

Abstract
Key points
With daily electrophysiological recordings and neurochemical analysis, we uncovered a transient period of synaptic imbalance between enhanced inhibition and suppressed excitation in rat visual cortical neurons from the end of the fourth toward the end of the fifth postnatal weeks.
The expression of brain‐derived neurotrophic factor (BDNF), which normally enhances excitation and suppresses inhibition, was down‐regulated during that time, suggesting that this may contribute to the inhibition/excitation imbalance.
An agonist of the BDNF receptor tropomyosin‐related kinase B (TrkB) partially reversed the imbalance, whereas a TrkB antagonist accentuated the imbalance during the transient period.
Monocular lid suture during the transient period is more detrimental to the function and neurochemical properties of visual cortical neurons than before or after this period.
We regard the period of synaptic imbalance as the peak critical period of vulnerability, and its existence is necessary for neurons to transition from immaturity to a more mature state of functioning.
Abstract
The mammalian visual cortex is immature at birth and undergoes postnatal structural and functional adjustments. The exact timing of the vulnerable period in rodents remains unclear. The critical period is characterized by inhibitory GABAergic maturation reportedly dependent on brain‐derived neurotrophic factor (BDNF). However, most of the studies were performed on experimental/transgenic animals, questioning the relationship in normal animals. The present study aimed to conduct in‐depth analyses of the synaptic and neurochemical development of visual cortical neurons in normal and monocularly‐deprived rats and to determine specific changes, if any, during the critical period. We found that (i) against a gradual increase in excitation and inhibition with age, a transient period of synaptic and neurochemical imbalance existed with suppressed excitation and enhanced inhibition at postnatal days 28 to 33/34; (ii) during this window, the expression of BDNF and tropomyosin‐related kinase B (TrkB) receptors decreased, along with glutamatergic GluN1 and GluA1 receptors and the metabolic marker cytochrome oxidase, whereas that of GABAARα1 receptors continued to rise; (iii) monocular deprivation reduced both excitatory and inhibitory synaptic activity and neurochemicals mainly during this period; and (iv) in vivo TrkB agonist partially reversed the synaptic imbalance in normal and monocularly‐deprived neurons during this time, whereas a TrkB antagonist accentuated the imbalance. Thus, our findings highlight a transitory period of synaptic imbalance with a negative relationship between BDNF and inhibitory GABA. This brief critical period may be necessary in transitioning from an immature to a more mature state of visual cortical functioning.
Keywords: BDNF, critical period, immunohistochemistry, monocular deprivation, patch‐clamp recording, visual cortex
Key points
With daily electrophysiological recordings and neurochemical analysis, we uncovered a transient period of synaptic imbalance between enhanced inhibition and suppressed excitation in rat visual cortical neurons from the end of the fourth toward the end of the fifth postnatal weeks.
The expression of brain‐derived neurotrophic factor (BDNF), which normally enhances excitation and suppresses inhibition, was down‐regulated during that time, suggesting that this may contribute to the inhibition/excitation imbalance.
An agonist of the BDNF receptor tropomyosin‐related kinase B (TrkB) partially reversed the imbalance, whereas a TrkB antagonist accentuated the imbalance during the transient period.
Monocular lid suture during the transient period is more detrimental to the function and neurochemical properties of visual cortical neurons than before or after this period.
We regard the period of synaptic imbalance as the peak critical period of vulnerability, and its existence is necessary for neurons to transition from immaturity to a more mature state of functioning.
Introduction
The mammalian visual cortex is not mature at birth and undergoes both structural and functional adjustments postnatally (Fagiolini et al. 1994). In the rat, within a few days after eye opening, visual acuity is only half its adult value, and both direction‐ and orientation‐selectivity are not apparent (Fagiolini et al. 1994). Very little is known about the progression of synaptic and neurochemical development in the normal rat visual cortex. Because abnormal development can lead to a host of visual impairments, it is crucial to understand the normal process.
Notable during postnatal visual cortical development is a critical period discovered in studies of monocularly‐deprived kittens and monkeys (Hubel & Wiesel, 1970; Hubel et al. 1977). Cortical neurons are particularly sensitive to environmental perturbations or experimental manipulations during that time. In rodents, dark rearing prolongs the critical period of plasticity and renders many neurons non‐selective for orientation and direction of movement (Fagiolini et al. 1994; Gianfranceschi et al. 2003) and monocular deprivation (MD) reduces the excitability and binocularity of neurons (Fagiolini et al. 1994; Nataraj et al. 2010). The extent of the critical period varies among species. In the rat, the exact timing is not well‐defined because, typically, only a few time points were examined, and deprivation periods varied widely among studies (Rothblat & Schwartz, 1979; Guire et al. 1999).
Currently, two widely accepted premises exist regarding the critical period in rodents: (i) the development and maturation of GABAergic inhibition are essential for the opening and closure of this period (Hensch et al. 1998; Fagiolini & Hensch, 2000) and (ii) the increase in GABA inhibition is dependent on an increase in brain‐derived neurotrophic factor (BDNF) during this time (Hanover et al. 1999; Huang et al. 1999; Gianfranceschi et al. 2003). BDNF is known to be involved in neuronal differentiation, survival, neurite outgrowth, synapse formation, stabilization and plasticity (Thoenen, 1995; Yoshii & Constantine‐Paton, 2010). In the visual cortex of rodents, the level of BDNF mRNAs reportedly increases after eye opening, peaks during the critical period, and plateaus until adulthood (Castren et al. 1992; Bozzi et al. 1995; Patz and Wahle, 2006). Relatively few studies have been performed on the expression of BDNF protein during development. Rossi et al. (1999) reported that BDNF immunoreactivity ‘peaks during the critical period’ (‘from P15 to P30’) and is ‘adult‐like after P20’. However, only five time points were examined [postnatal days (P)5, 10, 15, 20 and adult]. Tropea et al. (2001) found that BDNF‐immunoreactive neurons increased from P13 to P23, and plateaued from P23 to P90. Again, the period between P23 and P90 was not examined. Thus, both mRNA and protein expression of BDNF from P20/P23 to adulthood was assumed to be a plateau rather than empirically determined. The increase in GABAergic inhibition during the critical period and the proposed peaking of BDNF expression at that time form the basis for the assumption that the two are positively correlated. Over‐expressing BDNF also affected the timing of the critical period (Huang et al. 1999). Because most of the studies regarding the positive relationship between GABA and BDNF incorporated experimental perturbations, including dark rearing, visual deprivation or genetic manipulations (Hanover et al. 1999; Huang et al. 1999; Levelt & Hubener, 2012; Deidda et al. 2015), the temporal relationship between the two features during normal development remains unclear.
A critical period of development exists not only in the visual system, but also in the auditory, vestibular, respiratory (Filiano and Kinney, 1994; Robson, 2002; Kral, 2013) and perhaps other systems as well. In probing the physiological and neurochemical bases for such a period in the rat respiratory system, we found a 2 day window (postnatal days P12–P13) during which a distinct synaptic imbalance exists with suppressed excitation and enhanced inhibition, when the animal's response to hypoxia is at its weakest (Liu & Wong‐Riley, 2002; Liu et al. 2006; Wong‐Riley & Liu, 2008; Gao et al. 2011). Strikingly, the levels of BDNF and its high‐affinity tropomyosin‐related kinase (Trk) B receptors are reduced rather than elevated during this time in multiple brain stem respiratory‐related nuclei (Liu & Wong‐Riley, 2013; Gao et al. 2014, 2015). This negative relationship between inhibition and BDNF during a critical period of respiratory development is at odds with the existing model for the visual system. The possibility that BDNF may have opposing effects in the respiratory and visual systems during development deserves further scrutiny.
The present study aimed (i) to monitor the synaptic and neurochemical development of the rat visual cortex daily, from the day of eye opening (P14) to P36, when cortical neurons presumably have matured and (ii) to determine the relationship between BDNF and synaptic/neurochemical development under normal and monocularly‐deprived states. The neurochemicals tested included the metabolic marker of neuronal activity, cytochrome oxidase (CO), whose level is positively correlated with the energy demand of membrane repolarization after excitatory depolarization in neurons (Wong‐Riley, 1989), glutamatergic NMDA receptor GluN1 and α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptor (AMPAR) GluA1, inhibitory GABAergic receptor GABAARα1, brain‐derived neurotrophic factor BDNF and its TrkB receptor, and Cl− importer NKCC1 and the Cl− exporter KCC2 to indicate possible switches in dominance of the two during development (Liu and Wong‐Riley, 2012).
We tested our hypotheses that (i) synaptic and neurochemical development of the rat visual cortex do not follow a straight trajectory of incremental or decremental growth but rather, it will go through an abrupt and transient period of synaptic imbalance and (ii) that a decrease (rather than an increase) in BDNF expression correlates with an increase in GABAergic inhibition during the critical period of the rat visual cortical development.
Methods
Ethical approval
All animal experiments were performed in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) of the Medical College of Wisconsin (MCW, Milwaukee, WI, USA). Our ethics approval reference number granted by MCW's IACUC is AUA834. Our experiments also conform to the principles and regulations as described in Grundy (2015).
Animals
For visual cortical studies, a total of 235 Sprague–Dawley (SD) rats (Taconic Bioscience, Hudson, NY, USA) of both sexes from 27 litters were used. The distribution between male and female animals was roughly even for each group and each experiment. They were housed under a 12:12 h light/dark ambient photocycle with food and water available ad libitum. They were subdivided into five major groups: (i) normal synaptic development monitored by patch clamp recordings; (ii) normal neurochemical development; (iii) effect of in vivo TrkB agonist and antagonist on synaptic responses at three postnatal time points; (iv) effect of MD with or without TrkB agonist or antagonist on synaptic responses; and (v) effect of MD with or without TrkB agonist or antagonist on neurochemical expression. The bulk of the primary visual cortex examined was from the more medial portion that received input primarily, if not exclusively, from the contralateral eye.
In addition, two litters of hooded Long‐Evans (LE) rats (Charles River Laboratories, Inc., Hartford, CT, USA) of both sexes were used for normal neurochemical developmental studies. They were also housed under the same conditions as the SD rats.
For optical coherence tomography (OCT) studies, two SD rats (one male and one female) were scanned at P23, P30 and P37; and two LE rats (one male and one female) were scanned at 3½ months of age (adult).
Whole‐cell patch clamp recordings
SD rats of both sexes were used. For normal developmental studies, littermates were removed for recording on either odd days or even days, and each day from P14 to P36 was covered by animals from at least two litters. Procedures for patch clamp recordings were comparable to those reported previously (Gao et al. 2011). Briefly, animals were anaesthetized with isoflurane inhalation and decapitated. The brains were removed quickly and chilled in ice‐cold sucrose‐cerebrospinal fluid (sucrose‐CSF) that contained (in mm): 220 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgSO4, 26 NaHCO3, 25 glucose, 11.6 sodium ascorbate and 3.1 sodium pyruvate, pH 7.4. Visual cortices were dissected, and horizontal slices of 300 μm thickness were cut with a vibratome (Microslicer DTK‐1000; Ted Pella, Inc., Redding, CA, USA) in ice‐cold sucrose‐CSF gassed with carbogen (95% O2/5% CO2). The slices were transferred to an incubation chamber and maintained for 1 h in artificial CSF (aCSF) that contained (in mm): 119 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3 and 10 glucose. The aCSF was saturated with carbogen (95% O2/5% CO2) at room temperature (22.5°C).
Individual slices were transferred to a recording chamber on the microscope stage equipped with infrared‐differential interference contrast microscopy (Olympus BX51W1; Olympus America Inc., Center Valley, PA, USA). Slices were submerged with a constant flow of oxygenated aCSF and stabilized with platinum wire weights. Patch pipettes were pulled from borosilicate (KG‐33) glass capillary tubings (1.5 mm outer diameter; King Precision Glass, Claremont, CA, USA) with a Narishige PC‐10 two‐stage electrode puller (Narishige International Inc., Amityville, NY, USA). All recordings were performed at 32 ± 1°C using an automatic temperature controller (Warner Instruments, Hamden, CT, USA). All drugs were bath applied in the aCSF and the recordings to be analysed did not start until the responses were stabilized. In each 5 min of recording, we selected three continuous stabilized synaptic activities of 1 min in duration in the middle of the 5 min for analysis.
When recording basic electrophysiological characteristics (membrane capacitance, input resistance, rheobase current and series resistance), the pipette solution comprised (in mm): 140 K‐gluconate, 5 KCl, 2 MgCl2.6H2O, 10 Hepes, 0.2 EGTA‐Na, 4 MgATP, 0.3 Na2GTP and 10 Na2‐phosphocreatine, pH 7.2, with KOH. When recording spontaneous ESPCs (sEPSCs), the pipette solution comprised (in mm): 125 Cs‐gluconate, 10 CsCl, 10 Hepes, 10 EGTA, 1.2 MgCl2, 2 MgATP, 0.3 Na2GTP and 10 Na2‐phosphocreatine, pH 7.25. When recording spontaneous IPSCs (sIPSCs), the pipette solution comprised (in mm): 135 CsCl, 10 Hepes, 10 EGTA, 1.2 MgCl2, 2 MgATP, 0.3 Na2GTP and 10 Na2‐phosphocreatine, pH 7.25 with CsOH. The pipette resistance was 3–5 MΩ when filled with the above solution. For sEPSCs, 1 μm strychnine and 10 μm bicuculline were added to the aCSF to block glycinergic currents and GABAergic currents, respectively. For sIPSCs, 25 μm d‐2‐amino‐5‐phosphonopentanoic acid (d‐APV; a NMDA receptor antagonist) and 20 μm 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX; an AMPA/kainate receptor antagonist) were added to the aCSF to block NMDA and AMPA currents, respectively. All sEPSCs and sIPSCs were recorded at a V H of −70 mV in layer V visual cortical neurons.
