

Abstract
Key points
Chronic pain is disabling because sufferers form negative associations between pain and activities, such as work, leading to the sufferer limiting these activities. Pain information arriving in the amygdala is responsible for forming these associations and contributes to us feeling bad when we are in pain.
Ongoing injuries enhance the delivery of pain information to the amygdala. If we want to understand why chronic pain can continue without ongoing injury, it is important to know whether this facilitation continues once the injury has healed.
In the present study, we show that a 2 min noxious heat stimulus, without ongoing injury, is able to enhance delivery of pain information to the amygdala for 3 days. If the noxious heat stimulus is repeated, this enhancement persists even longer.
These changes may prime this information pathway so that subsequent injuries may feel even worse and the associative learning that results in pain‐related avoidance may be promoted.
Abstract
Pain is an important defence against dangers in our environment; however, some clinical conditions produce pain that outlasts this useful role and persists even after the injury has healed. The experience of pain consists of somatosensory elements of intensity and location, negative emotional/aversive feelings and subsequent restrictions on lifestyle as a result of a learned association between certain activities and pain. The amygdala contributes negative emotional value to nociceptive sensory information and forms the association between an aversive response and the environment in which it occurs. It is able to form this association because it receives nociceptive information via the spino‐parabrachio‐amygdaloid pathway and polymodal sensory information via cortical and thalamic inputs. Synaptic plasticity occurs at the parabrachial‐amygdala synapse and other brain regions in chronic pain conditions with ongoing injury; however, very little is known about how plasticity occurs in conditions with no ongoing injury. Using immunohistochemistry, electrophysiology and behavioural assays, we show that a brief nociceptive stimulus with no ongoing injury is able to produce long‐lasting synaptic plasticity at the rat parabrachial‐amygdala synapse. We show that this plasticity is caused by an increase in postsynaptic AMPA receptors with a transient change in the AMPA receptor subunit, similar to long‐term potentiation. Furthermore, this synaptic potentiation primes the synapse so that a subsequent noxious stimulus causes prolonged potentiation of the nociceptive information flow into the amygdala. As a result, a second injury could have an increased negative emotional value and promote associative learning that results in pain‐related avoidance.
Keywords: amygdala, pain, Synaptic plasticity, ampa receptor, parabrachial nucleus
Key points
Chronic pain is disabling because sufferers form negative associations between pain and activities, such as work, leading to the sufferer limiting these activities. Pain information arriving in the amygdala is responsible for forming these associations and contributes to us feeling bad when we are in pain.
Ongoing injuries enhance the delivery of pain information to the amygdala. If we want to understand why chronic pain can continue without ongoing injury, it is important to know whether this facilitation continues once the injury has healed.
In the present study, we show that a 2 min noxious heat stimulus, without ongoing injury, is able to enhance delivery of pain information to the amygdala for 3 days. If the noxious heat stimulus is repeated, this enhancement persists even longer.
These changes may prime this information pathway so that subsequent injuries may feel even worse and the associative learning that results in pain‐related avoidance may be promoted.
Introduction
Acute pain provides important warnings about dangers in our environment. However, some individuals experience chronic pain, such as lower back pain, that outlasts this useful role and continues after the injury has healed. Chronic pain is debilitating to the person (Gureje et al. 1998) and costly to society (Atkinson, 2004). Our experience of chronic pain comprises somatosensory elements of location and intensity, negative emotional/aversive feelings (Bushnell et al. 2013; Elman & Borsook, 2016) and associative learning of ‘dangerous activities’ (Gureje et al. 1998; Vlaeyen, 2015).
Acute and chronic pain activates the amygdala (Bornhovd et al. 2002; Baliki et al. 2006) and this contributes to the aversive response to nociceptive information (LeDoux, 2000; Cardinal et al. 2002) and the formation of an association between the aversive response and the environment (Tanimoto et al. 2003; Gao et al. 2004; Pedersen et al. 2007; Ansah et al. 2010). These learned associations, linked to sensory inputs such as smell, result in the chronic pain sufferer limiting activities they associate with pain, such as work or sport (Vlaeyen, 2015). In animals, this association can be measured as pain‐induced conditioned place aversion (Zhang et al. 2011) and relies on the central nucleus of the amygdala (CeA), including the laterocapsular CeA, for its expression (Tanimoto et al. 2003; Gao et al. 2004; Pedersen et al. 2007; Ansah et al. 2010). This associative learning in the CeA integrates polymodal sensory information (Moga et al. 1995; Sah et al. 2003; Vertes & Hoover, 2008; Pape & Pare, 2010; Marek et al. 2013; Neugebauer, 2015) and nociceptive information. The nociceptive information comes from the spinal cord via the external lateral parabrachial nucleus (PB) to the laterocapsular region of the CeA (CeLC) (Bernard et al. 1992; Bernard et al. 1993; Bester et al. 1997), with the final synapse required for pain‐induced associative learning (Watabe et al. 2013; Han et al. 2015; Sato et al. 2015). In addition to associating pain with the environment, the CeLC could also contribute to the negative emotional experience of pain through its projections via the cholingergic substantia innominata dorsalis (Bourgeais et al. 2001) to brain regions important for affective responses to pain (Rainville et al. 1997; Sah et al. 2003; Pape & Pare, 2010; Marek et al. 2013; Neugebauer, 2015). Therefore, if acute or chronic pain states alter this synapse delivering nociceptive information to the CeLC, this could influence both the learning and aversive aspects of our experience of pain. Chronic pain states without an ongoing injury, such as lower back pain, have been attributed to potentiation or sensitization of the neural circuits involved in pain (Ji et al. 2003; Basbaum et al. 2009; Latremoliere & Woolf, 2009; Woolf, 2011). Central sensitization describes a phenomenon where there is ‘increased responsiveness of nociceptive neurons in the central nervous system to their normal or subthreshold afferent input (Merskey & Bogduk 1994). In the context of pain states without an ongoing injury, potentiation or sensitization of the neural circuits may involve an acute injury that leaves the circuit in a heightened state even after it heals. As a result, subsequent nociceptive stimuli will more strongly activate the neural circuit and may manifest as hyperalgesia. Pain models with an ongoing injury potentiate synapses important for pain (Han & Neugebauer, 2004; Carrasquillo & Gereau, 2007; Ikeda et al. 2007; Cheng et al. 2011; Chen et al. 2014a) via increases in AMPA receptor expression (Ikeda et al. 2007; Cheng et al. 2011; Chen et al. 2014a). Indeed, a range of chronic pain states, such as arthritis, formalin inflammatory pain and colitis, potentiate the PB‐CeLC synapse (Han & Neugebauer, 2004; Carrasquillo & Gereau, 2007; Ikeda et al. 2007; Fu et al. 2008; Adedoyin et al. 2010). This potentiation correlates with increased pain hypersensitivity (Han & Neugebauer, 2004; Ikeda et al. 2007; Fu et al. 2008; Adedoyin et al. 2010) and inhibition of this synapse inhibits both synaptic plasticity and pain‐related behaviour (Fu et al. 2008; Adedoyin et al. 2010), suggesting that synaptic plasticity influences the experience of pain. The synaptic plasticity in chronic pain models with ongoing pain is initiated and maintained by a continuous peripheral activation and hence differs from conditions where there are no signs of peripheral injury. If potentiation in the latter is a result of sensitization of neural circuits caused by an injury, there is the need for an explanation regarding how the initial acute injury primes synapses for subsequent activation, as well as the mechanisms behind this. To answer this question in a pain pathway important for the learning and aversive aspects of pain, we used a single, brief nociceptive stimulus that activates the spinal‐parabrachial‐amygdala pathway, without damage, and therefore had a known ‘off point’ for peripheral nociceptor activity. This brief nociceptive stimulus increased AMPA receptors (AMPAR) at PB‐CeLC synapses for at least 3 days after the stimulus and showed mechanisms similar to long‐term potentiation. This first brief stimulus primed the synapse for more prolonged potentiation from further noxious stimuli. Given that the potentiation heightened postsynaptic responsiveness, the potentiation would not be expected to result in activation of the nociceptive amygdala and associated pain behaviours until a subsequent nociceptive stimulus occurs. However, when either a nociceptive stimulus, or previously subthreshold stimulus, prompts glutamate release from the parabrachial terminals, the increased responsiveness of nociceptive amygdala neurons could enhance the aversive response or the formation of associations between pain and the environment.
Methods
Ethical approval
Experimental procedures were approved by the University of Sydney Animal Ethics Committee and conform with the principles and regulations described in Grundy (2015).
Animals
Male Sprague–Dawley rats (aged 4–7 weeks old) were sourced from Animal Resources Centre, Perth. Rats were housed under a 12:12 h light/dark cycle in a temperature‐controlled environment and were provided with food and water ad libitum.
Nociceptive stimulus
Noxious heat was used as the nociceptive stimulus. Both hindpaws were immersed (up to the ankle) in a thermostatically controlled water bath at 44°C for 30 s, four times with 2 min intervals (Fig. 1 A). Control rats underwent the same protocol using a water bath at 33°C. For the priming experiments, this protocol was conducted on days 1 and 2. During the stimulus, rats were anaesthetized using 3.5% isoflurane in oxygen via inhalation. Initial experiments to establish the adequate depth of anaesthesia found that rats withdrew their hindpaws at 44°C when under lighter anaesthesia (<3.5%).
Figure 1. A brief nociceptive stimulus without inflammation or ongoing activation of the spino‐parabrachio‐amygdaloid pathway.
