

Abstract
Global protein translation as well as translation at the codon level can be regulated by tRNA modifications. In eukaryotes, levels of tRNA queuosinylation reflect the bioavailability of the precursor queuine, which is salvaged from the diet and gut microbiota. We show here that nutritionally determined Q‐tRNA levels promote Dnmt2‐mediated methylation of tRNA Asp and control translational speed of Q‐decoded codons as well as at near‐cognate codons. Deregulation of translation upon queuine depletion results in unfolded proteins that trigger endoplasmic reticulum stress and activation of the unfolded protein response, both in cultured human cell lines and in germ‐free mice fed with a queuosine‐deficient diet. Taken together, our findings comprehensively resolve the role of this anticodon tRNA modification in the context of native protein translation and describe a novel mechanism that links nutritionally determined modification levels to effective polypeptide synthesis and cellular homeostasis.
Keywords: cytosine‐5 methylation, protein translation, queuosine, tRNA modifications, unfolded protein response
Subject Categories: Metabolism, Protein Biosynthesis & Quality Control, RNA Biology
Introduction
The correct assignment of 20 amino acids to each of the 64 possible codon triplets within the messenger RNA (mRNA)‐coding sequence determines accurate protein synthesis. Codon:anticodon recognition is mediated by transfer RNAs (tRNAs), which physically link mRNAs to the amino acid sequence of the nascent polypeptide at the ribosome. The 1st and 2nd base of the codon and the 3rd and 2nd base of the anticodon, respectively, interact according to the Watson–Crick base‐pairing rules (A:U, U:A, G:C, and C:G). In contrast, the interaction between the 3rd base of the codon and the 1st base of the anticodon (position 34, so‐called wobble base) is less stringent, so that non‐standard base pairings are permitted (Crick, 1966). As a result, a given tRNA may read more than one synonymous codon. Post‐transcriptional modifications in the anticodon loop of tRNAs are critical for the translation process (Grosjean et al, 2010). In particular, position 34 is subject to various modifications (Agris et al, 2017), depending on the associated tRNA isoacceptor and the organism (Grosjean et al, 2010; El Yacoubi et al, 2012). Some of these modifications have been shown to be important for the fine‐tuning of protein translation and for the maintenance of proteome integrity in yeast and in C. elegans (Rezgui et al, 2013; Zinshteyn & Gilbert, 2013; Nedialkova & Leidel, 2015; Chou et al, 2017).
Queuosine is a hyper‐modified guanosine analog that comprises a 7‐deaza‐guanine core structure covalently linked to an amino‐methyl side chain and a cyclo‐pentanediol moiety (Fergus et al, 2015). The corresponding base is termed queuine (q), and the respective nucleotide is called queuosine (Q). Q occurs at the wobble position of tRNAs with GUN anticodons tRNAAsp GUC, tRNAHis GUG, tRNATyr GUA, and tRNAAsn GUU (Harada & Nishimura, 1972). In eukaryotes, both NAC/U codons are translated by tRNA genes with GUN anticodons. The translation of NAU is mediated by a non‐canonical G:U pairing. In eubacteria, lack of Q affects mRNA translation and reduces the virulence of certain pathogenic strains (Durand et al, 2000). In animal cells, changes in the abundance of Q have been shown to correlate with diverse phenomena including stress tolerance, cell proliferation, and tumor growth (Fergus et al, 2015). Nonetheless, the function of Q in mammals remains poorly understood. Animals obtain Q (or its analogs) as a micronutrient from dietary sources and from the gut microbiota (Farkas, 1980; Fergus et al, 2015). The difficulty of maintaining animals under bacteria‐free conditions and/or on Q‐deficient diets has severely hampered the study of Q metabolism and function in metazoans. Nevertheless, germ‐free mice fed with a Q‐free diet have been shown to be deficient in queuosine modifications of tRNA, while exogenous administration of queuine restores Q‐tRNA (Reyniers et al, 1981).
Cytosine‐5 methylation (m5C) is a widely known modification in the context of DNA methylation and epigenetic gene regulation (Jones, 2012). Interestingly, m5C also represents a conserved RNA modification (Gilbert et al, 2016), and several recent studies have provided evidence for a conserved role in the regulation of protein translation (Tuorto & Lyko, 2016). We have previously shown that Dnmt2‐mediated tRNA methylation affects the speed and accuracy of protein translation (Tuorto et al, 2015). In higher eukaryotes, m5C‐tRNA methylation is found at positions 48, 49, and 72, and in the anticodon loop at positions 34 and 38 (Motorin et al, 2010). The absence of a methyl group at these positions has been suggested to interfere with tRNA folding and stability, codon–anticodon interactions, and reading frame maintenance (Agris, 2004; Grosjean et al, 2010; El Yacoubi et al, 2012; Guy et al, 2014; Hori, 2014). Interestingly, tRNAAsp is modified with both m5C and Q, and it has been shown recently that DNMT2‐dependent tRNA methylation is enhanced by queuine in S. pombe and in D. discoideum (Muller et al, 2015).
Remarkably, the choice of C‐ versus U‐ending codons of certain developmental genes in Drosophila and the codon usage across Drosophila species at evolutionarily conserved Q codon sites relates to the level of Q‐tRNA modification (Zaborske et al, 2014). More specifically, it was observed that the level of Q‐modification provides an accuracy‐driven selective advantage of C‐ over U‐ending codons. This suggested a “kinetic competition model”, wherein the presence of Q34 leads to more accurate translation of the C‐ending codon as a result of increased binding affinity (Zaborske et al, 2014). This indicated that environmental conditions, such as the availability of a micronutrient from the gut microbiota, can influence the decoding of a genome. However, this concept is currently only supported by codon usage analysis in Drosophila, and direct experimental evidence has been lacking. Furthermore, the described preference of Q‐tRNAs for C‐ over U‐ending codons is in contrast with earlier findings that either did not detect any Q‐dependent changes in protein synthesis (Owenby et al, 1979) or suggested a mild preference for Q:U pairings over Q:C (Meier et al, 1985; Morris et al, 1999).
Here, we have obtained translatomes of Q‐containing and Q‐lacking human cells to comprehensively analyze the role of queuine in the native protein translation context and describe a novel mechanism that links nutritionally determined tRNA modification levels to the effective polypeptide synthesis and cellular homeostasis. Using ribosome profiling and SILAC, we show that nutritionally determined Q‐tRNA levels promote C38 methylation and control translational speed at codons decoded by Q‐tRNAs and their near‐cognate codons. The loss of Q‐modification results in pronounced changes in cellular and organismal phenotypes including the induction of the unfolded protein response. Our results establish a novel molecular link between microbial‐derived micronutrients and the coordinated decoding of the mammalian transcriptome, thus identifying a central nutrient‐controlled mechanism for the fine‐tuning of protein translation.