In total, 46 animals of both sexes from five litters were used at representative time points for recording miniature EPSCs (mEPSCs) and miniature IPSCs (mIPSCs). Recordings were made at a holding potential of −70 mV with 0.5 μm TTX added to the aCSF to block voltage‐dependent Na+ channels, thereby blocking the generation of action potentials. Moreover, 10 μm bicuculline and 1 μm strychnine were included for mEPSCs, whereas 20 μm CNQX and 25 μm d‐APV were added for mIPSCs. For mEPSCs, the pipette solution comprised (in mm): 130 Cs‐methylsulphate, 10 Hepes, 1.1 EGTA‐Na, 10 CsCl, 2.5 Mg‐ATP, 0.3 Na2GTP and 2 MgCl2, pH 7.2–7.4 with CsOH. For mIPSCs, the pipette solution comprised (in mm): 100 K‐gluconate, 50 KCl, 2 MgCl2·6H2O, 10 Hepes, 0.2 EGTA‐Na, 2 MgATP, 0.3 Na2GTP and 10 Na2‐phosphocreatine, pH 7.2–7.4 with KOH.
For recording evoked excitatory EPSCs and IPSCs (eEPSCs and eIPSCs) at different ages (P18, P25, P28, P36), the pipette solution comprised (in mm): 130 Cs‐methylsulphate, 10 CsCl, 10 Hepes, 1.1 EGTA‐Na, 2 MgCl2, 2.5 Mg‐ATP and 0.3 Na2GTP, pH 7.25 with CsOH. The pipette resistance was 3–5 MΩ when filled with the above solution. Electrical stimulation was delivered by a bipolar tungsten stimulation electrode that was placed at a fixed distance (∼100–150 μm) rostral to the somata of the recorded layer V neurons. Both excitatory and inhibitory currents were recorded for each neuron tested. The excitation–inhibition ratio (E/I ratio) was measured in the presence of the 25 μm NMDA receptor antagonist d‐APV. Neurons were voltage clamped at the reversal potentials for synaptic excitation (0 mV) and inhibition (–60 mV) to isolate eIPSCs and eEPSCs, respectively. The E/I ratios were calculated by dividing the amplitude of eEPSC by the amplitude of eIPSCs. For each neuron, there were six stimulations for either EPSCs or IPSCs, and the interval between each stimulation was 15 s. To determine AMPA‐ vs. NMDA‐mediated eEPSC ratio, bicuculline (10 μm) and strychnine (1 μm) were added to the aCSF to block inhibitory currents, and cells were voltage clamped at +40 mV. A stable baseline recording of total eEPSCs was achieved, followed by the application of d‐APV (25 μm) in the bath for 6–10 min to isolate the fast AMPAR‐mediated EPSCs. By subtracting the AMPAR‐EPSCs from the total EPSCs in the same neuron, the NMDAR‐EPSCs were obtained. The AMPA/NMDA ratio was calculated from the peaks of each. On average, 12–20 eEPSCs were collected for each type of EPSCs.
Whole‐cell recordings were made using a patch clamp amplifier (Multiclamp 700B; Molecular Devices, Union City, CA, USA). Data acquisition and analysis were performed using a digitizer (DigiData 1440A) and pClamp, version 10 (Molecular Devices). Signals were filtered at 2 kHz and sampled at 10 kHz. Series resistance (15–30 MΩ) was monitored throughout the recordings and data were discarded if the resistance changed by >20%. Membrane capacitance (Cm, in units of pF) was calculated from the function Cm = τ/Rs, where τ = time constant (μs) and Rs = series resistance (MΩ). Input resistance of neurons was measured by dividing the average voltage deflection (10 consecutive sweeps) of the membrane potential by a hyperpolarizing 50 pA current step (250 ms). Rheobase current was quantified as the minimal depolarizing current step (2 Hz, 25 ms in duration, intervals of 10 pA) sufficient to elicit an action potential.
All common chemicals were obtained from Sigma (St Louis, MO, USA). CNQX and all other drugs (e.g. antagonists) were from Tocris Bioscience (Ellisville, MO, USA).
Tissue preparation for histochemistry and immunohistochemistry
Pups were anaesthetized with sodium pentobarbital (60 mg kg−1) i.p. and initially perfused intracardially with warm saline (37°C) followed by cold 4% paraformaldehyde–4% sucrose (4°C) in 0.1 m sodium PBS, pH 7.4. Visual cortices were promptly removed, postfixed in the same fixative for 1 h at 4°C, cryoprotected with 30% sucrose in 0.1 m PBS at 4°C, and then frozen on dry ice and stored at −80°C until use.
Serial coronal sections (16 μm thickness) of visual cortices were cut with a Leica CM1850 cryostat (Leica Microsystems, Nussloch, Germany). They were each processed for (i) histochemical reaction for CO, as described previously (Wong‐Riley, 1979) or (ii) immunohistochemical reaction, as described previously (Liu & Wong‐Riley, 2002) for GluN1, GluA1, GABAARα1, BDNF, TrkB, NKCC1 and KCC2. Cytochrome oxidase is a metabolic marker for neuronal activity (Wong‐Riley, 1989). GluN1 (or NMDAR1) is an obligatory subunit 1 of NMDA receptor (Moriyoshi et al. 1991). GluA1 (or GluR1) is subunit 1, a major subunit of the AMPAR (Boulter et al. 1990). GABAARα1 is the α1, a major subunit of GABAA receptor (Levitan et al. 1988). BDNF is the second neurotrophic factor to be discovered (Barde et al. 1982). TrkB is tropomyosin receptor kinase or tyrosine protein kinase B, a high‐affinity receptor of BDNF (Klein et al. 1989). NKCC1 is Na+/K+/2Cl− cotransporter 1, a Cl− importer (Xu et al. 1994). KCC2 is a neuron‐specific K+/Cl− cotransporter, a Cl− exporter (Payne et al. 1996).
The primary antibodies used are listed in Table 1. The anti‐BDNF polyclonal antibody (sc‐546, N‐20) was a purified immunoglobulin raised against an internal region of human BDNF and did not cross‐react with neurotrophic factor 3 or nerve growth factor. By western blot analysis, this antibody yielded a single band at the correct molecular weight of 14 kDa (Ricci et al. 2004). It did not show any immune signals in brain tissues of BDNF knockout mice (Tongiorgi et al. 2004). This antibody has been used in many studies, including previous studies conducted by ourselves (Hwang et al. 2005; Galvão et al. 2008 ; Liu and Wong‐Riley, 2013). The anti‐TrkB rabbit polyclonal antibody (sc‐12) was a purified immunoglobulin raised against the intracellular C‐terminus (amino acids 794–808) of mouse TrkB and did not cross‐react with tropomyosin‐related kinase A or C. Its specificity was confirmed by a single band at the expected molecular size (∼145 kDa) in western blots (Ricci et al. 2004) and the manufacturer's datasheet), as well as the preadsorption test (Ricci et al. 2004). This antibody has also been used in several studies, including our own (Ricci et al. 2004 ; Hecht et al. 2005; Lee et al. 2007; Liu and Wong‐Riley, 2013). The anti‐GABAARα1 polyclonal antibody (G4416) was developed in rabbits using a highly purified peptide QPSQDELKDNTTVFTR, corresponding to amino acids 1–16 of the mouse or rat GABAA receptor α1 subunit (residues 28–43 of the precursor) with an additional C‐terminal cysteine as the immunogen. The epitope is specific for the GABAA receptor α1 subunit. It shows limited homology with α5 and α2 subunits but no homology with any other known proteins. Western blots showed a single band of the correct molecular weight (∼51 kDa) and pre‐adsorption with immunizing peptide removed the band (manufacturer's datasheet). The antibody has no known cross‐reactivity with other GABAA receptor subunits or other proteins. It has been used in a number of studies, including our own (Gao et al. 1993; Liu and Wong‐Riley, 2006; Broadbelt et al. 2010). The anti‐GluA1 antibody (AB1504) is an affinity‐purified rabbit polyclonal antibody against the C‐terminal peptide of the rat GluA1. Western blots showed a single band at 106 kDa, specific to GluA1 receptors. It has been used in numerous studies, including our own (Talos et al. 2006; Dhar et al. 2009; Adotevi and Leitch, 2017). The anti‐GluN1 monoclonal antibody (#1508‐NR1) was tested by the manufacturer (PhosphoSolutions, Aurora, CO, USA) as a single band of the correct molecular weight (120 kDa) against GluN1 from the rat hippocampus, and pre‐adsorption with the immunogen completely blocked the signal. It has been successfully used in a number of studies (Davies et al. 2008; Zhang et al. 2008; Hicklin et al. 2011). The anti‐neuron‐specific KCC2 mouse monoclonal antibody (#75‐013) was a purified immunoglobulin raised against the intracellular C‐terminus (amino acids 932–1043) of rat KCC2. Its specificity was confirmed by the manufacturer as a single band at the expected molecular size (140 kDa) in western blots. This antibody has also been used in many studies, including our own (Hedstrom et al. 2008; Horn et al. 2010 ; Liu and Wong‐Riley, 2012). The anti‐NKCC1 mouse monoclonal antibody (T4) was a purified immunoglobulin raised against a peptide at the C‐terminus (MET‐902 to SER‐1212) of human NKCC1. By western blot analysis of mouse brain tissue, this antibody recognized a band at the correct size (215 kDa) (Chen et al. 2005), and failed to show immune signals in the brain tissue of NKCC1 knockout mice (Chen et al. 2005; Liu and Wong‐Riley, 2012). This antibody has been used in a number of studies, including our own (Alvarez‐Leefmans et al. 2001; Gilbert et al. 2007; Liu and Wong‐Riley, 2012).
Table 1.
Primary antibodies used
| Antigen | Immunogen | Manufacturer, species, type Catalogue number | Dilution used | RRID |
|---|---|---|---|---|
| BDNF | Human BDNF, within an internal region of BDNF | Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA), rabbit polyclonal IgG, sc‐546, N‐20 | 1:100 | AB_630940 |
| GABAARα1 | Purified peptide containing residues 28–43 of rat GABAARα1 | Sigma (St Louis, MO, USA), rabbit polyclonal IgG, G4416 | 1: 1,000 | AB_477016 |
| GluA1 | C‐terminus peptide of rat GluR1 conjugated to BSA | Chemicon International (Temecula, CA, USA), rabbit polyclonal, AB1504 | 1:500 | AB_2113602 |
| GluN1 | Fusion protein containing amino acids 1–564 of the rat NMDA receptor NR1 | PhosphoSolutions (Aurora, CO, USA), mouse monoclonal, #1508‐NR1 | 1:1,000 | AB_2492165 |
| KCC2 | Fusion protein containing amino acids 932–1043 of rat KCC2 | UC Davis/NINDS/NIMH NeuroMab (Davis, CA, USA), mouse monoclonal, Clone N1/12, #75‐013 | 1:1,500 | AB_10672851 |
| NKCC1 | Human Na‐K‐Cl cotrans‐porter | DSHB (University of Iowa, Iowac City, IA, USA), mouse monoclonal, T4 | 1:400 | AB_528406 |
| TrkB receptor | Intracellular C‐terminus of mouse TrkB (amino acids 794–808) | Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; rabbit polyclonal IgG, sc‐12 (794) | 1:1,000 | AB_632557 |
Reaction products within individual neurons of layers II to VI visual cortex were semi‐quantified with optical densitometry by means of a Zonax MPM03 photometer (Carl Zeiss, Oberkochen, Germany), a 25× Zeiss PlanApo objective lens (numerical aperture of 0.65) and a 2 μm diameter measuring spot centred on the cytoplasm, as described previously (Liu & Wong‐Riley, 2002). In addition, an ∼1 μm diameter spot centred on the cell membrane was used with a 40× Neofluar objective lens (NA of 0.75) (Carl Zeiss) to assess the accumulation of immunoreaction product over the plasma membrane of labelled neurons (Fig. 5B), henceforth referred to as membrane labelling. In total, 50–150 neurons for each neurochemical in each cortical layer at each age from different animals were measured.
Figure 5. Neurochemical expression in the rat visual cortex.
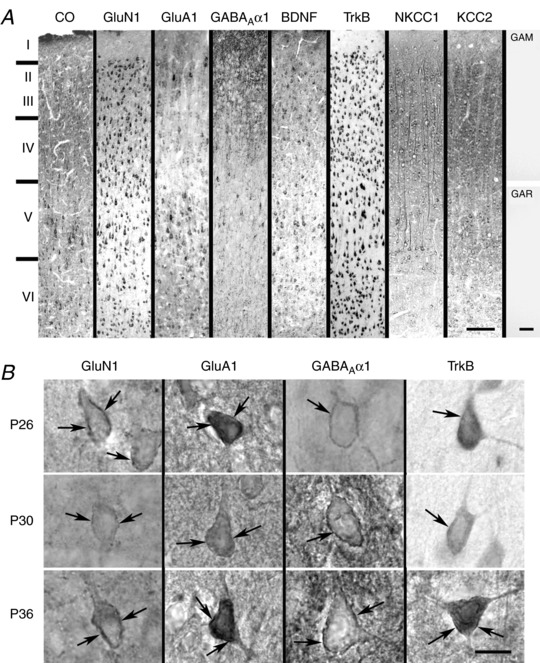
A, sample low magnification images of the primary visual cortex reacted histochemically for cytochrome oxidase (CO) and immunohistochemically for GluN1, GluA1, GABAARα1, BDNF, TrkB, NKCC1 and KCC2 at P26. Cortical layers are indicated on the left. Far right: two controls without primary antibodies but with secondary antibodies only: GAM, goat‐anti‐mouse; GAR, goat‐anti‐rabbit secondary antibodies. B, higher magnification images of membrane labelling (arrows) of GluN1, GluA1, GABAARα1 and TrkB in layer V pyramidal neurons at P26, P30 and P36. Scale bars in (A): 100 μm; in (B): 10 μm.