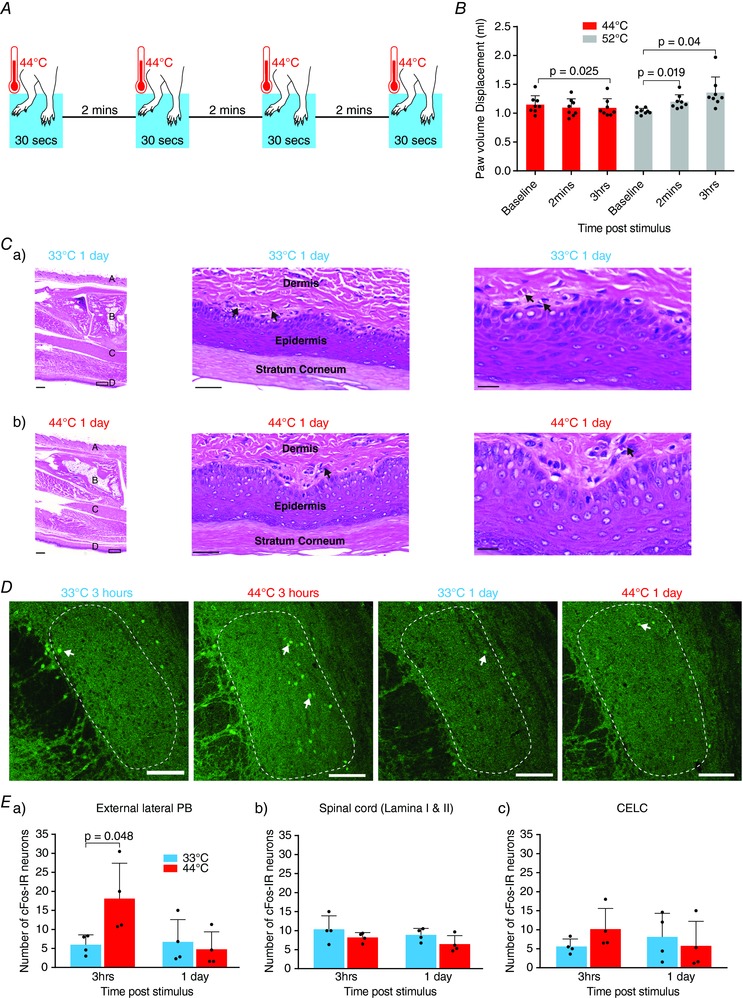
A, nociceptive stimulus procedure. Both hindpaws were immersed in a water bath at 44°C for 30 s. This was repeated four times with an interstimulus interval of 2 min. B, nociceptive stimulus does not cause immediate or early‐onset peripheral inflammation measured by paw volume displacement. Paw volume was not increased at 2 min and decreased 3 h after 44°C nociceptive stimulus, whereas paw volume was significantly increased 2 min and 3 h after the positive control treatment at 52°C. Statistical significance was tested with one‐way repeated measures ANOVA followed by a Sidak post hoc test. C, nociceptive stimulus does not cause late‐onset peripheral inflammation. There were no differences in footpad histology of rat hindpaws: Ca, 1 day after treatment with a control temperature of 33°C and Cb, 1 day after treatment with the nociceptive stimulus. Left images: 1× magnification images of hindpaw showing haired skin (A), bone (B), muscle (C) and non‐haired skin (D). Middle and right images: 10× and 20× magnification images of the boxed area on the 1× images showing some small blood vessels (arrows) throughout the superficial dermis. The small blood vessels were present in samples taken from control animals and nociceptive stimulus‐treated animals. Scale bars = 500, 50 and 20 μm, respectively (n = 4 for each group). D, confocal images of cFos‐immunoreactive (‐IR) neurons (arrow) in the PB following heat treatment. cFos‐IR neurons were counted in the external lateral portion of the PB (outlined). The rostrocaudal location of sections was: 33°C at 3 h = −9.68 mm from Bregma; 44°C at 3 h = −9.8, 33°C at 1 day = −9.8; 44°C at 1 day = −9.68. Scale bars = 100 μm. E, number of cFos‐IR neurons in the parabrachial, spinal cord and CeLC following heat treatment. Ea, in the external lateral parabrachial nucleus, the nociceptive stimulus significantly increased the number of cFos‐IR neurons after 3 h but had returned to baseline levels after 1 day. Eb, the nociceptive stimulus did not alter cFos‐IR in the spinal cord. Ec, the nociceptive stimulus did not alter cFos‐IR in the CeLC. Statistical significance was tested with an unpaired Student's t test. Dots show data from individual animals and the bar chart shows the mean ± SD. [Color figure can be viewed at http://wileyonlinelibrary.com]
Peripheral injury
Paw volume displacement
To determine whether the nociceptive stimulus produces peripheral injury, we used paw volume displacement as a measure of inflammation. Paw volume displacement was measured using a plethysmometer (Ugo Basile, Gemonio VA, Italy). For assessment of immediate and early onset injury, hind paw volume displacement was measured before the nociceptive stimulus, 2 min after the nociceptive stimulus and 3 h after the nociceptive stimulus. In these experiments, we used a water bath at 52°C as a positive control (Bester et al. 1997). Animals in the 52°C group were monitored for signs of distress, such as arching of the back, horizontal stretching, abdominal writhing, twitching and guarding. However, none of the animals displayed any of these signs during the 3 h period. For assessment of late‐onset injury, hindpaw volume displacement was measured 1 day after the nociceptive stimulus and compared to a negative control of a water bath at 33°C. The positive control of 52°C was not tested at the 1 day time point for ethical reasons.
Hindpaw histology
Animals were anaesthetized with isoflurane using the open‐drop method, decapitated and their hindpaws removed and placed in 10% neutral buffered formalin for 1 week. Samples were then transferred into a 10% formic acid solution for 1 week for bone decalcification. Samples were then processed through graded alcohols of 70%, 95% and 100% xylene and infiltrated with paraffin wax. Specimens were embedded and cut at 4 μm using a RM 2235 microtome (Leica Biosystems, Nussloch, Germany) onto slides. Following this, slides were deparaffinized using graded alcohols of 100%, 95% and 70% xylene then water and stained with a standard haemotoxylin and eosin (H&E) stain. Slides were imaged using the Zeiss Axio scan slide scanner (Carl Zeiss, Wetzlar, Germany) and assessed by a blinded specialist veterinary pathologist.
Immunohistochemistry
Three hours or 1 day after nociceptive stimulation, rats were deeply anaesthetized with sodium pentobarbitone (50 mg kg–1) and killed by transcardial perfusion with 3,000 IU L−1 heparin in a 0.5% NaNO2/0.9% saline (w/v) solution followed by a 4% paraformaldehyde (w/v) solution in 0.1 m PBS (pH 7.4). The brain and lumbar enlargement portion of the spinal cord were removed and postfixed overnight in 4% paraformaldehyde in PBS at 4°C. Brain and spinal cord were sectioned coronally into 50 μm sections using a Leica VT 1000S vibratome. Sections containing the amygdala, PB and L4/L5 regions of the spinal cord were collected in 0.1 m PBS. Sections were incubated in a 10% normal goat serum (NGS)/0.5% BSA/0.3% Triton X‐100 in PBS (w/v) for 30 min then washed using 0.1 m PBS. This was followed by an overnight incubation of sections in rabbit antibody to c‐Fos (dilution 1:100; SC‐52; Santa Cruz Biotechnology, Santa Cruz, CA, USA) primary antibody in 2% NGS in PBS at room temperature. c‐Fos primary antibody was washed off the following day with 0.1 m PBS. Sections were incubated for 1 h in guinea pig antibody to calcitonin gene‐related peptide (dilution 1:1000; T‐5027; Peninsula Laboratories, San Carlos, CA, USA) primary antibody in 2% NGS in PBS at room temperature. Sections were washed with 0.1 m PBS and then incubated in Alexa Fluor 488 goat anti‐rabbit (dilution 1:1000; A‐110088; Molecular Probes, Carlsbad, CA, USA) and CY3 conjugated donkey anti‐guinea pig (dilution 1:1000; 706‐165‐148; Jackson Laboratories, Bar Harbor, ME, USA) in 2% NGS in PBS for 2 h. Topro3 (nuclei stain) (dilution 1:500; T3605; Molecular Probes) was directly added to wells in the last 30 min of incubation. Sections were washed with 0.1 m PBS and mounted onto glass slides and cover slipped with Fluoromont‐G (ProSciTech, Kirwan, QLD, Australia). Sections were imaged with a LSM510 Meta confocal microscope (Carl Zeiss). A blinded observer counted the c‐Fos immunoreactive neurons in lamina I/II of the spinal cord, external lateral region of the PB and the CeLC. The rat brain atlas of Paxinos and Watson was used to identify the relevant regions (Paxinos & Watson, 1986). Calcitonin gene‐related peptide (Shimada et al. 1985; Kruger et al. 1988; Chieng et al. 2006) and Topro3 staining were used to help identify the relevant regions.
Electrophysiology
Rats were anaesthetized with isoflurane using the open‐drop method, decapitated and their brains removed into ice‐cold artificial cerebrospinal fluid (aCSF) containing (in mm) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4.2H2O, 2.5 MgCl2, 0.5 CaCl2, 25 NaHCO3 and 11 d‐glucose. Coronal slices (280 μm) containing the amygdala were obtained using a VT 1200s vibratome (Leica). Slices were transferred to a submerged chamber containing aCSF equilibrated to pH 7.4 with carbogen (95% O2/5% CO2) at 34°C for at least 1 h.