Results
C38 methylation depends on Q in mammalian cell lines
Standard cell culture medium contains 1–2 × 10−8 M q, which is provided through the fetal bovine serum (S) supplement and is sufficient for the quantitative modification of tRNAs (Katze et al, 1982; Fergus et al, 2015). Therefore, to obtain a q‐free cell culture system, human HeLa cells were grown in synthetic serum‐free medium (SF) (Rakovich et al, 2011), in the absence or presence of chemically synthesized q (Fig 1A). To quantify Q‐tRNA modification levels directly from total RNA, we used polyacrylamide gels that are covalently linked with N‐acryloyl‐3‐aminophenylboronic acid (APB; Igloi & Kossel, 1985; Zaborske et al, 2014). In APB gels, the additional ribose moiety of Q slows down Q‐tRNA migration compared to G‐tRNA, thus producing two bands, corresponding to Q‐tRNAHis QUG and G‐tRNAHis GUG (Fig 1A). Using APB Northern analysis, we confirmed a continuous reduction in Q‐tRNA in our SF cell culture at various time points over a 21‐day culture period with a probe specific to tRNAHis (Fig 1A). A similar loss of Q‐tRNA was observed using a probe detecting tRNAAsn (Fig EV1A). No separate Q and G bands were detected with tRNAAsp and tRNATyr (Fig EV1A), which is likely due to a secondary mannosyl modification of Q‐tRNAAsp and galactosyl modification of Q‐tRNATyr (Kasai et al, 1976; Zaborske et al, 2014).
Figure 1. m5C38‐tRNAAsp GUC dependency by Q in human cell culture.
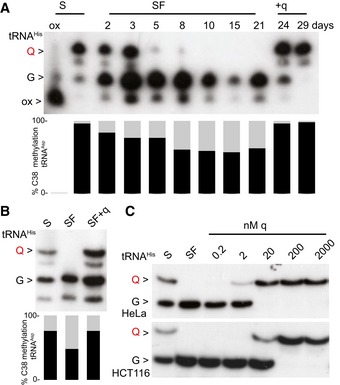
- APB Northern blot using a tRNAHis probe. m5C38 levels of tRNAAsp GUC were measured by 454 bisulfite sequencing at the indicated time points. Both queuosinylation and methylation levels could be restored by the addition of queuine. The slower migration of tRNAHis is eliminated by oxidizing the ribose with periodate, producing a single faster migrating band (ox).
- The addition of queuine to HCT116 cells cultured in serum‐free medium resulted in an increase in both queuosinylation and m5C38 levels.
- The addition of 20 nM queuine to the SF medium is sufficient to restore tRNAHis QUG in HeLa, whereas 200 nM of q is necessary for queuosinylation of tRNAHis GUG in HCT116 cells.
Figure EV1. m5C38‐tRNAA sp GUC dependency on Q in HeLa cell culture and tRNA level quantifications.
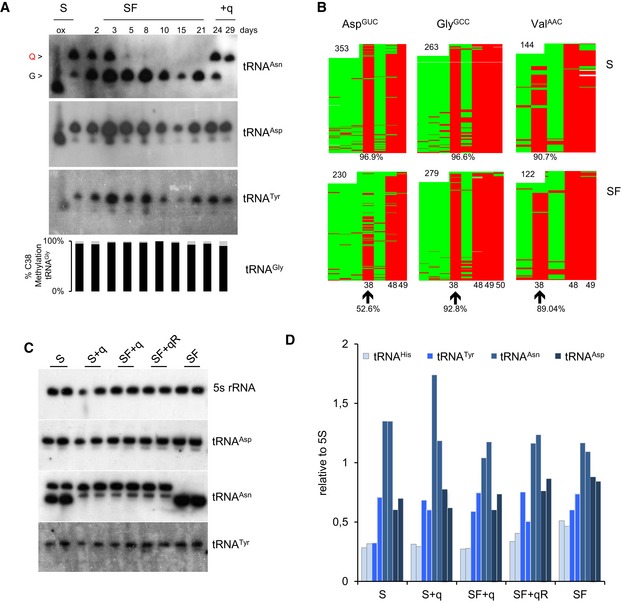
- APB Northern blot using tRNAAsn probe shows that SF medium depletes Q‐tRNA. No separate Q‐ and G‐tRNAAsp and Tyr bands were detected. m5C38 levels are measured by 454 bisulfite sequencing. A concomitant robust reduction in Q‐tRNAHis and m5C38‐tRNAAsp GUC is observed in HeLa cells cultivated in SF medium. Both queuosinylation and methylation levels could be restored by the addition of queuine to the SF medium for 3 or 8 days after 21 days in SF medium. The differential migration is eliminated by oxidizing the ribose with periodate, producing a single faster migrating band (ox).
- Bisulfite sequencing maps from a biological replicate culture under S and SF for 3 weeks are depicted. Each row represents one sequence read and each column a cytosine residue. Green boxes represent unmethylated cytosine residues, and red boxes indicate methylated cytosine residues. Sequencing gaps are shown in white. Numbers in the maps indicate the number of reads. The position of specific cytosine residues and level of C38 methylation are indicated at the bottom.
- APB Northern blot analysis that complete Fig 2B with tRNAAsp, tRNAAsn, tRNATyr, and 5S rRNA as a loading control.
- Quantitative analysis of signals for tRNAs at the indicated culture conditions. tRNA signal intensities were normalized to 5S rRNA levels for each of the two replicates.
In further experiments, we used tRNA bisulfite sequencing (Schaefer et al, 2009) to determine whether Dnmt2‐mediated tRNA methylation is affected by the presence of q in the medium, as shown previously for tRNAAsp GUC in fission yeast (Muller et al, 2015). A reduction in m5C38 tRNAAsp GUC from 96 to 57% was observed in SF medium (Fig 1A), while methylation levels of the non‐queuosinylated Dnmt2 targets tRNAGly GCC and tRNAVal AAC remained unaffected after culturing for 3 weeks in SF medium (Fig EV1A and B). To confirm that Q is necessary for physiological C38 methylation levels, we added synthetic q in rescue experiments. Indeed, C38 methylation of tRNAAsp GUC was completely restored (Figs 1A and EV1A and B). These findings demonstrate that Q enhances DNMT2 activity on tRNAAsp GUC in a mammalian cell line.
We also addressed the conservation of this mechanism in an additional cell culture model, and SF conditions strongly reduced the queuosinylation of tRNAHis in human colorectal carcinoma cells (HCT116; Fig 1B). When we analyzed tRNAHis queuosinylation at various concentrations of q, we observed that in HeLa cells quantitative queuosinylation was achieved at 20 nM (Fig 1C), which corresponds to the q levels of standard cell culture media (Katze et al, 1982; Fergus et al, 2015). Interestingly, HCT116 cells required higher q concentrations (200 nM) to quantitatively modify G‐tRNAHis. This indicates a role of cell‐type‐specific factors in queuine uptake and salvage (Fergus et al, 2015; Zallot et al, 2017). Taken together, our data thus suggest a conserved post‐transcriptional control mechanism of Q‐tRNA and m5C38‐tRNA modifications.
