

Abstract
Key points
Referring to the muscle memory theory, previously trained muscles acquire strength and volume much faster than naive muscles.
Using extreme experimental models such as synergist ablation or steroid administration, previous studies have demonstrated that the number of nuclei increases when a muscle becomes enlarged, which serves as a cellular muscle memory mechanism for the muscle.
In the present study, we found that, when rats were subjected to physiologically relevant resistance training, the number of myonuclei increased and was retained during a long‐term detraining period.
The acquired myonuclei were related to a greater degree of muscle hypertrophic and mitochondrial biogenesis processes following subsequent hypertrophic conditions.
Our data suggest a cellular mechanism supporting the notion that exposing young muscles to resistance training would help to restore age‐related muscle loss coupled with mitochondrial dysfunction in later life.
Abstract
Muscle hypertrophy induced by resistance training is accompanied by an increase in the number of myonuclei. The acquired myonuclei are viewed as a cellular component of muscle memory by which muscle enlargement is promoted during a re‐training period. In the present study, we investigated the effect of exercise preconditioning on mitochondrial remodelling induced by resistance training. Sprague–Dawley rats were divided into four groups: untrained control, training, pre‐training or re‐training. The training groups were subjected to weight loaded‐ladder climbing exercise training. Myonuclear numbers were significantly greater (up to 20%) in all trained muscles compared to untrained controls. Muscle mass was significantly higher in the re‐training group compared to the training group (∼2‐fold increase). Mitochondrial content, mitochondrial biogenesis gene expression levels and mitochondrial DNA copy numbers were significantly higher in re‐trained muscles compared to the others. Oxidative myofibres (type I) were significantly increased only in the re‐trained muscles. Furthermore, in vitro studies using insulin‐like growth factor‐1‐treated L6 rat myotubes demonstrated that myotubes with a higher myonuclear number confer greater expression levels of both mitochondrial and nuclear genes encoding for constitutive and regulatory mitochondrial proteins, which also showed a greater mitochondrial respiratory function. These data suggest that myonuclei acquired from previous training facilitate mitochondrial biogenesis in response to subsequent retraining by (at least in part) enhancing cross‐talk between mitochondria and myonuclei in the pre‐conditioned myofibres.
Keywords: muscle memory, myonuclei, mitochondrial biogenesis, resistance training
Key points
Referring to the muscle memory theory, previously trained muscles acquire strength and volume much faster than naive muscles.
Using extreme experimental models such as synergist ablation or steroid administration, previous studies have demonstrated that the number of nuclei increases when a muscle becomes enlarged, which serves as a cellular muscle memory mechanism for the muscle.
In the present study, we found that, when rats were subjected to physiologically relevant resistance training, the number of myonuclei increased and was retained during a long‐term detraining period.
The acquired myonuclei were related to a greater degree of muscle hypertrophic and mitochondrial biogenesis processes following subsequent hypertrophic conditions.
Our data suggest a cellular mechanism supporting the notion that exposing young muscles to resistance training would help to restore age‐related muscle loss coupled with mitochondrial dysfunction in later life.
Introduction
A muscle fibre represents one of the very few post‐mitotic syncytial cells found in vertebrates, in which each myofibre contains hundreds of nuclei distributed along the length of the cell (Bruusgaard et al. 2003). An individual myonucleus in the muscle cell is responsible for synthesis of the organelle, membrane and structural protein products localized in its proximal vicinity, suggesting nuclear mosaicism or a nuclear domain (Hall & Ralston, 1989; Pavlath et al. 1989; Ralston & Hall, 1992). During overload hypertrophy, extra nuclei are added from interstitial muscle stem cells (or satellite cells) that are activated and fused to existing muscle fibres. Referring to the karyoplasmic ratio hypothesis, the extra nuclei lead to the maintainance of the size of nuclear domain during muscle hypertrophy (Bruusgaard et al. 2010).
There have been indications that previously trained muscles reacquire some parameters (i.e. fibre area, maximal dynamic strength) at an accelerated rate when re‐subjected to exercise even after a prolonged detraining period (Staron et al. 1991; Taaffe & Marcus, 1997). This phenomenone, so‐called ‘muscle memory’, was originally perceived to be the result of a distinct motor learning capacity in the CNS (Rutherford & Jones, 1986). Recently, using an in vivo imaging technique, several studies have demonstrated that myonuclei are resistant to apoptosis and remain elevated in atrophied muscles (Bruusgaard & Gundersen, 2008; Bruusgaard et al. 2010; Egner et al. 2013) and also that they may serve as biological mediators of the muscle memory in previously trained myofibres (Bruusgaard et al. 2010). Nonetheless, the implications of muscle memory function in the context of physiologically feasible resistance exercise and its influence on mitochondrial adaptations remain unknown.
Mitochondria are dynamic organelles that are crucial not only for producing energy, but also for regulation of cell proliferation and growth (Liesa et al. 2009; Kim et al. 2013). We previously showed that proper mitochondrial remodelling is a necessary component for successful myogenic processes (Kim et al. 2013). More than 1000 mitochondrial‐localized proteins have been identified (Calvo et al. 2016). For successful mitochondrial biogenesis, these proteins may need to be co‐ordinately synthesized and imported into mitochondria. Thus, we speculated that increasing the myonuclei to muscle cell ratio in hypertrophic muscle would positively influence mitochondrial biogenesis by enhancing the mitochondria–nucleus network.
Skeletal muscle consists of multinucleated fibres with high plasticity, which responds to various types of overloading and unloading stimuli (Bolster et al. 2004; Harber et al. 2012). Resistance exercise increases myofibril expression levels (Chesley et al. 1992; Phillips et al. 1997; Kumar et al. 2009), inducing IIb to IIa fibre expression switching (Wang et al. 1993; Manfredi et al. 2013). Moreover, the expression of peroxisome proliferator‐activated receptor‐γ coactivator 1α (PGC‐1α), a key regulator of mitochondrial biogenesis, is also significantly up‐regulated after resistance exercise (Burd et al. 2012), suggesting mitochondrial adaptations. Therefore, the present study aimed to investigate the potential relevance of an increased number of myonuclei in previously trained muscle to the acquisition of muscle mass and mitochondrial remodelling following subsequent retraining. We hypothesized that: (i) resistance training would increase the number of myonuclei; (ii) myonuclei would be retained during a detraining period; and (iii) when re‐subjected to resistance training, the pre‐trained muscles would confer a greater degree of hypertrophic and mitochondrial adaptations compared to untrained muscles.
Methods
Ethical approval
The animal experiment was approved by Texas A&M International University's Animal Care and Use Committee.
Animals
Thirty‐two female Sprague–Dawley rats (Hilltop Lab Animals, Scottdale, PA, USA) were used. All animals were randomly assigned into four groups (n = 8 each group): control group (CON), training group (TR), pre‐training/detraining group (PRE), and retraining group (RT). A schematic diagram of each condition is provided in Fig. 1. Animals were housed in pairs and kept under a standard 12:12 h light/dark cycle. Animals were allowed access to water and food ad libitum.
Figure 1. Experimental design.
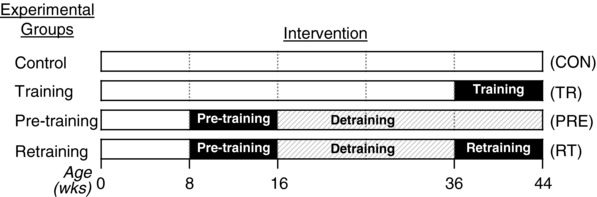
Schematic diagram of the experimental design.
