and thus the SR‐Ca2+ leak, is critically controlled by SR luminal Ca2+ (Nunn & Taylor, 1992; Terentyev et al. 2002). Spontaneous increased RyR2 P o under SR‐Ca2+ overload results in asynchronous SR‐Ca2+ release (e.g. Ca2+ waves) and may lead to delayed after‐depolarizations, a major cause of ventricular tachyarrhythmias and sudden cardiac death (Keller et al. 2007; Liu et al. 2013). Although largely outnumbered by RyR2s, overexpression of IP3R2s within cardiac pathology may render the latter receptor a more significant SR‐Ca2+ regulatory function. The present study focuses on potential mechanisms by which IP3R2 may have a Ca2+‐dependent anti‐arrhythmogenic function at the cellular level.
Our data in the present study reveal that stimulation of IP3ICR in freshly isolated ventricular myocytes from a cardiac‐specific IP3R2‐overexpressing mouse model leads to a decrease in asynchronous SR‐Ca2+ release, a decline in the SR‐Ca2+ content and subsequent decrease in pro‐arrhythmic Ca2+ wave occurrence, suggesting a potential anti‐arrhythmic role of IP3ICR. Although under cellular remodelling conditions the SR‐Ca2+ content appears to be under active control of IP3ICR, no additional IP3R2‐mediated local SR‐Ca2+ events (e.g. Ca2+ puffs) were identified. Interestingly, study of the SR‐Ca2+ leak revealed a more prominent IP3R2‐dependent component that controls the SR‐Ca2+ content. Thus, these results support the view that IP3ICR contributes to the total SR‐Ca2+ leak via “eventless” SR‐Ca2+ release. The underlying mechanism actively stabilizes the SR‐Ca2+ content and limits luminal RyR2 sensitization below the critical threshold for spontaneous Ca2+ release thereby offering a protective mechanism against arrhythmias in ventricular myocytes.
Methods
Ethical approval
All experiments were approved by the State Veterinary Office of Bern (Switzerland), in accordance with Swiss Federal Animal Protection Law and were approved by the in‐house ethical committee (Ref. BE96/16, Ref. EFWW0443). For single myocyte isolation, mice were killed by cervical dislocation, hearts were extracted and perfused using the Langendorff perfusion technique. For transthoracic echochardiography measurements, mice were anaesthetized with 1–1.5% isoflurane, maintaining heart rate at 450–500 beats min−1. Following data acquisition, mice were monitored until recovered from anaesthesia.
Cardiac‐specific IP3R2‐overexpressing mouse model
Mouse lines FVB‐Tg(Myh6/tet0‐Itpr2)3.11Jmol/J (The Jackson Laboratory, Bar Harbor, ME, USA) and FVB.CG‐Tg(MyH6‐tTA)6Smbf/J (Jacksons) were crossbred to generate the cardiac‐specific IP3R type 2 (IP3R2) overexpressing mouse model (TG; Nakayama et al. 2010). In the following, wild‐type (WT) will be a synonym for ventricular myocytes isolated from WT mice and transgene (TG) for cells from IP3R2‐overexpressing mice. Genotype was assessed via conventional polymerase chain reaction. Primers used include: for IP3R2 5′‐CTTTCATCGGATGCCTGCCT‐3′ and 5′‐TTGGGAGGCCAAGGTGGGTA‐3′ forward and reverse, respectively; and for tetracycline transactivator (tTA) 5′‐CGCTGTGGGGCATTTTACTTTAG‐3′ and 5′‐CATGTCCAGATCGAAATCGTC‐3′ forward and reverse, respectively. Heart and body weights (mg g−1) were determined at 5 months of age. All mice were fed with doxycycline‐containing food (0.2 g kg−1, Envigo, Füllinsdorf, Switzerland) during pregnancy and until 4 weeks of age.
Echocardiography
Transthoracic echocardiography was performed using a 30 MHz probe and the Vevo 2100 Ultrasound machine (VisualSonics, Bothell, WA, USA). All mice were anaesthetized with 1–1.5% isoflurane. The heart was imaged in 2‐D mode on a parasternal long‐axis view from where an M‐mode cursor was positioned perpendicular to the interventricular septum and the posterior wall of the left ventricle. Echocardiographies were performed at the Cardiovascular Assessment Facility (CAF, Lausanne, Switzerland).
RNA and protein analysis
Animals were killed by cervical dislocation and tissues from ventricle, atria, aortic smooth muscle, femoral skeletal muscle and brain were excised, washed and snap‐frozen in liquid nitrogen. For western blot analysis, ventricular tissue was ground in extraction buffer (1x Protease Inhibitor (04693159001, Sigma‐Aldrich Chemie, Buchs, Switzerland) in NP40 extraction buffer, Invitrogen, Thermo Fisher Scientific, Reinach, Switzerland) for 30 min. Samples were centrifuged at 1400 g for 10 min at 4°C to obtain the supernatant. Following quantification by the bicinchoninic acid assay (BCA) method, 25 μg per lane of protein in loading solution (10% glycerol, 5% β‐mercaptoethanol, 43% SDS, 62.5 mM Tris, 6 M urea) were loaded in 4–20% pre‐cast gel (Bio‐Rad, Basel, Switzerland) and run for 55 min at 200 V. Samples were transferred to PVDF membranes at 100 mA and 4°C overnight. After PBS washing steps, membranes were blocked for 1 h at room temperature (RT) in blocking solution (5% milk in PBS). Membranes were incubated with primary antibodies anti‐IP3R2 (1:5000 KM1083, kindly provided by Prof. Dr K. Mikoshiba; Sugiyama et al. 1994), anti‐RyR2 (1:2000 ab2827, Abcam, Cambridge, UK) or anti‐GAPDH (1:100,000 10R‐G109A, Fitzgerald, Acton, MA, USA) in primary antibody solution (PBS‐Tween, 0.5% milk). Secondary antibody anti‐mouse Alexa 790 (1:100,000, ThermoFisher Scientific, Reinach, Switzerland) was applied in secondary antibody solution (PBS‐Tween, 0.5% milk, 0.01% SDS) for 1 h at RT. Imaging was performed on an Odyssey set‐up and images were analysed using GelAnalyzer. Full western blot images are available upon request.
Total RNA was extracted from snap‐frozen mice tissues with NucleoSpin RNA Plus kit (Macherey‐Nagel, Oensingen, Switzerland) and concentrations were determined with a NanoDrop 2000c (ThermoFisher Scientific). RT‐qPCR was performed by loading 10 ng of sample in an Eco Real‐Time PCR system (Illumina, Cambridge, UK) using KAPA SYBR FAST One‐Step Universal kit (LabGene, Châtel‐Saint‐Denis, Switzerland) in accordance with the manufacturers’ instructions. Primers used include: ITPR2 primer pair (M_Itpr2_3, Sigma) and β‐2‐microglobulin primers (forward FM1_B2m, and reverse RM1_B2m, Sigma). ITPR2 gene relative expression was normalized to reference gene β‐2‐microglobulin. ΔΔCq was calculated and used for statistical analysis. TG data are shown normalized to their respective WT tissue (norm.).
Ventricular myocyte isolation
Animals were killed at 5 months of age by cervical dislocation. Ventricular cardiomyocytes were isolated in Tyrode Ca2+‐free solution with the Langendorff perfusion technique with collagenase type II (Worthington) and Protease type XIV (Sigma). Extracellular Ca2+ was slowly increased to physiological concentration (1.8 mM).
Solutions and chemicals
Ventricular cardiomyocytes were permeabilized with 0.05% escin (Sigma). Cells were kept in final solution containing (mM): 120 potassium aspartate (KAsp), 3 K‐ATP, 3.734 MgCl2, 0.5 EGTA, 10 phosphocreatine, 10 HEPES and 0.05 Fluo‐3 5K (Biotium), with the addition of 5 U L−1 creatine phosphokinase (pH 7.4, 310 mOsm L−1). Free‐[Ca2+] was measured using Indo‐1 AM (Molecular Probes, Eugene, OR, USA) in a NanoDrop fluorospectrometer and adjusted to 70 nM. Cells were plated using Cell‐Tak (Corning, Fisher Scientific, Schwerte, Germany)‐coated coverslips. Pharmacological interventions include 10 μM IP3‐salt (IP3, SiChem, Bremen, Germany), 2 μM 2‐aminoethoxydiphenyl borate (2‐APB, Calbiochem) and 20 mM caffeine (Sigma). Reports suggest that 2‐APB at higher concentrations than that used in the present study may have secondary effects in other intracellular components besides blocking IP3R (Maruyama et al. 1997; Ascher‐Landsberg et al. 1999; Peppiatt et al. 2003). In our hands, application of 2 μM 2‐APB in WT cells – in the absence of IP3 stimulation – returned no unspecific effects. To ensure that the responses seen with 2‐APB were due to IP3R block, 3 μM xestospongin C (XeC, Focus Biomolecules) was used as an alternative IP3R blocker. We are aware that both blockers used in our study do not have a 100% selectivity; however, the combination of both is currently the best option for a pharmacological separation of IP3 effects.
Intact myocytes were loaded at RT with 5 μM Fluo‐3 AM (Biotium, Fremont, CA, USA) for 30 min, plated in Cell‐Tak‐coated coverslips and kept in extracellular solution containing (mM): 140 NaCl, 5 HEPES, 1.1 MgCl2, 5.4 KCl, 10 glucose and 1.8 CaCl2. For Na+ and Ca2+ free solution, NaCl was replaced by 140 LiCl and 0.5 EGTA. To reach steady state cells were field stimulated for 30 s at a frequency of 1 Hz. The following pharmacological agents were used: 100 nM endothelin‐1 (ET‐1, Sigma), 2 μM 2‐APB, 3 μM XeC, 1 mM tetracaine (tet, Sigma) and 10 mM caffeine (Sigma).
[Ca]2+ measurements and protocols
For the analysis of the SR‐Ca2+ leak/load relationship, intact cardiomyocytes were kept at 1.8 mM Ca2+ and paced for 30 s at 1 Hz to reach steady state. External solution was rapidly exchanged to a Na+ and Ca2+ free solution containing 1 mM NiCl2 to ensure Na+/Ca2+ exchanger block (Kimura et al. 1987). After 20 s, 1 mM tetracaine was added to block RyR2. After 30 s, 10 mM caffeine was rapidly applied to determine the SR‐Ca2+ content. ΔCa2+ cyt is estimated as the difference (ΔCa2+ cyt = F tet – F 0) between fluorescence before (F 0) and after RyR2 block (F tet); as more Ca2+ is released to the cytosol via SR‐Ca2+ leak, the difference (ΔCa2+ cyt) between baselines shrinks and the SR‐Ca2+ leak becomes more positive.
For single cell arrhythmogenicity studies, intact cells were kept at 1.8 mM Ca2+ external solution and were field stimulated for 30 s at 1 Hz prior to a 10 s rest period to study Ca2+ wave occurrence as an estimate of cell propensity to aberrant SR‐Ca2+ release. Finally, SR‐Ca2+ load was assessed by application of 10 mM caffeine. Ca2+ wave occurrence (%) is calculated as the portion of cells analysed that showed Ca2+ waves during the 10 s resting period.