Q accelerates translational speed at Q‐decoded codons
In subsequent experiments, we aimed to determine how Q and the Q‐dependent m5C38 modification influence protein translation. To this end, we compared HeLa cells that were cultured 45 days under SF conditions with cells that were first cultured under SF conditions for 30 days and then received 20 nM queuine for 15 days (SF+qR) to rescue any q‐dependent defects (Fig 2A). Furthermore, a parallel control culture (45 days) was established using SF conditions supplemented with 20 nM queuine for the entire duration of the experiment (SF+q) (Fig 2A). HeLa cells grown in standard medium (S) and in standard medium supplemented with 20 nM queuine were included as additional controls (Fig 2A). Under all conditions, levels of Q‐tRNAHis and m5C38‐tRNA strictly depended on the availability of the supplemented q (Fig 2B). tRNA level measurements excluded an effect due to altered tRNA stability or tRNA availability by Q (Figs 2B and EV1C and D). We then examined genome‐wide codon occupancy during translation elongation, in different culture conditions (Fig 2A) using ribosome profiling. In this method, ribosome‐protected mRNA fragments are sequenced to obtain ribosomal position‐specific information at single‐nucleotide resolution (Ingolia et al, 2011). Two independent biological replicates for each culture condition were analyzed and showed a strong correlation (Fig EV2A). Quality controls (Figs 2C and D, and EV2A) suggested the generation of high‐quality datasets. Data analysis revealed that q‐free culture conditions resulted in a significant (P < 0.05, t‐test) increase in ribosome density at specific codons (Figs 3A and EV2B), which included all the queuosine‐decoded codons: tRNAAsp, tRNATyr, tRNAHis, and tRNAAsn (Figs 3A and EV2B), suggesting that translation of these codons was slower in the absence of the Q‐modification. We also observed reduced translation speed at specific near‐cognate Q‐decoded codons such as Glu (GAA, GAG), in agreement with earlier results from m5C38‐deficient mice (Tuorto et al, 2015). Hierarchical clustering of codons according to translational speed also resulted in the consistent separation of Q‐decoded and related near‐cognate codons from all the other codons (Fig EV2C). Notably, focusing on synonymous codon pairs, the U‐ending codons were slowed down more severely than the C‐ending codons, except for tRNAAsp GUC, where the concomitant reduction in m5C38 resulted in a slower translation of the GAC codon (Fig 3B).
Figure 2. Q‐depleted and quantitatively modified cell culture conditions.
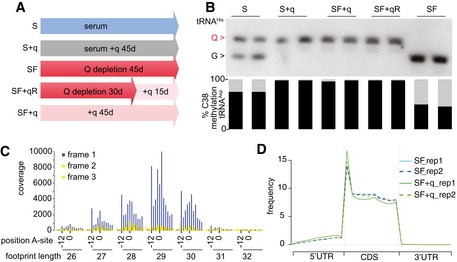
- Cell culture timing schedule.
- Corresponding queuosinylation and methylation levels, as determined by Northern blot using a tRNAHis probe and bisulfite sequencing of tRNAAsp GUC.
- Representative metaplot of SF cells. 25–32 mer ribosome footprint reads were summed across all annotated reading frames with the AUG staring codon at position zero. In‐frame 27–30 mer ribosome footprints showing the correct periodicity.
- Representative metagene plot of 27–30 mer ribosome footprints in S and SF culture conditions for two biological replicates (rep1 and rep2).
Figure EV2. Translation speed in Q‐free cells.
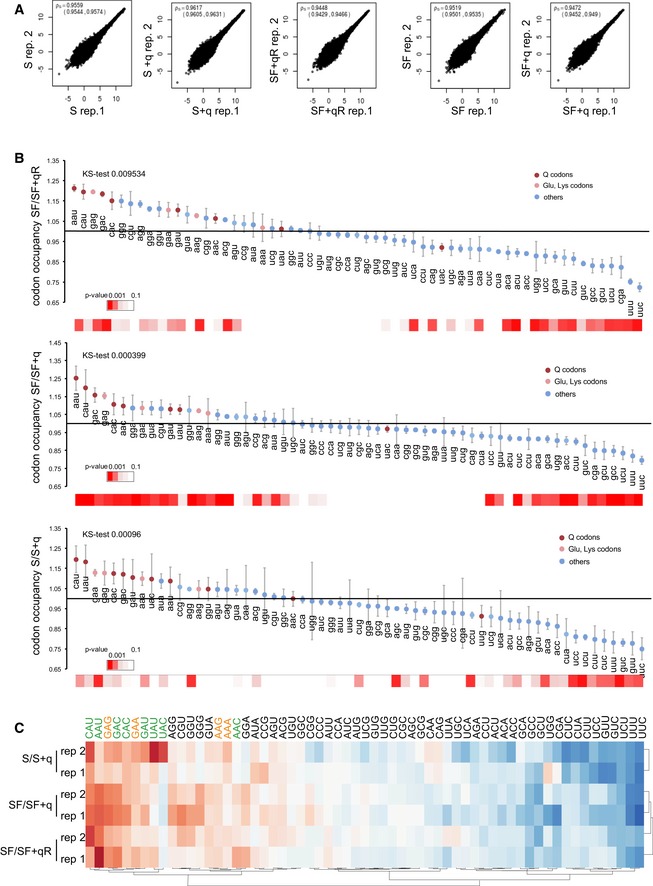
- Correlation between ribosome profiling dataset replicates. Spearman rho, 95% confidence interval for each pair of replicates.
- Changes in bulk codon occupancy in –Q cells compared to the relative rescue condition. All Q‐decoded codons and Glu/Lys codons are highlighted with the indicated colors. Error bars: ± SE of the permutated ratios of bulk codon occupancy for the indicated conditions; (n = 4). For each distribution, the statistical significance calculated using a Kolmogorov–Smirnov test is shown. P‐values (t‐test, adjusted for multiple testing) relative for each codon occupancy are indicated by a color scale heatmap at the bottom.
- The heatmap shows codon occupancy correlations between replicates using Ward's maximum distance clustering method. Q‐decoded codons are shown in green and Lys and Glu codons in orange. Note that hierarchical clustering grouped all Q‐decoded codons together.
Figure 3. Effects of Q depletion on translational speed.
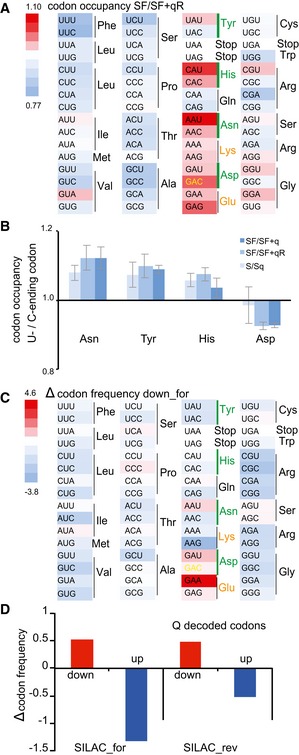
- Heatmap showing codon occupancy according to the color scale. Green bars indicate Q‐decoded codons. The Asp‐GAC codon translated by Q‐ and m5C38‐tRNAAsp GUC is shown in yellow and Lys and Glu codons are in orange. Lack of Q reduces ribosome translation speed at all Q‐dependent codons and at near‐cognate decoded codons.
- Translational speed of C‐ending codons relative to U‐ending codons. Error bars: ± SE of the permutated ratios of C‐ending codon occupancy relative to U for the indicated conditions; (n = 4).
- Heatmap displaying an increase or decrease in codon frequencies (‰) in SILAC down‐regulated proteins relative to the average frequency of that codon in unchanged proteins.
- Codons translated by Q‐tRNA are depleted in down‐regulated proteins and increased in up‐regulated proteins in both forward and reverse SILAC.
To confirm this finding, we used stable isotope labeling by amino acids (SILAC) in HeLa cell culture in the presence or absence of q for 3 weeks (Fig EV3A–C, Dataset EV1). Codon usage analysis of deregulated proteins confirmed an effect of Q on codons decoded by Q‐tRNA and near‐cognate codons (Fig 3C). Indeed, codons translated slower by Q‐tRNA in the absence of the Q‐modification were enriched in down‐regulated proteins and depleted in up‐regulated proteins in both forward and reverse labeling (Figs 3D and EV3D). These results confirm the ribosome profiling findings with an orthogonal approach and strongly indicate a role of Q and m5C38 in the decoding of the mammalian transcriptome.