Resistance training and detraining
A weight‐loaded ladder climbing exercise was performed. All training sessions were carried out by experienced investigators. The ladder was 1 m in length with a rung spacing of 2 cm and was inclined at 85°. Acclimation sessions were conducted on three separate days, which involved the animals climbing up the ladder without extra weight loading. For training, a cylindrical holder containing metal pieces was attached to the base of the tail of an animal with foam tape (Coban; 3M, Maplewood, MN, USA) fastened by Velcro stick back strips (VIL Ltd, London, UK). The amount of weight loaded was initially set at 50% of the body weight and gradually increased up to ∼300%. When needed, an investigator brushed the tail of an animal for motivation. The climbing was terminated when an animal neglected to climb up the ladder after three successive attempts. Each training session consisted of three sets of five climbing repetitions. There was a 1 min interval between repetitions and a 2 min interval between sets. Each animal was trained twice a day (09.00 h and 14.00 h) every third day for 8 weeks. An identical training protocol was applied for both pre‐training and re‐training sessions. For detraining, animals were kept in a cage for 20 weeks without training sessions.
Tissue collection
Approximately 48 h after their last training, muscles were collected as described previously (Lee et al. 2004). Prior to muscle collection, rats were initially anaesthetized with isoflurane (5% gas inhalation) followed by i.p. injection of sodium pentobarbital (60 mg kg–1). An incision was made through the skin around ankle area and the skin was reflected to expose the muscles of the lower leg. The Achilles tendon was cut and the plantar flexors reflected proximally when cutting the medial fascia of the posterior compartment. To dissect flexor hallucis longus (FHL) muscle, an incision was made with a scissor through the skin of the foot following a line from the tendon of the muscle 3–5 mm proximal of the third metatarsal. The skin of the foot was then gently reflected until the slips of the FHL muscle tendon became visible. The FHL was excised immediately from the leg, cleaned of excess fat and external connective tissue. To dissect tibialis anterior (TA) muscle, the fascia covering TA muscle was gently removed and the extensor ligament cut to release the distal TA tendon, which was used to peel off the TA muscle. The muscle samples were rapidly frozen in liquid nitrogen or embedded in OCT compound. The OCT tissue blocks were quickly frozen in isopentane (2‐methylbutane) pre‐chilled in liquid nitrogen. At the end of tissue collection, animals were killed with an intracardiac injection of an overdose of sodium pentobarbital (200 mg kg–1) and death was confirmed by incision of the chest cavity to induce a bilateral pneumothorax. All frozen samples were stored at –80°C until analysed.
Single muscle fibre isolation and the number of myonuclei
Single muscle fibres were prepared using a method previously described with slight modifications (Verma & Asakura, 2011). Briefly, FHL muscles were incubated in pre‐chilled isopropanol for 120 min and washed three times with PBS. Then, the tissues were incubated in dissociation solution containing 0.2% collagenase type I (Sigma‐Aldrich, St Louis, MO, USA) at 37°C until hair‐like single fibres were projected. After dissociation of the single muscle fibres, the enzymatic activity was quenched by adding 2% fetal bovine serum in Dulbecco's modified Eagle's medium. Single muscle fibres were then flushed out of the fibre bundles with heat‐polished, serum‐coated pipettes. Five to 11 single muscle fibres were analysed from each muscle (n = 8 each group). Numbers of myonuclei were determined by 4,6‐diamidino‐2‐phenylindole (DAPI) (300 nm) staining. To test for the existence of satellite cells in single myofibre samples, we also co‐stained for Pax7, a satellite cell‐specific marker, using a monoclonal mouse anti‐Pax7 antibody (dilution 1:50) (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA, USA) in some myofibres (n > 20). For Pax7 staining, myofibre was permeabilized (0.5% Triton X‐100 with 5% heat inactivated horse serum in PBS), then incubated with the Pax7 antibody overnight at 4°C. An Alexa 488 goat anti‐mouse secondary antibody (dilution 1:400) was used for visualization. As a positive staining, we also performed Pax7 immunostaining, co‐stained with laminin and DAPI, on cross‐sections of rat TA muscle with the same protocol described above. For counter‐staining, a rabbit polyclonal anti‐laminin primary antibody (dilution 1:500) (Sigma‐Aldrich) and Alexa 594 goat anti‐rabbit secondary antibody (dilution 1:400) were used. Microscopic images were acquired using a 20× or 63× objective lens (EC Plan‐Neofluar 20×/0.50, WD = 2 and LD Plan‐Neofluar 63×/0.75Ph2Corr, WD = 2.2) on a Zeiss Axio Imager A1 epifluorescence upright microscope system with an AxioCam MRm camera (Carl Zeiss, Oberkochen, Germany). Images were initially processed using the AxioVision image processing system and, when necessary, images were processed further using ImageJ (NIH, Bethesda, MD, USA).
Histochemistry
All histochemical analytic procedures were performed as described previously (Park et al. 2009). For haematoxylin and eosin (H&E) staining, OCT embedded FHL muscles were cut into 10 μm slices. The sections were fixed with 100% ethanol and rinsed with distilled water. After incubation in haematoxylin solution (Thermo Scientific, Waltham, MA, USA) for 8 min, the sections were rinsed with water and ethanol before incubation in eosin Y solution (Thermo Scientific) for 1 min. Subsequently, the sections were rinsed with water and ethanol. Then, the samples were cleared with xylene and mounted with mounting medium. For succinate dehydrogenase (SDH) staining, tissue slides were incubated in a solution containing 0.2 m phosphate buffer (pH 7.6), 100 mm sodium succinate and 1.2 mm nitro blue tetrazolium for 90 min at 37°C. After removing unbound nitro blue tetrazolium by washing with 30% acetone solution, slides were placed inside a chemical hood until dry, and then a cover glass was applied with a drop of Permount histological mounting medium (Fisher Scientific Co., Pittsburgh, PA, USA). For cytochrome c oxidase (COX) staining, sliced tissues were incubated 10 mL of distilled water containing 0.2 m sucrose, 0.2 m phosphate buffer (pH 7.6), 1.4 mm 3.3′‐diaminobenzidine (DAB), 10 mg of cytochrome c and 20 μg of catalase. The reaction was run at room temperature for 60 min and stopped with repeated washing steps using distilled water. Samples were then dehydrated in ascending isopropanol concentrations. Next, samples were cleared with xylene and mounted with mounting medium for microscopic analysis. For microscopic imaging, sample slides were placed on the stage of an upright microscope and images were captured with a 20× objective lens (LD A‐Plan 20×/0.3 Ph1, WD = 4.2) using AxioCam ICc1 and the AxioVision image processing system. Cross‐sectional area (CSA) assessment and densitometry analysis were performed using ImageJ (NIH).
mRNA isolation, cDNA synthesis and quantitative PCR
mRNAs were isolated using a Dynabeads direct kit (Invitrogen, Carlsbad, CA, USA) and cDNA synthesis was performed on poly‐dT magnetic beads by reverse transcription using superscript II (Invitrogen). mRNA expression levels were quantified by real‐time PCR using SYBR green fluorescence. Cycle threshold (Ct) values were normalized to a housekeeping gene, HPRT1. mRNA expression levels of Ndufa1, Ndufv1, Sdha, Cys1, Uqcrc1, Cox17, Atp5O, Nd1, Nd2, Nd5, Atp6, Cytb, Cox2, Myh1, Myh2, Myh4, Myh7 and Hprt genes were assessed. Primer sequences are listed in Table 1.