Confocal Ca2+ imaging
To assess intracellular Ca2+ changes, Fluo‐3 was excited at 488 nm wavelength of an argon laser and fluorescence was collected at 525 nm in line‐scan mode (2 ms per line) with a photomultiplier tube (PMT) on a MicroRadiance laser scanning confocal microscope (Bio‐Rad, Watford, UK) or alternatively a FluoView 1000 (Olympus, Volketswil, Switzerland) with a solid‐state laser (Sapphire, Coherent, Santa Clara, CA, USA).
Data analysis and statistics
Raw line‐scan data from permeabilized myocytes were analysed using the ImageJ (NIH) plug‐in SparkMaster to obtain Ca2+ spark frequency (Ca2+ sparks (100 μm)–1 s–1), amplitude (Amp, change in fluorescence relative to the fluorescence background (ΔF/F 0)), full‐width at amplitude half‐maximum (μm, FWHM), full duration at amplitude half‐maximum (ms, FDHM), full duration (ms), exponential time constant of the spark decay (τdecay, ms) and time to peak (ms). Spark mass was calculated as Amplitude × 1.206 × (FWHM)3 (Hollingworth et al. 2001); Ca2+ released via Ca2+ sparks was estimated as the product of the spark frequency and spark mass. Spark frequency data are presented as 100 sparks μm−1 s−1 or normalized to the vector where indicated (norm.). Data analysis was performed using the softwares ImageJ (NIH, Bethesda, MD, USA) and Igor Pro (WaveMetrics, Lake Oswego, OR, USA).
Data are presented as box (25–75 percentiles) and whisker (10 and 90 percentiles) plots; outliers are highlighted. Data within tables are presented as mean ± standard deviation (SD). Normal distribution of the data was assessed by D'Agostino's K 2 test. Statistical comparison was performed via one‐way ANOVA for multi‐parametric comparison followed by Dunnett's test or paired or unpaired Student's t test. Binary data were tested by the χ2 test. Number of animals (N), cells (n), Ca2+ events (n e) and statistical results are indicated in the figure legends.
Results
Cardiac IP3R2 overexpression induces cardiac remodelling both at cellular and organ levels
A cardiac‐specific IP3R2‐overexpressing mouse model (TG; Nakayama et al. 2010) has been used to mimic cardiac hypertrophy occurring in patients (Harzheim et al. 2009). In addition, the strategy to tune one element within a complex cellular remodelling situation to extreme could unmask an otherwise potentially invisible regulatory mechanism. The TG mice display a significantly higher heart weight to body weight ratio when compared to their wild‐type littermates (WT, 7.75 ± 0.92 mg g−1 vs. TG 10.6 ± 1.69 mg g−1; N = 5, 9; P = 0.001; Fig. 1 A). A cardiac hypertrophic phenotype was confirmed via transthoracic echocardiography (Table 1), which revealed greater interventricular septum (IVS, 0.78 ± 0.08 mm WT vs. 0.93 ± 0.09 mm TG; N = 4, 6; P = 0.039) and left ventricular posterior wall thickness (LVPW, 0.78 ± 0.07 mm WT vs. 0.94 ± 0.11 mm TG; P = 0.033) in TG hearts when compared to WT.
Figure 1. TG mice display cardiac remodelling at both cellular and organ levels.
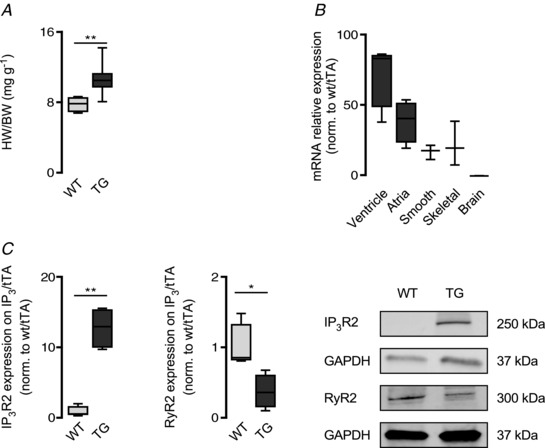
A, heart weight (mg)/body weight (g) of WT (N = 5) and TG (N = 9) hearts at 5 months of age (** P < 0.01; Student's t test). B, ITPR2 gene relative expression in various tissues depicted as ΔΔCq and normalized to WT. β‐2‐Microglobulin was used as reference gene (ventricle and atria N = 4 each; brain, smooth and skeletal muscle N = 3 each). C, relative IP3R2 and RyR2 protein expression in TG ventricular tissue when compared to WT and normalized to GAPDH. Box plots display IP3R2 (left) and RyR2 (right) (* P < 0.05, ** P < 0.01; N = 4 each; Student's t test).
Table 1.
Systolic and diastolic parameters measured by transthoracic echocardiography in WT and TG mice at 5 months of age
| WT | TG | |
|---|---|---|
| IVSd (mm) | 0.78 ± 0.08 | 0.93 ± 0.09* |
| IVSs (mm) | 1.07 ± 0.11 | 1.27 ± 0.13* |
| LVPWd (mm) | 0.78 ± 0.07 | 0.94 ± 0.11* |
| LVPWs (mm) | 1.09 ± 0.07 | 1.27 ± 0.16 |
| %FS | 33.35 ± 1.97 | 29.04 ± 4.06 |
| %EF | 62.45 ± 2.78 | 55.94 ± 6.21 |
| E‐wave deceleration time (ms) | 21.07 ± 6.84 | 12.38 ± 3.8* |
| ET (ms) | 49.29 ± 3.6 | 58.81 ± 6.42* |
TG mice display mild cardiac hypertrophy at 5 months of age. Values are means ± SD. Abbreviations correspond to: diastole (d), systole (s), interventricular septum (IVS), left ventricular posterior wall (LVPW), fractional shortening (%FS), ejection fraction (%EF) and ejection time (ET) (* P < 0.05; N = 4, 7; Student's t test).
Figure 1 B shows an increase in the relative expression of ITPR2 mRNA (IP3R2 encoding gene) levels in both ventricular and atrial tissue. A minor ITPR2 expression increase was detected in smooth and skeletal muscle but was absent in brain tissue. Accordingly, western blot analysis revealed a 12.72 (±2.97)‐fold increase in IP3R2 protein synthesis (N = 4 each; P = 0.001; Fig. 1 C, left) in ventricle. In addition and in line with other reports (Zhao et al. 2007), RyR2 protein level was significantly decreased in TG ventricular tissue (61.73 ± 0.24%; N = 4 each; P = 0.012; Fig. 1 C, right).
Global Ca2+ wave occurrence in response to IP3R2 pathway stimulation
Spontaneous Ca2+ waves are triggers for cardiac arrhythmias at the cellular level. To assess Ca2+‐dependent arrhythmogenicity in individual cells, IP3 synthesis was elicited via activation of the phosphatidylinositol 4,5‐bisphosphate (PIP2)–phospholipase C (PLC)–IP3 signalling cascade by endothelin‐1 (ET‐1, 100 nM) in intact ventricular myocytes (Table 2). Cells were field stimulated for 30 s at 1 Hz to ensure comparable SR‐Ca2+ loading conditions prior to a 10 s rest period to assess the occurrence of Ca2+ waves (Fig. 2 A). In control conditions, without ET‐1 stimulation and when compared to WT, TG myocytes displayed higher Ca2+ transient amplitude (1.9 ± 0.4 ΔF/F 0 WT vs. 2.32 ± 0.54 ΔF/F 0 TG; n = 13, 14; P = 0.014; Fig. 2 B) under conditions of comparable SR‐Ca2+ load (3.5 ± 0.79 ΔF/F 0 WT vs. 3.53 ± 0.78 ΔF/F 0 TG; P = 0.46) and consequently the fractional SR‐Ca2+ release in TG cells was increased (0.56 ± 0.1 a.u. WT vs. 0.65 ± 0.12 a.u. TG; P = 0.025). Furthermore, TG myocytes displayed a higher spontaneous Ca2+ wave occurrence (5.5% WT vs. 46% TG; P < 0.001; Fig. 2 C).
Table 2.
Parameters of field stimulated WT and TG intact ventricular myocytes upon IP3R2 pharmacological intervention
| WT | Ca2+ transient amplitude (ΔF/F 0) | τdecay (ms) | T50 (ms) | Fractional SR‐Ca2+ release (a.u.) | SR‐Ca2+ content (ΔF/F 0) |
|---|---|---|---|---|---|
| Control (13, 4) | 1.9 ± 0.4 | 156.92 ± 27.76 | 119.24 ± 19.25 | 0.56 ± 0.1 | 3.5 ± 0.79 |
| ET‐1 (11, 3) | 3.25 ± 1.17** | 162.26 ± 20.73 | 124.88 ± 12.6 | 0.66 ± 0.14* | 5.06 ± 1.81* |
| ET‐1 + 2‐APB (16, 3) | 2.69 ± 0.84 | 176.78 ± 40.56 | 132.62 ± 23.49 | 0.63 ± 0.11 | 4.17 ± 0.92 |
| 2‐APB (16, 3) | 2.09 ± 0.58 | 173.83 ± 42.17 | 129.04 ± 25.02 | 0.62 ± 0.15 | 3.47 ± 1.02 |
| XeC (15, 3) | 2.12 ± 0.73 | 168.44 ± 31.59 | 131.1 ± 21.19 | 0.62 ± 0.15 | 3.57 ± 1.46 |
| TG | Ca2+ transient amplitude (ΔF/F 0) | τdecay (ms) | T50 (ms) | Fractional SR‐Ca2+ release (a.u.) | SR‐Ca2+ content (ΔF/F 0) |
|---|---|---|---|---|---|
| Control (14, 4) | 2.32 ± 0.54‡ | 122.39 ± 22.3‡‡‡ | 97.01 ± 17.82‡‡ | 0.65 ± 0.12‡ | 3.53 ± 0.78 |
| ET‐1 (16, 4) | 2.63 ± 0.77 | 135.01 ± 35.5 | 106.82 ± 26.29 | 0.65 ± 0.13 | 4.07 ± 0.9 |
| ET‐1 + 2‐APB (14, 3) | 2.77 ± 0.86 | 126.68 ± 25.8 | 102.57 ± 20.22 | 0.71 ± 0.12 | 4.37 ± 0.65 |
| 2‐APB (14, 3) | 3.04 ± 1.28* | 131.9 ± 12.36 | 107.82 ± 13.74* | 0.65 ± 0.18 | 4.57 ± 1.18** |
| XeC (11, 3) | 2.95 ± 0.78* | 164.2 ± 44.4 | 128.5 ± 24.78 | 0.72 ± 0.1 | 4.11 ± 1.08 |
Values are means ± SD. Spatio‐temporal characteristics analysed include Ca2+ transient amplitude (ΔF/F 0), tau decay (τdecay, ms), time to 50% amplitude (T50, ms), fractional SR‐Ca2+ release (a.u.) and SR‐Ca2+ content (ΔF/F 0) from WT and TG (* P < 0.05, ** P < 0.01 vs. control; ‡ P < 0.05, ‡‡ P < 0.01, ‡‡‡ P < 0.001 vs. WT). Number of animals (N) and cells (n) are indicated below each condition as (n, N).