Figure EV3. SILAC.
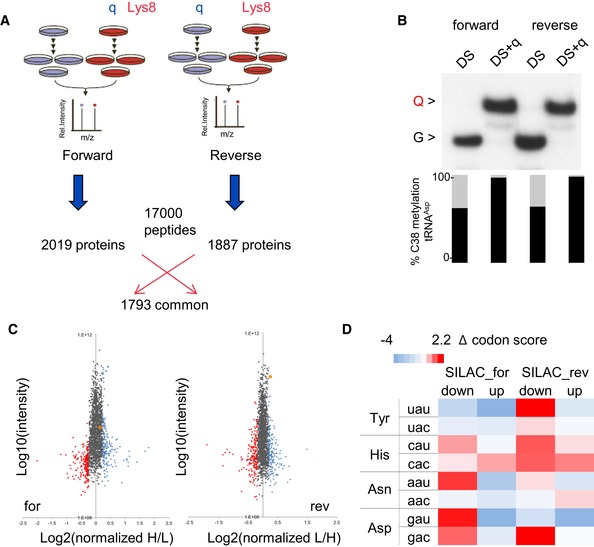
- Labeling culture conditions and numbers of identified proteins.
- APB Northern blot using tRNAHis probe and C38 methylation of tRNAAsp in SILAC labeled cells.
- SILAC analysis of deregulated proteins in the absence of Q. The top 10% of down‐regulated proteins are indicated in red, the top 10% of up‐regulated proteins are indicated in blue, HSPA5/BiP is indicated in orange.
- Increase or decrease in a codon frequency (‰) in up‐ or down‐regulated proteins relative to the average frequency of that codon in unchanged proteins is displayed as heatmap according to the color scale.
Q‐dependent phenotypes in cultured cell lines
To characterize the phenotypic consequences of Q depletion, we performed gene ontology (GO) enrichment analysis on mRNAs that were differentially translated in Q‐depleted cells (Figs 4A and EV4A–C, and Dataset EV2). Ingenuity pathway analysis revealed a strong enrichment for genes involved in eIF2 signaling (Fig 4B) a critical point of stress‐induced regulation of translation in eukaryotic cells (Bhat et al, 2015). Accordingly, we also identified several pathways related to stress signaling (Fig 4B). Other significantly enriched pathways, such as p70S6K signaling (Fig 4B), provided links between protein translation and a plethora of cellular functions (Holz et al, 2005).
Figure 4. Q‐dependent phenotypes in cultured cell lines.
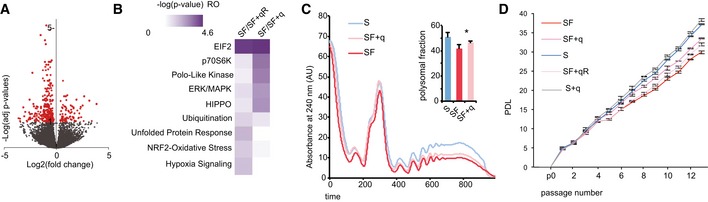
- The volcano plot shows differentially translated transcripts against adjusted P‐values for SF compared to SF + q. Red dots indicate P adj < 0.1 in multiple tests.
- Comparative analysis of differentially translated mRNAs of both experimental groups (SF compared to SF + q and SF compared to SF + qR) using ingenuity pathway analysis identified EIF2 signaling as the most significantly affected canonical pathway.
- Representative polysome profiles of HeLa cells grown in S and SF medium in the absence or presence of q. As a measurement for the global translation rate, the fraction of polysomal ribosomes was quantified; *P < 0.05 (t‐test); n = 5.
- Proliferation analysis of HeLa cells grown under the indicated culture conditions. Population doubling levels of three biological replicates were calculated for each time point. Error bars: ± SD; n = 3.
Figure EV4. Molecular functions affected by Q.
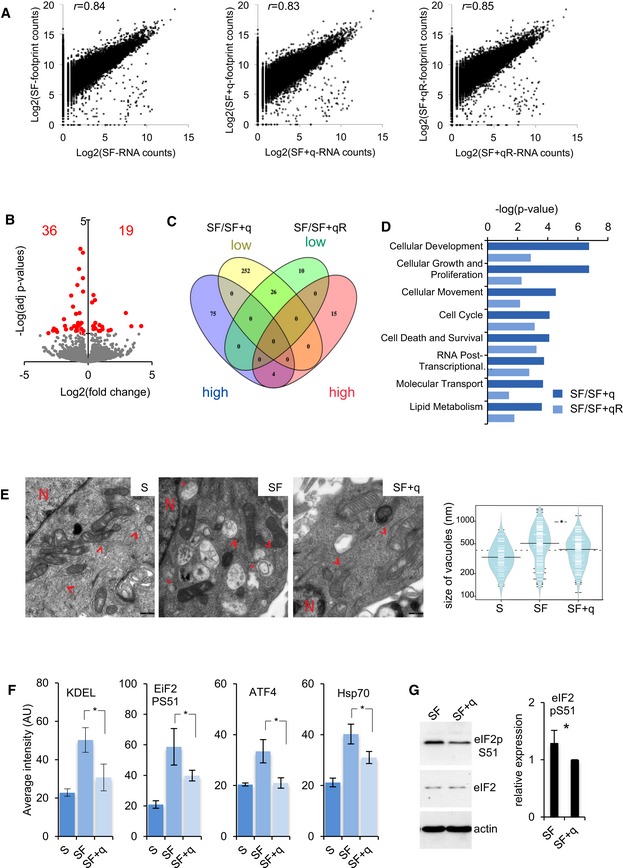
- Log2 fold changes in transcripts abundance and footprints at the indicated cell culture conditions. Pearson correlation coefficient (r) between footprints and mRNA abundance changes is shown.
- Volcano plot showing differentially translated mRNAs against adjusted P‐values for SF compared with SF + qR. Red dots indicate P adj < 0.1.
- Venn diagram showing the correlation of mRNA with high and low ribosome occupancy between the two rescue experiments (SF compared to SF + q and SF compared with SF + qR).
- Ingenuity molecular function analysis of differentially translated mRNAs. The top eight categories according to the ingenuity causal network P‐value are shown.
- Transmission electron microscopy images showing cellular overview with increased cystic vacuoles of the ER upon Q depletion. Arrowheads point to rough endoplasmic reticulum while expansions are indicated by asterisk. N, nucleus. Scale bar 500 nm. Automated quantification of the size of all vacuoles is presented in violin plot between the indicated grown conditions (5 optical fields). Black lines show the medians; white lines represent individual data points; polygons represent the estimated density of the data; *P < 0.05 (t‐test).
- Automated quantification of fluorescence intensity (AU: arbitrary units) relative to the immunofluorescence in Fig 6B. Error bars: ± SD (all cells in n ≥ 3 optical fields); *P < 0.05 (t‐test).
- Western blot showing increased eIF2α phosphorylation upon Q depletion. Actin and global eIF2 levels are used as loading controls. Quantification of the eIF2α phosphorylation signal normalized to β‐actin. Each bar represents three independent biological replicates. Error bars: ± SD; *P < 0.05 (t‐test).
The affected signaling pathways are strictly linked to the observed cellular phenotypes in the absence of Q: reduced global protein translation capacities (Fig 4C) and reduced cell proliferation rates (Fig 4D). Similarly, GO analysis of the differentially translated mRNAs showed enrichment for molecular functions that are closely related to the observed growth phenotype, such as cellular development, cell growth, and proliferation (Fig EV4D). Taken together, these results indicated that q‐dependent protein translation affects cell growth signaling and activated stress signaling.