Optical coherence tomography
Acquisition
To assess the progressive structural integrity of the retina from the third to the fifth week after birth, two SD littermates (one male, one female, oculus dextrus (OD)) were imaged with spectral‐domain OCT at P23, P30 and P37. In addition, two adult LE rats (one male, one female, OD) were also imaged for comparison between albino and pigmented retinas. An Envisu R2200 (Bioptigen Inc., Morrisville, NC, USA), InVivoVue, version 2.4.33 (Bioptigen Inc.) control software and a rat objective lens (Bioptigen Inc.) were used in conjunction with a custom animal stage for positioning the pupil. Anaesthesia was induced with 5% isoflurane in O2 at a flow‐rate of 1.0 L min−1, then maintained at 1–4% depending on breathing rate and eye motion. Dilatation and cycloplegia were induced with 1 drop of 1% tropicamide and 2.5% phenylephrine. GenTeal lubricant eye gel and Systane Ultra lubricating eye drops (Alcon, Fort Worth, TX, USA) were used as needed to maintain corneal hydration. Dispersion, reference arm position and polarization of the sample arm were optimized iteratively for each animal at each time point. All scans were displayed and acquired in logarithmic intensity mode and 2.5 mm B‐scans (650 A‐scans/B‐scan) were acquired at a position ∼1.25 mm temporal to the centre of the optic nerve head (ONH). The animal was then steered to pan around the retina and screen for any gross abnormalities. Axial length was measured via A‐scan ultrasonography (OTI‐Scan 1000; OTI Ophthalmic Technologies, Inc., Toronto, Ontario, Canada) at all time points.
Processing and analysis
The data were exported and processed with ImageJ (NIH, Bethesda, MD, USA). The 16‐bit greyscale frames of the B‐scans were aligned using the TurboReg plugin (http://bigwww.epfl.ch/thevenaz/turboreg) (Thévenaz et al. 1998) with rigid registration after manually selecting a representative template frame. After removing poorly registered frames, the 50 consecutive frames subjectively determined to be the best aligned were averaged. The processed images were imported to Photoshop CS6 (Adobe Systems Inc., San Jose, CA, USA) and scaled and resampled laterally with ‘bicubic smoother’ interpolation to account for ocular magnification and to obtain an isotropic pixel size. Ocular magnification calculations were based on the assumption that the nominal scan width was accurate for a rat with a 6.91 mm axial length (Lozano & Twa, 2013). The axial scale was determined using a group refractive index of 1.38 (Wojtkowski et al. 2002). The images were then registered manually using translation and vertical skew and cropped to a 0.25 mm region of interest (ROI) at a position 1.10 mm temporal to the ONH. This ROI was chosen to avoid large inner retinal vessels and because retinal thickness is relatively homogeneous at this eccentricity from the ONH.
MD
Under ketamine hydrochloride (40 mg kg−1), xylazine (2.5 mg kg−1) and acepromazine (0.6 mg kg−1) i.p. anaesthesia, pups at ages P19, P28 and P35 had one of their lids sutured under sterile conditions. Postoperatively, animals were given carprofen analgesic (Rimadyl; Zoetis Services LLC Parsippany, NJ, USA) (5 mg/kg) s.c. once a day for 2 days and were monitored daily for the duration of the lid suture. Four days after surgery, animals were anaesthetized with isoflurane inhalation and decapitated. Visual cortices from deprived (contralateral to lid suture) and non‐deprived (ipsilateral to lid suture) hemispheres were removed and prepared for whole‐cell patch clamp recordings. Visual cortical tissues for histochemical and immunohistochemical analyses were fixed by intracardiac perfusion.
In vivo administration of TrkB agonist and antagonist
At specified ages, animals were injected intraperitoneally with (i) 7,8‐DHF [a TrkB agonist (Sigma) dissolved first in 100% dimethylsulphoxide (DMSO) then diluted in 10% DMSO with 0.1 m phosphate buffer; 5 mg kg−1]; (ii) ANA‐12 [a TrkB antagonist (R&D Systems, Minneapolis, MN, USA) dissolved first in 100% DMSO then diluted in 10% DMSO with 0.1 m phosphate buffer; 1 mg kg−1]; or (iii) a comparable volume of vehicle (10% DMSO) i.p. The chosen dosages were based on previous studies (7,8‐DHF: Jang et al. 2010; ANA‐12: Cazorla et al. 2011), and the efficacy of the timeline (once a day for 2 days) was based on our previous report (Gao et al. 2015). The half‐life of 7,8‐DHF in primates was 4–8 h after oral administration, with a sustained presence in the circulation for 8–24 h (He et al. 2016). The half‐life of ANA‐12 is not known. However, the in vivo effect of BDNF/TrkB is much longer lasting as a result of their downstream signal cascading event (Yoshii and Constantine‐Paton, 2010). For normal animals, the above solutions were injected once a day for 2 days (P23–P24, P29–P30, or P35–P36) and recordings were performed 1 day after the second injection (i.e. P25, P31 and P37). For monocularly‐deprived animals with 4 days of lid suture, one of the above solutions was injected once on the first day and once on the third day of lid suture within three periods (P19–P23, P28–P32 and P35–P39). On the fourth day of deprivation (i.e. P23, P32 and P39), animals were either perfused for neurochemical processing or their fresh visual cortices were prepared for whole‐cell patch clamp recordings.
Statistical analysis
Data are presented as the mean ± SEM. Statistical comparisons of synaptic development or of neurochemical development among normal age groups were conducted using one‐way ANOVA (to control for the type I comparison‐wise error rate). When significant differences were found, comparisons were made between successive age groups (e.g. P14 vs. P15, P15 vs. P16, etc.) using Tukey's Studentized range test (a post hoc multiple comparisons to control for the type I experiment‐wise error rate). Regression analysis was used to determine the developmental trends of amplitudes and frequencies of sEPSCs and sIPSCs from P14 to P27. For comparing monocularly‐deprived and non‐deprived samples against those treated with or without TrkB agonist (7,8‐DHF) or antagonist (ANA‐12), two‐way ANOVA followed by Tukey's comparisons was used. Bonferroni corrections were performed for significance among groups. P < 0.01 was considered statistically significant for one way or two‐way ANOVA. P < 0.05 was considered statistically significant for Tukey's test.
Results
All of the results were obtained from SD rats, with the exception of those shown in Fig. 8 and part of Fig. 7, for which the neurochemical data were from LE rats and the retinal data were from both SD and LE rats.
Figure 8. Developmental comparisons between Long Evans and Sprague‐Dawley rats at the cortical and retinal levels.
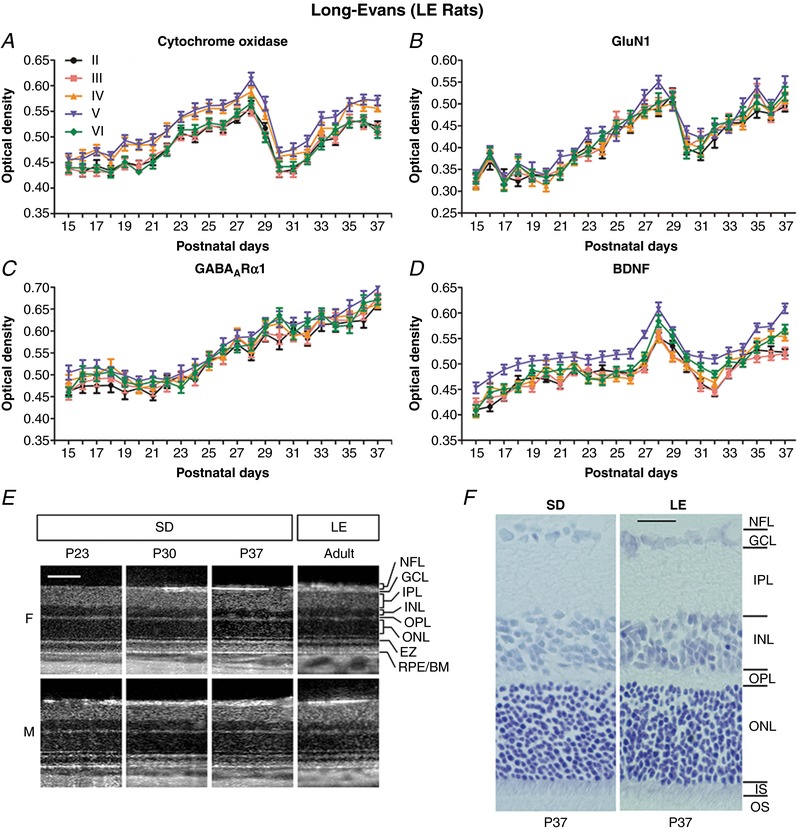
A–D, neurochemical development of layers II to VI visual cortical neurons in LE rats. A 1‐day delay in eye opening in these rats led to analysis from P15 to P37. The developmental trends of CO (A), GluN1 (B), GABAARα1 (C) and BDNF (D) in LE rats were virtually identical to those of SD rats (Fig. 6). N = 50–100 neurons from two litters for each layer at each time point tested. E, OCT of SD and adult LE rats. No evidence of major retinal changes was observed in SD rats from the third to fifth postnatal weeks. Log intensity OCT images from the same female (F) and male (M) SD rats at P23, P30 and P37 are shown. For comparison, images from an adult female and an adult male LE rats are included at approximately the same retinal location. For display purposes, images have been manually contrast stretched. Scale bar: 100 μm. F, Nissl‐stained retinal sections from SD and LE rats, both at P37. All retinal layers were comparable in thickness between the two strains. NFL, nerve fiber layer; GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; EZ, ellipsoid zone of the inner segment of photoreceptor cells; IS, inner segment; OS, outer segment of photoreceptors; RPE/BM, retinal pigmented epithelium/Bruch's membrane. Scale bar: 25 μm.
Figure 7. Comparisons between plasma membrane and cytoplasmic labeling during neurochemical development.
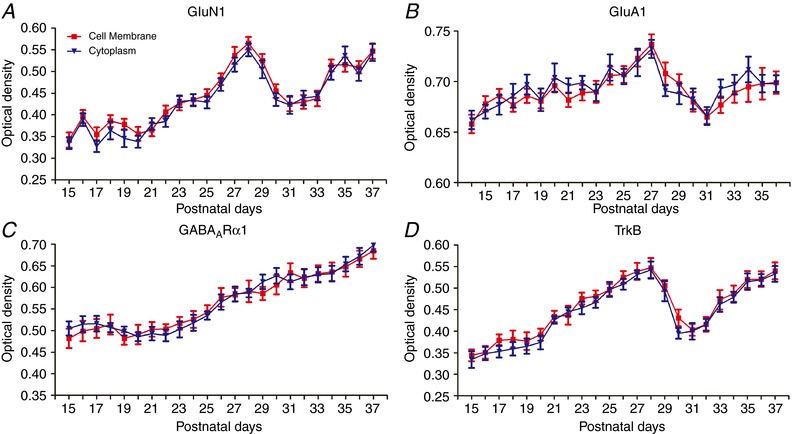
Optical densitometric analysis of the cell membrane vs. cytoplasmic labelling of GluN1 (A), GluA1 (B), GABAARα1 (C) and TrkB (D) in layer V pyramidal neurons daily, from P14/P15 to P36/P37. (A), (C) and (D) are from LE rats and (B) is from SD rats. The developmental trends are comparable, except that LE rats opened their eyes 1 day later (P15) than the SD rats. The developmental trends of cell membrane and cytoplasmic labelling are almost identical for each neurochemical tested. N = 100 neurons for neurochemical at each time point. The same neurons were analysed for both membrane and cytoplasmic labelling.
Electrophysiological analysis of normal visual cortical development
The basic electrophysiological characteristics of layer V pyramidal neurons (resting membrane potential, membrane capacitance, input resistance, rheobase current and series resistance) at representative time points are shown in Table 2. Tukey's tests, comparing the values of one age group with its adjacent younger age group in each category, revealed a significant increase in the rheobase current at P28–P30 compared to P23–P25. This indicates that it required significantly more current to elicit action potentials at P28–P30 than at P23–P25.
Table 2.
Electrophysiological characteristics of layer V pyramidal neurons during postnatal development
| Postnatal day | Resting membrane potential (mV) | Membrane capacitance (pF) | Input resistance (MΩ) | Rheobase current (pA) | Series resistance (MΩ) |
|---|---|---|---|---|---|
| P23–P25 | −61.57 ± 1.35 | 89.02 ± 4.67 | 141.65 ± 10.47 | 127.78 ± 12.22 | 18.94 ± 1.94 |
| P28–P30 | −63.71 ± 1.23 | 96.69 ± 3.98 | 148.81 ± 9.22 | 183.33 ± 13.48* | 24.92 ± 1.56 |
| P34–P36 | −63.37 ± 1.38 | 95.84 ± 5.24 | 142.30 ± 12.45 | 162.63 ± 12.37 | 23.86 ± 1.66 |
* P < 0.05 (Tukey's test, comparing one age group with the adjacent younger age group). N = 9–30 for each age group.