Figure 2. IP3R2 activation alters Ca2+ wave occurrence in intact cardiomyocytes.
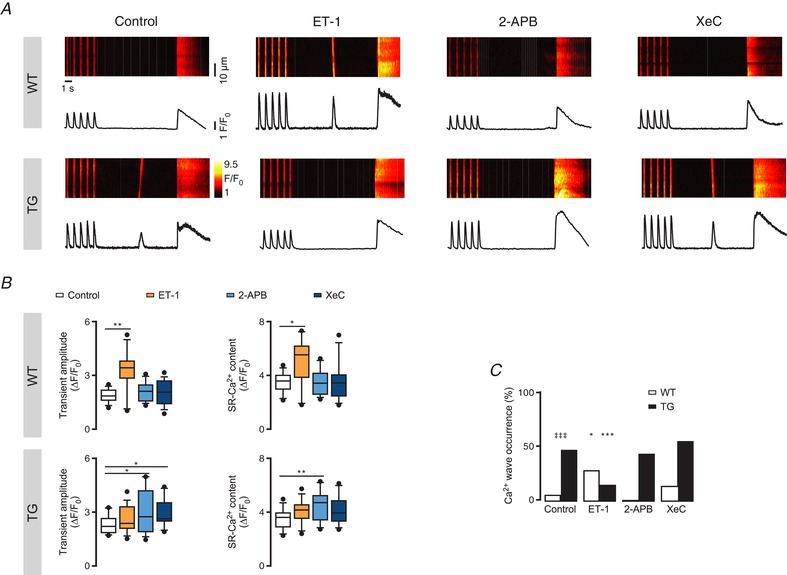
A, representative line‐scans of fluo‐3‐loaded intact cardiomyocytes with the corresponding fluorescence profiles upon pharmacological intervention with 100 nM ET‐1, 2 μM 2‐APB or 3 μM XeC. Cells were field stimulated for 30 s at 1 Hz to reach steady state prior to a 10 s rest period to assess Ca2+ wave occurrence as a measure for arrhythmogenicity at the single cell level. SR‐Ca2+ load was assessed by application of 10 mM caffeine. B, Ca2+ transient amplitude (ΔF/F 0) and SR‐Ca2+ content (ΔF/F 0) under the tested pharmacological interventions in WT (n of groups 1–4 = 13, 11, 16, 15; N of groups 1–4 = 4, 3, 3, 3) and TG myocytes (n of groups 1–4 = 14, 16, 14, 11; N of groups 1–4 = 4, 4, 3, 3; * P < 0.05, ** P < 0.01 vs. control; ANOVA followed by Dunnett's test). C, Ca2+ wave occurrence (%) calculated as the portion of cardiomyocytes displaying Ca2+ waves within the 10 s rest period (* P < 0.05, *** P < 0.001 vs. control; ‡‡‡ P < 0.001 vs. WT; N = 3–4, n = 9–21; χ2 test).
Compared to control, ET‐1 (100 nM, after 7–10 min application) triggered an increase in Ca2+ transient amplitude (3.25 ± 1.17 ΔF/F 0 ET‐1; n = 11; P = 0.001; Fig. 2 B, upper left), SR‐Ca2+ content (5.06 ± 1.81 ΔF/F 0 ET‐1; P = 0.013; Fig. 2 B, upper right) and Ca2+ wave occurrence (27.78% ET‐1; P = 0.035; Fig. 2 C) in WT cardiomyocytes, but surprisingly this effect was absent in IP3R2‐overexpressing cells (Fig. 2 B and C). In contrast, ET‐1 induced a 21.72% decrease in Ca2+ wave occurrence only in TG myocytes (Fig. 2 C; n = 16; P < 0.001).
In WT cardiomyocytes, block of IP3R2 by 2‐aminoethoxydiphenyl borate (2‐APB, 2 μM) or xestospongin C (XeC, 3 μM) returned no detectable changes in the analysed Ca2+ responses. In contrast, under IP3R2‐overexpressing conditions, blockade of IP3R2 led to increases in Ca2+ transient amplitude (3.04 ± 1.28 ΔF/F 0 2‐APB and 2.95 ± 0.78 ΔF/F 0 XeC; n = 14, 11; P = 0.033, 0.018; Fig. 2 B, lower left) with concomitantly higher SR‐Ca2+ content (4.57 ± 1.18 ΔF/F 0 2‐APB and 4.11 ± 1.08 ΔF/F 0 XeC; P = 0.007, 0.08; Fig. 2 B, lower right). Finally, block of IP3R2 by 2 μM 2‐APB abolished ET‐1‐mediated effects on cellular arrhythmogenicity both in WT and TG myocytes (Fig. 3). In other words, ET‐1 stimulation fails to alter the Ca2+ wave occurrence while IP3R remains blocked (Fig. 3).
Figure 3. Block of IP3R2 reverses ET‐1‐mediated arrhythmogenic effects.
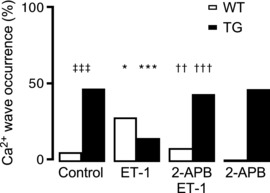
Intact ventricular myocytes were stimulated as indicated in Fig. 2. Application of 2 μM 2‐APB with 100 nM ET‐1 reverses the latter mediated arrhythmogenic effects in Ca2+ wave occurrence both in WT (n of groups 1–4 = 13, 11, 16, 16; N of groups 1–4 = 4, 3, 3, 3) and TG myocytes (n of groups 1–4 = 14, 16, 14, 14; N of groups 1–4 = 4, 4, 3, 3; * P < 0.05, *** P < 0.001 vs. control; †† P < 0.01, ††† P < 0.001 vs. ET‐1; ‡‡‡ P < 0.001 vs. WT; χ2 test).
To further study ET‐1‐mediated effects, estimation of sarco(endo)plasmic reticulum Ca2+‐ATPase (SERCA) and Na+–Ca2+ exchanger (NCX) relative contributions to the total Ca2+ transient decay was approached (Fig. 4). Fitting of a single exponential function to the action potential (AP)‐induced Ca2+ transient decay was used as a reference of the overall Ca2+ extrusion capacity of the cell, and to the Ca2+ transient (T) decay in the presence of 10 mM caffeine to assess NCX contribution (Fig. 4 A, left panel). SERCA and NCX (orange) contributions to the twitch decline (%) were calculated as: ((1/T twitch) − (1/T caffeine))/(1/T twitch) and (1/T caffeine)/(1/T twitch), respectively. In control conditions (Fig. 4 A, right panel), TG myocytes display a higher SERCA contribution when compared to WT (** P < 0.01; N = 4 each, n = 13, 14). An increase in SERCA contribution to the twitch Ca2+ decline upon 100 nM ET‐1 stimulation was seen only in WT ventricular myocytes (Fig. 4 Ba; ‡‡ P < 0.01; N = 4, 3; n = 13, 11).
Figure 4. ET‐1 alters SERCA/NCX balance in the cytosolic Ca2+ decay in WT myocytes.
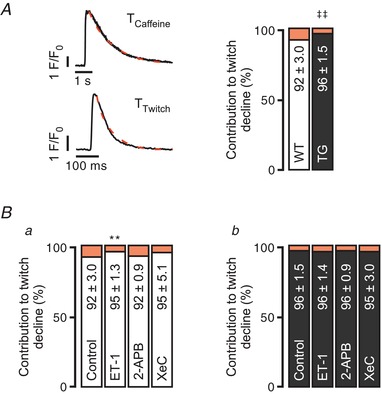
A, contribution to twitch decline of SERCA (%, white for WT and black for TG) and NCX (%, orange) in WT and TG myocytes (left panel). A single exponential function was fitted to the AP‐induced Ca2+ transient decay as a reference of the overall Ca2+ extrusion capacity of the cell; and to the Ca2+ transient decay in the presence of 10 mM caffeine to assess NCX contribution. SERCA and NCX contributions to twitch decline (%) were calculated as: ((1/T twitch) − (1/T caffeine))/(1/T twitch) and (1/T caffeine)/(1/T twitch), respectively (‡‡ P < 0.01; N = 4 each, n = 13, 14; Student's t test). B, contribution to twitch decline upon 100 nM ET‐1, 2 μM 2‐APB or 3 μM XeC intervention in WT (a; n of groups 1–4 = 13, 11, 16, 16; N of groups 1–4 = 4, 3, 3, 3) and TG ventricular myocytes (b; n of groups 1–4 = 14, 16, 14, 11; N of groups 1–4 = 4, 4, 3, 3; ** P < 0.01 vs. control; ANOVA followed by Dunnett's test).
Taken together, under conditions of IP3R2 overexpression, ET‐1 stimulation unexpectedly, and in contrast to the wild‐type situation, led to a decrease in Ca2+ wave frequency without altering Ca2+ transient amplitude nor SR‐Ca2+ content. However, block of IP3R2 results in a significant increase in SR‐Ca2+ content, which indicates that IP3ICR may play an intrinsic role in the regulation of the SR‐Ca2+ content and the propensity to arrhythmias.
Reduced SR‐Ca2+ content cannot be explained by Ca2+ sparks
The Ca2+ wave is a fundamental phenomenon that arises from unsynchronized SR‐Ca2+ release. Ca2+ sparks are elementary Ca2+ release events that can explain, at least in part, both the initiation and propagation of Ca2+ waves via the summation of individual Ca2+ release events and CICR. As was shown for atrial cells (Wullschleger et al. 2017), local bi‐directional crosstalk of IP3R2 and RyR2 may trigger and boost ventricular CICR. Under this view, pronounced IP3ICR may lead to an increased local Ca2+ events occurrence. Therefore, the effect of IP3ICR under IP3R2‐overexpressing conditions on the incidence of Ca2+ sparks was examined next.