Table 1.
Primer sequences used in quantitative PCR
| Target genes | Forward (5′‐ to 3′) | Reverse (5′‐ to 3′) |
|---|---|---|
| Ndufa1 | CATCCACAAGTTCACCAACG | TGCACAGCCTTCTAACAGGA |
| Ndufv1 | CATTTGTGCTCTGGGTGATG | GAGAGTGGGCAGCACTTGTT |
| Sdha | TACAAGGTGCGGATTGATGA | AGGAACGGATAGCAGGAGGT |
| Cys1 | GCCCTAATGAGGATGGGAAT | GTGGGAGGTTCACAGTAGCC |
| Uqcrc1 | GATCGAGAAGGAGCGAGATG | GGAGCTTTGTAGTGCCTGCT |
| Cox17 | GCGATCGTTACTGCTTTCCT | GCTCTCATGCACTCCTTGTG |
| Atp5O | CACAGTGACCACAGCGTTTC | GCCTTGCTGAGCTTCTGAAT |
| Nd1 | CTCCCTATTCGGAGCCCTAC | GGAGCTCGATTTGTTTCTGC |
| Nd2 | GAGCAATTATCTCCGAGCTTC | ACTTAATACTGTGAGGGTTGG |
| Nd5 | CCTCAGCTAACAATCTATTCC | CTGTGAGAGGGACTAGATTG |
| Atp6 | ACACCAAAAGGACGAACCTG | ACTGCTAGTGCTATCGGTTG |
| Cytb | CTACGGCTGACTAATCCGATA | CTGTAGCTCCTCAGAATGATA |
| Cox2 | GCTTACAAGACGCCACATCA | GAATGACAGCTGGGAGAATTG |
| Myh1 | GCAACATGGAGGTCATTTCC | TCCAGCTGGCGTGAATATTC |
| Myh2 | GAGACAGTCTCTAAGGCCAA | TCAGCTCCTCAATCTGTTGG |
| Myh4 | CTGCGCTTGATAAGAACCAG | TCTCCAGTTGATCCAGAGAC |
| Myh7 | AGATGGAGATCCAGCTCAGT | GATGGCGATGTTCTCCTTCA |
| Hprt | GACTTGCTCGAGATGTCATG | TACAGTCATAGGAATGGACC |
| CoxII | GCTTACAAGACGCCACATCA | GAATGACAGCTGGGAGAATTG |
| Cytb | CTACGGCTGACTAATCCGATA | CTGTAGCTCCTCAGAATGATA |
| 18s rRNA | CCCAACTTCTTAGAGGGACAAG | GCTGAGCCAGTCAGTGTAG |
Immunohistochemistry
Immunohistochemistry was performed as described previously (Park et al. 2009). Frozen tissue sections were submerged in PBS three times for 5 min and fixed in 10% neutral buffered formalin for 10 min. Following antigen retrieval with citrate buffer (sodium citrate, 10 mm, pH 6.0) in a boiling water bath for 10 min, the sections were washed with distilled water (three times for 3 min each). The sections were submerged in methanol containing 0.3% H2O2 for 20 min and washed twice with PBS for 3 min each and then with distilled water (once for 3 min). The sections were blocked with 5% normal goat serum for 60 min at room temperature in a humidified chamber. The primary monoclonal antibody (VDAC; Invitrogen) was applied to the sections overnight at 4°C in a humidified chamber. The sections were rinsed in PBS (three times for 5 min each) and then incubated for 30 min with an alkaline phosphatase‐streptavidin conjugate (Vector Laboratories, Inc., Burlingame, CA, USA). VDAC antibody binding was visualized using a DAB reaction (Vector kit; Vector Laboratories), which produces a brown precipitate. Following development, the sections were counter‐stained with haematoxylin. Microscopic images were acquired and processed with respect to histochemistry as described above.
Immunoblotting
Immunoblotting was performed as described previously (Park et al. 2009). Briefly, FHL muscles were homogenized and lysed in Ripa buffer (10 mm Tris‐HCl, 5 mm EDTA, 150 mm NaCl, 1% Triton X‐100, 0.1% SDS, 1% deoxycholate, pH 7.5). Following centrifugation (16,000 g for 15 min at 4°C), supernatants were collected and subjected to a Bradford assay to quantify protein concentrations. The resulting protein samples were subjected to SDS‐PAGE and transferred to a polyvinylidene difluoride membrane. Subsequently, the membrane was blocked with 5% non‐fat dry milk in Tris‐buffered saline‐Tween 20 (TBST) for 20 min at room temperature and incubated overnight with respective primary antibodies. Antibodies were purchased from the following sources: rabbit polyclonal anti‐PGC‐1α (Novus Biologicals, Littleton, CO, USA), mouse polyclonal DLP1 (BD Transduction Laboratories, Lexington, KT, USA), mouse monoclonal Mfn2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal Fis1 (Alexis Biochemicals, Lausen, Switzerland) and mouse monoclonal α‐tubulin (Sigma‐Aldrich). The membranes were then washed twice in TBST and incubated with HRP‐conjugated secondary antibodies for 1 h followed by washing three times with TBST. Then, membranes were subjected to standard enhanced chemiluminescence (Thermo Fisher Scientific Inc., Waltham, MA, USA) for visualization.
Mitichondrial DNA (mtDNA) copy number quantification
mtDNA copy numbers were assessed as described previously (Kim et al. 2013). Briefly, nuclear and mtDNA were isolated from FHL muscles using the QIAamp DNA Mini Kit (Qiagen, Valancia, CA, USA). mtDNA copy number was assessed by real‐time RT‐PCR. The relative ratio between mitochondrial DNA (COX II, cytochrome c oxidase II; Cytb, cytochrome b) compared to nuclear DNA (18s rRNA) was calculated. Primer sequences are listed in Table 1.
L6 differentiation and generation of myonuclei‐enriched myotubes
L6 rat myoblasts were grown in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum at 37°C (5% CO2). When L6 myoblasts were confluent (95%), myotube differentiation was induced by serum deprivation (2% horse serum) in differentiation media (DM) for 48 h. DM was changed every 24 h. To generate myonuclei‐enriched myotubes, the myotubes were treated further with insulin‐like growth factor (IGF‐1) (range 0–100 ng mL–1) in DM for 48 h. After discarding IGF‐1, the myotubes were treated with 1 mm 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) (or DMSO control) for 6 or 24 h. Myonuclear density was calculated by myonuclei counts within a given area covered by myotubes and expressed as the percentage value adjusted to no treatment control.