Slices were then transferred to a recording chamber and superfused continuously at 2.5 mL min–1 with aCSF containing (in mm) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4.2H2O, 1 MgCl2, 2 CaCl2, 25 NaHCO3 and 11 d‐glucose saturated with carbogen. The temperature was maintained between 33 and 34°C using an inline heater and monitored using a thermistor. Slices were visualized using a BX51 microscope (Olympus, Tokyo, Japan) equipped with 40× water immersion objective and Dodt gradient contrast optics. Whole cell, patch clamp recordings were made from neurons in the CeLC. Patch electrodes (2–4 MΩ) were filled with internal solution containing (in mm) 140 CsCl, 5 Hepes, 10 EGTA, 2 CaCl2, 2 Mg2ATP, 0.3 NaGTP and 3 QX‐314.Cl (pH 7.3, osmolarity 280–285 mOsm L–1). Neurons were voltage clamped using a patch clamp amplifier (MultiClamp 700B; Axon Instruments, Foster City, CA, USA). Current signals were filtered at 5 kHz and sampled at 10 kHz. Series resistance (≤12 MΩ) was compensated by 60% and continuously monitored throughout the experiment. Data was discarded if series resistance fluctuated by more than 20% during recording. Recordings were not corrected for liquid junction potentials. EPSCs were evoked via concentric bipolar stimulating electrodes (rate, 0.05 Hz; stimuli, 2–99 V, 100 μs) (FHC Inc., Bowdoin, ME, USA). Stimulus intensity was set to yield subthreshold EPSCs amplitudes. All EPSCs were recorded in the presence of the GABAA receptor antagonist picrotoxin (100 μm). CeLC neurons were voltage‐clamped at −70 mV or +40 mV for AMPA/NMDA ratio recordings. The AMPAR EPSC amplitude was determined by measuring the peak amplitude of the EPSC at −70 mV (average of at least five EPSCs). The NMDAR EPSC amplitude was determined by taking the average amplitude between 70 and 90 ms after the stimulus at +40 mV (average of at least five EPSCs). The AMPA/NMDA ratio was calculated by dividing the AMPAR EPSC amplitude by the NMDAR EPSC amplitude (Fig. 2 Ab). To determine the voltage dependence of the AMPAR EPSC, the membrane potential was stepped from −70 mV to +40 mV (in 10 mV steps) during superfusion of the NMDAR antagonist dl‐2‐amino‐5‐phosphonopentanoic acid (DL‐APV) (100 μm) and after the addition of spermine (100 μm) to the internal solution. The rectification index is peak EPSC+40 mV divided by peak EPSC−60 mV. For comparison of AMPAR deactivation kinetics, EPSCs were recorded at −70 mV, averaged and the current decay fitted to a double exponential function. The weighted time constant was calculated using the equation:where Af = amplitude of the fast decay component, As = amplitude of the slow decay component, τf = decay time constant of fast decay component, τs = decay time constant of slow decay component.
where A f is the amplitude of the fast decay component, A s is the amplitude of the slow decay component, τf is the decay time constant of fast decay component and τs is the decay time constant of slow decay component.
Figure 2. A brief nociceptive stimulus induces long lasting synaptic plasticity specifically at the PB‐CeLC synapse.
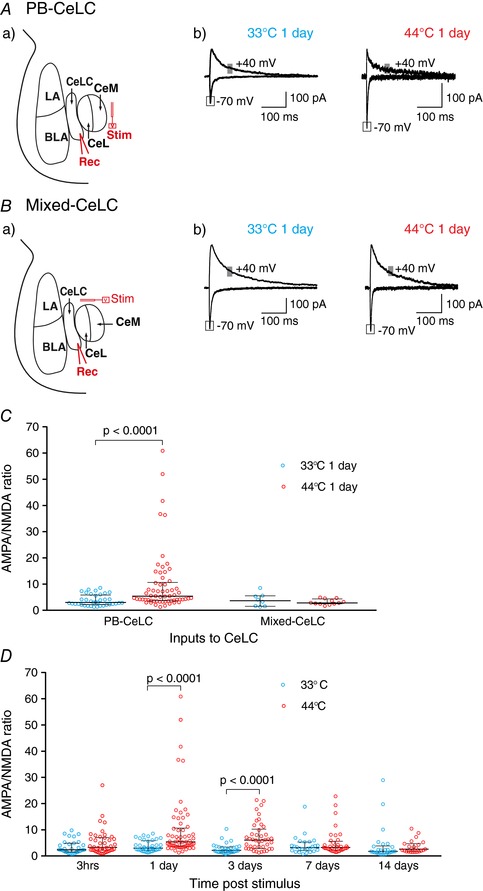
A, nociceptive stimulus potentiates the PB‐CeLC synapse. Aa, schematic diagram of stimulation and recording site. Stimulating electrodes were placed dorsomedial to the CeA to stimulate PB fibers. The response of the CeLC neurons to this stimulation was recorded. Ab, example traces of EPSCs from control and nociceptive treated rats 1 day after treatment. The amplitude of the AMPAR component of the EPSC was measured at the peak current recorded at −70 mV and the amplitude of the NMDAR component of the EPSC was measured as the average amplitude 70–90 ms after stimulation recorded at +40 mV (grey box). B, nociceptive stimulus does not potentiate the mixed synaptic input. Ba, schematic diagram of stimulation and recording site. Stimulating electrodes were placed dorsal to CeA to stimulate fibres coming from but not limited to the cortex, hypothalamus, thalamus and PB. The response of the CeLC neurons to this stimulation was recorded. Bb, example traces of EPSCs from control and nociceptive treated rats 1 day after treatment. The amplitude of AMPAR and NMDAR EPSCs was measured as above. C, scatter plots of AMPA/NMDA ratio at the PB‐CeLC and mixed‐CeLC synapses for individual neurons. Nociceptive stimulus increased the AMPA/NMDA ratio at the PB‐CeLC synapse but not at the mixed input‐CeLC synapse. D, nociceptive stimulus causes long‐lasting changes at the PB‐CeLC synapse. Scatter plot showing AMPA/NMDA over the 2 weeks following the nociceptive stimulus. The nociceptive stimulus increased the AMPA/NMDA ratio for at least 3 days. Statistical significance was tested with two‐tailed Mann–Whitney test. Each circle shows the data from an individual neuron and the graph also shows the median ± interquartile range. [Color figure can be viewed at http://wileyonlinelibrary.com]
The paired pulse ratio (PPR) of AMPAR‐mediated EPSCs was obtained by evoking two consecutive stimuli of identical stimulus strength (interstimulus interval of 30 ms). PPR was calculated by dividing the second EPSC amplitude by the amplitude of the first (EPSC2/EPSC1). All data were acquired and analysed using Axograph software (Molecular Devices, Sunnyvale, CA, USA).
Behavioural testing
Thermal hyperalgesia
The experimenter was blinded to rat treatment group. To measure thermal paw withdrawal latency (PWL), rats were placed in perspex enclosures (15 × 15 × 18 cm) and given 10–15 min to acclimatize to the test environment. The testing was conducted using a plantar tester (Ugo Basile) in accordance with the method of Hargreaves et al. (1988). Focal infrared heat was applied through the plastic bottom of the enclosure to the rear left hindpaw and the latency for the rat to respond by moving its hindpaw away from the noxious heat source was recorded.
Mechanical allodynia
To measure mechanical allodynia, mechanical paw withdrawal thresholds (PWTs) were determined with a series of Von Frey hairs (range 0.4–15 g). Rats were placed in elevated perspex enclosures (28 × 15 × 18 cm) with wire mesh bases and given 15–20 min to acclimatize to this environment. Each Von Frey hair was tested six times at random locations on the plantar surface of the left hindpaw. Von Frey hairs were pressed perpendicularly against the hindpaw and held for ∼2 s. Testing began with the 2.0 g Von Frey hair. A withdrawal response was recorded if the hindpaw was sharply withdrawn, if any paw licking took place or if the animal flinched upon removal of the Von Frey hair. If the animal responded, then the next heavier hair was tested. If the animal did not respond, then the next lighter hair was tested. Once there was a change in response, four more hairs were tested and the mechanical PWT was calculated using the up–down paradigm (Chaplan et al. 1994). If the animals did or did not respond to any hairs, then the mechanical PWT was assigned as 0.2 or 15 g, respectively.
Drugs
Picrotoxin and spermine were purchased from Sigma‐Aldrich (St Louis, MO, USA). DL‐APV was purchased from Tocris Bioscience (Bristol, UK). Picrotoxin was added directly to aCSF. Spermine was added to internal solution. Distilled water was used to make a stock solution for APV. The stock solution was diluted to working a concentration in ACSF immediately before use and applied by gravity driven superfusion.
Statistical analysis
Prism, version 7 (GraphPad, San Diego, CA, USA) was used for the statistical analysis. Data with normal distribution are expressed as the mean ± SD. Data with a non‐normal distribution are expressed as the median. The interquartile range (difference between the 75th and 25th percentile) was used to quantify variability in non‐normal data. Normality was tested using the Kolmogorov–Smirnov test. A Mann–Whitney test (two‐tailed), ANOVA or Student's unpaired t test (two‐tailed) were used to test significance as appropriate. P < 0.05 was considered statistically significant.
Results
A brief nociceptive stimulus
To investigate synaptic plasticity that outlasts the noxious stimulus, we wanted to use a brief stimulus that activates the spino‐parabrachio‐amygdaloid pathway but does not produce ongoing activation of this neural pathway. Briefly immersing the hindpaws of rats in water at 44°C activates the spino‐parabrachial pathway (Bester et al. 1997). A 44°C heat stimulus probably does not produce ongoing activation of the nociceptor because nociceptor firing dissipates quickly after stimulation of nociceptive afferents by noxious heat (LaMotte & Campbell, 1978; Adriaensen et al. 1984; Martin et al. 1988; Yeomans & Proudfit, 1996). In rats, C‐fibre firing in response to heating the skin to 43°C dissipates within 2 s of stopping the heat (Yeomans & Proudfit, 1996). Even when skin was heated to higher temperatures than used in the present study, such as 50°C, the potentiation of C‐fibre firing still dissipates within 3–15 min of stopping the heat (Schouenborg, 1984). Our stimulus is repeated four times with an interstimulus interval of 2 min. This repeated stimulus probably does not produce a ‘wind‐up’ effect on C‐fibre firing, as the magnitude of C‐fibre firing actually decreases with repeated heat stimuli (LaMotte & Campbell, 1978; Adriaensen et al. 1984). Thus, because nociceptive afferents do not produce prolonged firing after noxious heat stimulus and repeated heat stimuli does not produce ‘wind‐up’, our stimulus probably does not produce ongoing activation of this pathway. However, to test this, we determined whether there was inflammation of the paw or ongoing c‐Fos production in the spino‐parabrachio‐amygdala neural pathway.