Daily (from P14 to P36) whole‐cell patch clamp recordings of layer V pyramidal neurons in the primary visual cortex revealed a significant difference among the ages (P < 0.0001; one‐way ANOVA). From P14 to P27, the frequency showed a greater increase with age than the amplitude (regression coefficient of 0.1784 vs. 0.0864) (Fig. 1 A and B) (Table 3). From P28 to P33, however, both parameters fell precipitously and significantly below those of the previous period (P < 0.001 for amplitudes). At P34, both increased significantly (P < 0.001) to levels comparable to those of P27, and remained so for the following 2 days examined. Sample tracings of sEPSCs at P24, P30 and P36 are shown in Fig. 1 C. Clearly, both the amplitudes and frequencies of sEPSCs were strikingly lower at P30 than those at the other two time points tested. Quantitative analysis indicated statistical significance for all pairwise comparisons (P < 0.05 to P < 0.001) (Fig. 1 D). The 10–90% rise time of sEPSCs did not vary significantly with age (Fig. 1 E), although the decay time accelerated significantly with age (P14 vs. P36: P < 0.001; one‐way ANOVA followed by Tukey's test) (Fig. 1 F).
Figure 1. Development of sEPSCs in the rat visual cortex.
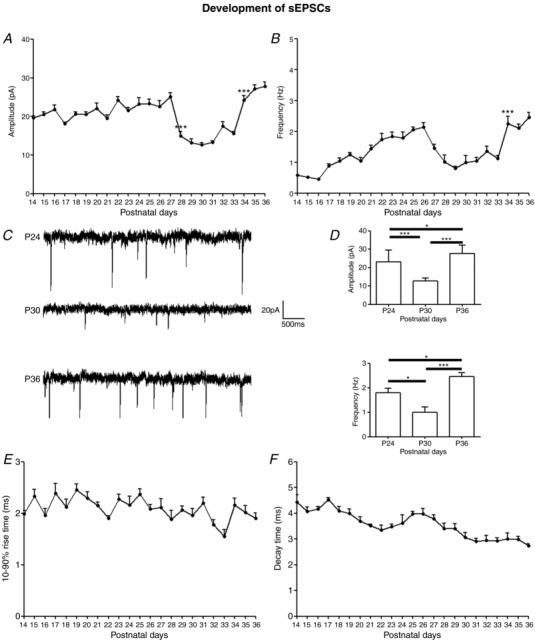
A and B, mean amplitudes and frequencies of sEPSCs in layer V pyramidal neurons recorded daily from P14 to P36. The amplitudes decreased significantly at P28 (P < 0.001) and remained low until rising at P34 (P < 0.001) to P27 levels. The frequencies decreased gradually from P27 to P29 and stayed low until increasing significantly at P34 (P < 0.001). N ≥ 15 from at least two litters for each time point tested.C, sample traces of sEPSCs at P24, P30 and P36. D, histograms and comparisons of mean ± SEM amplitudes and frequencies of sEPSC at P24, P30 and P36. All pairwise comparisons are significant. For amplitudes, P24 vs. P30: *** P < 0.001; P30 vs. P36: *** P < 0.001; P24 vs. P36: * P = 0.0312. For frequencies, P24 vs. P30: * P = 0.0112; P30 vs. P36: *** P < 0.001; P24 vs. P36: * P = 0.0111. E, 10–90% rise time of sEPSCs remained stable with age (mean ± SEM). F, decay time of sEPSCs (mean ± SEM) accelerated with age (P14 vs. P36: P < 0.001).
Table 3.
Regression coefficients and correlation coefficients of developmental trends for sEPSC and eIPSC amplitudes and frequencies from P14 to P27
| P14–P27 | Correlation coefficient | r 2 | Y‐intercept | Regression coefficienta (slope) | Significance (P value) |
|---|---|---|---|---|---|
| sEPSC amplitude | 0.3471 | 0.1205 | −1.762 | 0.0864 | 3.65 × 10−7 |
| sEPSC frequency | 0.7166 | 0.5135 | −3.6377 | 0.1784 | 1.92 × 10−33 |
| sIPSC amplitude | 0.4248 | 0.1805 | −2.1321 | 0.1043 | 5.44 × 10−10 |
| sIPSC frequency | 0.8024 | 0.6438 | −4.0181 | 0.1958 | 4.92 × 10−46 |
For regression coefficient analysis, the data for amplitudes and frequencies of sEPSCs have been normalized to the same scale to enable better comparison. The same procedure was carried out for sIPSCs.
The developmental trend of sIPSCs from P14 to P27 also showed an increase with age, with a steeper slope for the frequency than for the amplitude (regression coefficient of 0.1958 vs. 0.1043) (Fig. 2 A and B) (Table 3). At P28, however, both parameters rose suddenly and significantly (P < 0.001) and were sustained until P33, before falling significantly at P34 (P < 0.001) to levels comparable to those at P27, and then remained so for the next 2 days examined. Sample traces of sIPSCs at P24, P30 and P36 are shown in Fig. 2 C and quantitative analysis indicated significantly higher values at P30 than those at P24 or P36 (P < 0.001) (Fig. 2D). Both the 10–90% rise time and decay kinetics of sIPSCs accelerated with age (P14 vs. P36: P < 0.001 for both; one‐way ANOVA followed by Tukey's test) (Fig. 2 E and F).
Figure 2. Development of sIPSCs in the rat visual cortex.
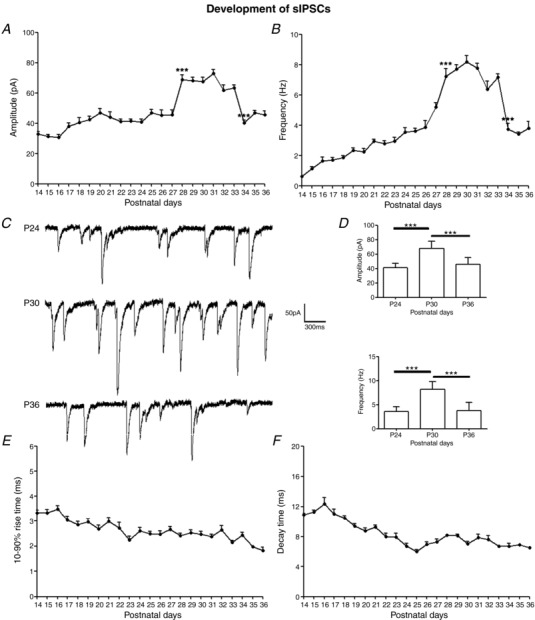
A and B, mean amplitudes and frequencies of sIPSCs in layer V pyramidal neurons daily from P14 to P36. Both parameters increased significantly at P28 (P < 0.001 for both) and stayed high until falling abruptly at P34 (P < 0.001 for both). N ≥ 15 from at least two litters for each time point tested. C, sample traces of sIPSCs recorded at P24, P30 and P36. D, histograms and comparisons of mean ± SEM amplitudes and frequencies of sIPSCs at P24, P30 and P36. Pairwise comparisons reaching significance are indicated with asterisks. For amplitudes, P24 vs. P30: *** P < 0.001; P30 vs. P36: *** P < 0.001. For frequencies, P24 vs. P30: *** P < 0.001; P30 vs. P36: *** P < 0.001. E, 10–90% rise time of sIPSCs (mean ± SEM) with age. F, decay time of sIPSCs (mean ± SEM) with age. Both rise time and decay time accelerated with age (P14 vs. P36: P < 0.001 for both).
Figure 3 illustrates the development of mEPSCs (Fig. 3 A and B) and mIPSCs (Fig. 3 C and D) in layer V pyramidal neurons at representative time points from P23 to P36. One‐way ANOVA indicated significant differences among the ages for the amplitudes of mEPSCs (Fig. 3 A) and for both the amplitudes and frequencies of mIPSCs (Fig. 3 C and D) (P < 0.01 for all). There was a significant reduction in the amplitude of mEPSCs at P28 compared to P27 (P < 0.05, Tukey's test) and it remained low at P30, P32 and P33, although it was significantly increased at P34 (P < 0.05) (Fig. 3 A). A downward trend was found for the frequency of mEPSCs at P28 and P30, although it did not reach statistical significance (Fig. 3 B). On the other hand, an upward trend in both the amplitude and frequency of mIPSCs was revealed from P23 to P32, although day‐to‐day comparisons by the Tukey's test did not yield a significant difference (Fig. 3 C and D).
Figure 3. Development of mEPSCs and mIPSCs in the rat visual cortex.
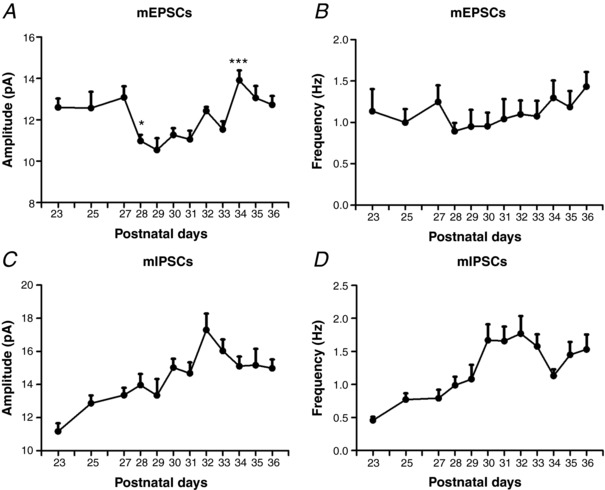
Amplitudes and frequencies of mEPSCs (A and B) and IPSCs (C and D) in layer V pyramidal neurons at representative time points from P23 to P36. One‐way ANOVA indicated significant differences among the ages for the amplitudes of mEPSCs (A) and for both the amplitudes and frequencies of mIPSCs (C and D) (P < 0.01 for all). Tukey's tests comparing one age group with the immediately adjacent younger age group revealed a significant decrease in the amplitude of mEPSCs at P28 (P < 0.05) and a significant increase in the amplitude of mEPSCs at P34 (P < 0.05). N for mEPSCs at each time point between P23 and P27 = 13–30; at P28–P33 = 26–37; and at P34–P36 = 21–37; N for mIPSCs at each time point between P23 and P27 = 19–46; at P28–P33 = 29–40; and at P34–P36 = 16–45.
E/I ratio and AMPA/NMDA receptor‐mediated eEPSC ratio
Evoked EPSCs and IPSCs were recorded from the same visual cortical neurons at each of the four ages (P18, P25, P28 and P36). Representative tracings at P18 and P28 are shown in Fig. 4 A. One‐way ANOVA and Tukey's test indicate that the E/I ratio was significantly reduced at P28, a time of synaptic imbalance, compared to the other three time points (P18: 0.6521 ± 0.0978; P25: 0.7086 ± 0.0845; P28: 0.2976 ± 0.08253; P36: 0.708 ± 0.07449; P18 vs. P28, P < 0.0001; P25 vs. P28, P < 0.0001; P28 vs. P36, P < 0.0001) (Fig. 4 B). No significance was found when the other three time points were compared with each other.
Figure 4. E/I ratio and AMPA/NMDA receptor‐mediated eEPSC ratio.

A, sample tracings of eIPSCs (voltage clamped at 0 mV) and eEPSCs (voltage clamped at ‐60 mV) recorded at P18 and P28. The amplitude of eIPSC was greater and that of eEPSC was less at P28 compared to those at P18. B, E/I ratio of P18, P25, P28 and P36, respectively. The E/I at P28 (N = 9; 6 stimulations for each cell) was significantly less than those at P18 (N = 9), P25 (N = 9) and P36 (N = 5). P < 0.0001 for all comparisons. C, representative tracings of AMPA and NMDA receptor‐mediated eEPSCs at P23–P24, P28 and P35. D, AMPA/NMDA receptor‐mediated synaptic ratio at P23–P24 (N = 8), P28 (N = 6) and P35–P36 (N = 6). No significant differences were found among the three groups.
The AMPA/NMDA receptor‐mediated eEPSC ratio was analysed before, during and after the period of synaptic imbalance. Figure 4 C shows typical tracings of AMPA and NMDA receptor‐mediated eEPSCs at P35. One‐way ANOVA did not reveal any statistically significant differences in their ratio among the three age groups tested (the mean ± SEM values for each group are: P23–P24: 1.8181 ± 0.3584; P28: 1.1565 ± 0.1251; P35–P36: 1.1682 ± 0.1204) (Fig. 4 D).
Neurochemical development
To probe for the neurochemical basis of synaptic imbalance, we examined developmental trends of CO, GluN1, GluA1, GABAARα1, BDNF, TrkB, NKCC1 and KCC2 in individual visual cortical neurons within layers II to VI daily, from P14 to P36. Representative low magnification images of labelled neurons at P26 are shown in Fig. 5 A. In agreement with our previous study (Wong‐Riley, 1988), cytochrome oxidase was rich particularly in the neuropil of lower layers III and IV, and it highlighted neuronal cell bodies of layers V and VI. GluN1 was prominent in the neuropil in the granular and supragranular layers, as well as in neuronal somata throughout the cortex, especially in pyramidal neurons of layer V. GluA1 immunoreactivity was observed in the somata and plasma membrane of many neurons, especially pyramidal neurons, but also some non‐pyramidal neurons, as well as the neuropil throughout the cortex. GABAARα1 was also more prominent in both the neuropil and neurons in the upper layers, although labelling was also found in both non‐pyramidal and pyramidal neurons throughout the cortex. BDNF was present in the neuropil, as well as in the perikarya of pyramidal and many non‐pyramidal neurons throughout the cortex. TrkB immunoreactivity was distinct in cell bodies of all neurons throughout the cortex. NKCC1 and KCC2 were clearly observed in the cell bodies and neuropil (mainly in proximal dendrites) throughout the cortex, and immunoreaction product was also clearly present in the plasma membrane of many labelled neurons. Membrane labelling of GluN1, GluA1, GABAARα1 and TrkB in layer V pyramidal neurons at P26, P30 and P36 are shown at higher magnification in Fig. 5 B. Control sections reacted without any primary antibodies but yielded no labelling with either the goat‐anti‐mouse or the goat‐anti‐rabbit secondary antibody (Fig. 5 A, right).