Figure 5 A shows spontaneous Ca2+ sparks recorded in permeabilized cardiomyocytes upon diverse pharmacological interventions targeting the IP3R2 pathway. Under resting conditions, IP3R2‐overexpressing cells exhibit a higher spontaneous Ca2+ spark frequency (CaSpF) than WT (3.67 ± 1.8 sparks (100 μm)−1 s−1 TG vs. 1.76 ± 1 sparks (100 μm)−1 s−1 WT; n = 25 each; P < 0.001; Fig. 5 C, left panel) under similar SR‐Ca2+ loading conditions (Fig. 5 B, right, and C). In line with the concept that IP3ICR can boost CICR (e.g. Ca2+ sparks), in WT ventricular cardiomyocytes IP3‐salt (10 μM) evoked a significant increase in CaSpF (24.2%; n = 22; P = 0.006; Fig. 5 Da, left) together with a statistically significant decrease of the SR‐Ca2+ content (3.73 ± 0.89 ΔF/F 0 control vs. 3.3 ± 0.69 ΔF/F 0 IP3; P = 0.041; Fig. 5 Da, right) with no further changes in Ca2+ spark mass (Table 3). This response was sensitive both to 2 μM 2‐APB and 3 μM XeC (Fig. 5 Da). In contrast to the native situation, in IP3R2 overexpressing cardiomyocytes, IP3 induced a 25.17% decrease of the CaSpF (P = 0.015; n = 25, 13; P = 0.015; Fig. 5 Db, right) along with a reduction of the SR‐Ca2+ content (4.17 ± 0.48 ΔF/F 0 control vs. 3.56 ± 0.57 ΔF/F 0 IP3; P = 0.012; Fig. 5 Db, left) 2–4 min after application. Both effects were sensitive to XeC and 2‐APB.
Figure 5. IP3 evokes a divergent response in CaSpF in WT and TG permeabilized myocytes.
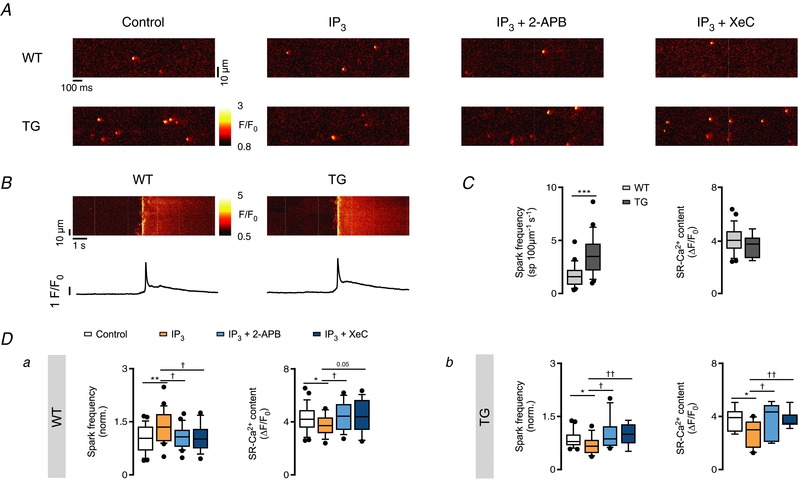
A, representative line‐scan images of Ca2+ events in permeabilized cardiomyocytes in control conditions and in the presence of 10 μM IP3‐salt and/or 2 μM 2‐APB, 3 μM XeC in WT and TG. After permeabilization and plating, each cell was scanned before and 2–4 min after pharmacological intervention. B, representative line‐scan images with the corresponding fluorescence profiles of de‐skewed caffeine (20 mM)‐induced Ca2+ transients under control conditions in WT and TG permeabilized myocytes. C, Ca2+ spark frequency (Ca2+ sparks (100 μm)−1 s−1) and SR‐Ca2+ content (ΔF/F 0) in control conditions in WT and TG cells (*** P < 0.001 vs. WT; n = 25 each, N = 8 each; Student's t test). D, normalized Ca2+ spark frequencies (norm.) and SR‐Ca2+ content (20 mM caffeine, ΔF/F 0) after Ca2+ event occurrence assessment in control conditions and in the presence of IP3‐salt and/or 2‐APB, XeC in WT (n of groups 1–4 = 25, 22, 20, 16; N of groups 1–4 = 8, 8, 3, 3) and TG myocytes (n of groups 1–4 = 25, 13, 11, 10; N of groups 1–4 = 8, 3, 3, 3; * P < 0.05, ** P < 0.01 vs. control; † P < 0.05, †† P < 0.01 vs. IP3; ANOVA followed by Dunnett's test).
Table 3.
Spatio‐temporal characteristics of Ca2+ events in permeabilized ventricular myocytes upon pharmacological intervention of the IP3R2 pathway
| WT | Amplitude (ΔF/F 0) | FDHM (ms) | Full duration (ms) | FWHM (μm) | Spark mass (a.u.) | τdecay (ms) | Time to peak (ms) |
|---|---|---|---|---|---|---|---|
| Control (25, 8) | 1.33 ± 0.12 | 18.17 ± 1.35 | 45.62 ± 5.08 | 1.95 ± 0.16 | 13.41 ± 4.44 | 18.12 ± 2.9 | 10.8 ± 1.28 |
| IP3 (22, 8) | 1.34 ± 0.15 | 18.22 ± 2.32 | 44.71 ± 7.26 | 1.96 ± 0.16 | 14 ± 4.26 | 19.82 ± 8.38 | 10.76 ± 1.14 |
| 2‐APB + IP3 (20, 3) | 1.37 ± 0.22 | 18.51 ± 2.15 | 48 ± 9 | 2.01 ± 0.14 | 15.3 ± 4.47 | 17.56 ± 4.54 | 11.5 ± 1.94 |
| XeC + IP3 (16, 3) | 1.27 ± 0.11 | 22.75 ± 3.41††† | 50.69 ± 10.35††† | 2.04 ± 0.2 | 16.55 ± 5.37 | 24.67 ± 9.48† | 13.32 ± 3.31 |
| 2‐APB (12, 3) | 1.32 ± 0.13 | 19.5 ± 3.53 | 50.25 ± 10.07 | 1.98 ± 0.2 | 13.96 ± 3.63 | 20.81 ± 8.87 | 11.83 ± 2.28 |
| XeC (16, 3) | 1.33 ± 0.25 | 22.54 ± 2.54*** | 55.28 ± 8.66*** | 2.03 ± 0.24 | 15.31 ± 5.02 | 22.34 ± 4.8** | 13.9 ± 2.88*** |
| TG | Amplitude (ΔF/F 0) | FDHM (ms) | Full duration (ms) | FWHM (μm) | Spark mass (a.u.) | τdecay (ms) | Time to peak (ms) |
|---|---|---|---|---|---|---|---|
| Control (25, 8) | 1.32 ± 0.14 | 26.22 ± 6.53 | 62.13 ± 14.89 | 1.96 ± 0.22 | 13.89 ± 4.34 | 26.86 ± 8.38 | 13.6 ± 2.71 |
| IP3 (13, 3) | 1.41 ± 0.15 | 24.79 ± 7.5 | 57.94 ± 13.57 | 1.98 ± 0.15 | 16 ± 5.22 | 32.49 ± 17.1 | 13.38 ± 5.01 |
| 2‐APB + IP3 (11, 3) | 1.32 ± 0.25 | 29.95 ± 12.8 | 68.63 ± 27.89 | 2 ± 0.12 | 15.04 ± 4.58 | 38.29 ± 27.57 | 15.64 ± 7.37 |
| XeC + IP3 (10, 3) | 1.31 ± 0.08† | 27.16 ± 8.57 | 61.99 ± 16.85 | 1.91 ± 0.17 | 13.27 ± 4.76 | 36.91 ± 19.59 | 12.35 ± 1.57 |
| 2‐APB (17, 4) | 1.31 ± 0.17 | 26.1 ± 5.27 | 57.12 ± 10.58 | 2.05 ± 0.23 | 16.34 ± 6.52 | 30.12 ± 11.52 | 13.03 ± 2.58 |
| XeC (14, 3) | 1.28 ± 0.16 | 26.93 ± 7.47 | 62.62 ± 14.65 | 2.02 ± 0.2 | 15.69 ± 5.17 | 42.3 ± 12.72 | 15.4 ± 4.73 |
Following cell permeabilization and plating, a representative line‐scan was recorded to assess Ca2+ events in control conditions. Pharmacological effects were assessed 2–4 min post‐intervention (10 μM IP3‐salt, 2 μM 2‐APB, 3 μM XeC). Parameters analysed include Ca2+ spark amplitude (ΔF/F 0), full duration at amplitude half‐maximum (FDHM, ms), full duration (ms), full width at amplitude half‐maximum (FWHM, μm), spark mass (a.u.), tau decay (τdecay, ms) and time to peak (ms) for WT and TG (** P < 0.01, *** P < 0.001 vs. control; † P < 0.05, ††† P < 0.001 vs. IP3‐salt; ANOVA followed by Dunnett's test). N and n are indicated below each condition as (n, N).
To elucidate how the SR‐Ca2+ content can be reduced even with a lower number of Ca2+ sparks, individual Ca2+ events were analysed. Spatio‐temporal Ca2+ event analysis revealed no differences in individual parameters between Ca2+ events observed in WT and TG cells upon direct IP3 stimulation (Table 3). Recently, we reported that multi‐parametric analysis of single Ca2+ events upon similar pharmacological interventions as presented in the current study can be used as a tool to differentiate among Ca2+ sparks and Ca2+ puffs in permeabilized atrial myocytes (Wullschleger et al. 2017). Hence, we repeated the analysis for the data presented here. Figure 6 shows single Ca2+ events in control conditions and upon IP3 application plotted according to their individual fluorescence amplitude and full duration at amplitude half‐maximum (FDHM). IP3 application did not induce any detectable differences either in WT (Fig. 6 Aa; n e = 195, 374) or TG myocytes (Fig. 6 Ab; n e = 443, 269). The Ca2+ event analysis suggests that only one population of Ca2+ events was detected and can be identified as Ca2+ sparks. In other words, a reduced SR‐Ca2+ content in TG cardiomyocytes cannot be explained by RyR2 activity alone, given the reduced Ca2+ sparks occurrence.
Figure 6. Multi‐parametric analysis of single Ca2+ event properties failed to differentiate IP3R2‐mediated Ca2+ events.
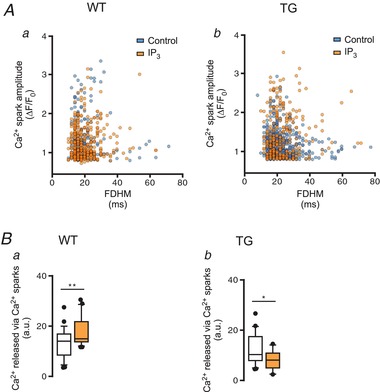
A, scatter plot of single Ca2+ events (from Fig. 3) according to amplitude (ΔF/F 0) and full‐duration at half‐maximum amplitude (FDHM, ms) in WT (a, n e for control and IP3 = 195, 374, N = 8 each) and TG (b, n e = 443, 269; N = 8, 3) myocytes. B, box and scatter plots display Ca2+ released via Ca2+ sparks (a.u.) estimated as the product of spark frequency and mass per cell in WT (a) and TG (b) myocytes in control conditions and upon stimulation with 10 μM IP3‐salt (data from Fig. 3; * P < 0.05, ** P < 0.01 vs. control; Student's t test).
In addition, we estimated the Ca2+ released via Ca2+ sparks under the tested conditions as the product of the Ca2+ spark mass and the CaSpF. Figure 6 B shows that IP3 stimulation increased Ca2+ release via Ca2+ sparks in WT (Fig. 6 Ba), but evoked a reduction in TG cells (Fig. 6 Bb). Both effects are presumably due to the differences in CaSpF induced by the application of IP3.