To test whether the noxious stimulus causes peripheral injury, we used paw volume displacement as a measure of inflammation and histological assessment of damage. To determine immediate and early onset injury, hindpaw volume displacement was measured before, 2 min and 3 h after heat treatment at 44°C and 52°C. We found that immersing the hindpaws of anaesthetized rats in water at 44°C for a total of 2 min (Fig. 1 A) did not increase paw volume (Fig. 1 B). Indeed, there was a small but significant decrease in paw volume 3 h after the noxious heat treatment (one‐way repeated measures ANOVA, Sidak post hoc test, F 2,14 = 3.277, P = 0.025, n = 8 animals) (Fig. 1 B). By contrast, the heat treatment at 52°C significantly increased paw volume 2 min (one‐way repeated measures ANOVA, Sidak post hoc test, F 2,14 = 6.452, P = 0.019, n = 8 animals) and 3 h (one‐way repeated measures ANOVA, Sidak post hoc test, F 2,14 = 6.452, P = 0.04, n = 8 animals) after the heat treatment (Fig. 1 B). Therefore, heat treatment of the paws at 44°C does not cause immediate or early onset injury, although higher temperatures do. We then assessed whether the nociceptive stimulus causes injury with a late onset by comparing the paw volume displaced 1 day after the nociceptive stimulus with the volume displaced 1 day after the control stimulus. We found no difference in paw volume between animals that had undergone treatment with the nociceptive stimulus and control (mean ± SD: nociceptive stimulus group: 1.88 ± 0.139 mL, n = 4 animals vs. control group: 1.92 ± 0.152 mL, n = 4 animals, P = 0.69, two‐tailed Student's unpaired t test). For further assessment of late onset injury, we performed a histological assessment (H&E stain) of hindpaws 1 day after treatment with the nociceptive stimulus and control stimulus of 33°C. We examined the footpad (ventral hairless side of the limb) and also the haired skin of the limb. When looking at the footpad, we considered the nature of the epidermis and keratin layer, the superficial dermis vascularity and cellularity in particular. In all animals, the dorsal haired skin, bone, muscle and epidermis of the footpad were normal (Fig. 1 C). There were some scattered small blood vessels throughout the superficial dermis in all samples (Fig. 1 C). However, there were no significant differences between animals that had undergone treatment with the nociceptive stimulus and control animals in terms of superficial dermal cellularity and superficial dermal vascularity (Fig. 1 C). Therefore, at 3 h or 1 day at the brief nociceptive stimulus, we found no evidence of paw volume increases, indicative of inflammation, or histological signs of damage to the paw.
We used c‐Fos, a marker for neuronal activation (Suwanprathes et al. 2003; Gao & Ji, 2009), to assess whether our nociceptive stimulus produces acute or ongoing activation of the spino‐parabrachio‐amygdaloid neural pathway. C‐Fos protein levels peak 1–2 h after a stimulus and return to basal levels 6–8 h after the stimulus (Suwanprathes et al. 2003; Gao & Ji, 2009). Thus, we hypothesized that the brief nociceptive stimulus would increase c‐Fos expression at the 3 h time point but that, without ongoing activation of the nociceptors, new c‐Fos expression would not be stimulated and, thus, at the 1 day time point, c‐Fos expression would not differ from control conditions. If the nociceptive stimulus does cause increase in activity at this pathway either via a change in neurotransmitter release or change in receptor numbers, then we might also expect to see an increase in c‐Fos expression at the day 1 time point if a second nociceptive stimulus is delivered. We counted the number of c‐Fos positive neurons in the spinal cord, parabrachial nucleus and laterocapsular amygdala 3 h and 1 day after the heat treatment. We compared rats treated with noxious heat at 44°C vs. control innocuous heat at 33°C (Fig. 1 A). Noxious heat significantly increased c‐Fos‐IR in the external lateral parabrachial nucleus 3 h after the stimulus (Fig. 1 D and Ea), although this increase in c‐Fos expression was not ongoing because it had returned to control 1 day later (Fig. 1 D and Ea). The noxious stimulus did not increase c‐Fos expression in the spinal cord or amygdala (Fig. 1 Eb and Ec). This is consistent with previous experiments using this noxious stimulus where, at all noxious temperatures tested, c‐Fos protein expression was greater in the parabrachial than the spinal cord (Bester et al. 1997). Therefore, the increased c‐Fos immunoreactivity observed in the parabrachial nucleus 3 h after the nociceptive stimulus is probably in response to the activation of this neural pathway by the noxious stimulus 3 h earlier. Because c‐Fos expression is not increased 24 h later, this suggests that, at least 6–8 h before, this neural pathway has returned to control levels of activity. Therefore, the lack of paw inflammation, normal histology and brief elevations in c‐Fos expression are consistent with the heat stimulus at 44°C providing a brief noxious stimulus.
Nociceptive stimulus induces long‐lasting synaptic plasticity specifically at the PB‐CeLC synapse
We examined whether the brief nociceptive stimulus produces synaptic plasticity at the PB‐CeLC synapse. A well characterized hallmark of synaptic plasticity is changes in postsynaptic AMPARs and NMDARs (Rao & Finkbeiner, 2007). Thus, our first experiment determined the relative contribution of postsynaptic AMPARs and NMDARs (AMPA/NMDA ratio) to synaptic transmission at the PB‐CeLC synapse. We placed stimulating electrodes dorsomedial to the CeA to stimulate PB inputs (Bernard et al. 1993; Han & Neugebauer, 2004). We then made whole‐cell, patch clamp recordings from CeLC neurons (Fig. 2 A) and recorded the EPSC amplitude at −70 mV and + 40 mV. We found that the nociceptive stimulus significantly increased the AMPA/NMDA ratio at day 1 (Fig. 2 A and C). These data suggest that the nociceptive stimulus increases AMPA receptors at the PB‐CeLC synapses.
To determine whether the nociceptive stimulus specifically changes synapses in the PB‐CeLC, we tested whether the stimulus also changes a mixed synaptic input. These mixed inputs include polymodal sensory information from the thalamus (Moga et al. 1995; Vertes & Hoover, 2008), hypothalamus (Canteras et al. 1994), entorhinal cortex (McDonald & Mascagni, 1997) and lateral occipital area (McDonald et al. 1996). They also include inputs from areas delivering affective information, such as the prefrontal cortex, insular cortex and anterior cingulate cortex (McDonald et al. 1996). Stimulating electrodes were placed dorsal to the CeA to stimulate this mixed input (Canteras et al. 1994; Moga et al. 1995; McDonald et al. 1996; McDonald & Mascagni, 1997; Vertes & Hoover, 2008) (Fig. 2 Ba). The nociceptive stimulus did not alter the AMPA/NMDA ratio at this mixed input‐CeLC synapse at 1 day (Fig. 2 B and C). This indicates that the nociceptive stimulus does not induce synaptic potentiation at all synapses onto CeLC neurons.
The noxious stimulus only increased the AMPA/NMDA ratio in a subpopulation of CeLC neurons (Fig. 2 C). The variability in the response may result from whether individual CeLC neurons receive noxious information in response to the stimulus because only ∼40% of CeLC neurons are excited by cutaneous inputs from the PB (Bernard et al. 1990, 1992). The variability in response may also result from innate differences between CeLC neurons. Although the CGRP receptor is widely expressed in CeLC neurons (van Rossum et al. 1997; Oliver et al. 1998; Han et al. 2015), the selective expression of corticotrophin‐releasing factor (Harrigan et al. 1994; Ji et al. 2013), somatostatin (Li et al. 2013; Han et al. 2015) and protein kinase C (PKC)‐δ (Haubensak et al. 2010; Han et al. 2015) in subpopulations of CeLC neurons raises the possibility of functional subgroups of neurons. Indeed, the expression profile of PKC‐δ defines the direction of fear conditioning induced synaptic plasticity, with enhanced excitatory responses in PKC‐δ negative neurons but reduced excitatory responses in PKC‐δ positive neurons (Ciocchi et al. 2010; Haubensak et al. 2010). We also found that the plasticity at the PB‐CeLC synapse was bilateral (44°C, 1 day right hemisphere: mean ± SD: 4.69 ± 4.688, n = 8 cells vs. 44°C, 1 day left hemisphere: 6.51 ± 7.161, n = 23 cells, P = 0.49, two‐tailed Student's unpaired t test) and was not influenced by the rostrocaudal location of the neuron or the time elapsed following dissection (up to 7 h).
Because our nociceptive stimulus is brief and does not produce ongoing activation or inflammation, we can track how long this pain‐induced change in synaptic glutamate receptors persists after the stimulus. We found that, 3 h after the nociceptive stimulus, the AMPA/NMDA ratio was unchanged from control (Fig. 2 D). The biggest increase in AMPA/NMDA ratio occurs 1 day and 3 days after the nociceptive stimulus (Fig. 2 D). By 7 days, the AMPA/NMDA ratio had returned to control levels (Fig. 2 D). These data show that a brief nociceptive stimulus that does not produce ongoing peripheral activation can induce long lasting synaptic changes at a central synapse important for pain.
Nociceptive stimulus produces a transient change in AMPAR subunit composition at the PB‐CeLC synapse
The increase in AMPA/NMDA ratio at the PB‐CeLC synapse may be the result of either an increase in postsynaptic AMPARs or a decrease in NMDARs. AMPARs are heterotetramers composed of GluA1–4 subunits and the composition of synaptic AMPA receptors changes during some forms of synaptic plasticity (Chater & Goda, 2014). In some forms of in vitro long‐term potentiation (LTP), a model for learning and memory (Chater & Goda, 2014), synaptic AMPARs undergo a change in subunit from GluA2‐containing to GluA2‐lacking (Plant et al. 2006; Guire et al. 2008; Morita et al. 2014). Unlike GluA2‐containing AMPARs, GluA2‐lacking AMPARs are Ca2+ permeable (Chater & Goda, 2014), have faster decay kinetics (Chater & Goda, 2014) and display an inwardly rectifying current–voltage (I–V) relationship as a result of blockade by intracellular polyamines (Chater & Goda, 2014). To clarify whether the increase in AMPA/NMDA ratio was associated with a change in AMPAR subunits, we examined the I–V relationships of AMPAR EPSCs at the 1 and 3 day time points. To isolate the AMPAR synaptic response, experiments were conducted in the presence of the GABAA antagonist pictrotoxin (100 μm) and NMDA receptor antagonist DL‐APV(100 μm). Spermine (100 μm) was added to the intracellular solution to compensate for possible dialysis of intracellular polyamines during whole‐cell recording. We found that the nociceptive stimulus significantly increased the inward rectification of AMPAR‐mediated EPSCs at 1 day (Fig. 3 A). This is consistent with the nociceptive stimulus increasing the incorporation of GluA2‐lacking AMPARs at the PB‐CeLC synapse. Interestingly, although there was a significant increase in AMPA/NMDA ratio in the nociceptive group at 3 days, the rectification index of the nociceptive stimulus group at 3 days did not differ from the control, suggesting that, 3 days after the stimulus, the AMPAR subunit composition had reverted to GluA2‐containing AMPARs (Fig. 3 A), even though the increase in AMPA/NMDA receptor persists.