Optical densitometric analysis of single neurons in layers II to VI revealed that the developmental trends of CO, GluN1, GluA1, BDNF and TrkB, did not follow continuous, linear paths (Fig. 6 A, B, C, E and F). Instead, against a general increase with age, their expression fell abruptly between P28 and P33/P34, before returning to P27 levels at P34/P35. This trend was similar among neurons of layers II to VI. On the other hand, the expression of GABAARα1 exhibited a steady increase with age from P14 to P36 in all five cellular layers (Fig. 6 D). There was a sharper rise in GABAARα1 expression within layers II, III and VI neurons between P27 and P31, before this levelled off at P32. This increase from P27 to P31 was statistically significant in all five cellular layers (P < 0.001 for all). The expression of NKCC1 decreased with age, whereas that of KCC2 increased with age (Fig. 6 G and H) and the two trends intercepted at around P27 (Fig. 6 H, inset).
Figure 6. Neurochemical development of visual cortical neurons in each of the five cellular layers (II–VI) daily from P14 to P36.
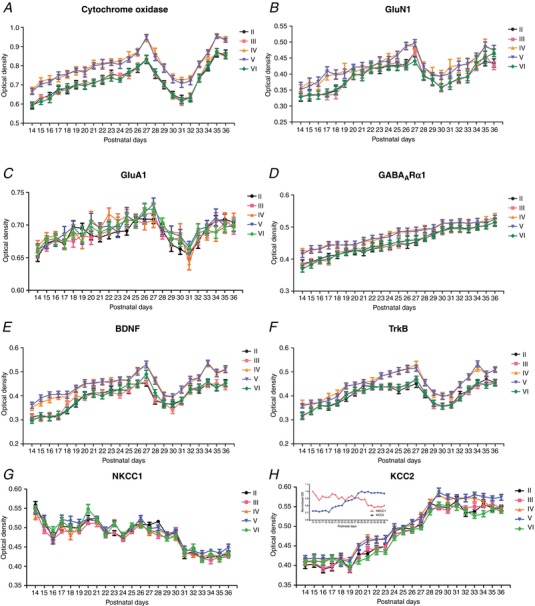
A–H, optical densitometric analysis of reaction product of CO histochemistry (A) or of immunohistochemistry against GluN1 (B), GluA1 (C), GABAARα1 (D), BDNF (E), TrkB (F), NKCC1 (G) and KCC2 (H) in single neurons of each of the five cellular layers. Note the fall in the expression of CO, GluN1, GluA1, BDNF and TrkB between P28 and P33/P34. On the other hand, the expression of GABAARα1 continued to rise with age. The expression of NKCC1 decreased with age, whereas that of KCC2 increased with age. The two trends intersected at around P27 (H, inset). N = 100–150 from at least two litters for each time point tested.
To compare the developmental trends of cytoplasmic vs. membrane labelling of neurotransmitter receptors GluN1, GluA1 and GABAARα1, as well as the BDNF receptor TrkB, we performed optical densitometric measurements of immunoreaction product localized to the cytoplasm vs. the cell membrane in the same cells (layer V pyramidal neurons) daily, from P15 to P37. Figure 7 shows that the labelling of each receptor in the two locations followed almost the same developmental trend.
Neurochemical development in LE rats
To ensure that neurochemical changes were not unique to SD rats, we analysed the developmental trends of CO, GluN1, GABAARα1 and BDNF in the hooded LE rats. As shown in Figure 8 A–D, the trends in these rats were almost identical to those of SD rats (Fig. 6 A, B, D and E), except that there was a 1 day shift because the eye opened 1 day later (P15 vs. P14). Again, a distinct dip occurred in the expression of CO, GluN1 and BDNF from the end of the fourth to the end of the fifth postnatal weeks, before returning to high levels thereafter (Fig. 8 A, B and D). As in the SD rats, the expression of GABAARα1 increased in LE rats with age (Fig. 8 C).
Retinal OCT and histology of SD and LE rats
To ensure that unexpected cortical changes did not result from any unforeseen retinal changes, OCT was performed on a male and a female SD rat before, during and after the period of synaptic imbalance (P23, P30 and P37, respectively). No remarkable difference was found in retinal layering and retinal thickness among the three time points in both animals (Fig. 8 E). For comparison, OCT was performed on a pair of adult male and female LE rats. The images were almost identical to those of the SD rats (Fig. 8 E). No signs of abnormality were observed at any time point in any animal when steered to pan around the retina (data not shown). The average rate of change in eye axial length between the two SD rats was approximately linear at 77.6 μm day−1 (female: 68.3 μm day−1, r 2 = 0.930; male: 86.8 μm day−1, r 2 = 1.00). By P37, the male SD rat reached an axial length similar to that of the adult male LE rat (6.84 mm vs. 7.0 mm). Nissl‐stained retinal sections also showed an almost identical retinal layer thickness between the SD and LE rats at P37 (Fig. 8 F).
Effect of 7,8‐DHF on synaptic activity
To determine whether exogenous BDNF would affect cortical synaptic development, and because BDNF does not readily cross the blood–brain barrier (Makar et al. 2008), we administered a TrkB agonist, 7,8‐DHF, which does cross the blood–brain barrier (Jang et al. 2010), once a day for 2 days, in vivo, at P23 and P24; P29 and P30; or P35 and P36. Responses in layer V pyramidal neurons 1 day after the second dose indicated that 7,8‐DHF significantly enhanced the amplitudes and frequencies of sEPSCs only when administered during the period of synaptic imbalance (P29 and P30) (P < 0.001) and not at the other two time points tested (Fig. 9 A and B). On the other hand, 7,8‐DHF significantly suppressed the amplitudes (P = 0.0036) and frequencies (P = 0.0297) of sIPSC also when given at P29–P30, and not at the other two time points tested (Fig. 9 C and D). No significance was found for either sEPSCs or sIPSCs when 7,8‐DHF was given at P15 and P16 and recorded at P17 (data not shown).
Figure 9. Effects of 7,8‐DHF and ANA‐12 on sEPSCs and sIPSCs.
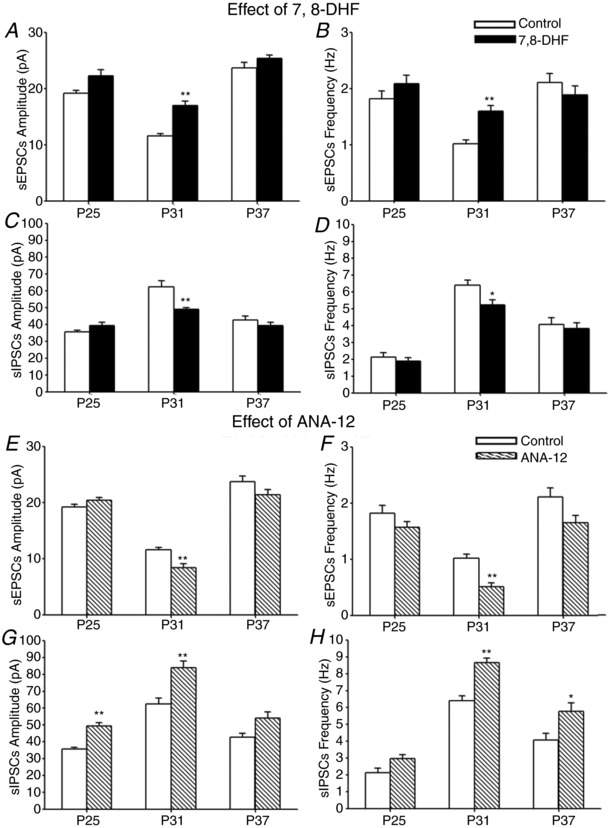
Effect of in vivo 7,8‐DHF or ANA‐12 on amplitudes and frequencies of sEPSCs (A, B, E and F) and of sIPSCs (C, D, G and H) at three time points (injections at P23–P24, P29–P30 or P35–P36, and recordings 1 day later at P25, P31 and P37, respectively). Note that 7,8‐DHF significantly increased the amplitudes and frequencies of sEPSC but decreased those of sIPSCs only at P31. N = 14 for controls at P25 and at P37; and N = 15 for controls at P31 and for 7,8‐DHF‐injected at each of the three time points tested for sEPSCs and sIPSCs, respectively. On the other hand, ANA‐12 significantly decreased the amplitudes and frequencies of sEPSC only at P31, whereas it increased those of sIPSCs mainly at P31, and also increased the amplitudes of sIPSCs at P25 and the frequencies at P37. For controls: N = 14 at P25 and at P37, and 15 at P31; for ANA‐12‐treated: N = 15 at P25, 16 at P31 and 14 at P37 for each of sEPSCs and sIPSCs. A, *** P < 0.0003. B, *** P < 0.0003. C, ** P = 0.0036. D, * P = 0.0297. E, *** P < 0.0003. F, *** P < 0.0003. G, *** P < 0.0003 for P25 and ** P = 0.0012 for P31. H, *** P < 0.0003 for P31 and * P = 0.0414 for P37.
Effect of ANA‐12 on synaptic activity
Comparable experiments were performed with a TrkB antagonist, ANA‐12, once a day for 2 days, in vivo, at the same three time points (P23–P24, P29–P30 and P35–P36) and recorded from layer V pyramidal neurons 1 day later (at P25, P31 and P37, respectively). As shown in Figure 9 E and F, ANA‐12 reduced both the amplitudes and frequencies of sEPSCs, reaching significance only during the period of synaptic imbalance (P31) (P < 0.0003) and not at the other two time points tested. On the other hand, ANA‐12 significantly enhanced the amplitudes of sIPSCs at P25 (P < 0.0003) and P31 (P = 0.0012), as well as the frequencies of sIPSCs at P31 (P < 0.0003) and P37 (P = 0.0414)) (Fig. 9 G and H). No significant difference was found for either sEPSCs or sIPSCs when ANA‐12 was given at P15 and P16 and recorded at P17 (data not shown).
Effect of MD on synaptic activity with and without in vivo 7,8‐DHF
Monocular lid suture (MD) is known to have its greatest effect during the critical period (Hubel & Wiesel, 1970). To determine whether MD led to a different synaptic response before, during and after the period of synaptic imbalance, as well as whether 7,8‐DHF in vivo would induce a different effect, we monocularly lid sutured SD rats at P19, P28 or P35 for 4 days, and 7,8‐DHF was given on the first and third days of MD. Patch clamp recordings were performed on layer V pyramidal neurons on the fourth day (i.e. at P23, P32 and P39).
As shown in Figure 10 A and B, MD reduced the amplitudes and frequencies of sEPSCs at P23 and P32 (P < 0.05 to P < 0.01), although it had relatively little effect at P39. It also decreased the amplitudes and frequencies of sIPSCs, reaching significance only at P32 (P < 0.05 to P < 0.01) (Fig. 10 C and D).
Figure 10. Effect of MD on sEPSCs and sIPSCs without or with in vivo 7,8‐DHF.
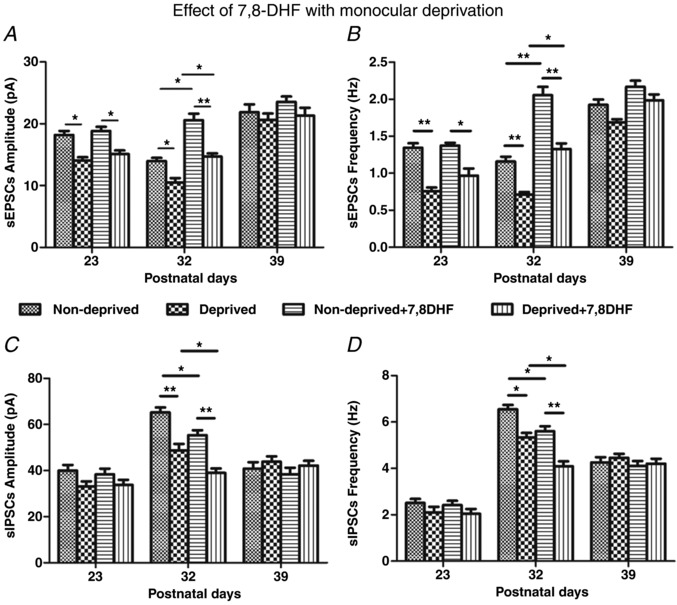
Amplitudes and frequencies of sEPSCs (A and B) and sIPSCs (C and D) in layer V pyramidal neurons of non‐deprived (ipsilateral) and deprived (contralateral) visual cortices recorded 4 days after MD at three time points (P23, P32 or P39). Note that MD reduced the amplitudes and frequencies of sEPSCs but significantly reduced the amplitudes and frequencies of sIPSCs in both non‐deprived and deprived cortical neurons only at P32. N = 8 for non‐deprived neurons at each of the three age groups for sEPSCs or sIPSCs. For deprived neurons, N = 8 at P32 and at P39, and N = 6 at P23 for sEPSCs or sIPSCs. N = 6 for non‐deprived + 7,8‐DHF neurons at each of the three age groups for sEPSCs or sIPSCs. For deprived + 7,8‐DHF neurons, N = 8 at P23 for sEPSCs or sIPSCs, and N = 6 at P32 and at P39 for sEPSCs or sIPSCs. A, at P23: * P = 0.046 for ND vs. D; * P = 0.030 for ND + DHF vs. D + DHF. At P32: * P = 0.040 for ND vs. D; ** P = 0.006 for ND + DHF vs. D + DHF; * P = 0.015 for ND vs. ND + DHF; * P = 0.045 for D vs. D + DHF. B, at P23: ** P = 0.001 for ND vs. D; * P = 0.020 for ND + DHF vs. D + DHF. At P32: ** P = 0.001 for ND vs. D; ** P = 0.002 for ND + DHF vs. D + DHF; ** P = 0.002 for ND vs. ND + DHF; * P = 0.020 for D vs. D + DHF. C, at P32: ** P = 0.002 for ND vs. D; ** P = 0.002 for ND + DHF vs. D + DHF; * P = 0.034 for ND vs. ND + DHF; * P = 0.040 for D vs. D + DHF. D, at P32: * P = 0.010 for ND vs. D; ** P = 0.001 for ND + DHF vs. D + DHF; * P = 0.030 for ND vs. ND + DHF; * P = 0.025 for D vs. D + DHF.