Mitochondrial oxygen consumption measures
Mitochondrial oxygen consumption rate was measured using the Seahorse Bioscience XF96 Analyzer (Agilent Technologies, Santa Clara, CA, USA). All procedures were performed in accordance with the manufacturer's instructions (Miller et al. 2014). Briefly, L6 myoblasts were seeded in XF96 cell culture microplates at an optimized seeding density of 2 × 103 cells per well. Myotube differentiation and myonuclei‐enriched myotubes generation were performed as described above. Then, a subset of myotubes was incubated with 1 mm AICAR, an exercise mimetic, for 24 h. Prior to the XF assays, 175 μL of XF assay medium supplemented with 25 mm glucose and 1 mm sodium pyruvate (pH 7.4) was added to each well and then the plates were placed in a 37°C incubator without CO2 for 2 h. For the XF assays, after measuring the basal oxygen consumption rate, cells were sequentially exposed to antimycin A plus rotenone (complexes III and I inhibitors, respectively; 0.5 μm each), carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone (uncoupler; 1.25 μm) and oligomycin (complex V inhibitor; 1 μm). Protein concentrations measured by the BCA method (Thermo Fisher Scientific Inc.) were used to adjust mitochondrial oxygen consumption rates to cell density. Batch‐to‐batch variation was adjusted by basal respiration rates.
Statistical analysis
Analysis was performed using SAS, version 9.4 (SAS Institute, Cary, NC, USA). All data are reported as the mean ± SD. Repeated measure analysis of variance (ANOVA) was performed to determine a significant overall group effect on the number of myonuclei per single myofibre. A linear mixed effect model was chosen to fit, with the consideration of clustering of fibre measures within each muscle. Compound symmetry covariance structure within muscle was specified to account for the repeated fibre measures (Fitzmaurice, 2004). All other outcome measures were assessed using one‐way ANOVA to determine significant overall main effects. Once an overall effect was confirmed, statistical significance of the measured difference between groups was assessed further by Tukey's post hoc analysis, and adjusted P values with the corresponding 95% confidence interval are reported (Tukey, 1949). P < 0.05 was considered statistically significant. As a result of multiple outcome assessements, the findings regarding between group differences should be interpreted with caution unless they are highly significant (P < 0.001)
Results
Myonuclei acquired by resistance training are not lost during detraining
Previous studies have elegantly demonstrated that additional myonuclei in overloaded muscle are retained during severe atrophic episodes caused by either denervation, pharmacological nerve block or unloading (Bruusgaard & Gundersen, 2008; Bruusgaard et al. 2010; Egner et al. 2013). In the present study, we assessed changes in the number of myonuclei of singly isolated muscle fibres in conjunction with physiologically relevant resistance exercise training and detraining. The number of myonuclei was significantly higher in pre‐trained/detrained (36.84 ± 8.98), trained (37.17 ± 8.70) and re‐trained (38.20 ± 9.20) muscles compared to controls (30.14 ± 8.52) [Tukey's adjusted P values are 0.0029, 0.0017 and 0.0002, with corresponding 95% confidence intervals (CIs) of 2.048–10.981, 2.395–11.337 and 3.704–12.549, respectively] (Fig. 2 A and B). Note that there was no loss of the elevated number of myonuclei induced by previous resistance training after 20 weeks of detraining. A Pax7 staining on cross‐section of skeletal muscle from the same animal indicated resident satellite cells within the muscle tissues (Fig. 2 C); however, we confirmed that there were no Pax7‐positive cells in the single muscle fibre staining, suggesting that satellite cells were completely washed off during single cell preparation steps.
Figure 2. Pre‐trained myofibres present greater number of myonuclei compared to untrained fibres.
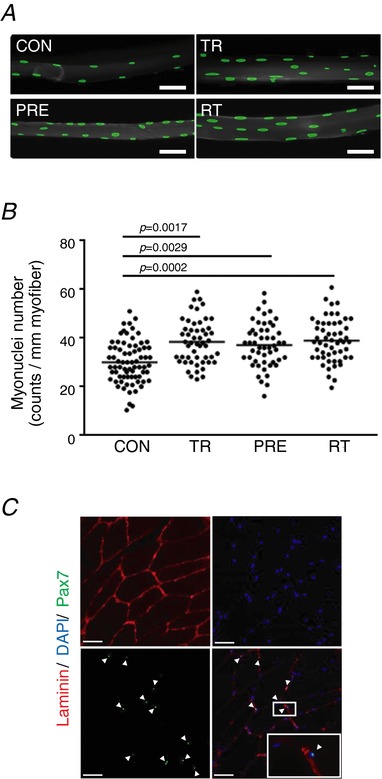
A, single myofibres isolated from FHL muscle were stained with DAPI (green). Scale bar = 50 μm (1:200 magnification). B, myonuclei numbers per single myofibre (counts per mm myofibre length) (n = 8). Data are presented as individual data points. C, positive Pax7 staining on a cross‐section of tibialis anterior muscle. Magnifications are 1:200 (left) and 1:630 (right).
Pre‐trained muscle confers a greater hypertrophy and increases oxidative myofibres on re‐training
Skeletal muscles comprise a highly plastic organ that changes size, contractile properties and myofibrillar protein composition primarily dependent on how much it is used acutely and chronically. Studies have shown that, once hypertrophy occurs, the muscle tends to regain muscle mass relatively quickly toward the largest size that it ever has, even after a prolonged lay‐off period (Staron et al. 1991; Taaffe & Marcus, 1997). We therefore investigated muscle hypertrophy in our unique experimental model including pre‐training/detraining, training and re‐training conditions (Fig. 1). CSA (μm2) was significantly larger in the training (3316.2 ± 168.8) and re‐training (3551.0 ± 162.1) groups compared to the control (3044.8 ± 246.3) and pre‐training (3092.1 ± 206.3) groups by 271.4 μm2 (95% CI = 0.076–542.724; P = 0.050) and 459.0 μm2 (95% CI = 187.551–730.199; P < 0.001), respectively. Also, in comparison with untrained muscle, trained muscle increased CSA to a greater extent after resisitance training [8.9% increase (CON vs. TR) vs. 14.8% increase (PRE vs. RT)] (Fig. 3 B). Similarly, the relative muscle weight/body weight ratio was greater in retrained (RT: 5.74 ± 0.62) animals compared to that of the once trained group (TR: 4.97 ± 0.57) by 0.77 (95% CI = 0.060–1.478; P = 0.0297) (Fig. 3 D). A 20 week detraining caused a reduction of both CSA and muscle mass to the untrained level. Together, the previously trained muscles showed a significantly greater muscle size increase when re‐trained compared to muscle without a pre‐training history.
Figure 3. Muscle mass and muscle fibre typing.