Figure 3. A brief nociceptive stimulus produces a transient change in AMPAR subunit composition at the PB‐CeLC synapse.
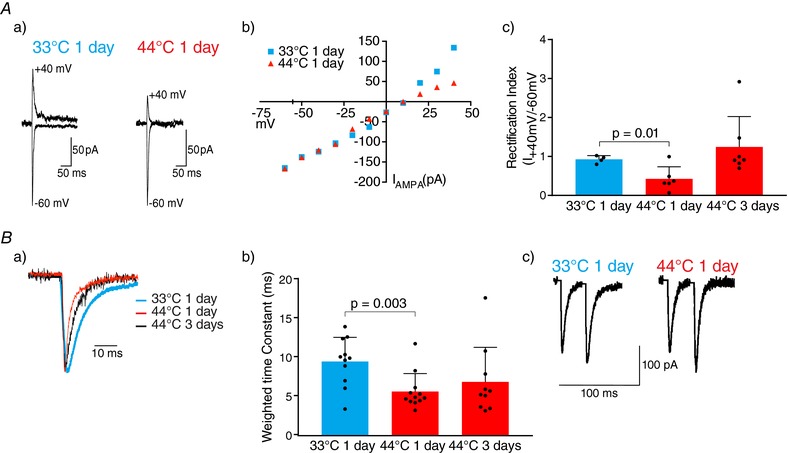
A, nociceptive stimulus increases inward rectification of AMPAR EPSCs at 1 day. Aa, example traces of AMPAR EPSCs recorded at −60 mV and +40 mV after control or nociceptive treatment. Recordings were made in the presence of APV (100 μm) and spermine (100 μm) was included in the internal solution. Ab, I–V plot showing greater inward rectification of AMPAR EPSCs 1 day after the nociceptive stimulus. Ac, rectification index (I +40 mV/I −60 mV) showing a significantly lower rectification index in nociceptive stimulus group at 1 day. Rectification index returns to control levels at 3 days. B, AMPAR‐mediated EPSCs have faster decay kinetics in nociceptive group at 1 day and return to control levels after 3 days. Ba, normalized example traces of AMPAR EPSCs after control and nociceptive treatments. Bb, weighted time constants showing that the nociceptive stimulus speeds decay of the AMPAR synaptic at 1 day. Bc, PPR was not changed by the nociceptive stimulus. Example traces of two consecutive EPSCs of identical intensity (interstimulus interval of 30 ms) showing that the PPR was not changed by the nociceptive stimulus. PPR was calculated by dividing the second EPSC amplitude by the amplitude of the first. Statistical significance was tested using a two‐tailed unpaired Student's t test. Dots show data from individual neurons and the bar chart shows the mean ± SD. [Color figure can be viewed at http://wileyonlinelibrary.com]
The nociceptive stimulus quickened the AMPAR decay at 1 day (Fig. 3 B), although the decay did not differ from the control group by 3 days after stimulus (Fig. 3 B). Both the increase in inward rectification and quicker decay of the AMPAR synaptic response after the nociceptive stimulus suggests that the stimulus triggers the transient insertion of GluA2‐lacking AMPAR at the PB‐CeLC synapse for 1 day. After this, although the AMPA/NMDA receptor ratio is elevated, the AMPAR subunit composition of the PB‐CeLC synapse returns to a baseline composition similar to LTP (Plant et al. 2006; Guire et al. 2008; Morita et al. 2014).
We next examined whether the nociceptive stimulus causes changes in presynaptic glutamate release at the PB‐CeLC synapse. The nociceptive stimulus did not change the PPR 1 day after the stimulus (nociceptive stimulus group: mean ± SD: 1.01 ± 0.3831, n = 49 cells vs. control group: 1.13 ± 0.6942, n = 36 cells, P = 0.36, two‐tailed unpaired Student's t test) (Fig. 3 C), suggesting that the nociceptive stimulus does not alter glutamate release.
PB‐CeLC synapse undergoes metaplastic‐like changes
Metaplasticity denotes a higher order form of synaptic plasticity, where a ‘primer’ synaptic activity at a particular point in time alters or changes the ability of neurons or synapses to generate subsequent plasticity (Abraham, 2008). It is possible that the synaptic plasticity produced by the nociceptive stimulus at 1 day could influence the nature of subsequent plasticity. In particular, we aimed to investigate whether the synaptic plasticity would be prolonged by a second nociceptive stimulus administered 1 day later when there is more calcium permeable GluA2‐lacking AMPAR at the synapse. After a single stimulus, the AMPA/NMDA ratio returned to control levels within 7 days of the nociceptive stimulus (Fig. 2 D). Therefore, if the first nociceptive stimulus primes the synapse to produce longer lasting plasticity to a second nociceptive stimulus, the plasticity should persist 7 days after the second nociceptive stimulus. To test this, we compared the plasticity produced by two consecutive nociceptive stimuli on day 1 (44°C) and on day 2 (44°C) (Fig. 4 A) vs. a nociceptive stimulus on day 1 (44°C) and a control stimulus on day 2 (33°C) (Fig. 4 A). The nociceptive stimulus protocol was conducted as before (4 × 30 s with 2 min interval between) (Fig. 1 A). We examined the AMPA/NMDA ratio 7 days after the final nociceptive stimulus and found that, 7 days after the second nociceptive stimuli (day 8), the AMPA/NMDA ratio was still significantly higher than 7 days after a single nociceptive stimulus (day 7) (Fig. 4 A and B). This suggests that, when a second nociceptive stimulus is delivered during a time of prior synaptic potentiation or priming, this prolongs the synaptic plasticity. To ensure that the results obtained above were not a result of peripheral damage caused by the consecutive treatment with the nociceptive stimulus, we measured paw volume displacement and performed histological assessment of the hindpaw of animals in the two groups (44°C and 44°C at 8 days vs. 44°C and 33°C at 7 days). We also measured the paw volume and examined histology in animals that were killed immediately after treatment with the second nociceptive stimulus on day 2. We found no significant differences in paw volume displacement across the three groups (F 2,9 = 0.122, P = 0.886, n = 4 animals for all groups, one‐way ANOVA). The areas of the hindpaw examined in the histology were the same as described previously. We found no significant differences between the samples across the three groups (Fig. 4 C). These results confirmed that consecutive treatment with the nociceptive stimulus does not cause inflammation or peripheral damage and the lengthening of the synaptic plasticity is probably a result of metaplastic‐like changes at the PB‐CeLC synapse.
Figure 4. PB‐CeLC synapse undergoes metaplastic‐like changes following synaptic plasticity.
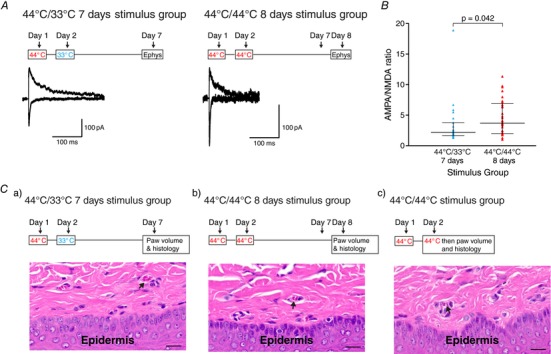
A, timeline of treatment (above) and example traces of EPSCs (below) in the control two stimuli 7 days group (44°C/33°C) and increased EPSC in the nociceptive two stimuli 8 days group (44°C/44°C). The amplitude of the AMPA and NMDA EPSC was measured as in Fig. 2. B, AMPA/NMDA ratio showing the increase in AMPA/NMDA ratio in the nociceptive two stimuli 8 days group. C, consecutive treatment with the nociceptive stimulus protocol on days 1 and 2 does not cause peripheral damage. The timeline of treatment (above) and histology images of (below) the three groups. Paw volume displacement measurements and histological samples were taken on day 7 for the 44°C/33°C at 7 days group (Ca), day 8 for the 44°C/44°C at 8 days group (Cb) and immediately after the completion of the second nociceptive stimulus protocol on day 2 for the 44°C/44°C group (Cc). Similar to single treatment with the nociceptive stimulus protocol, small blood vessels were found throughout the superficial dermis in all groups (arrows). Scale bar = 20 μm (n = 4 for each group). Statistical significance was tested using a two‐tailed Mann–Whitney test. Dots show data from individual neurons and the graph also shows the median ± interquartile range. [Color figure can be viewed at http://wileyonlinelibrary.com]
Nociceptive stimulus produces mechanical but not thermal hyperalgesia
The PB‐CeLC synapse contributes to development of hyperalgesia in numerous pain states (Hebert et al. 1999; Han et al. 2005; Carrasquillo & Gereau, 2007; Pedersen et al. 2007; Fu & Neugebauer, 2008; Ansah et al. 2010; Ji et al. 2010). Therefore, we examined whether the single nociceptive stimulus induces mechanical and thermal hyperalgesia. The nociceptive stimulus significantly reduced the mechanical threshold at 1 day (Fig. 5 A). At all other time points, the mechanical threshold was unchanged by the nociceptive stimulus (Fig. 5 A). The thermal threshold was unchanged at all time points (Fig. 5 B). Given that the nociceptive stimulus itself is a noxious thermal stimulus, it may be somewhat unexpected that it produced mechanical rather than thermal hyperalgesia. However, it is consistent with secondary hyperalgesia (i.e. hyperalgesia outside of the site of injury) that is produced through central sensitization only resulting in mechanical hyperalgesia (Raja et al. 1984; Coderre et al. 1993; Dahl et al. 1993; Woolf, 2011).
Figure 5. Nociceptive stimulus produces mechanical but not thermal hyperalgesia.