When 7,8‐DHF was given in conjunction with MD, even though it did not prevent the reduction of sEPSC amplitudes and frequencies by MD at P23 and P32 (P < 0.05 to P < 0.01), it did raise both values in both non‐deprived and deprived neurons above those of their respective, non‐treated counterparts only at P32 (P < 0.05 to P < 0.01) and not at the other two ages examined (Fig. 10 A and B). As for sIPSCs, 7,8‐DHF down‐regulated further the amplitudes and frequencies in both non‐deprived and deprived cortical neurons only at P32 (P < 0.05) and not at the other two time points tested (Fig. 10 C and D).
In vivo application of the vehicle (DMSO) did not result in any changes different from MD alone (data not shown).
Effect of MD on synaptic activity with and without in vivo ANA‐12
To ensure that our observations were indeed specific to BDNF/TrkB, we ran a parallel study with the TrkB antagonist, ANA‐12. The effects of MD alone were comparable to those described above (Fig. 11). ANA‐12 suppressed further the amplitudes of sEPSCs in both non‐deprived and deprived neurons at P32 (P < 0.05 to P < 0.01) and not at the other two time points tested (Fig. 11 A and B). On the other hand, ANA‐12 significantly up‐regulated further both the amplitudes and frequencies of sIPSCs in both non‐deprived and deprived neurons only at P32 (P < 0.05 to P < 0.01) and not at the other two time points tested (Fig. 11 C and D).
Figure 11. Effect of MD on sEPSCs and sIPSCs without or with in vivo ANA‐12.
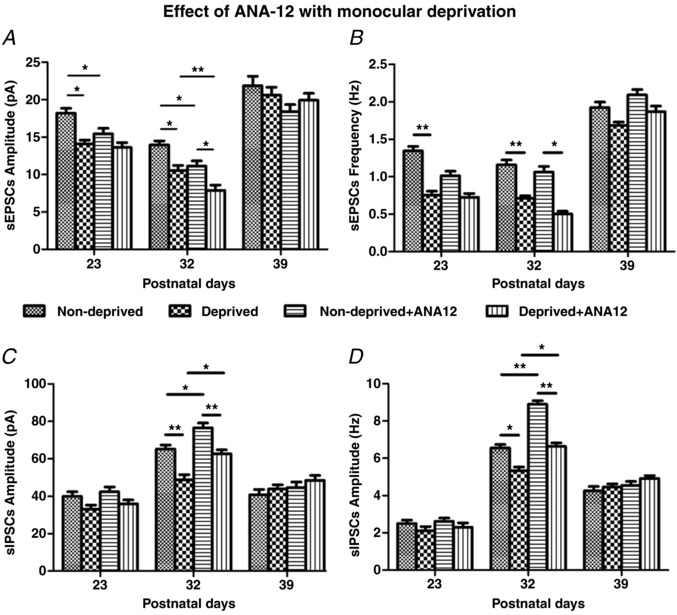
Comparable experiments as those shown in Fig. 10 were performed, except that 7,8‐DHF was replaced by ANA‐12. Note that MD reduced the amplitudes and frequencies of sEPSCs before and during, but not after the period of synaptic imbalance (A and B). However, it reduced the amplitudes and frequencies of sIPSCs only during the period of synaptic imbalance (P32) (C and D). In vivo ANA‐12 significantly decreased the amplitudes and frequencies of sEPSCs but significantly increased those of sIPSCs in both non‐deprived and deprived cortical neurons only at P32. N = 8 for non‐deprived neurons at each of the three age groups for sEPSCs or sIPSCs. For deprived neurons, N = 8 at P32 and at P39, and N = 6 at P23 for sEPSCs or sIPSCs. For non‐deprived + ANA‐12 neurons, N = 8 at P23, and N = 6 at P32 and at P39 for sEPSCs or sIPSCs. For deprived + ANA‐12 neurons, N = 8 at P23, and N = 6 at P32 and at P39 for sEPSCs or sIPSCs. A, at P23: * P = 0.041 for ND vs. D; * P = 0.046 for ND vs. ND + ANA. At P32: * P = 0.040 for ND vs. D; * P = 0.030 for ND + ANA vs. D + ANA; * P = 0.01538 for ND vs. ND + ANA; ** P = 0.009 for D vs. D + ANA. B, at P23: ** P = 0.001 for ND vs. D. At P32: ** P = 0.001 for ND vs. D; * P = 0.015 for ND + ANA vs. D + ANA. C, at P32: ** P = 0.002 for ND vs. D; ** P = 0.009 for ND + ANA vs. D + ANA; * P = 0.020 for ND vs. ND + ANA; * P = 0.015 for D vs. D + ANA. D, at P32: * P = 0.010 for ND vs. D; ** P = 0.001 for ND + ANA vs. D + ANA; ** P = 0.007 for ND vs. ND + ANA; * P = 0.032 for D vs. D + ANA.
Again, in vivo application of the vehicle (DMSO) did not result in any changes different from MD alone (data not shown).
Effect of MD on neurochemical expression with and without in vivo 7,8‐DHF or ANA‐12
To determine whether synaptic changes observed above had a neurochemical counterpart, we analysed the expression of CO, GluN1, GABAARα1, BDNF and TrkB in layer V neurons monocularly deprived for 4 days during the same three time periods with or without 7,8‐DHF or ANA‐12. Figure 12 shows that MD reduced the expression of all neurochemicals tested at P32, with or without 7,8‐DHF or ANA‐12 (P < 0.001 for all). However, 7,8‐DHF up‐regulated, whereas ANA‐12 down‐regulated, all of these neurochemicals (except for GABAARα1) in both non‐deprived and deprived cortical neurons during this period (P < 0.001 for all, except for CO, where P = 0.006 when ‘Deprived’ was compared with ‘Deprived + 7,8‐DHF’) (Fig. 12 A, B, D–G, I and J). The effect was opposite for GABAARα1, which was down‐regulated by 7,8‐DHF but up‐regulated by ANA‐12 in both non‐deprived and deprived neurons at P32 (P < 0.001 for both) (Fig. 12 C and H).
Figure 12. Effect of MD on neurochemical expression without or with in vivo 7,8‐DHF or ANA‐12.
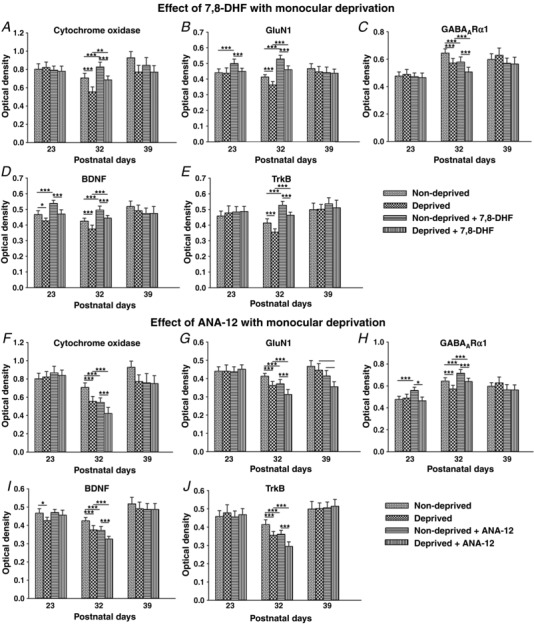
Optical densitometric measurements of reaction product of CO (A and F) and immunoreaction product of GluN1 (B and G), GABAARα1 (C and H), BDNF (D and I) and TrkB (E and J) in single, non‐deprived and deprived layer V neurons are shown for the three time points (P23, P32 and P39) after 4 days of MD without or with in vivo 7,8‐DHF or ANA‐12. Comparisons that have reached statistical significance are indicated by asterisks. Changes were most striking at P32. All *** P < 0.001. A, ** P = 0.006 for D vs. D + DHF. B, ** P = 0.005 for ND vs. ND + DHF at P23; ** P = 0.004 for ND + DHF vs. D + ANA at P23. D, * P = 0.023 for ND vs. D at P23. G, * P = 0.016 for D vs. D + ANA at P39; ** P = 0.003 for ND + ANA vs. D + ANA at P39. H, * P = 0.017 for ND + ANA vs. D + ANA. I, * P = 0.023. N = 150 neurons from at least two litters for each bar shown (i.e. for each animal group at each time point and for each neurochemical tested).
MD had relatively little effect outside of the period of synaptic imbalance with the following exceptions: (i) GluN1 was significantly up‐regulated by 7,8‐DHF in non‐deprived neurons at P23 (P < 0.001) (Fig. 12 B) but was significantly down‐regulated by ANA‐12 in deprived neurons at P39 (P < 0.01) (Fig. 12 G); (ii) BDNF was down‐regulated in deprived cortical neurons at P23 with or without 7,8‐DHF (P < 0.001 and P = 0.023, respectively) but was up‐regulated by 7,8‐DHF in non‐deprived neurons at this time (P < 0.001) (Fig. 12 D); and (iii) GABAARα1 level was raised by ANA‐12 in non‐deprived neurons at P23 (P < 0.001) (Fig 12 H) and a difference was seen between non‐deprived and deprived neurons at P23, both treated with ANA‐12 (P = 0.017) (Fig. 12 H).
In vivo DMSO yielded results not statistically different from those of MD alone (data not shown).
Discussion
We have uncovered four striking features of visual cortical development not previously recognized in normal and monocularly‐deprived rats: (i) against a gradual increase in excitatory and inhibitory development with age, an abrupt and transient period of synaptic imbalance existed within a defined time, from P28 to P33/P34; (ii) during this period, not only was inhibition markedly enhanced, but also excitation was abruptly depressed, as indicated by a significant reduction in the E/I ratio; (iii) the expression of BDNF and its TrkB receptors was decreased rather than elevated as previously assumed, and this may comprise the underlying mechanism of the transient imbalance. Exogenous TrkB agonist (7,8‐DHF) partially reversed, whereas a TrkB antagonist (ANA‐12) accentuated, the imbalance; and (iv) this transitory phase created a critical period of vulnerability, when functional perturbation such as MD is more detrimental than before or after this period. These findings challenge the prevailing view that development trends follow linear paths, and that the relationship between inhibition and BDNF is a positive one during the critical period of visual cortical development. It also revealed that MD suppressed not only excitatory, but also inhibitory synapses, especially during the critical period.
The transient synaptic imbalance in the cortex is probbaly not a result of unforeseen retinal degeneration. The retinal thickness analysed with OCT remained relatively constant from P23 to P37 and was comparable to that of the adult LE rats (Fig. 8 E), in agreement with a previous OCT study of similar thickness between adult LE and albino Wistar rats (Berger et al. 2014). Our animals were exposed only to normal room lighting, which is not known to cause any retinal pathology in SD rats (O'Steen et al. 1974), unlike the case of Royal College of Surgeons rats (Prokosch et al. 2011). The neurochemical development of the LE visual cortex was also almost identical to that of the SD rats (Fig. 8 A–D).
The existence of a critical period of visual cortical development is well‐recognized subsequent to the pioneering work of Hubel & Wiesel (1970). However, the exact timing of this period in the rat is variable among the reported studies, ranging from the time of eye opening to as late as P50 (Rothblat & Schwartz, 1979; Guire et al. 1999). Typically, only a few time points were examined, and intervening times were assumed to be a continuum. Such assumptions can be misleading. The length and timing of MD also varied among studies (Rothblat & Schwartz, 1979; Deidda et al. 2015). A ‘peak’ in the critical period has been reported for rats (Fagiolini et al. 1994) and mice (Gordon & Stryker, 1996) and falls within our period of synaptic imbalance.
The opening and closure of this period have been attributed to the maturational process of inhibitory GABAergic transmission (Hensch et al. 1998; Jiang et al. 2005; Levelt & Hübener, 2012; Li et al. 2012). In mice whose gene for GABA synthesizing enzyme (GAD65) has been mutated, the onset of the critical period is halted but can be triggered by an exogenous GABAA receptor agonist, diazepam (Fagiolini & Hensch, 2000). Our findings of a sudden rise in both the amplitudes and frequencies of sIPSCs at the start of the transient period (P28) and a sustained inhibitory enhancement with a continuous rise in GABAARα1 expression through P33/P34 are consistent with the critical period being characterized by augmented inhibition. Although the rise in GABAARα1 immunoreactivity was a steady incline, the synaptic sIPSCs showed a sharp rise at P28, which correlated with a switch in dominance from NKCC1 to KCC2, 1 day earlier (around P27) (Fig. 6 H), indicating that a decline in intracellular Cl− favoured a more inhibitory hyperpolarizing action of GABA, regardless of whether the density of receptors was greatly increased or not. Besides enhanced inhibition; however, our data also revealed a definitive suppression of excitatory synaptic transmission during that time, with a significant reduction in both the amplitudes and frequencies of sEPSCs, and a down‐regulation of excitatory neurotransmission involving both NMDA and AMPA receptors.