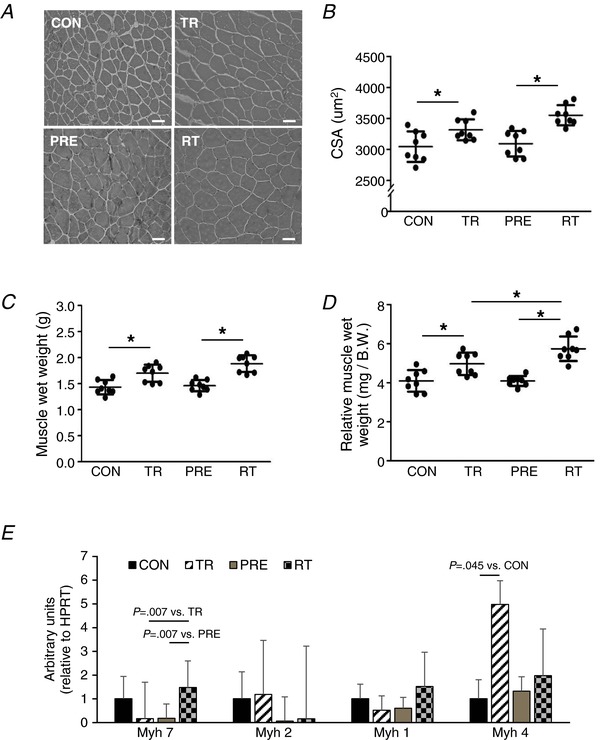
A, myofibres in FHL muscle were visualized by H&E staining. B, CSA of myofibres and (C and D) muscle wet weights with or without normalization to body weight were quantified (n = 8). E, mRNA expression of myosin heavy chain (Myh) isoforms (n = 4). Scale bar = 50 μm. 1:200 magnification. Data are the mean ± SD. * P < 0.05. [Color figure can be viewed at http://wileyonlinelibrary.com]
Previous studies reported that resistance training increases oxidative phosphorylation capacity in conjunction with an increase in aerobically‐oriented oxidative fibres switched from glycolytic fast twitch fibres (Wang et al. 1993; Manfredi et al. 2013). Thus, we measured mRNA expression levels of four muscle myosin heavy chain isoforms. We observed that only Myh 7 (type I, oxidative), and not Myh 4 (type IIb, glycolytic), expression level was significantly increased in the re‐trained muscle (Fig. 3 E), where non‐pretrained muscle significantly increased only the Myh 4 expression level following resistance training. There were significant differences in SDH and COX activities between control vs. trained and pre‐trained/detrained vs. retrained groups (Fig. 4). SDH activity was significantly higher in trained (33.09 ± 5.83; P < 0.001 vs. CON) and re‐trained (38.66 ± 3.98; P < 0.001 vs. CON) groups compared to the control group (20.86 ± 3.32). COX activity was also significantly greater in trained (41.07 ± 5.42; P < 0.001 vs. CON) and re‐trained (49.54 ± 4.77; P < 0.001 vs. CON) groups compared to the control group (27.34 ± 7.44).
Figure 4. Mitochondrial respiratory complex enzymatic activities.
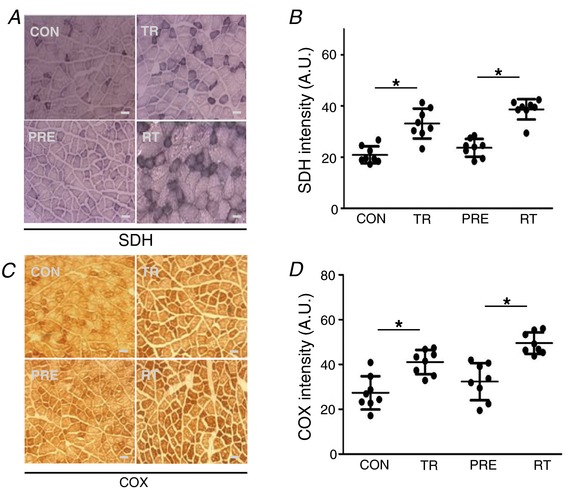
A and B, SDH staining on FHL muscle. C and D, COX staining on the FHL muscle (n = 8). Scale bar = 50 μm. 1:200 magnification. Data are the mean ± SD. * P < 0.05.
Previous resistance exercise training facilitates mitochondrial biogenesis on retraining
Mitochondrial biogenesis is a complex event that may involve more than ∼1000 nuclear genome‐encoded mitochondrial proteins and the regulatory signalling molecules orchestrating these processes (Ryan & Hoogenraad, 2007; Calvo et al. 2016). As described above, the re‐trained muscle represented the characteristics of more oxidative features, as well as the highest mitochondrial enzyme activities, compared to the trained yet non‐preconditioned muscle, which suggests a potential enhancement of mitochondrial biogenesis processes in the pre‐trained/re‐trained muscle. Therefore, considering the nature of skeletal muscle in which the nucleus‐to‐cytoplasm ratio is very low, we hypothesized that the large number of myonuclei retained in previously trained muscle would facilitate mitochondrial biogenesis on re‐training. We observed the highest abundance of mitochondrial biogenesis regulatory proteins in the re‐trained muscle (Fig. 5 A–E). Accordingly, mitochondrial DNA copy number was significantly greater in retrained muscle compared to once trained muscle (Fig. 5 F). Similarly, we further investigated mitochondrial content using immunohistochemistry to quantify the mitochondrial marker protein VDAC (porin) (Fig. 5 G and H). Mitochondrial content was greater in trained (15.23 ± 1.92) and retrained (18.12 ± 2.64) muscles compared to control muscle (10.16 ± 1.26) (95% CI = 2.253–7.886; P = 0.002 and 95% CI = 5.148–10.780; P < 0.001, respectively). The retrained muscle showed a significantly higher level of mitochondrial content compared to the once trained muscle by 2.89 AU (95% CI = 0.078–5.710; P = 0.0423).
Figure 5. Previously trained muscle showed greater mitochondrial adaptations following re‐training compared to non‐pretrained muscles.
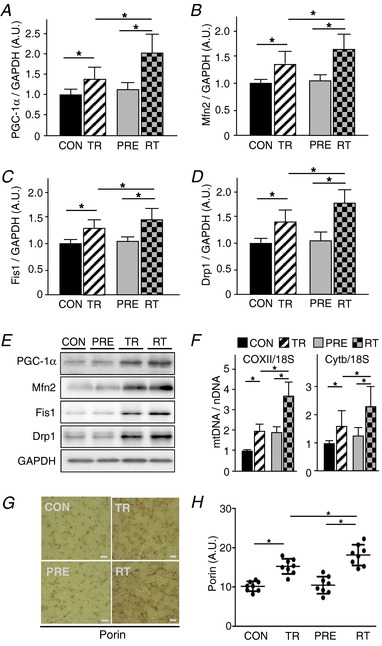
A–E, western blots for PGC‐1α, Mfn2, Fis1 and Drp1. Plots are the results of quantification. GAPDH was used as a loading control. F, mitochondrial copy number. G and H, mitochondrial content determined by immuohistochemisty using anti‐porin antibody (n = 8). 1:200 magnification. Staining intensity of porin was quantified. FHL muscles were used for all the experiments. Data are the mean ± SD. * P < 0.05.
Cumulative myonuclei augments expression levels of the genes related to the electron transport chain and enhances mitochondrial respiratory function with AICAR treatment
To investigate the potential cellular mechanisms responsible for the relationship between the number of myonuclei and the degree of mitochondrial biogenesis processes, we conducted in vitro studies using myonuclear ‘dense’ myotubes. To generate the myonuclear‐enriched myotubes, we treated myotubes with IGF‐1 at various concentrations for 48 h (Fig. 6 A). As shown in Fig. 6 B and C, myonuclear density (myonuclei count per myotube area) was gradually increased as a function of the IGF‐1 concentration and peaked at 50 ng mL–1. Myonuclear density was increased up to ∼2‐fold (∼200%) in IGF‐1 treated myotubes compared to non‐treated myotubes. After discarding IGF‐1 from the culture media, the myotubes were further treated with AICAR (1 mm), an AMP‐dependent protein kinase‐specific activator (often referred to as an exercise mimetic), to induce mitochondrial biogenesis. DMSO treatment was used as a control. As shown in Fig. 6 D, the myonuclei‐enriched myotubes (IGF‐1 treated) showed greater mRNA expression levels for the selected mitochondrial‐ and nuclear‐genome‐encoded genes related to the electron transport chain with AICAR treatment. Moreover, both basal and maximal oxygen consumption rates were significantly higher in the myotubes with myonuclei‐enriched myotubes compared to the control after AICAR treatment (Fig. 6 E).