To further characterize the effects of q depletion, we also used a destabilized variant of firefly luciferase (FlucR188Q) with fused enhanced GFP (EGFP; Gupta et al, 2011). The luminescence activity of Fluc mutation reflects imbalances in cellular protein homeostasis, and it requires chaperone surveillance to maintain soluble and enzymatically active state (Gupta et al, 2011). The GFP‐tagged Fluc variant could efficiently be used to measure the formation of protein aggregates in the absence of Q (Fig 5A and B). Furthermore, and consistent with a decreased folding capacity, the destabilized luciferase had a decreased luminescence activity in the absence of Q (Fig 5C). Importantly, the presence of aggregates and the luminescence activity of Fluc in Q‐depleted cells were consistently lower compared to Q‐rescue in the absence of any stress stimulus (Fig 5), which is likely due to the altered translational speed in Q‐free cells.
Figure 5. Translational stress results in unfolded proteins.
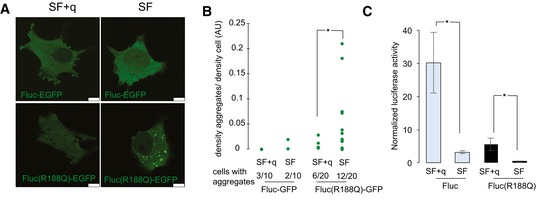
- Representative fluorescence confocal images of live HeLa cells expressing Fluc‐EGFP and FlucR188Q‐EGFP maintained in SF compared to SF + q. Scale bars, 10 μm.
- Fluorescence intensity of aggregates was quantified as a fraction of fluorescence of the entire cell. The number of cells showing at least one aggregate is indicated. Ten cells were imaged for Fluc‐EGFP and 20 cells for FlucR188Q‐EGFP from three independent transfections; *P < 0.05 (t‐test).
- Specific luminescence activity of EGFP‐tagged sensor proteins maintained in SF compared to SF + q. Luciferase activity was normalized to the luciferase protein content in the sample. Error bars: ± SD; *P < 0.05 (t‐test); n = 8.
We also used transmission electron microscopy (TEM) to characterize the cellular morphology and ultrastructure of Q‐deficient cells. Remarkably, TEM analysis revealed a strong dilatation of the endoplasmic reticulum (ER; Ghadially, 1998), with irregular vacuolization in Q‐depleted cells (Figs 6A and EV4E), suggesting pronounced ER stress. This finding was confirmed by immunostaining for the ER stress marker KDEL, which represents the Lys‐Asp‐Glu‐Leu (KDEL) ER retrieval sequence and suggested an accumulation of ER chaperones due to misfolded proteins (Yamamoto et al, 2001) specifically in Q‐depleted cells (Figs 6B and EV4F). Of note, the observed increase in ER size has also been shown to be an integral part of the cellular program to overcome ER stress and is closely associated with activation of the unfolded protein response (UPR) (Schuck et al, 2009).
Figure 6. ER stress and UPR response upon Q depletion.
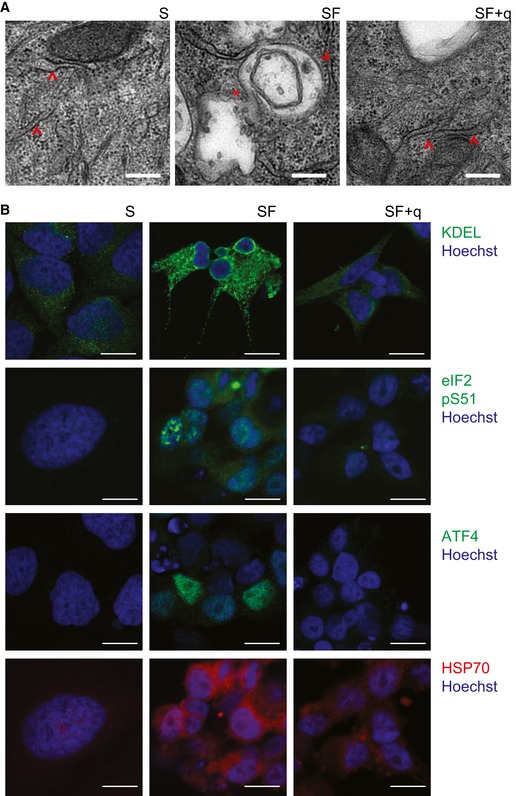
- Transmission electron microscopy images showing the increase in cystic ER vacuole upon q depletion. Arrowheads indicate rough endoplasmic reticulum while expansions are indicated by asterisks. Scale bar 250 nm.
- Representative images showing immunofluorescence of HeLa cells grown in S and SF medium in the absence or presence of q. Anti‐KDEL (ER stress marker), anti‐eIF2‐pS51 (ER stress‐induced inhibitor of translation), anti‐ATF4 (transcription factor that activates ER stress‐responsive genes), and anti‐Hps70 (chaperone). Nuclei were stained with Hoechst. Scale bar 10 μm.
Activation of the UPR leads to the phosphorylation of the eukaryotic translation initiation factor 2 alpha (eIF2α; Ron & Walter, 2007). Indeed, we observed an increase in phosphorylated eIF2α in Q‐depleted cells (Figs 6B and EV4F and G), which was in agreement with the observed enrichment of eIF2 signaling genes among the mRNA that is differentially translated upon Q depletion (Fig 4B). In addition, we also detected increased staining and expression for the ATF4 transcription factor and the HSP70 chaperone, two key components of the UPR, that are known to increase the protein folding capacity of the ER under stress conditions (Figs 6B and EV4F; Buchberger et al, 2010), as well as HSPA5 a major ER chaperone protein (Wang et al, 2009), which is up‐regulated in the absence of Q (Fig EV3C). It should be noted that the observed UPR activation is compatible with the adaptation of the cells to Q‐free conditions, as this is not an acute stress response. Importantly, expression of all markers could be restored to baseline levels by the addition of 20 nM q to the cell culture medium (Figs 6B and EV4F). Taken together, these findings strongly suggest that translation defects in Q‐deficient cells lead to the accumulation of misfolded proteins, which in turn trigger ER stress and the UPR.
Q‐dependent phenotypes in mice
To analyze the effects of Q depletion in vivo, we used a germ‐free mouse model (axenic NMRI mice, Fig EV5A). Axenic mice fed with conventional sterilized food showed physiological levels of both Q‐tRNA (48–58% of total tRNAHis in liver) and m5C38 (94% of tRNAAsp in liver) (Figs 7A and EV5B–D). In order to deplete Q in live animals, axenic mice were fed with a q‐free synthetic diet. After 60 days, various tissues were analyzed for tRNAHis queuosinylation and for m5C38‐tRNAAsp GUC (Figs 7A and EV5B–D). The results showed that highly proliferative tissues, such as the liver and small intestine, had very low levels of Q‐tRNA (21% and 19% of total tRNAHis), whereas brain, which represents a tissue with very low turnover rates, retained high levels of Q‐tRNA (60% of total tRNAHis; Figs 7A and EV5B–D). This is likely due to the efficient salvage of Q in non‐proliferating tissues.
Figure EV5. Q‐dependent phenotypes in Q‐deficient mice.