Changes in sEPSCs and sIPSCs were paralleled by similar developmental trends in mEPSCs and mIPSCs, albeit with some subtle differences. A significant fall at P28 and a significant rise at P34 in the amplitudes of both sEPSCs (Fig. 1 A) and mEPSCs (Fig. 3 A) indicate that such a fall during the transient period was mainly the result of a postsynaptic adjustment of excitatory receptors, in agreement with an abrupt down‐regulation of GluN1 and GluA1 at that time (Fig. 6 B and C). The decline in the frequencies of sEPSCs from P28 to P33 (Fig. 1 B) probably resulted from both a reduction in excitatory drive and an increase in inhibitory input. The removal of action potentials by the addition of TTX in mEPSCs led not only to less excitatory, but also less inhibitory input, resulting in a lower reduction in the frequencies of mEPSC than that of sEPSCs (Fig. 3 B). In the case of IPSCs, the amplitudes of sIPSCs showed a significant rise between P28 and P33 (Fig. 2 A), whereas those of the mIPSCs had a more gradual increase (Fig. 3 C). This suggests that the density of postsynaptic inhibitory (GABAergic) receptors increased steadily during that time (mIPSCs), in agreement with a gradual increase in GABAARα1 expression with age (Fig. 6 D). The development of the Cl− importer KCC2 also showed a steady increase with age (Fig. 6 H). However, strong inhibitory spiking input within that period might induce a longer and/or more frequent opening of Cl− channels, leading to a striking increase of sIPSC amplitudes. A more prominent rise in the frequencies of sIPSCs (Fig. 2 B) than that of mIPSCs (Fig. 3 D) during this period indicates that the presynaptic inhibitory spiking activity generated a strong response in sIPSCs, and its removal in mIPSCs resulted in a lesser, although still pronounced response.
Reduced excitation was also accompanied by a reduced level of cytochrome oxidase, a marker of primarily postsynaptic excitatory activity that requires energy to repolarize the membrane after depolarization (Wong‐Riley, 1989). Less excitatory activity during the transient period resulted in less energy demand in visual cortical neurons. The synaptic imbalance most probably renders this period more vulnerable to functional perturbations, such as MD. Indeed, MD consistently disrupts both synaptic and neurochemical development much more so during this transient period than before or after the period (Figs 10, 11, 12).
A major difference between past interpretations of the critical period and the findings of the present study concerns the relationship between BDNF and GABA inhibition. BDNF has been proposed and widely accepted to be necessary for the maturation of GABAergic inhibition during the critical period (Hanover et al. 1999; Huang et al. 1999). BDNF transcript levels rise after eye opening (Bozzi et al. 1995; Bracken & Turrigiano, 2009), as does the maturation of GABAergic synaptic transmission (Hensch et al. 1998; Jiang et al. 2005). Over‐expressing BDNF in mice ‘hastens the maturation of cortical inhibition’ and ‘induces an earlier closure of the critical period’ (Hanover et al. 1999; Huang et al. 1999). A reduced level of BDNF has also been proposed to contribute to ‘retarded maturation of GABAergic inhibition’ (Gianfranceschi et al. 2003). The findings of the present study, however, suggest a different scenario: a reduction rather than a rise in the expression of BDNF (and its TrkB receptors) may be necessary for the normal maturation of inhibitory synaptic transmission during the critical period. With regard to the underlying rationale, first, there is a temporal coincidence between a precipitous fall in BDNF/TrkB expression, a surge in amplitudes and frequencies of sIPSCs, and a continual rise in GABAARα1 expression during the transient period. Second, exogenous TrkB agonist (7,8‐DHF) significantly suppressed inhibitory synaptic transmission and neurochemical expressions but enhanced excitatory ones specifically during the transient period. A TrkB antagonist (ANA‐12) had the opposite effect. These findings are consistent with the known action of BDNF in enhancing excitation and attenuating inhibition (Levine et al. 1995; Tanaka et al. 1997; Poo, 2001; Martin & Finsterwald, 2011). In the hippocampus, BDNF potentiates evoked glutamate release when the postsynaptic neuron is glutamatergic but not when it is GABAergic (Schinder et al. 2000). Moreover, BDNF may reduce the efficacy of inhibitory transmission by down‐regulating Cl− transport (Wardle & Poo, 2003). Third, with MD, exogenous 7,8‐DHF significantly enhanced the amplitudes and frequencies of sEPSCs in both non‐deprived and deprived cortical neurons, but it significantly reduced those of sIPSCs primarily within the transient period, and ANA‐12 had the opposite effect (Figs 9). Thus, if the level of BDNF/TrkB is reduced during the transient period, it facilitates the growth and maturation of inhibitory synaptic transmission at the same time as suppressing the influence of excitatory ones. Exogenous TrkB agonist and antagonist have less of an effect immediately outside of the transient period, presumably because endogenous BDNF and TrkB levels are already high at those times.
How can the present results be reconciled with previous conclusions? Several possibilities exist. First, over‐expressing BDNF may indeed hasten maturation, although it probably hastens the establishment of excitatory synaptic connections first before inhibitory ones can flourish. Second, it is not known whether BDNF and TrkB undergo a transient period of down‐regulation at the same time as inhibitory GABAergic synapses mature in transgenic mice over‐expressing BDNF. This remains to be determined. Third, there may be a species difference. This necessitates closely timed comparisons between BDNF/TrkB and GABAergic expression in mice. Comparable developmental trends in neurochemical expression are found in LE rats (Fig. 8; the present study). Fourth, it is possible that BDNF facilitates early GABAergic synaptic growth, when GABA mediates excitatory depolarization as a result of the dominant action of the Cl− importer NKCC1 (Ben‐Ari, 2002). However, BDNF is known to inhibit the Cl− exporter KCC2 (Rivera et al. 2002), which aids in the conversion of GABAergic action from depolarizing to hyperpolarizing during neuronal maturation (Rivera et al. 1999). Finally, as yet unidentified factor(s) and/or neurochemical(s) may also facilitate the maturation of inhibitory GABAergic synapses in the visual cortex.
The existence of a critical period in visual cortical development is known and well accepted for decades, yet the rationale behind such a period has remained elusive. Is a transitory period of synaptic imbalance inevitable during development? It is known that excitatory synapses develop and mature earlier than inhibitory ones in the rat cortex (Sutor & Luhmann, 1995). This is essential for establishing key extrinsic and intrinsic excitatory connections within the visual cortex. BDNF most probably supports and enhances the growth and establishment of these excitatory connections. BDNF is known to affect neurite outgrowth, synapse formation and synaptic efficacy. In turn, neuronal activity augments BDNF expression (Thoenen, 1995; Yoshii & Constantine‐Paton, 2010). However, the refinement of this system requires the development and maturation of inhibitory modulation. GABA‐mediated inhibition is involved in the development of ocular dominance, orientation sensitivity, directional selectivity and receptive field substructure in visual cortical neurons (Wolf et al. 1986; Hensch & Stryker, 2004). Such enhancement of GABAergic inhibition would benefit from a transient down‐regulation of BDNF/TrkB expression. The critical period is a time of experience‐dependent synaptic refinement and, during this period, the orchestration of suppressed excitation and enhanced inhibition probably involves many players. In the respiratory system, excitatory synapses also develop earlier than inhibitory ones (Gao et al. 2011). There, we found a similar, albeit earlier and briefer critical period (P12–P13 in the rat) of synaptic imbalance, with enhanced inhibition and reduced excitation, and with suppressed expression of BDNF and TrkB, when the response of the animal to hypoxia is at its weakest (Liu & Wong‐Riley, 2002, 2013; Liu et al. 2006; Gao et al. 2011, 2014, 2015). We regard the period of synaptic imbalance as the peak critical period of vulnerability that is necessary for visual cortical (or other systems) neurons to transition from an immature to a more mature state of functioning. BDNF may be an important mechanism underlying such an imbalance during the critical period of neuronal development. Adult‐like visual cortical physiology is reportedly in place after the end of the fifth postnatal week in the rat (Fagiolini et al. 1994). At that time, a state of balanced excitation and inhibition is presumably reached for mature visual processing.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
The work was performed in laboratories of the Department of Cell Biology, Neurobiology and Anatomy at the Medical College of Wisconsin. HZ, LM, DW and DX contributed equally to the conception, acquisition, analysis and interpretation of data, as well as drafting part of the work. AS and QL contributed to acquisition of data and drafting of part of the work. MTTWR contributed to the conception and design of the work, analysis and interpretation of data, and completion of the writing of the manuscript. All authors have approved the final version of the manuscript submitted for publication and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was partially supported by NIH NEI Grants R01 EY018441, T32 EY014537 and P30 EY001931.
Acknowledgements
We thank Drs Qing‐Song Liu and Xiaojie Liu for their expert advice on the patch clamp recordings; Dr Joseph Carroll for his valuable discussion of optical coherence tomography; and Dr Xiaoqing Pan for her professional assistance in statistical analysis.
Biographies
Hanmeng Zhang received her BS degree (MD equivalent) from the Sport Science College of Beijing Sport University, Beijing, China, and is currently a graduate student at the Medical College of Wisconsin, Milwaukee, WI, USA.

Lianwei Mu received his BS degree from Weifang Medical University, Weifang, China, and is currently a PhD student at the Sport Science College of Beijing Sport University, Beijing, China.
Dandan Wang received her BS and Master of Medicine degrees from the Shanxi Medical University, Shanxi, China, and is currently enrolled as a graduate student at the Medical College of Wisconsin, Milwaukee, WI, USA.
Dongdong Xia received her BS degree from Beijing Sport University, Beijing, China, where she is pursuing a master's degree working in the Wong‐Riley laboratory.
Edited by: Ole Paulsen & Jesper Sjöström
This is an Editor's Choice article from the 15 September 2018 issue.
References
- Alvarez‐Leefmans FJ, León‐Olea M, Mendoza‐Sotelo J, Alvarez FJ, Antón B & Garduño R (2001). Immunolocalization of the Na(+)‐K(+)‐2Cl(–) cotransporter in peripheral nervous tissue of vertebrates. Neurosci 104, 569–582. [DOI] [PubMed] [Google Scholar]
- Adotevi NK & Leitch B (2017). Synaptic changes in AMPA receptor subunit expression in cortical parvalbumin interneurons in the Stargazer model of absence epilepsy. Front Mol Neurosci 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde YA, Edgar D & Thoenen H (1982). Purification of a new neurotrophic factor from mammalian brain. EMBO J 1, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Ari Y (2002). Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci 3, 728–739. [DOI] [PubMed] [Google Scholar]
- Berger A, Cavallero S, Dominguez E, Barber P, Simonutti M, Sahel JA, Sennlaub F, Raoul W, Paques M & Bemelmans AP (2014). Spectral‐domain optical coherence tomography of the rodent eye: highlighting layers of the outer retina using signal averaging and comparison with histology. PLoS ONE 9, e96494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter J, Hollmann M, O'Shea‐Greenfield A, Hartley M, Deneris E, Maron C & Heinemann S (1990). Molecular cloning and functional expression of glutamate receptor subunit genes. Science 31, 1033–1037. [DOI] [PubMed] [Google Scholar]
- Bozzi Y, Pizzorusso T, Cremisi F, Rossi FM, Barsacchi G & Maffei L (1995). Monocular deprivation decreases the expression of messenger RNA for brain‐derived neurotrophic factor in the rat visual cortex. Neurosci 69, 1133–1144. [DOI] [PubMed] [Google Scholar]
- Brachen BK & Turrigiano GG (2009). Experience‐dependent regulation of TrkB isoforms in rodent visual cortex. Dev Neurosci 69, 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbelt KG, Paterson DS, Rivera KD, Trachtenberg FL & Kinney HC (2010) Neuroanatomic relationships between the GABAergic and serotonergic systems in the developing human medulla. Auton Neurosci 154, 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castren E, Zafra F, Thoenen H & Lindholm D (1992). Light regulates expression of brain‐derived neurotrophic factor mRNA in rat visual cortex. Proc Natl Acad Sci U S A 89, 9444–9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M, Premont J, Mann A, Girard N, Kellendonk C & Rognan D (2011). Identification of a low‐molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest 121, 1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Luo J, Kintner DB, Shull GE & Sun D (2005). Na(+)‐dependent chloride transporter (NKCC1)‐null mice exhibit less gray and white matter damage after focal cerebral ischemia. J Cereb Blood Flow Metab 25, 54–66. [DOI] [PubMed] [Google Scholar]
- Davies KD, Goebel‐Goody SM, Coultrap SJ & Browning MD (2008). Long term synaptic depression that is associated with GluR1 dephosphorylation but not α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptor internalization. J Biol Chem 283, 33138–33146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deidda G, Allegra M, Cerri C, Naskar S, Bony G, Zunino G, Bozzi Y, Caleo M & Cancedda L (2015). Early depolarizing GABA controls critical‐period plasticity in the rat visual cortex. Nat Neurosci 18, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Liang HL & Wong‐Riley MTT (2009). Nuclear respiratory factor 1 co‐regulates AMPA glutamate receptor subunit 2 and cytochrome c oxidase: tight coupling of glutamatergic transmission and energy metabolism in neurons. J Neurochem 108, 1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M & Hensch TK (2000). Inhibitory threshold for critical‐period activation in primary visual cortex. Nature 404, 183–196. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Pizzorusso T, Berardi N, Domenici L & Maffei L (1994). Functional postnatal development of the rat primary visual cortex and the role of visual experience: dark rearing and monocular deprivation. Vision Res 34, 709–720. [DOI] [PubMed] [Google Scholar]