Figure 6. Enhanced mitochondrial gene expression response in myonuclear‐enriched myotubes after AICAR treatment.
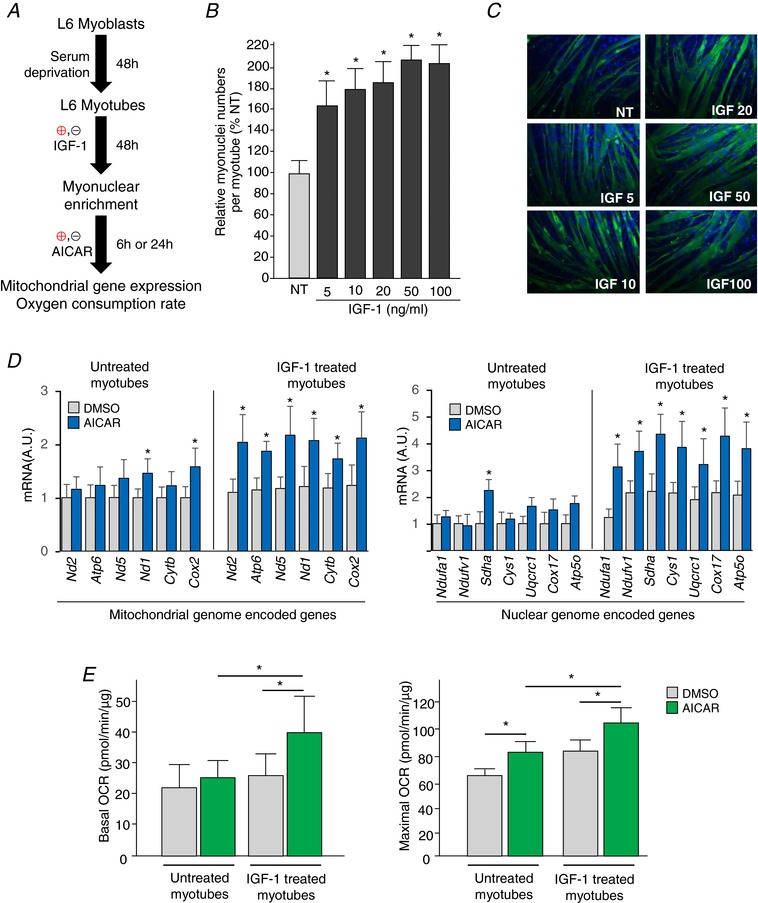
A, experimental design for in vitro studies. L6 myoblast differentiation was induced by serum deprivation for 48 h, and then the myotubes were further treated with (or without) IGF‐1 (50 ng mL–1) for an additional 48 h. Myonuclear enriched myotubes were incubated with 1 mm AICAR for either 6 or 24 h. B, relative myonuclei numbers per myotubes. Note that myonuclei numbers peaks at 50 ng mL–1. * P < 0.05 vs. non‐treated. C, representative images of myotubes (green) with nuclei staining (blue). 1:200 magnification. D, mitochondrial genome‐encoded (left) and nuclear genome‐encoded (right) mitochondrial gene expression levels. * P < 0.05 vs. DMSO. E, basal (left) and maximal oxygen consumption rate (OCR) (right). Data are the mean ± SD. * P < 0.05.
Discussion
In the past two decades, a nuclear domain theory has emerged in which each myonucleus governs a certain designated area in the sarcoplasm (Cavalier‐Smith, 1978; Hall & Ralston, 1989). Accordingly, when a myofibre becomes larger, additional nuclei appear to be incorporated into the pre‐existing muscle fibre, which originate from activated satellite cells in the surrounding area. The overarching premise of the present study was that the acquired myonuclei are sustained during atrophy and facilitate, as a part of cellular muscle memory mechanisms, the re‐gaining of muscle mass and mitochondrial biogenesis when the muscles are subjected to retraining. We report a positive role of the acquired myonuclei on mitochondrial biogenesis in hypertrophic skeletal muscles. The primary findings of the present study are that: (i) resistance exercise training increases the number of myonuclei retained during atrophy in skeletal muscles over a long‐term detraining period; and (ii) the acquired myonuclei are associated with a greater degree of adaptations with respect to muscle hypertrophy and mitochondrial biogenesis following subsequent retraining.
A role of muscle memory in re‐gaining muscle mass
Many studies showed that high intensity exercise leads to satellite cell recruitment (Allen et al. 1995; Kadi et al. 1999). In the present study, we show that previously trained high‐nuclei muscles have a biological predisposition to hypertrophy in response to subsequent retraining after a long‐intervening period. This finding is consistent with previous studies using surgical (Bruusgaard et al. 2010) and pharmacological (Egner et al. 2013) means for inducing muscle overload, although, to the best of our knowledge, this is the first study to use physiologically relevant resistance training as a modality of muscle overload when investigating muscle memory. We used a rodent version of ‘Jacobs ladder climbing’ as an exercise modality. There were concomitant increases in muscle mass and the number of myonuclei in trained muscles suggesting that weight‐loaded ladder climbing training provided an adequate stimulus for muscle adaptation. Furthermore, following 20 weeks of detraining (>15 years in humans), there was no significant reduction in the number of myonuclei where significant atrophy (∼20%) occurred, which is in agreement with previous studies (Bruusgaard & Gundersen, 2008; Bruusgaard et al. 2010).
A role of cellular mechanism of muscle memory on mitochondrial remodelling
The most prominent finding of the present study is that previously trained muscles confer greater mitochondrial adaptations when subjected to resistance exercise training at a later time. We also observed that the protein expression of various mitochondrial regulatory proteins, such as PGC‐1α and mitochondrial fusion/fission proteins (i.e. Mfn2, Fis1 and Drp1), was upregulated to a greater extent in retrained muscles compared to naive muscles. Furthermore, there was a tendency to express more oxidative fibre‐like properties in the retained muscles. These findings support the notion that pre‐trained muscles would be prone to greater metabolic adaptations in response to training. We speculate that the greater mitochondrial adaptations observed in the pretrained muscle may be attributed to a reduced nuclear domain size because of the increased myonuclear number within muscle fibre (Fig. 7).
Figure 7. Enhanced nuclear‐mitochondrial cross‐talk in pre‐trained skeletal muscle.
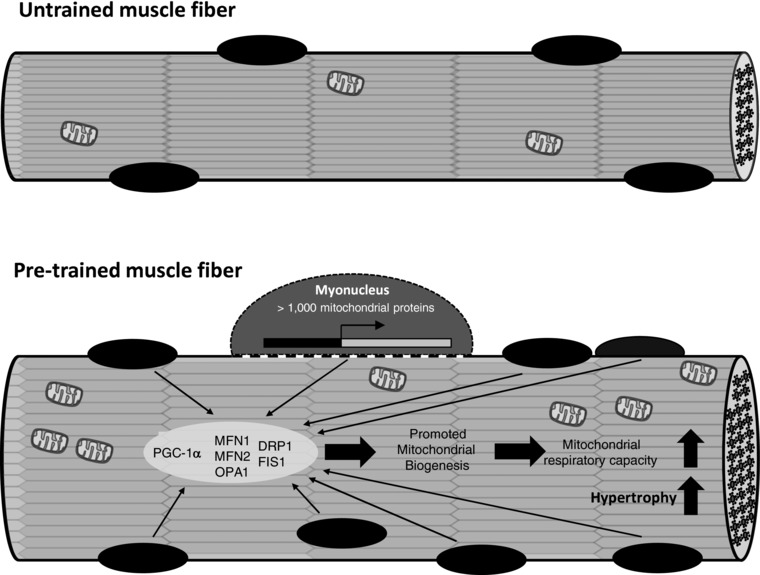
Schematic diagram of the proposed mechanism on enhanced nuclear‐mitochondrial cross‐talk in pre‐trained skeletal muscle.