The discrepancy between reduced SR‐Ca2+ content and the total amount of Ca2+ released observed during IP3 stimulation in TG cardiomyocytes can neither be explained by the number of detected events nor the spatio‐temporal characteristics of individual Ca2+ events.
Intrinsically active IP3ICR modulates SR‐Ca2+ content via SR‐Ca2+ leak
Enhanced luminal Ca2+ increases RyR2 channel open probability (P o; Györke & Györke, 1998). Thus aberrant SR‐Ca2+ overload may result in spontaneous SR‐Ca2+ release (Chen et al. 2014). Since a discrepancy between CaSpF and SR‐Ca2+ content was evident, we hypothesized a contribution of IP3ICR via “eventless” SR‐Ca2+ release. We performed a series of SR‐Ca2+ leak protocols (Fig. 7 A) in the presence of RyR2 antagonists and/or IP3R2 agonists/antagonists to assess a potential contribution of “eventless” IP3ICR on the total SR‐Ca2+ leak (Shannon et al. 2002). Figure 7 B shows zoomed‐in examples with the corresponding fluorescence profiles for WT and TG myocytes (left and right, respectively) showing the RyR2 contribution to the total SR‐Ca2+ leak estimated as the difference (ΔCa2+ cyt = F tet − F 0) between the fluorescence before (F 0) and after RyR2 block (F tet). In principle, an increase in the “tetracaine‐insensitive” SR‐Ca2+ leak (e.g. via IP3ICR) may result in an apparent elevation in cytosolic [Ca2+] despite the presence of tetracaine (F tet), thus the difference between baselines (ΔCa2+ cyt) may become smaller, implying an increase in the total SR‐Ca2+ leak. Upon NCX block, the apparent ΔCa2+ cyt significantly depends on the SR‐Ca2+ leak/load balance (e.g. SR‐Ca2+ release and re‐uptake) and steady state prior to RyR2 block (F 0). In other words, the expected ΔCa2+ cyt decrease may not fully reflect large increases in SR‐Ca2+ leak above steady state. This is seen in Figs 7 and 8.
Figure 7. TG intact myocytes display altered basal SR‐Ca2+ leak.

A, representative confocal line‐scan images of WT and TG cell responses (left and right, respectively). Intact ventricular myocytes were electrically paced for 30 s at 1 Hz to reach steady state. Tyrode solution was rapidly switched to a 0 Na+, 0 Ca2+ solution for 15 s prior to addition of 1 mM tetracaine (tet). Finally, SR‐Ca2+ content was assessed (10 mM caffeine, Caff). B, representative zoomed‐in confocal line‐scan images (from dotted box in A) of WT and TG cell responses (right) in control conditions with the corresponding fluorescence profiles (left). For illustrative purposes each plot is shown overlapping with a trace corresponding to the fit of a single exponential decay function. SR‐Ca2+ leak (as ΔCa2+ cyt) was estimated by approaching the difference between fluorescence before (F 0) and after RyR2 block (F tet, ΔCa2+ cyt = F tet − F 0). C, compared to WT, TG cells display a smaller ΔCa2+ cyt (−0.16 ± 0.06 ΔF/F 0 WT vs. −0.05 ± 0.07 ΔF/F 0 TG; P < 0.001), larger Ca2+ transients (1.95 ± 0.78 ΔF/F 0 WT vs. 3.18 ± 0.99 ΔF/F 0 TG; P = 0.002) with enhanced fractional SR‐Ca2+ release (0.51 ± 0.13 a.u. WT vs. 0.69 ± 0.22 a.u. TG; P = 0.011) and SR‐Ca2+ content (3.84 ± 1.62 ΔF/F 0 WT vs. 4.75 ± 1.39 ΔF/F 0 TG; P = 0.075; * P < 0.05, ** P < 0.01, *** P < 0.001 vs. WT; n = 10, 16, N = 4 each; Student's t test).
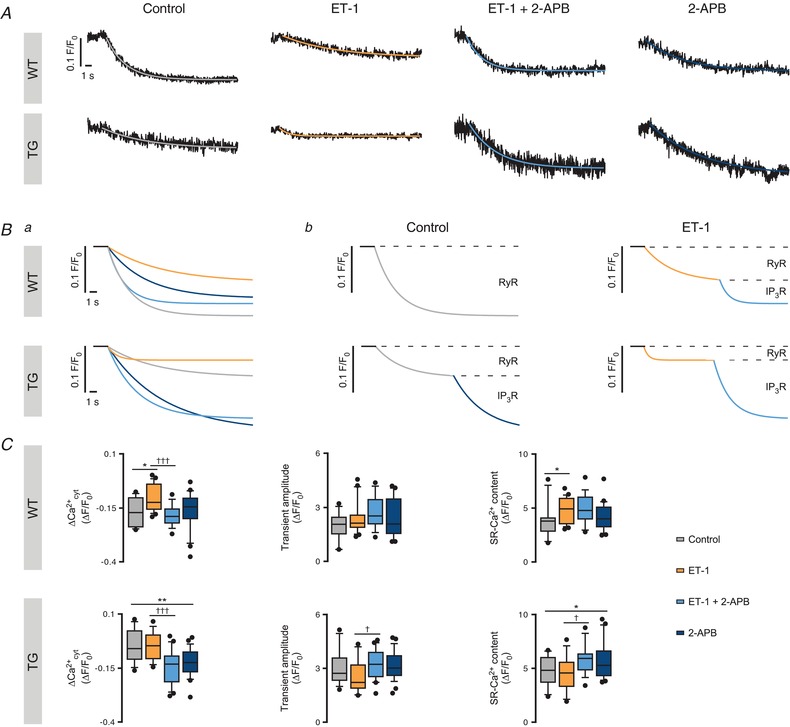
Figure 8. IP3ICR modulation of the SR‐Ca2+ leak diverges upon IP3R2 overexpression.
A, representative SR‐Ca2+ leak profiles in the tested conditions for WT and TG ventricular myocytes. Individual cells were separately treated with 100 nM ET‐1 and/or 2 μM 2‐APB to study the contribution of IP3ICR in the total SR‐Ca2+ leak. For illustrative purposes each trace is fitted with a single exponential decay function shown as an overlapped coloured trace. Ba, illustrative representation of the SR‐Ca2+ leak protocol responses shown in A (WT and TG, above and below, respectively). Bb, illustrative montage of SR‐Ca2+ leak curves in the tested conditions showing the contribution of IP3R and RyR Ca2+ leak to the total leak in WT and TG myocytes (above and below, respectively). Application of 1 mM tetracaine reveals the RyR‐mediated SR‐Ca2+ leak while addition of 2‐APB – in the presence of tetracaine – unmasks the IP3R2 component. C, ΔCa2+ cyt (ΔF/F 0), Ca2+ transient amplitude (ΔF/F 0) and SR‐Ca2+ content (ΔF/F 0) of WT (a, n of groups 1–4 = 10, 20, 18, 21; N = 4 each) and TG (b, n of groups 1–4 = 16, 14, 21, 28; N of groups 1–4 = 4, 3, 3, 4) groups upon pharmacological intervention (* P < 0.05, ** P < 0.01 vs. control; † P < 0.05, ††† P < 0.001 vs. ET‐1; ANOVA followed by Dunnett's test).
Figure 7 C shows that compared to WT, TG cells display a lower ΔCa2+ cyt (−0.16 ± 0.06 ΔF/F 0 WT vs. −0.05 ± 0.07 ΔF/F 0 TG; n = 10, 16; P < 0.001), larger Ca2+ transients with enhanced fractional SR‐Ca2+ release and SR‐Ca2+ content (3.84 ± 1.62 ΔF/F 0 WT vs. 4.75 ± 1.39 ΔF/F 0 TG; P = 0.075).
Figure 8 A shows representative SR‐Ca2+ leak profiles in the tested conditions (control, ET‐1, ET‐1 + 2‐APB and 2‐APB) for WT and TG ventricular myocytes. Figure 8 Ba exhibits the aforementioned traces from the different conditions overlapped for each animal group (WT and TG, above and below, respectively). Finally, Fig. 8 Bb displays the fitted samples according to cell type (WT or TG, above and below, respectively) and pharmacological intervention (control vs. ET‐1, left and right, respectively) and indicates the SR‐Ca2+ leak portion corresponding to RyR or IP3R.
In WT myocytes, 100 nM ET‐1 induced a 31.25% decrease in ΔCa2+ cyt (n = 20; P = 0.011; Fig. 8 C, top left panel), which was sensitive to 2 μM 2‐APB. 2‐APB alone did not significantly affect any of the tested parameters. In addition, ET‐1 induced IP3‐independent increases of the SR‐Ca2+ content but not the Ca2+ transient amplitude (Fig. 8 C, top middle and right panels, respectively).
Surprisingly, in TG cells, ET‐1 application resulted in no detectable ΔCa2+ cyt changes (Fig. 8 C, bottom left panel) and non‐significant decreases of the Ca2+ transient amplitude and the SR‐Ca2+ content (Fig. 8 C, bottom middle and left panels, respectively). In contrast, IP3R2 block by 2‐APB resulted in a large decrease of the ΔCa2+ cyt both in the presence and absence of ET‐1 (Fig. 8 C, bottom left panel) together with increases in the SR‐Ca2+ content (Fig. 8 C, bottom right panel). No further changes were detected in Ca2+ transient amplitude (Fig. 8 C, bottom middle panel) upon IP3R2 block. However, the “hidden” IP3‐dependent ΔCa2+ cyt effect in the leak protocol is unmasked by IP3R2 block.
Taken together, in IP3R2‐overexpressing myocytes IP3ICR modulates the SR‐Ca2+ content via sustained SR‐Ca2+ leak driven by “eventless” IP3ICR rather than by localized Ca2+ release events, e.g. Ca2+ puffs. In addition, under IP3R2‐overexpressing conditions, IP3ICR is intrinsically active and thereby fine‐tunes the SR‐Ca2+ load and cytosolic Ca2+, subsequently modulating Ca2+ wave occurrence.
Discussion
Our results obtained in ventricular cardiomyocytes reveal that, upon IP3R2 overexpression, IP3ICR stimulation leads to a decrease in RyR2 activity together with a decline in SR‐Ca2+ content, indicating that IP3R2 activation can act as a SR‐Ca2+ gateway mechanism to escape ominous SR‐Ca2+ overload and subsequent unsynchronized SR‐Ca2+ release. Our approach unmasks a so far unrecognized mechanism by which IP3ICR modulates SR‐Ca2+ content via “eventless” SR‐Ca2+ leak and effectively restores cardiomyocyte Ca2+ homeostasis.
These observations are based on a cardiac‐specific IP3R2‐overexpressing mouse model that mimics at least in part cardiac hypertrophy in patients (Nakayama et al. 2010). Cardiac remodelling is a complex process that affects a broad range of intracellular Ca2+ signalling components. TG myocytes are characterized by increases in IP3R2 expression together with RyR2 down‐regulation, a phenomenon known for various cardiac diseases (Go et al. 1995; Domeier et al. 2012; Zima et al. 2014).