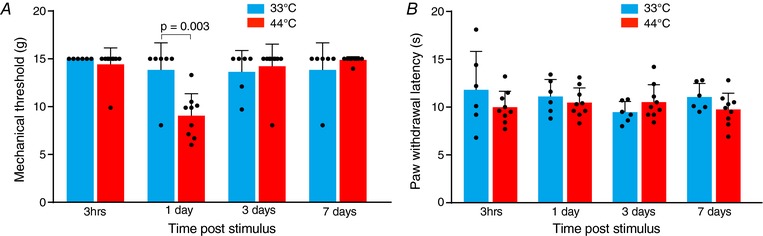
A, mechanical threshold showing that the single nociceptive stimulus (44°C) causes a reduction in mechanical threshold at 1 day. B, PWL following the Hargreaves test showing that the single nociceptive stimulus (44°C) does not produce thermal hyperalgesia at any time point. Statistical significance was tested using a two‐tailed unpaired Student's t test. Dots show data from individual neurons and the bar chart shows the mean ± SD. [Color figure can be viewed at http://wileyonlinelibrary.com]
Discussion
In the present study, we used a brief nociceptive stimulus that does not produce ongoing activation of the spino‐parabrachio‐amygdaloid pathway to investigate synaptic plasticity that persists beyond the stimulus. We found that a 2 min nociceptive stimulus potentiates the PB‐CeLC synapse. Our precise control of the nociceptive timing allowed us to determine that the increase in AMPAR response at the PB‐CELC synapse developed over 1 day and lasted for at least 3 days. Additionally, the AMPAR potentiation was biphasic, with incorporation of more GluA2‐lacking AMPARs at 1 day, which were subsequently replaced by GluA2‐containing AMPARs at 3 days. We found that, during the period of high GluA2‐lacking AMPAR activity, an additional nociceptive stimulus produced prolonged synaptic plasticity. We also found that rats experienced mechanical hyperalgesia following the nociceptive stimulus.
We used several approaches to confirm that our stimulus was truly brief. If our stimulus produced inflammation or damage, then the activation of the neural pathways carrying nociceptive information could have persisted beyond the termination of the heat stimulus. However, neither the 44°C single heat, nor the double heat stimulus inflamed the paw, even though higher temperatures did. Additionally, heat treatment did not produce histological signs of damage to the paw. Consistent with this, expression of c‐Fos, a marker for neuronal activation, was elevated in parabrachial neurons 3 h after the stimulus but not at 1 day. This suggests that, although this neural pathway is briefly activated by the heat stimulus, ongoing activation of the afferent neurons to the CeLC did not occur. The lack of elevated c‐Fos expression in the nociceptive amgydala neurons is consistent with the heightened postsynaptic responsiveness found in the present study. This plasticity would not activate nociceptive amygdala neurons, and thus c‐Fos expression, until a subsequent nociceptive stimulus prompted glutamate release from parabrachial neurons. Therefore, this is a measure of activity of neurons in this nociceptive pathway, rather than a measure of neuronal plasticity, and, together with the lack of inflammation or pathological changes in the paw, strongly indicates that our nociceptive stimulus was truly brief.
We know that a two‐phase noxious stimulation of deep tissue (Cheng et al. 2011) or prolonged noxious stimulation of joints over several hours (Neugebauer & Li, 2003; Neugebauer et al. 2003; Han et al. 2005) strengthens the PB‐CELC synapse for 2–10 h. The mechanism of this potentiation is not well defined, although it may be the result of an increase in postsynaptic AMPARs (Cheng et al. 2011). Our findings have allowed us to define how the glutamate receptors change over days in response to simple nociceptive activation over which we have temporal control because it does not cause paw damage, inflammation or ongoing activation of the pain neural pathway. In vitro long‐term potentiation mechanisms are considered to model synaptic modifications that underlie learning (Chater & Goda, 2014). The synaptic changes produced by the nociceptive stimulation are reminiscent of some forms of LTP (Plant et al. 2006; Guire et al. 2008; Morita et al. 2014) but stretched over a much longer time frame. Although in vitro high frequency stimulation increases GluA2‐lacking AMPA receptors over minutes, these receptors only remain at the synapse for 20–30 min and the potentiation is then maintained by subsequent replacement with GluA2‐containing AMPA receptors (Plant et al. 2006; Guire et al. 2008; Morita et al. 2014). The nociceptive stimulus increases the AMPA/NMDA ratio during the first day after stimulus and, similar to the early phase of LTP, the potentiation of the AMPA response is associated with greater insertion of GluA2‐lacking AMPARs. By 3 days, the synapses are still potentiated but the GluA2‐lacking AMPARs seen at 1 day are replaced by GluA2‐containing AMPARs by 3 days. The timing of the plasticity induced by the brief nociceptive stimulus is longer than LTP, although this is consistent with arthritis‐induced and possibly acid‐induced plasticity at the PB‐CeLC synapse. An arthritis model increases the responsiveness of CeA neurons to parabrachial stimulation progressively, with increased responsiveness of CeA neurons by 3–4 h and maximal increases 6 h after initiation of arthritis (Neugebauer & Li, 2003). Whether acid‐induced plasticity of this synapse is also slowly developing is less clear. PB‐CeLC plasticity occurs 2 h after the second of two acid injections spaced 3 days apart (Cheng et al. 2011). This synapse was not potentiated after a single acid injection, presumably also measured 2 h after the acid injection (Chen et al. 2014b). It is possible that the acid‐induced plasticity requires two stimuli or that, as observed in the present study and as also occurs with arthritis, it develops slowly after a single acid injection and needs longer than 2 h to develop. This arthritis pain model, which presumably stimulates ongoing activation of the SC‐PB‐CeLC pathway, and the acid induced nociceptive stimuli are very different from the brief nociceptive stimulus used in the present study. Therefore, this makes comparisons of the time course or intensity of stimulation difficult; however, the slow developing nature of the plasticity in response to arthritis and the brief nociceptive stimulus (and possibly acid) suggests that pain‐induced synaptic plasticity at the PB‐CeLC synapse follows a slower time course than LTP. Given this slower development, it is possible that the plasticity relies of synthesis of new proteins, similar to corticosterone induced increase in AMPA receptors in stress (Groc et al. 2008; Martin et al. 2009). If the pain hypersensitivity seen after 1 day is a result of the plastic changes in the CeLC, this may also explain why it is also delayed. At 1 day after the pain stimulus, GluA2‐lacking receptors are present at the PB‐CeLC synapse but, between 1 and 3 days after the heat stimulus, the AMPA receptor subunits revert to the GluA2‐containing receptors present under control conditions. This is consistent with GluA2‐lacking receptors being replaced with GluA2‐containing receptors as the cell surface receptors undergo regular turnover over 18–43 h (Mammen et al. 1997; Archibald et al. 1998; O'Brien et al. 1998)
The role of the PB‐CeLC neural pathway in acute behavioural responses to nociceptive stimuli, such as mechanical and thermal thresholds (Han et al. 2015; Sato et al. 2015), is limited but the CeA is important for the development of hyperalgesia in various pain states (Hebert et al. 1999; Han et al. 2005; Carrasquillo & Gereau, 2007; Pedersen et al. 2007; Fu & Neugebauer, 2008; Ansah et al. 2010; Ji et al. 2010). This could be via CeLC projections to the CeM, (Jolkkonen & Pitkanen, 1998; Ciocchi et al. 2010; Haubensak et al. 2010), which in turn projects to the periaqueductal gray (Haubensak et al. 2010). The periaqueductal gray controls the descending analgesic pathway (Bushnell et al. 2013; Veinante et al. 2013) and thus PB‐CeLC synaptic plasticity could also mediate/alter the descending modulation of pain. Therefore, the hyperalgesia that we observed 1 day after the nociceptive stimulus may result from differential activation of these pain modulatory pathways when the PB‐CeLC potentiation is in place. However, the hyperalgesia could also be a result of changes at other points in the pain pathway, such as the spinal cord.
CeA (including CeLC) neurons have large bilateral receptive fields and display a sigmoid‐like stimulus–response curve to noxious stimuli (Bernard et al. 1992; Neugebauer & Li, 2002). They initially produce a graded response to increased stimulus intensity, which reaches a plateau, where increases in stimulus intensity do not produce a subsequent increase in response. Although these characteristics would not allow appropriate sensory‐discrimination of pain, they are consistent with the contribution of the PB‐CeLC neural pathway to pain‐induced negative affect (Han et al. 2015), associative learning (Han et al. 2015; Sato et al. 2015) or regulation of pain hypersensitivity (Hebert et al. 1999; Han et al. 2005; Carrasquillo & Gereau, 2007; Pedersen et al. 2007; Fu & Neugebauer, 2008; Ansah et al. 2010; Ji et al. 2010). During the 3 days when we have shown that there are more AMPA receptors at the PB‐CELC synapses, a given nociceptive stimulus would produce an unchanged release of glutamate from PB terminals in the CeLC but a stronger postsynaptic depolarization of CeLC. In addition, during the first phase after the stimulus, the increased incorporation of calcium permeable GluA2‐lacking AMPAR could act as an additional source for activity‐dependent calcium entry and thus facilitate subsequent synaptic plasticity (Plant et al. 2006; Guire et al. 2008; Chater & Goda, 2014; Morita et al. 2014). Thus, if a second nociceptive stimulus was delivered during that period, greater activation of this pathway would be expected. Consistent with this, we found that a second nociceptive stimulus delivered when there was high GluA2‐lacking AMPAR incorporation prolongs the potentiation of the nociceptive information flow into the amygdala for at least 1 week. Given the role of the PB‐CeLC synapse in pain associated negative affect and pain‐induced associative learning, both of these aspects of the pain experience could be enhanced during this week. It remains an open question whether additional nociceptive stimuli during this time could prolong this synaptic potentiation even further. The associative learning connecting a nociceptive/noxious stimulus with the environment relies in part on the PB‐CeLC neural pathway (Han et al. 2015; Sato et al. 2015) and the CeA (Tanimoto et al. 2003; Gao et al. 2004; Pedersen et al. 2007; Ansah et al. 2010) but the underlying neural plasticity is not defined. One possibility is PB‐CeLC assisted potentiation of other polymodal sensory inputs to the CeA (Canteras et al. 1994; Moga et al. 1995; McDonald et al. 1996; McDonald & Mascagni, 1997; Sah et al. 2003; Vertes & Hoover, 2008). However, we found that the nociceptive stimulus did not change the AMPA/NMDA ratio for mixed inputs entering dorsal to the CeA, which contains some of the polymodal inputs to the CeLC (Canteras et al. 1994; Moga et al. 1995; McDonald et al. 1996; McDonald & Mascagni, 1997; Vertes & Hoover, 2008). This may be because the relevant synaptic plasticity for associative learning does not comprise change in AMPA/NMDA ratio, or it occurs at other synapses or even did not happen in these experiments because the animals had minimal sensory input during the nociceptive stimulus as a result of anaesthesia.