Enhanced mitochondrial gene expression response in myonuclear‐enriched myotubes
We also found that myotubes with a greater number of nuclei contribute to mitochondrial gene expression (from both nuclear and mitochondrial genomes) when treated with AICAR. Thus, it is plausible to propose that a higher mitochondrial adaptability may be the result of a higher rate of gene expression that serves as a crucial mediator for mitochondrial biogenesis and mitochondrial dynamics. Furthermore, considering that AICAR‐mediated expression of both nucleus‐encoded and mitochondrial‐encoded genes related to mitochondrial biogenesis was greater in the IGF‐1 pre‐conditioned, myonuclei‐enriched, myotubes, this suggests that mitochondrial‐nuclear communication might be improved. In recent years, cross‐talk between nuclei and mitochondria via antegrade (nucleus to mitochondria) and retrograde signalling (mitochondria to nucleus) has been suggested in the proper cellular adaptation to a certain stimulus (Whelan & Zuckerbraun, 2013). It may be attributed to an activated cross‐talk between the nucleus and mitochondrion not only for modulating mitochondrial remodelling, but also for eliciting various cellular adaptations such as hypertrophic responses (Butow & Avadhani, 2004; Liu & Butow, 2006; da Cunha et al. 2015).
Other potential muscle memory mechanisms
In the present study, we observed an association between the number of myonuclei and muscle adaptability to exercise (or AICAR, an exercise mimetic). Thus, acquired myonuclei in pre‐trained muscles may be an important part of the cellular muscle memory. However, recent studies have demonstrated that, in some genetically engineered mouse models, a hypertrophic phenotype was observed in the absence of a concurrent increase in the number of nuclei (Amthor et al. 2009; Blaauw et al. 2009; Raffaello et al. 2010), suggesting the possibility that myonuclei might not be a sole contributor to the cellular muscle memory mechanism. Accordingly, recently, epigenetic modifications have been viewed as a potential mechanism of muscle memory. For example, skeletal muscle cells preconditioned with tumor necrosis factor‐α retained elevated MyoD methylation even after 30 population doublings (Sharples et al. 2016). In addition, there is accumulating evidence indicating that various types of exercise modulate histone acetylation and/or DNA methylation (Barres et al. 2009; Lavratti et al. 2017). However, it remains unclear whether these exercise‐induced epigenetic modifications are retained in later life. Therefore, mechanistic studies are warranted to identify the specific skeletal muscle memory mechanisms in the future.
In conclusion, we report that, in muscles subjected to physiologically relevant resistance training, the number of myonuclei increased and this was retained during a long‐term detraining period. Furthermore, the data showed that the acquired myonuclei may assist with muscle hypertrophy and mitochondrial biogenesis following subsequent re‐training. Taken together, the results of the present study suggest a potential cellular mechanism supporting the notion that exposing young muscles to resistance exercise training may have benefits with respect to regaining metabolically active muscles in later life. It is noteworthy that the animal studies were conducted with a relatively small sample size; thus, these findings must be replicated in a larger group. Nonetheless, we consider that the results of the present study provide a proof‐of‐concept of the biological processes underlying the importance of nuclear‐mitochondrial interplay during muscle growth. Future studies are warranted that aim to identify a novel target for treating muscle degenerative conditions such as sarcopaenia.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
HJL wrote the first draft of manuscript. HJL, KJK, SHL and JYP participated in the study design. HJL, KJK, BK, JCS and SR performed the experiments. HJL, MDB, SHL, JW, XWC and JYP conducted the data interpretation and analyses. MDB, SHL and JYP reviewed the manuscript submitted for publication. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by National Institutes of Health Grant R01HL126952 and American Heart Association Grants 12SDG12070327 (to J‐Y. Park), University Fellowship Program from Temple University (to B. Kim and H. Lee), and National Research Foundation of South Korea Grant NRF‐2011‐356‐G00013 (to K. Kim).
Biography
Hojun Lee received MS Degree (2009) in Kinesiology from Seoul National University and a PhD (2016) in Integrative Exercise Physiology from Temple University, Philadelphia. He worked as a Postdoctoral research fellow at the Korea Mouse Phenotyping Center in Seoul National University, then moved to Kyungsung University as a research professor of Kinesiology with a dual appointment in Seoul National University Bundang Hospital. His main research interests are in the areas of the physiology of exercise in skeletal muscle.

Edited by: Scott Powers & Paul Greenhaff
Linked articles This article is highlighted in a Perspectives article by Gundersen et al. To read this article, visit http://doi.org/10.1113/JP276354.
References
- Allen DL, Monke SR, Talmadge RJ, Roy RR & Edgerton VR. (1995). Plasticity of myonuclear number in hypertrophied and atrophied mammalian skeletal muscle fibers. J Appl Physiol 78, 1969–1976. [DOI] [PubMed] [Google Scholar]
- Amthor H, Otto A, Vulin A, Rochat A, Dumonceaux J, Garcia L, Mouisel E, Hourde C, Macharia R, Friedrichs M, Relaix F, Zammit PS, Matsakas A, Patel K & Partridge T. (2009). Muscle hypertrophy driven by myostatin blockade does not require stem/precursor‐cell activity. Proc Natl Acad Sci U S A 106, 7479–7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres R, Osler ME, Yan J, Rune A, Fritz T, Caidahl K, Krook A & Zierath JR. (2009). Non‐CpG methylation of the PGC‐1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab 10, 189–198. [DOI] [PubMed] [Google Scholar]
- Blaauw B, Canato M, Agatea L, Toniolo L, Mammucari C, Masiero E, Abraham R, Sandri M, Schiaffino S & Reggiani C. (2009). Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J 23, 3896–3905. [DOI] [PubMed] [Google Scholar]
- Bolster DR, Jefferson LS & Kimball SR. (2004). Regulation of protein synthesis associated with skeletal muscle hypertrophy by insulin‐, amino acid‐ and exercise‐induced signalling. Proc Nutr Soc 63, 351–356. [DOI] [PubMed] [Google Scholar]