IP3 is known to significantly contribute to a variety of intracellular Ca2+ signalling mechanisms by functionally exercising as a secondary messenger that interconnects extracellular signals to numerous subcellular processes. In ventricular myocytes, IP3Rs are preferentially located within the nuclear envelope and are found at a lower extent in the SR membrane (Bare et al. 2005). While nuclear IP3ICR has been linked to the modulation of gene expression (Higazi et al. 2009), the contribution of SR‐IP3Rs in ventricular ECC under physiological conditions remains controversial (Zhang et al. 2002; Harzheim et al. 2009, 2010; Nakayama et al. 2010; Cooley et al. 2013; Signore et al. 2013). Whether and in which direction enhanced IP3ICR may effectively modulate cardiac ECC in pathophysiological conditions warrants further investigation.
Ca2+ wave occurrence is controlled by IP3ICR
In the myocardium, the amplitude of global Ca2+ signals is locally controlled by Ca2+ influx through L‐type Ca2+ channels (LTCCs) and subsequently amplified by SR‐Ca2+ release via CICR. Because of its positive feedback, CICR has an inherent tendency to oscillate, particularly under pathological conditions. Based on spontaneous SR‐Ca2+ release, these oscillations have been found to play a fundamental role in the generation of cardiac arrhythmias. At the cellular level, Ca2+ oscillations originating from spontaneous SR‐Ca2+ release channel openings often manifest as Ca2+ waves that travel from release junction to release junction in an unsynchronized manner. This process can be described as a “fire‐diffuse‐fire” mechanism (Keizer et al. 1998). In this scenario, aberrant SR‐Ca2+ releases can substantially increase cytosolic Ca2+ levels and trigger delayed afterdepolarizations, a major source for ventricular tachyarrhythmias (Lipp & Niggli, 1993). A contribution of IP3ICR to the genesis of Ca2+‐dependent arrhythmias has been suggested for some time (Zhang et al. 2002; Zima & Blatter, 2004; Kockskämper et al. 2008; Harzheim et al. 2009; Nakayama et al. 2010). Our observations in wild‐type cells (WT) are in agreement with this concept. Under control conditions we found an increase in SR‐Ca2+ release activity triggered by stimulation of the IP3 pathway in the shape of Ca2+ waves or Ca2+ sparks.
The use of G‐protein coupled receptor (GPCR) agonists (e.g. ET‐1) leads to the intracellular synthesis of IP3 together with activation of protein kinase C (PKC). Although the positive inotropic action of ET‐1 in ventricular myocytes is generally accepted, the underlying mechanisms are controversial, while numerous cellular targets are involved (Braz et al. 2004; Li et al. 2005; Domeier et al. 2012; Smyrnias et al. 2018). In our hands, the inotropic responses triggered by ET‐1 in WT cells (Figs 4 and 7) might be partially mediated by PKC during the ET‐1/GPCR pathway stimulation. In the presence of ET‐1, the SR‐Ca2+ leak increases together with the SR‐Ca2+ content; this discrepancy could be explained by the positive inotropic PKC‐related effects on the LTCCs. As a consequence, increased Ca2+ influx and SERCA activity during the pacing protocol may lead to the observed increased SR‐Ca2+ content (Fig. 2). This may also have some relevance for the leak protocol (Fig. 7). Interestingly, application of 2‐APB in the presence of ET‐1 restores the SR‐Ca2+ leak to basal levels, while the SR‐Ca2+ content remains elevated (Fig. 8). In other words, the leak effect trigged by ET‐1 is IP3R dependent. In addition, the ET‐1‐triggered arrhythmogenic responses on a cellular level can be reversed by IP3R2 block excluding significant PKC‐dependent side effects (e.g. LTCCs, NCX) and indicating that IP3ICR seems to be the main mechanism responsible for the induced arrhythmic responses.
Analogously, the increase in Ca2+ spark and Ca2+ wave occurrence upon IP3 stimulation may be partly explained by a functional bi‐directional interaction between IP3R2 and RyR2 (Wullschleger et al. 2017). Therefore, IP3ICR may directly trigger and/or amplify CICR resulting in the initiation and/or propagation of Ca2+ waves.
Under conditions of pathophysiological stress and the associated increased IP3R2 expression, the scenario given above may be fundamentally altered. Neurohumoral GPCR stimulation (e.g. via ET‐1) serves as a positive inotropic reserve that partially functions via IP3ICR activation. This may include sensitization of RyR2 to activating (“triggering”) cytosolic Ca2+ together with increases in SR‐Ca2+ load, events that cooperatively increase single RyR2 channel open probability (P o) and hence the tendency to develop arrhythmogenic Ca2+ waves (Domeier et al. 2008). Hence activation of GPCR via ET‐1 unfolds a positive inotropic reserve at the expense of increased spontaneous SR‐Ca2+ release events. In line with this scenario, under increased IP3R2 expression, an even higher probability of Ca2+ wave occurrence compared to the wild‐type situation would be expected. Surprisingly, the opposite response was evident: activation of IP3ICR in conditions of IP3R2 overexpression (TG) resulted in a decrease rather than an increase in Ca2+ wave occurrence. Furthermore, block of IP3R2 increased the SR‐Ca2+ content, suggesting an inherent regulatory role of IP3ICR in the control of the SR‐Ca2+ content. Thus, under conditions of enhanced IP3ICR, we hypothesized a secondary regulatory mechanism operating through IP3ICR activation and resulting in the reduction of Ca2+ wave occurrence.
Global cytosolic Ca2+ waves are composed of the temporal and spatial summation of individual CICR events (Ca2+ sparks) that occur by spontaneous but concerted openings of RyR2 clusters (Cheng et al. 1993). Analogously, IP3R2s are organized in clusters prerequisite to the generation of local Ca2+ events known as Ca2+ puffs (Yao et al. 1995). Analysis of individual Ca2+ events may reveal the contribution of IP3ICR (e.g. Ca2+ puffs) to the Ca2+ wave occurrence during IP3R stimulation. Under equal cytosolic Ca2+ levels, TG cells displayed a higher Ca2+ spark frequency (Fig. 5 C) than cells from WT littermates, reflecting a higher basal RyR2 sensitization. In agreement with others (Domeier et al. 2008), IP3 stimulation in WT cells induced an expected increase in RyR activity. Surprisingly, the effect was reversed in TG myocytes, in which IP3R2 stimulation returned not an increase but a decrease in Ca2+ event occurrence and SR‐Ca2+ content. The results in CaSpF in permeabilized myocytes are thus in line with those obtained in Ca2+ wave occurrence within intact ventricular myocytes (Fig. 2), suggesting that (1) cell permeabilization did not disrupt the mechanism by which IP3 may be altering SR‐Ca2+ release, and (2) discarding sarcolemmal components (e.g. LTCCs, Na+–Ca2+ exchanger) as the mechanisms directly involved in the reduction of Ca2+ wave occurrence.
IP3 stimulation of TG permeabilized ventricular myocytes induced a decrease in Ca2+ event frequency together with a reduction of the SR‐Ca2+ content. To explain such discrepancy, we hypothesized that IP3 may induce a change in single Ca2+ event properties, so that larger Ca2+ events carrying more Ca2+ may keep CaSpF steady but yet reduce the SR‐Ca2+ content. In this context, multi‐parametric analysis of Ca2+ event profiles has been recently used to distinguish between Ca2+ sparks and Ca2+ puffs (Hohendanner et al. 2015; Wullschleger et al. 2017). Recently we showed that, under similar experimental conditions, IP3 application to permeabilized atrial myocytes triggered the appearance of a group of Ca2+ events that can be identified and classified – via multi‐parametric analysis of single Ca2+ event properties – as IP3R2‐mediated Ca2+ puffs. In the present study, however, multi‐parametric analysis of Ca2+ event characteristics from permeabilized ventricular myocytes (Fig. 6 A) failed to identify and distinguish between two classes of events and thus the sole group detected was classified as RyR2 Ca2+ sparks. Hence, as no differences in Ca2+ event parameters were detectable and IP3 application triggered a decrease in Ca2+ event occurrence, RyR2 Ca2+ sparks cannot account for the decrease in SR‐Ca2+ content.
SR‐Ca2+ leak and “eventless” SR‐Ca2+ release
The local SR‐Ca2+ release carried via stochastic opening of SR‐Ca2+ release channels independent from the beat‐to‐beat ECC mechanism is known as SR‐Ca2+ leak. The luminal SR‐Ca2+ has been shown to be a major regulatory factor controlling Ca2+ release channel P o, hence the SR‐Ca2+ leak (Terentyev et al. 2002; Keller et al. 2007). Although SR‐Ca2+ release channels are grouped in clusters from which detectable Ca2+ events arise, a differential channel distribution has been proposed (Sobie et al. 2006). Individual RyR2s that do not belong in a cluster or small RyR clusters can still release Ca2+ from the SR in a manner that does not generate a Ca2+ event detectable by confocal microscopy (Lipp & Niggli, 1996; Brochet et al. 2011). Single or “rogue” RyR2s may modulate SR‐Ca2+ content with no apparent changes in Ca2+ event occurrence (Song et al. 2006). Analogously, singular or “rogue” IP3Rs may modulate SR‐Ca2+ content in an “eventless” manner. Thus the SR‐Ca2+ leak mechanism includes both SR‐Ca2+ release events (Ca2+ sparks and Ca2+ puffs) and non‐detectable or “eventless” SR‐Ca2+ release. Our data in CaSpF showing a decrease in Ca2+ spark occurrence with no detectable changes in Ca2+ event characteristics suggest that the triggered IP3ICR occurs in an “eventless” manner and thus contributes to the total SR‐Ca2+ leak without generating distinct Ca2+ events. In this scenario, we hypothesized that “eventless” IP3ICR reduces SR‐Ca2+ content and ultimately modulates the cardiomyocytes’ propensity to Ca2+ waves. Indeed, by approaching an adapted SR‐Ca2+ leak/load protocol (Shannon et al. 2002) we revealed a significant IP3R2‐dependent component of the SR‐Ca2+ leak suggesting that albeit being largely outnumbered by RyR2s, IP3R2s in wild‐type ventricular myocytes can still contribute to the total SR‐Ca2+ efflux when recruited by GPCR agonists.