Chronic pain states without an ongoing injury, such as lower back pain, are a significant clinical problem (Atkinson, 2004) and this probably is a result of complex change in multiple neural circuits. The present study used a brief temporally controlled stimulus without ongoing damage to investigate why pain persists without an ongoing injury. The tight temporal control of the nociceptive stimulus allowed us to demonstrate that noxious stimuli potentiated the delivery of nociceptive information into the amygdala for several days after the stimulus. Without a subsequent nociceptive stimulus, this postsynaptic plasticity will not increase activation of the nociceptive amygdala and associated pain behaviours. However, when a subsequent injury activates the spinal cord‐parabrachial‐amygdala pathway, the increased responsiveness of the nociceptive amygdala neurons that we observed could enhance the pain responses, as seen with synaptic potentiation of this synapse in chronic pain states (Han & Neugebauer, 2004; Ikeda et al. 2007; Fu et al. 2008; Adedoyin et al. 2010)
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
EEB conceived the study. EEB and SAK designed the study. EEB and SAK conducted the electrophysiology experiments. SAK conducted the immunohistochemistry assays and confocal microscopy. SAK conducted the paw volume displacement experiments. SAK and EEB wrote and edited the manuscript. All authors approved the final version of the manuscript submitted for publication.
Funding
SAK was supported by the Australian Pain Society/Australian Pain Relief Association/Janssen‐Cilag PhD scholarship. SAK and EEB were supported by National Health and Medical Research Council (NHMRC) project grants APP1047372, APP1077806, Bosch Institute Bishop Fellowship and USYD Thompson Fellowship.
Acknowledgements
We would like to acknowledge the support received from the Bosch Institute Advanced Microscope Facility and the expert help of facility staff, especially Dr Louise Cole. We would also like to acknowledge the veterinary pathology diagnostic service at the University of Sydney particularly Elaine Chew for all their help with the histology experiments. We also thank Dr Mark Krockenberger for taking the time to analyse the histology images.
Biography
Sarah A Kissiwaa graduated from Charles Sturt University with a Bachelor of Pharmacy (Hons) degree. After completing her degree, she undertook a PhD at the University of Sydney, which she completed in 2018. The research reported in the present study is her first research article originating from her PhD. Her goal is to increase our understanding of how the brain functions and changes in different disease states and it is hoped that the work presented here will further our knowledge of how the brain changes in pain.

Edited by: Jaideep Bains & David Adams
References
- Abraham WC. (2008). Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci 9, 387. [DOI] [PubMed] [Google Scholar]
- Adedoyin MO, Vicini S & Neale JH. (2010). Endogenous N‐acetylaspartylglutamate (NAAG) inhibits synaptic plasticity/transmission in the amygdala in a mouse inflammatory pain model. Mol Pain 6, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adriaensen H, Gybels J, Handwerker HO & Van Hees J. (1984). Suppression of C‐fibre discharges upon repeated heat stimulation may explain characteristics of concomitant pain sensations. Brain Res 302, 203–211. [DOI] [PubMed] [Google Scholar]
- Ansah OB, Bourbia N, Goncalves L, Almeida A & Pertovaara A. (2010). Influence of amygdaloid glutamatergic receptors on sensory and emotional pain‐related behavior in the neuropathic rat. Behav Brain Res 209, 174–178. [DOI] [PubMed] [Google Scholar]
- Archibald K, Perry MJ, Molnar E & Henley JM. (1998). Surface expression and metabolic half‐life of AMPA receptors in cultured rat cerebellar granule cells. Neuropharmacology 37, 1345–1353. [DOI] [PubMed] [Google Scholar]
- Atkinson JH. (2004). Chronic back pain: searching for causes and cures. J Rheumatol 31, 2323–2325. [PubMed] [Google Scholar]
- Baliki MN, Chialvo DR, Geha PY, Levy RM, Harden RN, Parrish TB & Apkarian AV. (2006). Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J Neurosci 26, 12165–12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G & Julius D. (2009). Cellular and molecular mechanisms of pain. Cell 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard JF, Alden M & Besson JM. (1993). The organization of the efferent projections from the pontine parabrachial area to the amygdaloid complex: a Phaseolus vulgaris leucoagglutinin (PHA‐L) study in the rat. J Comp Neurol 329, 201–229. [DOI] [PubMed] [Google Scholar]
- Bernard JF, Huang GF & Besson JM. (1990). Effect of noxious somesthetic stimulation on the activity of neurons of the nucleus centralis of the amygdala. Brain Res 523, 347–350. [DOI] [PubMed] [Google Scholar]
- Bernard JF, Huang GF & Besson JM. (1992). Nucleus centralis of the amygdala and the globus pallidus ventralis: electrophysiological evidence for an involvement in pain processes. J Neurophysiol 68, 551–569. [DOI] [PubMed] [Google Scholar]
- Bester H, Matsumoto N, Besson JM & Bernard JF. (1997). Further evidence for the involvement of the spinoparabrachial pathway in nociceptive processes: a c‐Fos study in the rat. J Comp Neurol 383, 439–458. [PubMed] [Google Scholar]
- Bornhovd K, Quante M, Glauche V, Bromm B, Weiller C & Buchel C. (2002). Painful stimuli evoke different stimulus‐response functions in the amygdala, prefrontal, insula and somatosensory cortex: a single‐trial fMRI study. Brain 125, 1326–1336. [DOI] [PubMed] [Google Scholar]
- Bourgeais L, Gauriau C & Bernard JF. (2001). Projections from the nociceptive area of the central nucleus of the amygdala to the forebrain: a PHA‐L study in the rat. Eur J Neurosci 14, 229–255. [DOI] [PubMed] [Google Scholar]
- Bushnell MC, Ceko M & Low LA. (2013). Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci 14, 502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canteras NS, Simerly RB & Swanson LW. (1994). Organization of projections from the ventromedial nucleus of the hypothalamus: a Phaseolus vulgaris‐leucoagglutinin study in the rat. J Comp Neurol 348, 41–79. [DOI] [PubMed] [Google Scholar]
- Cardinal RN, Parkinson JA, Hall J & Everitt BJ. (2002). Emotion and motivation: the role of the amygdala, ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev 26, 321–352. [DOI] [PubMed] [Google Scholar]
- Carrasquillo Y & Gereau RWt. (2007). Activation of the extracellular signal‐regulated kinase in the amygdala modulates pain perception. J Neurosci 27, 1543–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM & Yaksh TL. (1994). Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53, 55–63. [DOI] [PubMed] [Google Scholar]
- Chater TE & Goda Y. (2014). The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front Cell Neurosci 8, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Koga K, Descalzi G, Qiu S, Wang J, Zhang LS, Zhang ZJ, He XB, Qin X, Xu FQ, Hu J, Wei F, Huganir RL, Li YQ & Zhuo M. (2014a). Postsynaptic potentiation of corticospinal projecting neurons in the anterior cingulate cortex after nerve injury. Mol Pain 10, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WN, Lee CH, Lin SH, Wong CW, Sun WH, Wood JN & Chen CC. (2014b). Roles of ASIC3, TRPV1, and NaV1.8 in the transition from acute to chronic pain in a mouse model of fibromyalgia. Mol Pain 10, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SJ, Chen CC, Yang HW, Chang YT, Bai SW, Chen CC, Yen CT & Min MY. (2011). Role of extracellular signal‐regulated kinase in synaptic transmission and plasticity of a nociceptive input on capsular central amygdaloid neurons in normal and acid‐induced muscle pain mice. J Neurosci 31, 2258–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng BC, Christie MJ & Osborne PB. (2006). Characterization of neurons in the rat central nucleus of the amygdala: cellular physiology, morphology, and opioid sensitivity. J Comp Neurol 497, 910–927. [DOI] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SB, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, Muller C & Luthi A. (2010). Encoding of conditioned fear in central amygdala inhibitory circuits. Nature 468, 277–282. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Katz J, Vaccarino AL & Melzack R. (1993). Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain 52, 259–285. [DOI] [PubMed] [Google Scholar]
- Dahl JB, Brennum J, Arendt‐Nielsen L, Jensen TS & Kehlet H. (1993). The effect of pre‐ versus postinjury infiltration with lidocaine on thermal and mechanical hyperalgesia after heat injury to the skin. Pain 53, 43–51. [DOI] [PubMed] [Google Scholar]
- Elman I & Borsook D. (2016). Common brain mechanisms of chronic pain and addiction. Neuron 89, 11–36. [DOI] [PubMed] [Google Scholar]
- Fu Y, Han J, Ishola T, Scerbo M, Adwanikar H, Ramsey C & Neugebauer V. (2008). PKA and ERK, but not PKC, in the amygdala contribute to pain‐related synaptic plasticity and behavior. Mol Pain 4, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y & Neugebauer V. (2008). Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain‐related synaptic facilitation and behavior. J Neurosci 28, 3861–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ & Ji RR. (2009). c‐Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? Open Pain J 2, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ren WH, Zhang YQ & Zhao ZQ. (2004). Contributions of the anterior cingulate cortex and amygdala to pain‐ and fear‐conditioned place avoidance in rats. Pain 110, 343–353. [DOI] [PubMed] [Google Scholar]
- Groc L, Choquet D & Chaouloff F. (2008). The stress hormone corticosterone conditions AMPAR surface trafficking and synaptic potentiation. Nat Neurosci 11, 868–870. [DOI] [PubMed] [Google Scholar]
- Grundy D. (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guire ES, Oh MC, Soderling TR & Derkach VA. (2008). Recruitment of calcium‐permeable AMPA receptors during synaptic potentiation is regulated by CaM‐kinase I. J Neurosci 28, 6000–6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gureje O, Von Korff M, Simon GE & Gater R. (1998). Persistent pain and well‐being: a World Health Organization Study in Primary Care. JAMA 280, 147–151. [DOI] [PubMed] [Google Scholar]