- Filiano JJ & Kinney HC (1994). A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple risk model. Biol Neonate 65, 194–197. [DOI] [PubMed] [Google Scholar]
- Galvão RP, Garcia‐Verdugo JM & Alvarez‐Buylla A (2008). Brain‐derived neurotrophic factor signaling does not stimulate subventricular zone neurogenesis in adult mice and rats. J Neurosci 28, 13368–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Fritschy JM, Benke D & Mohler H (1993). Neuron‐specific expression of GABAA‐receptor subtypes: differential association of the alpha 1‐ and alpha 3‐subunits with serotonergic and GABAergic neurons. Neurosci 54, 881–892. [DOI] [PubMed] [Google Scholar]
- Gao XP, Liu QS, Liu Q & Wong‐Riley MTT (2011). Excitatory‐inhibitory imbalance in hypoglossal neurons during the critical period of postnatal development in the rat. J Physiol 589, 1991–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XP, Liu Q, Nair B & Wong‐Riley MTT (2014). Reduced levels of brain‐derived neurotrophic factor contribute to synaptic imbalance during the critical period of respiratory development in rats. Eur J Neurosci 40, 2183–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XP, Zhang H & Wong‐Riley MTT (2015). Role of brain‐derived neurotrophic factor in the excitatory‐inhibitory imbalance during the critical period of postnatal respiratory development in the rat. Physiol Rep 3, e12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianfranceschi L, Siciliano R, Walls J, Morales B, Kirkwood A, Huang ZJ, Tonegawa S & Maffei L (2003). Visual cortex is rescued from the effects of dark rearing by overexpression of BDNF. Proc Natl Acad Sci U S A 100, 12486–12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert D, Franjic‐Würtz C, Funk K, Gensch T, Frings S & Möhrlen F (2007). Differential maturation of chloride homeostasis in primary afferent neurons of the somatosensory system. Int J Dev Neurosci 25, 479–489. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in the Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guire ES, Lickey ME & Gordon B (1999). Critical period for the monocular deprivation effects in rats: assessment with sweep visually evoked potentials. J Neurophysiol 81, 121–128. [DOI] [PubMed] [Google Scholar]
- Gordon JA & Stryker MP (1996). Experience‐dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci 16, 3274–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover JL, Huang ZJ, Tonegawa S & Stryker MP (1999). Brain‐derived neurotrophic factor overexpression induces precocious critical period in mouse visual cortex. J Neurosci 19, RC40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Xiang Z, Zhu X, Ai Z, Shen J, Huang T, Liu L, Ji W & Li T (2016). Neuroprotective effects of 7,8‐dihydroxyflavone on midbrain dopaminergic neurons in MPP+‐treated monkeys. Sci Rep 6, 34339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht M, Schulte JH, Eggert A, Wilting J & Schweigerer L (2005). The neurotrophin receptor TrkB cooperates with c‐Met in enhancing neuroblastoma invasiveness. Carcinogenesis 26, 2105–2115. [DOI] [PubMed] [Google Scholar]
- Hedstrom KL, Ogawa Y & Rasband MN (2008). AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol 183, 635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK & Stryker MP (2004). Columnar architecture sculpted by GABA circuits in developing cat visual cortex. Science 303, 1678–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S & Kash SF (1998). Local GABA circuit control of experience‐dependent plasticity in developing visual cortex. Science 282, 1504–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicklin TR, Wu PH, Radcliffe RA, Freund RK, Goebel‐Goody SM, Correa PR, Proctor WR, Lombroso PJ & Browning MD (2011). Alcohol inhibition of the NMDA receptor function, long‐term potentiation, and fear learning requires striatal‐enriched protein tyrosine phosphatase. Proc Natl Acad Sci U S A 108, 6650–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn Z, Ringstedt T, Blaesse P, Kaila K & Herlenius E (2010). Premature expression of KCC2 in embryonic mice perturbs neural development by an ion transport‐independent mechanism. Eur J Neurosci 31, 2142–2155. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L & Tonegawa S (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98, 739–755. [DOI] [PubMed] [Google Scholar]
- Hubel DH & Wiesel TN (1970). The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol 206, 419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN & LeVay S (1977). Plasticity of ocular dominance columns in monkey striate cortex. Philos Trans R Soc Lond B Biol Sci 278, 377–409. [DOI] [PubMed] [Google Scholar]
- Hwang JJ, Park MH, Choi SY & Koh JY (2005). Activation of the Trk signaling pathway by extracellular zinc. Role of metalloproteinases. J Biol Chem 280, 11995–12001. [DOI] [PubMed] [Google Scholar]
- Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sum YE & Ye K (2010). A selective TrkB agonist with potent neurotrophic activities by 7,8‐dihydroxyflavone. Proc Natl Acad Sci U S A 107, 2687–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Huang ZJ, Morales B & Kirkwood A (2005). Maturation of GABAergic transmission and the timing of plasticity in visual cortex. Brain Res Brain Res Rev 50, 126–133. [DOI] [PubMed] [Google Scholar]
- Klein R, Parada LF, Coulier F & Barbacid M (1989). TrkB, a novel tyrosine protein kinase receptor expression during mouse neural development. EMBO J 8, 3701–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kral A (2013). Auditory critical period: a review from system's perspective. Neurosci 247, 117–133. [DOI] [PubMed] [Google Scholar]
- Lee E, Jeong YI, Park SM, Lee JY, Kim JH, Park SW, Hossein MS, Jeong YW, Kim S, Hyun SH & Hwang WS (2007). Beneficial effects of brain‐derived neurotropic factor on in vitro maturation of porcine oocytes. Reproduction 134, 405–414. [DOI] [PubMed] [Google Scholar]
- Levelt CN & Hübener M (2012). Critical‐period plasticity in the visual cortex. Annu Rev Neurosci 35, 309–330. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB & Plummer MR (1995). Brain‐derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci U S A 92, 8074–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan ES, Schofield PR, Burt DR, Rhee LM, Wisden W, Köhler M, Fujita N, Rodriguez HF, Stephenson A, Darlison MG, Barnard EA & Seeburg PH (1988). Structural and functional basis for GABAA receptor heterogeneity. Nature 335, 76–79. [DOI] [PubMed] [Google Scholar]
- Li YT, Ma WP, Pan CJ, Zhang LI & Tao HW (2012). Broadening of cortical inhibition mediates developmental sharpening of orientation selectivity. J Neurosci 32, 3981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q & Wong‐Riley MTT (2002). Postnatal expression of neurotransmitter, receptors, and cytochrome oxidase in the rat pre‐Bötzinger complex. J Appl Physiol 92, 923–934. [DOI] [PubMed] [Google Scholar]
- Liu Q & Wong‐Riley MTT (2006). Developmental changes in the expression of GABAA receptor subunits α1, α2, and α3 in brain stem nuclei of rats. Brain Res 1098, 129–138. [DOI] [PubMed] [Google Scholar]
- Liu Q & Wong‐Riley MTT (2012). Postnatal development of NKCC1 and KCC2 immunoreactivity in seven brain stem respiratory nuclei of the rat. Neurosci 210, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q & Wong‐Riley MTT (2013). Postnatal development of brain‐derived neurotrophic factor (BDNF) and tyrosine protein kinase (TrkB) receptor immunoreactivity in multiple brain stem respiratory nuclei of the rat. J Comp Neurol 521, 109–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Lowry TF & Wong‐Riley MTT (2006). Postnatal changes in ventilation during normoxia and acute hypoxia in the rat: implication for a sensitive period. J Physiol 577, 957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano DC & Twa MD (2013). Development of rat schematic eye from in vivo biometry and the correction of lateral magnification in SD‐OCT imaging. Invest Ophthalmol Vis Sci 54, 6446–6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makar TK, Trisler D, Sura KT, Sultana S, Patel N & Bever CT (2008). Brain derived neurotrophic factor treatment reduces inflammation and apoptosis in experimental allergic encephalomyelitis. J Neurolog Sci 270, 70–76. [DOI] [PubMed] [Google Scholar]
- Martin JL & Finsterwald C (2011). Cooperation between BDNF and glutamate in the regulation of synaptic transmission and neuronal development. Commun Integr Biol 4, 14–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N & Nakanishi S (1991). Molecular cloning and characterization of the rat NMDA receptor. Nature 354, 31–37. [DOI] [PubMed] [Google Scholar]
- Nataraj K, LeRoux N, Nahmani M, Lefort S & Turrigiano G (2010). Visual deprivation suppresses L5 pyramidal neuron excitability by preventing the induction of intrinsic plasticity. Neuron 68, 750–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Steen WK, Anderson KV & Shear CR (1974). Photoreceptor degeneration in albino rats: dependency on age. Invest Ophthalmol 13, 334–339. [PubMed] [Google Scholar]
- Patz S & Wahle P (2006). Developmental changes of neurotrophin mRNA expression in the layers of rat visual cortex. Eur J Neurosci 24, 2453–2460. [DOI] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ & Donaldson LF (1996). Molecular characterization of a putative K‐Cl cotransporter in rat brain. J Biol Chem 271, 16245–16252. [DOI] [PubMed] [Google Scholar]
- Poo MM (2001). Neurotrophins as synaptic modulators. Nat Rev Neurosci 2, 24–32. [DOI] [PubMed] [Google Scholar]
- Prokosch V, Fink J, Heiduschka P, Melkonyan H & Thanos S (2011). VEGF, Ang‐2 and SRIF associated abnormal postnatal development of the retinal capillary network in the Royal College of Surgeons rat. Exp Eye Res 92, 128–137. [DOI] [PubMed] [Google Scholar]
- Ricci A, Felici L, Mariotta S, Mannino F, Schmid G, Terzano C, Cardillo G, Amenta F & Bronzetti E (2004). Neurotrophin and neurotrophin receptor protein expression in the human lung. Am J Respir Cell Mol Biol 30, 12–19. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M & Kaila K (1999). The K+/Cl− co‐transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas‐Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K & Saarma M (2002). BDNF‐induced TrkB activation down‐regulates the K+‐Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol 159, 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson AL (2002). Critical/sensitive periods. In Child Development, ed. Salkind NJ, pp. 101–103. Macmillan Reference, New York, NY. [Google Scholar]
- Rossi FM, Bozzi Y, Pizzorusso T & Maffei L (1999). Monocular deprivation decreases brain‐derived neurotrophic factor immunoreactivity in the rat visual cortex. Neurosci 90, 363–368. [DOI] [PubMed] [Google Scholar]
- Rothblat LA & Schwartz ML (1979). The effect of monocular deprivation on dendritic spines in visual cortex of young and adult albino rats: evidence for a sensitive period. Brain Res 161, 156–161. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Berninger B & Poo M (2000). Postsynaptic target specificity of neurotrophin‐induced presynaptic potentiation. Neuron 25, 151–163. [DOI] [PubMed] [Google Scholar]
- Sutor B & Luhmann HJ (1995). Development of excitatory and inhibitory postsynaptic potentials in the rat neocortex. Perspect Dev Neurobiol 2, 409–419. [PubMed] [Google Scholar]
- Talos DM, Fishman RE, Park H, Folkerth RD, Follett PL, Volpe JJ & Jensen FE (2006). Developmental regulation of α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole‐propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. I. Rodent cerebral white matter and cortex. J Comp Neurol 497, 42–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Saito H & Matsuki N (1997). Inhibition of GABAA synaptic responses by brain‐derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci 17, 2959–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenaz P, Ruttimann UE & Unser M (1998). A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process 7, 27–41. [DOI] [PubMed] [Google Scholar]
- Thoenen H (1995). Neurotrophins and neuronal plasticity. Science 270, 593–598. [DOI] [PubMed] [Google Scholar]
- Tongiorgi E, Armellin M, Giulianini PG, Bregola G, Zucchini S, Paradiso B, Steward O, Cattaneo A & Simonato M (2004). Brain‐derived neurotrophic factor mRNA and protein are targeted to discrete dendritic laminas by events that trigger epileptogenesis. J Neurosci 24, 6842–6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea D, Capsoni S, Tongiorgi E, Giannotta S, Cattaneo A & Domenici L (2001). Mismatch between mRNA and protein expression in the developing visual cortex: the role of visual experience. Eur J Neurosci 13, 709–721. [DOI] [PubMed] [Google Scholar]
- Wardle RA & Poo MM (2003). Brain‐derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. J Neurosci 23, 8722–8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtkowski M, Leitgeb R, Kowalczyk A, Bajraszewski T & Fercher AF (2002). In vivo human retinal imaging by Fourier domain optical coherence tomography. J. Biomed Opt 7, 457–463. [DOI] [PubMed] [Google Scholar]
- Wolf W, Hicks TP & Albus K (1986). The contribution of GABA‐mediated inhibitory mechanisms to visual response properties of neurons in the kitten's striate cortex. J Neurosci 6, 2779–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong‐Riley M (1979). Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res 171, 11–28. [DOI] [PubMed] [Google Scholar]
- Wong‐Riley MTT (1988). Comparative study of the mammalian primary visual cortex with cytochrome oxidase histochemistry In Vision: Structure and Function, ed. Yew DT, So KF, & Tsang DSC, pp. 450–486. World Scientific Press, Hoboken, NJ. [Google Scholar]
- Wong‐Riley MTT (1989). Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci 12, 94–101. [DOI] [PubMed] [Google Scholar]
- Wong‐Riley MTT & Liu Q (2008). Neurochemical and physiological correlates of a critical period of respiratory development in the rat. Invited review. In Special Issue on Neurochemistry of Respiratory Control. Ed. McCrimmon D, Mitchell G, & Alheid G, Resp Physiol Neurobiol 164, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J‐C, Lytle C, Zhu TT, Payne JA, Benz Jr E & Forbush B III (1994). Molecular cloning and functional expression of the bumetanide‐sensitive Na‐K‐Cl cotransporter. Proc Natl Acad Sci U S A 91, 2201–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii A & Constantine‐Paton M (2010). Postsynaptic BDNF‐TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol 70, 304–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Dong Y, Zhang B, Ichinose F, Wu X, Culley DJ, Crosby G, Tanzi RE & Xie Z (2008) Isoflurane‐induced caspase‐3 activation is dependent on cytosolic calcium and can be attenuated by memantine. J Neurosci 28, 4551–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]