Regarding the leak/load protocol, interpretation of the data is less straightforward than expected. Strong evidence of pronounced SR‐Ca2+ leak mediated by IP3ICR is shown by IP3R block (2‐APB) in TG cells, where 2‐APB triggers a pronounced decay of the total SR‐Ca2+ leak, especially when compared to the antagonists’ effect in WT (Fig. 8 C). In the latter, no differences to the control are detected whereas in the former a strong decrease in the basal SR‐Ca2+ leak is seen, indicative of increased IP3ICR under IP3R2‐overexpression conditions. Now, in TG cells, an intuitively expected SR‐Ca2+ leak increase was virtually absent upon ET‐1 stimulation. When compared to WT, in TG cells, tetracaine revealed a much smaller basal RyR2‐dependent SR‐Ca2+ leak (Fig. 6). Under these specific conditions SERCA works close to the cytosolic steady‐state [Ca2+] (F 0) and thus, additional or excess IP3R‐mediated SR‐Ca2+ leak is immediately compensated by SERCA activity. Because of the closed system (NCX block), cytosolic Ca2+ levels above the steady‐state (F 0) cannot be expected, as also shown by others (Shannon et al. 2002; Curran et al. 2007; Gonzalez et al. 2007; Santiago et al. 2010; Bers, 2014; Tzimas et al. 2017). Hence the ET‐1 effect is minor or not detectable in TG myocytes as it is masked by SERCA activity. On the contrary, in WT cells, as tetracaine application sharply decreases the cytosolic Ca2+ (F tet) far from steady‐state levels (F 0), extra SR‐Ca2+ release (triggered by ET‐1) is readily detectable. In other words, a direct comparison of ΔCa2+ cyt WT versus TG is inapplicable and only feasible in combination with IP3R2 blocking conditions, as suggested.
In addition, our data in TG cells show that the RyR2 protein down‐regulation detected concomitantly diminishes the receptors’ contribution to the basal SR‐Ca2+ leak. One would expect that the RyR2 down‐regulation seen under remodelling conditions goes in parallel with an increased ECC gain in order to stabilize the CICR as long as possible. Indeed, that was the case. The “remaining” RyR2 Ca2+ release units are more sensitive to the trigger Ca2+ resulting in a higher P o. That would imply a significantly higher RyR2 Ca2+ leak and/or a significant increase in Ca2+ spark occurrence. However, that was not observed under IP3R2 overxpression conditions and can be used as additional evidence that the observed SR‐Ca2+ leak was not due to increased RyR2 P o alone. Moreover IP3R block induces a large reduction of the total SR‐Ca2+ leak suggesting that IP3R2 P o compensates at least in part the loss of RyR2‐mediated SR‐Ca2+ leak. This view is supported by increases in the SR‐Ca2+ content only seen upon IP3R inhibition in IP3R2‐overexpressing cells.
Taken together our results indicate that the contribution of IP3ICR to the total SR‐Ca2+ release is mainly driven by “eventless” SR‐Ca2+ leak rather than by localized Ca2+ events. We hypothesize that this may be because the low expression of IP3R2s, even in pathologically high expressing conditions, may not suffice to group the receptors in clusters large enough to form discernible Ca2+ events (e.g. Ca2+ puffs). By this mechanism, activation of enhanced IP3ICR reduces the SR‐Ca2+ content below the threshold for Ca2+ spark generation, which prevents aberrant SR‐Ca2+ release and subsequently stabilizes intracellular Ca2+ homeostasis. In conclusion, the data presented reveal a novel mechanism by which activation of IP3R2s in a model of cardiac pathophysiology serves as an anti‐arrhythmic mechanism that fine‐tunes the SR‐Ca2+ content via “eventless” SR‐Ca2+ release.
Limitations of the study
At present, elucidating cardiac IP3‐dependent regulatory mechanisms is mainly limited to observations at the level of global Ca2+ events (e.g. of Ca2+ waves or Ca2+ transients) or by indirect proof of interaction by counting changes in Ca2+ event frequencies (Yamasaki et al. 2013). The reasons for this are: (1) in contrast to RyR Ca2+ signals, the smaller and rare IP3ICR events are difficult to identify and to discern among the larger Ca2+ responses that originate from CICR, and (2) identifying the specific contribution of IP3R2s in local and/or global Ca2+ responses is impeded by a deficiency of highly selective pharmacological tools (Gafni et al. 1997; Bootman et al. 2002). Although studies have fostered a contribution of IP3ICR on local Ca2+ signalling in cardiomyocytes, the observation of individual functional interactions of Ca2+ sparks and Ca2+ puffs was just recently successful (Wullschleger et al. 2017). In addition, immunocytochemistry largely failed to identify the precise localization of individual IP3R2s in mouse cardiac preparations, presumably because of limited antibody specificity. This limitation thus impedes the confirmation of the “rogue” IP3R2 function hypothesis (data not shown; Mohler et al. 2005; Kockskämper et al. 2008; Nakayama et al. 2010; Wullschleger et al. 2017).
Transgenic mouse models are sufficient and appropriate tools to examine regulatory mechanisms of CICR in cardiomyocytes. However, there is no doubt that a 1:1 extrapolation of findings emerging from animal studies to the human situation has to be critically evaluated. There are only a few studies comparing cellular remodelling situations in human cardiac disease with TG animal models used for specific aspects of cardiac remodelling both on global and cellular levels. Not only that, at present ECC under physiological conditions in human cardiomyocytes is only rudimentarily characterized (Signore et al. 2013). The TG mouse model used in the present study reveals cardiac hypertrophy with several similarities to a human pathology. However, it was used as an appropriate tool to study a potential regulatory function of IP3ICR in CICR. A modulatory mechanism based on IP3‐dependent SR‐Ca2+ leak has been unmasked by overexpression of IP3R2 which would otherwise have been potentially overlooked. Although several studies focus on IP3ICR as a potential pro‐arrhythmogenic factor (Mackenzie et al. 2002; Proven et al. 2006) this model shows that higher expression of the IP3R2 might actually be beneficial for the preservation of ECC within pathology.
Additional information
Competing interests
None declared.
Author contributions
All experiments were performed at the Department of Physiology, University of Bern, Switzerland. M.E. conceived and designed the experiments. J.B. collected and analysed the experimental data. M.E. and J.B. drafted and revised the article. All authors approved the final version for publication and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the Swiss National Science Foundation (31‐149301) and Novartis Research Foundation to M.E.
Acknowledgements
The authors thank Ernst Niggli, Radoslav Janicek, Miguel Fernandez‐Tenorio, Jin Li and Ange Maguy for their helpful comments on the manuscript and Marianne Courtehoux for her technical support.
Biography
Joaquim Blanch i Salvador received his BS and MS degrees in Biomedical Sciences and Biotechnology from the University of Barcelona. After graduating, he joined Professor Dr M. Egger's group in the Department of Physiology of the University of Bern as a PhD student. Combining electrophysiology techniques with confocal Ca2+ imaging and molecular biology approaches, his research focuses on the study of intracellular Ca2+ oscillations within cardiomyocytes in a framework of cardiac pathology.

Edited by: Don Bers & Fernando Santana
Linked articles This article is highlighted in a Perspectives article by Dixon. To read this article, visit http://doi.org/10.1113/JP276793.
References
- Ascher‐Landsberg J, Saunders T, Elovitz M & Phillippe M (1999). The effects of 2‐aminoethoxydiphenyl borate, a novel inositol 1,4,5‐trisphosphate receptor modulator on myometrial contractions. Biochem Biophys Res Commun 264, 979–982. [DOI] [PubMed] [Google Scholar]
- Bare DJ, Kettlun CS, Liang M, Bers DM & Mignery GA (2005). Reversible phosphorylation as a controlling factor for sustaining calcium oscillations in HeLa cells: Involvement of calmodulin‐dependent kinase II and a calyculin A‐inhibitable phosphatase. J Biol Chem 280, 15912–15920.15710625 [Google Scholar]
- Berridge MJ (2009). Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta 1793, 933–940. [DOI] [PubMed] [Google Scholar]
- Bers DM (2002). Cardiac excitation‐contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Bers DM (2014). Cardiac sarcoplasmic reticulum calcium leak: Basis and roles in cardiac dysfunction. Annu Rev Physiol 76, 107–127. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ & Peppiatt CM (2002). 2‐Aminoethoxydiphenyl borate (2‐APB) is a reliable blocker of store‐operated Ca2+ entry but an inconsistent inhibitor of InsP3‐induced Ca2+ release. FASEB J 16, 1145–1150. [DOI] [PubMed] [Google Scholar]
- Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli‐Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG & Molkentin JD (2004). PKC‐α regulates cardiac contractility and propensity toward heart failure. Nat Med 10, 248–254. [DOI] [PubMed] [Google Scholar]
- Brochet DXP, Xie W, Yang D, Cheng H & Lederer WJ (2011). Quarky calcium release in the heart. Circ Res 108, 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y, Zhou Q, Xie C, Zhang J, Guo A, Tian X, Jones PP, O'Mara ML, Liu Y, Mi T, Zhang L, Bolstad J, Semeniuk L, Cheng H, Zhang J, Chen J, Tieleman DP, Gillis AM, Duff HJ, Fill M, Song LS & Chen W (2014). The ryanodine receptor store‐sensing gate controls Ca2+ waves and Ca2+‐triggered arrhythmias. Nat Med 20, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ & Cannell MB (1993). Calcium sparks: elementary events underlying excitation‐contraction coupling in heart muscle. Science 262, 740–744. [DOI] [PubMed] [Google Scholar]
- Cooley N, Ouyang K, McMullen JR, Kiriazis H, Sheikh F, Wu W, Mu Y, Du X‐J, Chen J & Woodcock EA (2013). No contribution of IP3‐R(2) to disease phenotype in models of dilated cardiomyopathy or pressure overload hypertrophy. Circ Heart Fail 6, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran J, Hinton MJ, Rios E, Bers DM & Shannon TR (2007). β‐Adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium calmodulin‐dependent protein kinase. Circ Res 100, 391–398. [DOI] [PubMed] [Google Scholar]
- Domeier TL, Blatter LA & Zima AV (2010). Changes in intra‐luminal calcium during spontaneous calcium waves following sensitization of ryanodine receptor channels. Channels 4, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA & Blatter LA (2008). IP3 receptor‐dependent Ca2+ release modulates excitation‐contraction coupling in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol 294, H596–H604. [DOI] [PubMed] [Google Scholar]