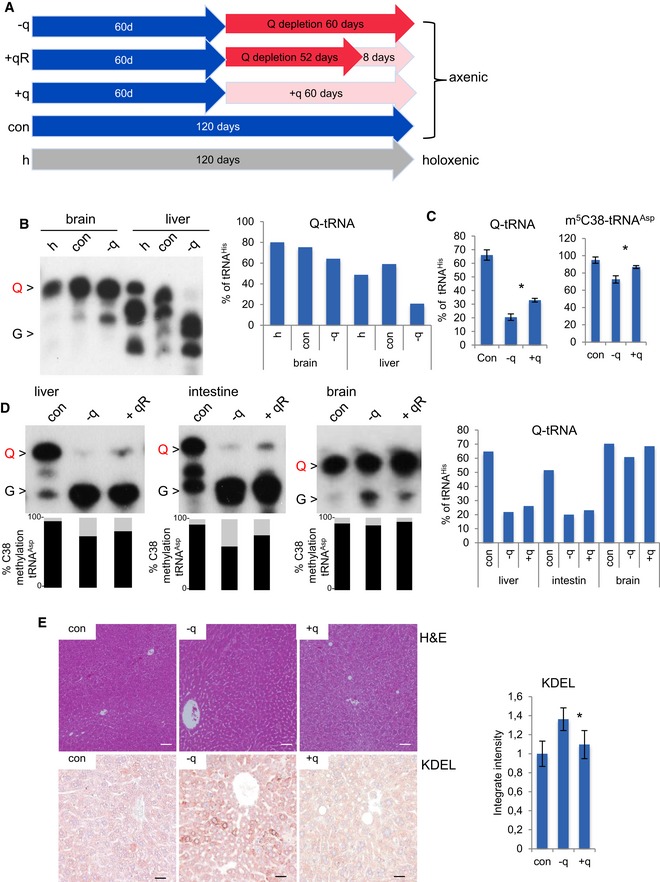
- Mouse feeding timing schedule.
- APB Northern blot using tRNAHis probe indicates that liver and brain axenic mouse tissues show physiological levels of Q‐tRNA and that q‐free diet depletes Q‐tRNA. Q‐tRNA level is obtained by quantification of Q and G signals and is presented on the right graph.
- Liver C38 methylation of tRNAAsp and Q‐tRNA percentage obtained by quantification of the Q and G signals in Fig 7A. Error bars: ± SD; (n = 3).
- APB Northern blot using tRNAHis probe shows that q‐free diet depletes Q‐tRNA. m5C38 state measured by 454 bisulfite sequencing in the same RNA. Both queuosinylation and methylation levels could be slightly restored by the addition of q to the diet for 8 days. Q‐tRNA percentage is obtained by quantification of the Q and G signals.
- The upper panels show liver sections from indicated mice stained with hematoxylin and eosin, whereas the lower panels show an overview of the immunostaining with antibodies against KDEL. Scale bar 50 μm. Automated quantification of KDEL intensity. Error bars: ± SD; *P < 0.05 (t‐test); n ≥ 3 optical fields; for three mice for condition.
Figure 7. Q‐dependent phenotypes in mice.
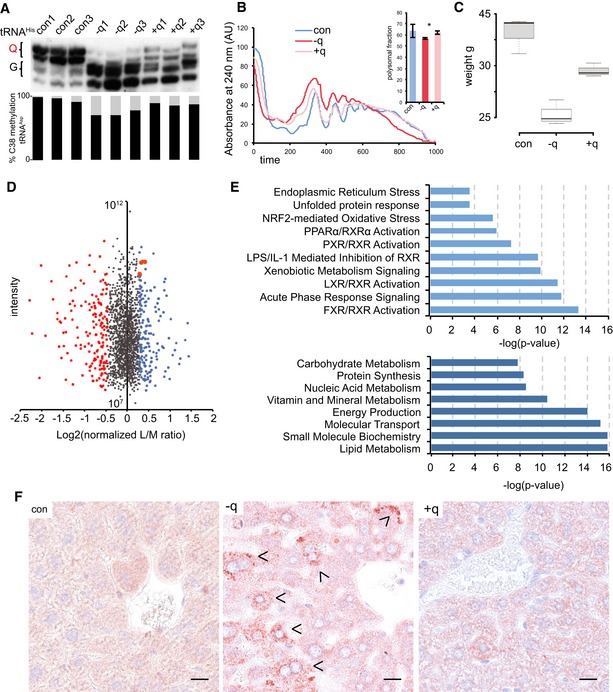
- APB Northern blot using a tRNAHis probe. m5C38 levels were determined by 454 bisulfite sequencing of RNA from the same liver tissue. Both queuosinylation and methylation levels could be restored by the addition of q to the feed. The results are shown for three biological replicates.
- Representative polysome profiles showing a reduction in the protein translation rate in the liver of axenic mice fed with a q‐free synthetic diet. Addition of q to the q‐free diet significantly rescued the translation rate. n = 3. Asterisks indicate statistically significant (P < 0.05, t‐test) differences.
- Box plot showing the body weight of mice fed a q‐free synthetic diet for 60 days, center lines show the medians; box limits indicate the 25th and 75th percentiles, whiskers extend 1.5 times the interquartile range; n = 3. Addition of q to the q‐free diet significantly rescued body weights.
- Dimethyl‐labeling analysis of −q versus +q liver tissue. Top 10% of deregulated proteins are indicated in red and blue. UPR effectors: HSPA5/BiP, HSP90b1, and CALR are in orange.
- Gene ontology analysis of deregulated proteins. Signaling pathways and biological functions are presented
- Liver sections from indicated mice were stained with anti‐KDEL antibody. Arrowheads indicate ER‐stressed hepatocytes. Scale bar 25 μm.
Reduced levels of Q‐tRNA in mice were consistently associated with lower levels of C38 methylation (Figs 7A and EV5B–D). Importantly, both effects were partially reversed by the addition of synthetic q to the diet for 8 days (21–23% of tRNAHis for liver and intestine, 60% of tRNAHis for brain) and more efficiently for 60 days (33% of total tRNAHis for liver) (Figs 7A and EV5B–D). Taken together, these results confirm the Q dependency of m5C38‐tRNAAsp GUC in vivo.
Next, we asked whether protein translation was affected in Q‐depleted mouse tissues. We therefore measured the protein translation rate in freshly dissected livers by polysome profiling. The results showed that low levels of Q‐tRNA and m5C‐tRNA were consistently associated with reduced levels of ribosomes engaged in translation (Fig 7A and B). Addition of q to the diet significantly (P < 0.05, t‐test) reversed the translation rate (Fig 7B). Consistent with reduced rates of protein translation, Q‐deficient mice had a substantially reduced body weight (Fig 7C). This phenotype could again be effectively reversed by the addition of 40 nM synthetic q to the diet (Fig 7C).
We also performed proteome‐wide profiling of Q‐depleted liver samples using dimethyl‐labeling analysis. We identified 1,690 common proteins in two biological replicates (Dataset EV3). Ingenuity pathway analysis of differentially expressed proteins identified all the nuclear retinoid X family of receptors signaling (LXR, PPARs, FXR, and RXR). RXR receptors act as “master regulators” of a number of different nuclear receptor‐based signal transduction pathways, with roles in development, cell growth and differentiation, metabolism, and cell death (Fig 7E; Szanto et al, 2004). Concordantly, GO term analysis of the deregulated proteins showed enrichment for molecular functions such as lipid, carbohydrate, nucleic acid metabolism, and protein synthesis (Fig 7E). In addition, signaling pathway analysis confirmed UPR, ER stress, and oxidative stress signaling as deregulated pathways (Fig 7E), according, three well‐established effectors of the UPR: HSPA5/Bip, HSP90b1, and CALR were up‐regulated in the absence of Q (Fig 7D).