- Bruusgaard JC & Gundersen K. (2008). In vivo time‐lapse microscopy reveals no loss of murine myonuclei during weeks of muscle atrophy. J Clin Invest 118, 1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruusgaard JC, Johansen IB, Egner IM, Rana ZA & Gundersen K. (2010). Myonuclei acquired by overload exercise precede hypertrophy and are not lost on detraining. Proc Natl Acad Sci U S A 107, 15111–15116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruusgaard JC, Liestol K, Ekmark M, Kollstad K & Gundersen K. (2003). Number and spatial distribution of nuclei in the muscle fibres of normal mice studied in vivo. J Physiol 551, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd NA, Andrews RJ, West DW, Little JP, Cochran AJ, Hector AJ, Cashaback JG, Gibala MJ, Potvin JR, Baker SK & Phillips SM. (2012). Muscle time under tension during resistance exercise stimulates differential muscle protein sub‐fractional synthetic responses in men. J Physiol 590, 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butow RA & Avadhani NG. (2004). Mitochondrial signaling: the retrograde response. Mol Cell 14, 1–15. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR & Mootha VK. (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1251–D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier‐Smith T. (1978). Nuclear volume control by nucleoskeletal DNA, selection for cell volume and cell growth rate, and the solution of the DNA C‐value paradox. J Cell Sci 34, 247–278. [DOI] [PubMed] [Google Scholar]
- Chesley A, MacDougall JD, Tarnopolsky MA, Atkinson SA & Smith K. (1992). Changes in human muscle protein synthesis after resistance exercise. J Appl Physiol 73, 1383–1388. [DOI] [PubMed] [Google Scholar]
- da Cunha FM, Torelli NQ & Kowaltowski AJ. (2015). Mitochondrial retrograde signaling: triggers, pathways, and outcomes. Oxid Med Cell Longev 2015, 482582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egner IM, Bruusgaard JC, Eftestol E & Gundersen K. (2013). A cellular memory mechanism aids overload hypertrophy in muscle long after an episodic exposure to anabolic steroids. J Physiol 591, 6221–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzmaurice G, Laird NM & Ware JH (2004). Applied Longitudinal Analysis. John Wiley & Sons, Inc., Hoboken, NJ. [Google Scholar]
- Hall ZW & Ralston E. (1989). Nuclear domains in muscle cells. Cell 59, 771–772. [DOI] [PubMed] [Google Scholar]
- Harber MP, Konopka AR, Undem MK, Hinkley JM, Minchev K, Kaminsky LA, Trappe TA & Trappe S. (2012). Aerobic exercise training induces skeletal muscle hypertrophy and age‐dependent adaptations in myofiber function in young and older men. J Appl Physiol 113, 1495–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadi F, Eriksson A, Holmner S, Butler‐Browne GS & Thornell LE. (1999). Cellular adaptation of the trapezius muscle in strength‐trained athletes. Histochem Cell Biol 111, 189–195. [DOI] [PubMed] [Google Scholar]
- Kim B, Kim JS, Yoon Y, Santiago MC, Brown MD & Park JY. (2013). Inhibition of Drp1‐dependent mitochondrial division impairs myogenic differentiation. Am J Physiol Regul Integr Comp Physiol 305, R927–R938. [DOI] [PubMed] [Google Scholar]
- Kumar V, Selby A, Rankin D, Patel R, Atherton P, Hildebrandt W, Williams J, Smith K, Seynnes O, Hiscock N & Rennie MJ. (2009). Age‐related differences in the dose‐response relationship of muscle protein synthesis to resistance exercise in young and old men. J Physiol 587, 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavratti C, Dorneles G, Pochmann D, Peres A, Bard A, de Lima Schipper L, Dal Lago P, Wagner LC & Elsner VR. (2017). Exercise‐induced modulation of histone H4 acetylation status and cytokines levels in patients with schizophrenia. Physiol Behav 168, 84–90. [DOI] [PubMed] [Google Scholar]
- Lee S, Barton ER, Sweeney HL & Farrar RP. (2004). Viral expression of insulin‐like growth factor‐I enhances muscle hypertrophy in resistance‐trained rats. J Appl Physiol 96, 1097–1104. [DOI] [PubMed] [Google Scholar]
- Liesa M, Palacin M & Zorzano A. (2009). Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89, 799–845. [DOI] [PubMed] [Google Scholar]
- Liu Z & Butow RA. (2006). Mitochondrial retrograde signaling. Annu Rev Genet 40, 159–185. [DOI] [PubMed] [Google Scholar]
- Manfredi TG, Monteiro MA, Lamont LS, Singh MF, Foldvari M, White S, Cosmas AC & Urso ML. (2013). Postmenopausal effects of resistance training on muscle damage and mitochondria. J Strength Cond Res 27, 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BA, Hoffman NE, Merali S, Zhang XQ, Wang J, Rajan S, Shanmughapriya S, Gao E, Barrero CA, Mallilankaraman K, Song J, Gu T, Hirschler‐Laszkiewicz I, Koch WJ, Feldman AM, Madesh M & Cheung JY. (2014). TRPM2 channels protect against cardiac ischemia‐reperfusion injury: role of mitochondria. J Biol Chem 289, 7615–7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Wang PY, Matsumoto T, Sung HJ, Ma W, Choi JW, Anderson SA, Leary SC, Balaban RS, Kang JG & Hwang PM. (2009). p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ Res 105, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlath GK, Rich K, Webster SG & Blau HM. (1989). Localization of muscle gene products in nuclear domains. Nature 337, 570–573. [DOI] [PubMed] [Google Scholar]
- Phillips SM, Tipton KD, Aarsland A, Wolf SE & Wolfe RR. (1997). Mixed muscle protein synthesis and breakdown after resistance exercise in humans. Am J Physiol Endocrinol Metab 273, E99–E107. [DOI] [PubMed] [Google Scholar]
- Raffaello A, Milan G, Masiero E, Carnio S, Lee D, Lanfranchi G, Goldberg AL & Sandri M. (2010). JunB transcription factor maintains skeletal muscle mass and promotes hypertrophy. J Cell Biol 191, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston E & Hall ZW. (1992). Restricted distribution of mRNA produced from a single nucleus in hybrid myotubes. J Cell Biol 119, 1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford OM & Jones DA. (1986). The role of learning and coordination in strength training. Eur J Appl Physiol 55, 100–105. [DOI] [PubMed] [Google Scholar]
- Ryan MT & Hoogenraad NJ. (2007). Mitochondrial‐nuclear communications. Annu Rev Biochem 76, 701–722. [DOI] [PubMed] [Google Scholar]
- Sharples AP, Polydorou I, Hughes DC, Owens DJ, Hughes TM & Stewart CE. (2016). Skeletal muscle cells possess a ‘memory’ of acute early life TNF‐alpha exposure: role of epigenetic adaptation. Biogerontology 17, 603–617. [DOI] [PubMed] [Google Scholar]
- Staron RS, Leonardi MJ, Karapondo DL, Malicky ES, Falkel JE, Hagerman FC & Hikida RS. (1991). Strength and skeletal muscle adaptations in heavy‐resistance‐trained women after detraining and retraining. J Appl Physiol 70, 631–640. [DOI] [PubMed] [Google Scholar]
- Taaffe DR & Marcus R. (1997). Dynamic muscle strength alterations to detraining and retraining in elderly men. Clin Physiol 17, 311–324. [DOI] [PubMed] [Google Scholar]
- Tukey JW. (1949). Comparing individual means in the analysis of variance. Biometrics 5, 99–114. [PubMed] [Google Scholar]
- Verma M & Asakura A. (2011). Efficient single muscle fiber isolation from alcohol‐fixed adult muscle following beta‐galactosidase staining for satellite cell detection. J Histochem Cytochem 59, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Hikida RS, Staron RS & Simoneau JA. (1993). Muscle fiber types of women after resistance training – quantitative ultrastructure and enzyme activity. Pflugers Arch 424, 494–502. [DOI] [PubMed] [Google Scholar]
- Whelan SP & Zuckerbraun BS. (2013). Mitochondrial signaling: forwards, backwards, and in between. Oxid Med Cell Longev 2013, 351613. [DOI] [PMC free article] [PubMed] [Google Scholar]