- Drawnel FM, Wachten D, Molkentin JD, Maillet M, Aronsen JM, Swift F, Sjaastad I, Liu N, Catalucci D, Mikoshiba K, Hisatsune C, Okkenhaug H, Andrews SR, Bootman MD & Roderick HL (2012). Mutual antagonism between IP3RII and miRNA‐133a regulates calcium signals and cardiac hypertrophy. J Cell Biol 199, 783–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF & Pessah IN (1997). Xestospongins: potent membrane permeable blockers of the inositol 1,4,5‐trisphosphate receptor. Neuron 19, 723–733. [DOI] [PubMed] [Google Scholar]
- Go L, Moschella MC, Watras J, Handa KK, Fyfe BS & Marks AR (1995). Differential regulation of two types of intracellular calcium release channels during end‐stage heart failure. J Clin Invest 95, 888–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez DR, Beigi F, Treuer AV & Hare JM (2007). Deficient ryanodine receptor S‐nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A 104, 20612–20617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev K, Domenighetti AA, Delbridge LMD, Pedrazzini T, Niggli E & Egger M (2009). Angiotensin II‐mediated adaptive and maladaptive remodeling of cardiomyocyte excitation‐contraction coupling. Circ Res 105, 42–50. [DOI] [PubMed] [Google Scholar]
- Györke I & Györke S (1998). Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J 75, 2801–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harzheim D, Foo RS‐Y, Ritterc O, Tashfeen A, Conway SJ, Bootman MD & Roderick HL (2009). Increased InsP3Rs in the junctional sarcoplasmic reticulum augment Ca2+ transients and arrhythmias associated with cardiac hypertrophy. Proc Natl Acad Sci U S A 106, 11406–11411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harzheim D, Talasila A, Movassagh M, Foo RS‐Y, Figg N, Bootman MD & Roderick HL (2010). Elevated InsP3R expression underlies enhanced calcium fluxes and spontaneous extra‐systolic calcium release events in hypertrophic cardiac myocytes. Channels 4, 67–71. [DOI] [PubMed] [Google Scholar]
- Higazi DR, Fearnley CJ, Drawnel FM, Talasila A, Corps EM, Ritter O, McDonald F, Mikoshiba K, Bootman MD & Roderick HL (2009). Endothelin‐1‐stimulated InsP3‐induced Ca2+ release is a nexus for hypertrophic signaling in cardiac myocytes. Mol Cell 33, 472–482. [DOI] [PubMed] [Google Scholar]
- Hohendanner F, McCulloch AD, Blatter LA & Michailova AP (2014). Calcium and IP3 dynamics in cardiac myocytes: experimental and computational perspectives and approaches. Front Pharmacol 5, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohendanner F, Walther S, Maxwell JT, Kettlewell S, Awad S, Smith GL, Lonchyna VA & Blatter LA (2015). Inositol‐1,4,5‐trisphosphate induced Ca2+ release and excitation‐contraction coupling in atrial myocytes from normal and failing hearts. J Physiol 593, 1459–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth S, Peet J, Chandler WK & Baylor SM (2001). Calcium sparks in intact skeletal muscle fibers of the frog. J Gen Physiol 118, 653–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keizer J, Smith GD, Ponce‐Dawson S & Pearson JE (1998). Saltatory propagation of Ca2+ waves by Ca2+ sparks. Biophys J 75, 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M, Kao J, Egger M & Niggli E (2007). Calcium waves driven by “sensitization” wave‐fronts. Cardiovasc Res 74, 39–45. [DOI] [PubMed] [Google Scholar]
- Kimura J, Miyamae S & Noma A (1987). Identification of sodium‐calcium exchange current in single ventricular cells of guinea‐pig. J Physiol 384, 199–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockskämper J, Zima AV, Roderick HL, Pieske B, Blatter LA & Bootman MD (2008). Emerging roles of inositol 1,4,5‐trisphosphate signaling in cardiac myocytes. J Mol Cell Cardiol 45, 128–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zima AV, Sheikh F, Blatter LA & Chen J (2005). Endothelin‐1‐induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol‐1,4,5‐trisphosphate(IP3)‐receptor type 2‐deficient mice. Circ Res 96, 1274–1281. [DOI] [PubMed] [Google Scholar]
- Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W & Bootman MD (2000). Functional InsP3 receptors that may modulate excitation‐contraction coupling in the heart. Curr Biol 10, 939–942. [DOI] [PubMed] [Google Scholar]
- Lipp P & Niggli E (1993). Microscopic spiral waves reveal positive feedback in subcellular calcium signaling. Biophys J 65, 2272–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipp P & Niggli E (1996). Submicroscopic calcium signals as fundamental events of excitation‐contraction coupling in guinea‐pig cardiac myocytes. J Physiol 492, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y & Kimlicka L, Hiess F, Tian X, Wang R, Zhang L, Jones PP, Van Petegem F & Chen W (2013). The CPVT‐associated RyR2 mutation G230C enhances store overload induced Ca2+ release and destabilizes the N‐terminal domains. Biochem J 454, 123–131. [DOI] [PubMed] [Google Scholar]
- Mackenzie L, Bootman MD, Laine M, Berridge MJ, Holmes A, Li W‐H & Lipp P (2002). The role of inositol 1,4,5‐trisphosphate receptors in Ca2+ signalling and the generation of arrhythmias in rat atrial myocytes. J Physiol 541, 395–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T & Mikoshiba K (1997). 2APB, 2‐aminoethoxydiphenyl borate, a membrane‐penetrable modulator of Ins(1,4,5)P3‐induced Ca2+ release. J Biochem 122, 498–505. [DOI] [PubMed] [Google Scholar]
- Mohler PJ, Davis JQ & Bennett V (2005). Ankyrin‐B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T‐tubule/SR microdomain. PLoS Biol 3, 2158–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Bodi I, Maillet M, DeSantiago J, Domeier TL, Mikoshiba K, Lorenz JN, Blatter LA, Bers DM & Molkentin JD (2010). The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ Res 107, 1385–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunn DL & Taylor CW (1992). Luminal Ca2+ increases the sensitivity of Ca2+ stores to inositol 1,4,5‐trisphosphate. Mol Pharmacol 41, 115–119. [PubMed] [Google Scholar]
- Peppiatt CM, Collins TJ, Mackenzie L, Conway SJ, Holmes AB, Bootman MD, Berridge MJ, Seo JT & Roderick HL (2003). 2‐Aminoethoxydiphenyl borate (2‐APB) antagonises inositol 1,4,5‐trisphosphate‐induced calcium release, inhibits calcium pumps and has a use‐dependent and slowly reversible action on store‐operated calcium entry channels. Cell Calcium 34, 97–108. [DOI] [PubMed] [Google Scholar]
- Proven A, Roderick L, Conway SJ, Berridge MJ, Horton JK, Capper SJ & Bootman MD (2006). Inositol 1,4,5‐trisphosphate supports the arrhythmogenic action of endothelin‐1 on ventricular cardiac myocytes. J Cell Sci 119, 3363–3375. [DOI] [PubMed] [Google Scholar]
- Santiago DJ, Curran JW, Bers DM, Lederer WJ, Stern MD, Ríos E & Shannon TR (2010). Ca sparks do not explain all ryanodine receptor‐mediated SR Ca leak in mouse ventricular myocytes. Biophys J 98, 2111–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS & Bers DM (2002). Quantitative assessment of the SR Ca2+ leak‐load relationship. Circ Res 91, 594–600. [DOI] [PubMed] [Google Scholar]
- Signore S, Sorrentino MA, Ferreira‐Martins MJ, Kannappan PR, Shafaie PM, Del Ben BF, Isobe MK, Arranto PC, Wybieralska ME, Webster PA, Sanada F, Ogorek PB, Zheng PH, Liu PX, del Monte MF, DAlessandro PDA, Wunimenghe MO, Michler MRE, Hosoda MT, Goichberg PP, Leri PA, Kajstura MJ, Anversa PP & Rota MM (2013). Inositol 1,4,5‐trisphosphate receptors and human left ventricular myocytes. Circulation 128, 1286–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyrnias I, Goodwin N, Wachten D, Skogestad J, Aronsen JM, Robinson EL, Demydenko K, Segonds‐Pichon A, Oxley D, Sadayappan S, Sipido K, Bootman MD & Roderick HL (2018). Contractile responses to endothelin‐1 are regulated by PKC phosphorylation of cardiac myosin binding protein‐C in rat ventricular myocytes. J Mol Cell Cardiol 117, 1–18. [DOI] [PubMed] [Google Scholar]
- Sobie EA, Guatimosim S, Gomez‐Viquez L, Song LS, Hartmann H, Jafri MS & Lederer WJ (2006). The Ca2+ leak paradox and “rogue ryanodine receptors”: SR Ca2+ efflux theory and practice. Prog Biophys Mol Biol 90, 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L‐S, Sobie EA, McCulle S, Lederer WJ, Balke CW & Cheng H (2006). Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A 103, 4305–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Furuya A, Monkawa T, Yamamoto‐Hino M, Satoh S, Ohmori K, Miyawaki A, Hanai N, Mikoshiba K & Hasegawa M (1994). Monoclonal antibodies distinctively recognizing the subtypes of inositol 1,4,5‐trisphosphate receptor: Application to the studies on inflammatory cells. FEBS Lett 354, 149–154. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko‐Karpinski S, Valdivia HH, Escobar AL & Gyorke S (2002). Luminal Ca2+ controls termination and refractory behavior of Ca2+‐induced Ca2+ release in cardiac myocytes. Circ Res 91, 414–420. [DOI] [PubMed] [Google Scholar]
- Tzimas C, Johnson DM, Santiago DJ, Vafiadaki E, Arvanitis DA, Davos CH, Varela A, Athanasiadis NC, Dimitriou C, Katsimpoulas M, Sonntag S, Kryzhanovska M, Shmerling D, Lehnart SE, Sipido KR, Kranias EG & Sanoudou D (2017). Impaired calcium homeostasis is associated with sudden cardiac death and arrhythmias in a genetic equivalent mouse model of the human HRC‐Ser96Ala variant. Cardiovasc Res 113, 1403–1417. [DOI] [PubMed] [Google Scholar]
- Ullal AJ, Abdelfattah RS, Ashley EA & Froelicher VF (2016). Hypertrophic cardiomyopathy as a cause of sudden cardiac death in the young: a meta‐analysis. Am J Med 129, 486–496. [DOI] [PubMed] [Google Scholar]
- Wu X & Bers DM (2006). Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circ Res 99, 283–291. [DOI] [PubMed] [Google Scholar]
- Wullschleger M, Blanch J & Egger M (2017). Functional local crosstalk of inositol 1,4,5‐trisphosphate receptor‐ and ryanodine receptor‐dependent Ca2+ release in atrial cardiomyocytes. Cardiovasc Res 113, 542–552. [DOI] [PubMed] [Google Scholar]
- Yamasaki‐Mann M, Demuro A & Parker I (2013). Cytosolic [Ca2+] regulation of InsP3‐evoked puffs. Biochem J 449, 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Choi J & Parker I (1995). Quantal puffs of intracellular Ca2+ evoked by inositol trisphosphate in Xenopus oocytes. J Physiol 482, 533–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA & Olson EN (2002). Class II histone deacetylases act as signal‐responsive repressors of cardiac hypertrophy. Cell 110, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZH, Zhang HC, Xu Y, Zhang P, Li XB, Liu Y‐S & Guo JH (2007). Inositol 1,4,5‐trisphosphate and ryanodine‐dependent Ca2+ signaling in a chronic dog model of atrial fibrillation. Cardiology 107, 269–276. [DOI] [PubMed] [Google Scholar]
- Zima AV & Blatter LA (2004). Inositol‐1,4,5‐trisphosphate‐dependent Ca2+ signalling in cat atrial excitation–contraction coupling and arrhythmias. J Physiol 555, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W & Terentyev D (2014). Ca2+ handling during excitation–contraction coupling in heart failure. Eur J Physiol 466, 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]