Finally, we used immunohistochemistry to analyze ER stress markers in mouse tissues to determine whether the limited availability of q leads to activation of the UPR in vivo. Indeed, the depletion of Q induced a strong increase in the ER stress marker KDEL in mouse liver (Figs 7F and EV5E). Again, the expression of this ER stress marker was reversed to physiological levels by the addition of q in the diet (Figs 7F and EV5E). Together, these results confirm our cell‐based observations in vivo.
Discussion
In spite of various described Q‐dependent phenotypes in different organisms (Vinayak & Pathak, 2010; Fergus et al, 2015), a mechanistic understanding for the role of Q in protein translation and cellular homeostasis is still missing. Our results show that nutrition‐dependent levels of tRNA modifications control protein translation on the translatome scale. We find that reduced Q‐tRNA and m5C38‐tRNAAsp GUC modifications affect translational speed at Q‐decoded codons and near‐cognate codons. Their cumulative effects deregulate protein translation by modifying the amino acid composition of the proteome, resulting in unfolded proteins and protein aggregates, altered cell growth signaling and ultimately induce the activation of the unfolded protein response in cell culture and in vivo.
We have previously shown that the loss of C38 tRNA methylation in Dnmt2 knockout mice affects the discrimination of near‐cognate codons, resulting in highly specific amino acid exchanges (Tuorto et al, 2015). Our present study identifies queuosinylation as a key example for the concept of modification‐dependent genome recoding (Tuorto & Lyko, 2016). Interestingly, our findings provide evidence that nutritional factors are involved in this regulatory mechanism.
At present, our understanding of how nutrients control protein expression in mammalian cells and in vivo is relatively limited, although emerging evidence suggests that multiple mechanisms exist (Kilberg et al, 2012). Our findings uncover a direct link between micronutrient availability and protein translation based on the dynamics of tRNA modifications. Variations in translation speed are increasingly recognized as a mechanism that couples the decoding of genetic information to nascent polypeptide maturation, altered folding, membrane targeting, and modifying enzyme interactions (Yu et al, 2015; Rodnina & Wintermeyer, 2016). Our results show that Q‐tRNA and m5C38‐tRNAAsp GUC affect the protein translation speed at specific codons. In our Q‐free cell culture system and mouse model, dysregulation of translation resulted in the accumulation of misfolded proteins and aggregates that trigger the activation of ER stress and the UPR. This mechanism is in agreement with the phenotypes recently described in mouse and yeast that lack certain modifications at the wobble position (Laguesse et al, 2015; Nedialkova & Leidel, 2015; Chou et al, 2017).
Anticodon loop tRNA modifications are known to play a key role in the codon:anticodon pairing and decoding process (Agris et al, 2017). Recently, an alternative view of the genetic code has been proposed that considers the decoding process as an interplay of interactions between mRNA, tRNA, and rRNA taking place during protein synthesis (Grosjean & Westhof, 2016; Komar, 2016). Accordingly, decoding is explained by G + C “strong” or A + U “weak” binding capability of codon‐anticodon pairs, in the context of the ribosome shape (Grosjean & Westhof, 2016). In this model, tRNA modifications have been proposed to equilibrate the codon–anticodon strength by increasing the binding of weak codons and destabilizing the strong ones (Grosjean & Westhof, 2016). In agreement with this model, our ribosome profiling data show that Q alters the cellular translatome by accelerating the comparably weak Asn/Tyr and the intermediate His/Asp codons. In addition, Q increased the rate of U‐ending cognate recognition, which is in agreement with an earlier study on single Drosophila tRNAHis (Meier et al, 1985). In contrast, G‐tRNAAsp GUC became simultaneously reduced of m5C upon Q depletion and GAC speed thus became more affected than GAU.
Reduced translation speed was also observed at Glu (GAA, GAG) and Lys (AAA, AAG) codons, which is most likely caused by near‐cognate G‐tRNAs misreading. We suggest that G‐tRNAs slow down the elongation rate of near‐cognate triplets, reducing the ability of the ribosome to discriminate incorrect aminoacyl‐tRNAs. Thus, the observed translational delay and ribosome stalling can be explained by the error‐correcting action of the ribosome. The effect of G‐tRNA on near‐cognate codons is in agreement with recent findings showing that tRNA modifications do not solely serve to restrict the decoding capacity of the tRNA to its cognate codon, but also for the suppression of near‐cognate stop codons (Blanchet et al, 2018). Finally, bulky tRNA modifications have also been proposed to avoid non‐synonymous misreading by blocking second‐position errors (Manickam et al, 2014; Rozov et al, 2015). Consistently, we observed reduced translation speed on Gly (GGU), Arg (CGU), or Ser (AGU) codons in the absence of Q. Altogether, our findings define a role for Q in the decoding of the mammalian transcriptome by modulating of translation speed at Q codons and near‐cognate Q codons.
The frequencies of alternative synonymous codons vary both among species and among genes from the same species. However, the mechanisms determining codon usage bias, as well as the biological significances of biased codon composition, are only beginning to be deciphered (Nabiyouni et al, 2013; Zaborske et al, 2014). Positive selection for optimal codons due to benefits in translational efficiency and fidelity is a major evolutionary driver, and tRNA modifications have been suggested to play a central role by regulating the speed of ribosomal translation (Novoa & Ribas de Pouplana, 2012). Among eukaryotes, yeasts and mammals display distinct codon usage bias. For the codons translated by Q‐modified tRNAs, the human preferences are for C‐ending codons, whereas in yeast, U‐ending codons are preferred (Nakamura et al, 2000). It will be interesting to further explore the impact of physiological and pathological changes in queuine bioavailability on the plasticity of protein translation.
Materials and Methods
Cell culture
HeLa and HCT116 cell lines were obtained from ATCC and authenticated by Multiplex Human Cell Line Authentication Test (Multiplexion). HeLa cell line was grown in Dulbecco's modified Eagle's medium (DMEM), while HCT116 cell line was cultured in McCoy`s 5A. The cultures were supplemented with 10% heat‐inactivated FBS, 2 mM l‐glutamine, and a commercial cocktail of antibiotics (Invitrogen).
For all Q conditions, UltraCULTURE serum‐free medium (Lonza) was supplemented with 2 mM L‐glutamine and 100 units/ml penicillin/streptomycin. In order to dissociate cells, StemPro Accutase (Gibco) was used instead of trypsin, since trypsin is of animal source and may contain significant amount of Q. Synthetic queuine, kindly provided by Hans‐Dieter Gerber and Gerhard Klebe (Universität Marburg) (Gerber & Klebe, 2012), was used at 20 nM unless otherwise stated. Viable cells were counted using a CASY Cell Counter (Roche Diagnostics).
Immunofluorescence, immunohistochemistry, and Western blotting
Immunofluorescence was performed on 2% PFA‐fixed cells blocked with 10% NGS, 0.1% Triton X‐100 in PBS. Secondary antibodies (Alexa Fluor‐conjugated, Invitrogen) were added at a dilution of 1:500 for 45 min at room temperature. Cells were finally stained with DAPI and mounted with Fluoromount‐G (SouthernBiotech). Confocal images were acquired on a Leica TCS SP5 microscope. All the images were processed with Photoshop CS5 (Adobe) software. Automated quantification of average fluorescence intensity was performed using ImageJ.