- Han JS, Li W & Neugebauer V. (2005). Critical role of calcitonin gene‐related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J Neurosci 25, 10717–10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS & Neugebauer V. (2004). Synaptic plasticity in the amygdala in a visceral pain model in rats. Neurosci Lett 361, 254–257. [DOI] [PubMed] [Google Scholar]
- Han S, Soleiman MT, Soden ME, Zweifel LS & Palmiter RD. (2015). Elucidating an affective pain circuit that creates a threat memory. Cell 162, 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C & Joris J. (1988). A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32, 77–88. [DOI] [PubMed] [Google Scholar]
- Harrigan EA, Magnuson DJ, Thunstedt GM & Gray TS. (1994). Corticotropin releasing factor neurons are innervated by calcitonin gene‐related peptide terminals in the rat central amygdaloid nucleus. Brain Res Bull 33, 529–534. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong HW, Deisseroth K, Callaway EM, Fanselow MS, Luthi A & Anderson DJ. (2010). Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature 468, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert MA, Ardid D, Henrie JA, Tamashiro K, Blanchard DC & Blanchard RJ. (1999). Amygdala lesions produce analgesia in a novel, ethologically relevant acute pain test. Physiol Behav 67, 99–105. [DOI] [PubMed] [Google Scholar]
- Ikeda R, Takahashi Y, Inoue K & Kato F. (2007). NMDA receptor‐independent synaptic plasticity in the central amygdala in the rat model of neuropathic pain. Pain 127, 161–172. [DOI] [PubMed] [Google Scholar]
- Ji G, Fu Y, Adwanikar H & Neugebauer V. (2013). Non‐pain‐related CRF1 activation in the amygdala facilitates synaptic transmission and pain responses. Mol Pain 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Sun H, Fu Y, Li Z, Pais‐Vieira M, Galhardo V & Neugebauer V. (2010). Cognitive impairment in pain through amygdala‐driven prefrontal cortical deactivation. J Neurosci 30, 5451–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA & Woolf CJ. (2003). Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci 26, 696–705. [DOI] [PubMed] [Google Scholar]
- Jolkkonen E & Pitkanen A. (1998). Intrinsic connections of the rat amygdaloid complex: projections originating in the central nucleus. J Comp Neurol 395, 53–72. [DOI] [PubMed] [Google Scholar]
- Kruger L, Sternini C, Brecha NC & Mantyh PW. (1988). Distribution of calcitonin gene‐related peptide immunoreactivity in relation to the rat central somatosensory projection. J Comp Neurol 273, 149–162. [DOI] [PubMed] [Google Scholar]
- LaMotte RH & Campbell JN. (1978). Comparison of responses of warm and nociceptive C‐fiber afferents in monkey with human judgments of thermal pain. J Neurophysiol 41, 509–528. [DOI] [PubMed] [Google Scholar]
- Latremoliere A & Woolf CJ. (2009). Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 10, 895–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE. (2000). Emotion circuits in the brain. Annu Rev Neurosci 23, 155–184. [DOI] [PubMed] [Google Scholar]
- Li H, Penzo MA, Taniguchi H, Kopec CD, Huang ZJ & Li B. (2013). Experience‐dependent modification of a central amygdala fear circuit. Nat Neurosci 16, 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Huganir RL & O'Brien RJ. (1997). Redistribution and stabilization of cell surface glutamate receptors during synapse formation. J Neurosci 17, 7351–7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek R, Strobel C, Bredy TW & Sah P. (2013). The amygdala and medial prefrontal cortex: partners in the fear circuit. J Physiol 591, 2381–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin HA, Basbaum AI, Goetzl EJ & Levine JD. (1988). Leukotriene B4 decreases the mechanical and thermal thresholds of C‐fiber nociceptors in the hairy skin of the rat. J Neurophysiol 60, 438–445. [DOI] [PubMed] [Google Scholar]
- Martin S, Henley JM, Holman D, Zhou M, Wiegert O, van Spronsen M, Joels M, Hoogenraad CC & Krugers HJ. (2009). Corticosterone alters AMPAR mobility and facilitates bidirectional synaptic plasticity. PLoS ONE 4, e4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ & Mascagni F. (1997). Projections of the lateral entorhinal cortex to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience 77, 445–459. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F & Guo L. (1996). Projections of the medial and lateral prefrontal cortices to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience 71, 55–75. [DOI] [PubMed] [Google Scholar]
- Merskey H & Bogduk N (1994) Part III: Pain terms, A Current List with Definitions and Notes on Usage In Classification of Chronic Pain, Merskey H. & Bogduk N. (Eds.) (2nd edn, pp 209–214), IASP Press, Seattle. [Google Scholar]
- Moga MM, Weis RP & Moore RY. (1995). Efferent projections of the paraventricular thalamic nucleus in the rat. J Comp Neurol 359, 221–238. [DOI] [PubMed] [Google Scholar]
- Morita D, Rah JC & Isaac JT. (2014). Incorporation of inwardly rectifying AMPA receptors at silent synapses during hippocampal long‐term potentiation. Philos Trans R Soc Lond B Biol Sci 369, 20130156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V. (2015). Amygdala pain mechanisms. Handb Exp Pharmacol 227, 261–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V & Li W. (2002). Processing of nociceptive mechanical and thermal information in central amygdala neurons with knee‐joint input. J Neurophysiol 87, 103–112. [DOI] [PubMed] [Google Scholar]
- Neugebauer V & Li W. (2003). Differential sensitization of amygdala neurons to afferent inputs in a model of arthritic pain. J Neurophysiol 89, 716–727. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Bhave G & RWt Gereau. (2003). Synaptic plasticity in the amygdala in a model of arthritic pain: differential roles of metabotropic glutamate receptors 1 and 5. J Neurosci 23, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD & Huganir RL. (1998). Activity‐dependent modulation of synaptic AMPA receptor accumulation. Neuron 21, 1067–1078. [DOI] [PubMed] [Google Scholar]
- Oliver KR, Wainwright A, Heavens RP, Hill RG & Sirinathsinghji DJ. (1998). Distribution of novel CGRP1 receptor and adrenomedullin receptor mRNAs in the rat central nervous system. Mol Brain Res 57, 149–154. [DOI] [PubMed] [Google Scholar]
- Pape HC & Pare D. (2010). Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev 90, 419–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G & Watson C. (1986). The Rat Brain in Stereotaxic Coordinates. Academic Press, Sydney. [Google Scholar]
- Pedersen LH, Scheel‐Kruger J & Blackburn‐Munro G. (2007). Amygdala GABA‐A receptor involvement in mediating sensory‐discriminative and affective‐motivational pain responses in a rat model of peripheral nerve injury. Pain 127, 17–26. [DOI] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL & Isaac JT. (2006). Transient incorporation of native GluR2‐lacking AMPA receptors during hippocampal long‐term potentiation. Nat Neurosci 9, 602–604. [DOI] [PubMed] [Google Scholar]
- Rainville P, Duncan GH, Price DD, Carrier B & Bushnell MC. (1997). Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science 277, 968–971. [DOI] [PubMed] [Google Scholar]
- Raja SN, Campbell JN & Meyer RA. (1984). Evidence for different mechanisms of primary and secondary hyperalgesia following heat injury to the glabrous skin. Brain 107, 1179–1188. [DOI] [PubMed] [Google Scholar]
- Rao VR & Finkbeiner S. (2007). NMDA and AMPA receptors: old channels, new tricks. Trends Neurosci 30, 284–291. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M & Power J. (2003). The amygdaloid complex: anatomy and physiology. Physiol Rev 83, 803–834. [DOI] [PubMed] [Google Scholar]
- Sato M, Ito M, Nagase M, Sugimura YK, Takahashi Y, Watabe AM & Kato F. (2015). The lateral parabrachial nucleus is actively involved in the acquisition of fear memory in mice. Mol Brain 8, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouenborg J. (1984). Functional and topographical properties of field potentials evoked in rat dorsal horn by cutaneous C‐fibre stimulation. J Physiol 356, 169–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada S, Shiosaka S, Emson PC, Hillyard CJ, Girgis S, MacIntyre I & Tohyama M. (1985). Calcitonin gene‐related peptidergic projection from the parabrachial area to the forebrain and diencephalon in the rat: an immunohistochemical analysis. Neuroscience 16, 607–616. [DOI] [PubMed] [Google Scholar]
- Suwanprathes P, Ngu M, Ing A, Hunt G & Seow F. (2003). c‐Fos immunoreactivity in the brain after esophageal acid stimulation. Am J Med 115, 31s‐38s. [DOI] [PubMed] [Google Scholar]
- Tanimoto S, Nakagawa T, Yamauchi Y, Minami M & Satoh M. (2003). Differential contributions of the basolateral and central nuclei of the amygdala in the negative affective component of chemical somatic and visceral pains in rats. Eur J Neurosci 18, 2343–2350. [DOI] [PubMed] [Google Scholar]
- van Rossum D, Hanisch UK & Quirion R. (1997). Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci Biobehav Rev 21, 649–678. [DOI] [PubMed] [Google Scholar]
- Veinante P, Yalcin I & Barrot M. (2013). The amygdala between sensation and affect: a role in pain. J Mol Psychiatry 1, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP & Hoover WB. (2008). Projections of the paraventricular and paratenial nuclei of the dorsal midline thalamus in the rat. J Comp Neurol 508, 212–237. [DOI] [PubMed] [Google Scholar]
- Vlaeyen JW. (2015). Learning to predict and control harmful events: chronic pain and conditioning. Pain 156, S86‐93. [DOI] [PubMed] [Google Scholar]
- Watabe AM, Ochiai T, Nagase M, Takahashi Y, Sato M & Kato F. (2013). Synaptic potentiation in the nociceptive amygdala following fear learning in mice. Mol Brain 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ. (2011). Central sensitization: implications for the diagnosis and treatment of pain. Pain 152, S2‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeomans DC & Proudfit HK. (1996). Nociceptive responses to high and low rates of noxious cutaneous heating are mediated by different nociceptors in the rat: electrophysiological evidence. Pain 68, 141–150. [DOI] [PubMed] [Google Scholar]
- Zhang XJ, Zhang TW, Hu SJ & Xu H. (2011). Behavioral assessments of the aversive quality of pain in animals. Neurosci Bull 27, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]