Tissue slices of mouse liver were fixed in 4% formaldehyde in PBS for histological and immunohistological analyses. Paraffin‐embedded liver sections (4 μm) were stained with hematoxylin and eosin or alternatively processed with anti‐KDEL using a standard immunohistochemistry protocol. Control sections were made by the omission of the primary antibody. The pictures were taken with the Mirax Viewer imaging system (Carl Zeiss MicroImaging GmbH).
Western blotting was performed according to the standard protocols using 100 μg of total proteins. Protein expression of EIF2S1 was analyzed using ImageJ.
Antibodies such as anti‐KDEL (Abcam ab176333), anti‐EIF2S1 (Abcam ab32157), anti‐ATF4 (Cell Signaling 11815), anti‐HSP70 (Abcam ab5439), and anti‐EIF2 (Enzo ADI‐KAP‐CP130) were used.
Electron microscopy
Cells were fixed with 2.5% (w/v) glutaraldehyde (Plano, Wetzlar)/0.05 M cacodylate buffer (Serva). A second fixation using 2.5% (w/v) glutaraldehyde/2% (w/v) osmium tetroxide (ChemPur) in 0.05 M cacodylate buffer was performed. Subsequently, cells were embedded in araldite (Serva). Ultrathin sections (70 nm) were cut using an ultramicrotome (UCT Leica) and then contrasted applying uranyl acetate (West Chester) and citric lead (Serva). The analysis was performed with a Zeiss EM 910 microscope (Carl Zeiss), and micrographs were taken with a CCD K2 camera (TRS). Automated quantification of vacuole size was performed using camera System CCD 2K (Tröndle) and program EM Images SP (Tröndle).
Aggregate and luciferase assays
Cells were seeded in 96‐well plates or in Chambered Coverglass (Thermo Scientific Nunc Lab‐Tek II) 24 h before transfection and grown at appropriate conditions. 70–80% confluent cells were transfected with Fluc‐GFP or FlucR188Q‐GFP plasmids obtained by Addgene (Gupta et al, 2011) using jetPRIME Transfection Reagent (PolyPlus) according to the manufacturer's instructions. After 24 h, cells were directly assessed for GFP aggregation or luciferase activity. To assess GFP aggregation, confocal images of live cells were acquired using Leica TCS SP5 microscope. All the images were processed with Photoshop CS5 (Adobe) software. Automated quantification of fluorescence intensity of aggregates was obtained as a fraction of fluorescence of the entire cell using ImageJ. For Fluc activity measurements, ~10,000 cells were seeded in 96‐well plates, and luciferase assay was performed according to manufacturer's instructions (Promega). Luminescence activity was then recorded in a luminometer (FLUOStar Optima) with acquisition time of 10 s. To determine relative specific luciferase activities, luciferase luminescence values were normalized to the luciferase protein content in the sample. After the measurements, aliquots were withdrawn and analyzed by immunoblotting with anti‐GFP antibody (Roche 118114460001).
RNA isolation
50–100 mg of each tissue or 105–107 cells were homogenized in 1 ml of TRIzol (Invitrogen), and RNA was extracted according to the procedural guidelines. The nucleic acid concentrations and purity for further analysis were analyzed on the NanoDrop ND‐1000. RNA integrity was measured on a TapeStation (Agilent). Samples were stored at −80°C.
Northern blot—acryloyl aminophenylboronic acid (APB) gels
Acryloyl aminophenylboronic acid gels were prepared and run with a few modifications according to Igloi and Kossel (1985) and Zaborske et al (2014). 15 μg of RNA was deacetylated in 100 mM Tris–HCl pH 9 for 30 min at 37°C. RNA was ethanol‐precipitated and resuspended in 1× RNA‐loading dye (Fermentas). Periodate‐oxidated samples were prepared as a control. Samples were then denatured for 10 min at 70°C and run at 4°C on TAE, 8 M urea, 15% acrylamide, and 5 mg/ml aminophenylboronic acid (Sigma) gels. Gels were blotted, hybridized, and quantified as described previously (Tuorto et al, 2012). Northern probes are listed in Appendix Table S1.
RNA bisulfite sequencing
Bisulfite conversion was performed using the EZ RNA Methylation™ Kit—Zymo. Amplicons for 454 (Roche) sequencing were generated by PCR as described in Tuorto et al (2015). The primer sequences are listed in Appendix Table S1. For each amplicon, sequences showing a conversion > 95% and a lower threshold identity > 90% were aligned and aggregated in heatmaps. The methylation rate was calculated at each position as the fraction of non‐converted cytosines.
Polysome analysis
107 cells were treated with cycloheximide (100 μg/ml) for 5 minutes washed once in cold PBS/cycloheximide (100 μg/ml) and lysed in 400 μl buffer (20 mM Tris–HCl, pH 7.4, 5 mM MgCl2, 150 mM NaCl, 1% Triton X‐100, 100 μg/ml cycloheximide, 1× complete protease inhibitors (Roche)). The lysates were centrifuged at 9,300 g for 10 min at 4°C, and the supernatants were applied to linear 17.5–50% sucrose gradients in 20 mM Tris–HCl (pH 7.4), 5 mM MgCl2, and 150 mM NaCl. Centrifugation was carried out at 35,000 rpm for 2.5 h at 4°C in a Beckmann SW60 rotor. Gradients were eluted with an ISCO UA‐6 gradient fractionator, and polysome profiles were recorded by continuously monitoring the absorbance at 254 nm. In order to calculate the fraction of ribosomes engaged in translation, the area under the polysomal part of the curve was divided by the area below the entire curve.
Ribosome footprinting
Ribosome footprinting was performed according to Ingolia et al (2011) and as previously described in Tuorto et al (2015). Briefly, 400 μl aliquot of lysate prepared as described above was treated with 4 U of DNase I (Thermo Scientific) and 800 U of RNase I (Ambion) for 45 min at room temperature with gentle shaking. 800 U of RNasin ribonuclease inhibitor (Promega) was added to quench the reaction, and the samples were run on a 17.5–50% sucrose gradient to isolate monosomes. Monosome‐protected RNA fragments were end‐repaired with T4 polynucleotide kinase (Takara) and size‐selected at 28–31 nucleotides on a 15% polyacrylamide TBE‐urea gel. Sequencing libraries were prepared according to protocol of the NEB NEXT Small RNA Library Prep Set for Illumina (Multiplex Compatible) E7330.
RNA sequencing
RNA Sequencing libraries were prepared using the TruSeq RNA Sample Preparation Kit v2 (Illumina) according to manufacturer's instructions. Briefly, poly (A)+ RNA was purified from 1 μg of total RNA using oligo(dT) beads, fragmented, and converted to cDNA. The double‐stranded cDNA fragments were then end‐repaired, adenylated on the 3′ end, adapter‐ligated and amplified with 12 cycles of PCR. The final libraries were validated using Qubit Fluorometer (Life Technologies—Invitrogen) and Agilent TapeStation 4200 (Agilent Technologies). After size validation, libraries were normalized, pooled, and clustered on the cBot (Illumina, Inc.) with a final concentration of 10 pM (spiked with 1% PhiX control v3, Cat No. FC‐110‐3001) using the TruSeq SR Cluster Kit v3 (Cat no. GD‐401‐3001). Sequencing on HiSeq 2000 (Illumina) was performed using the 50 cycles TruSeq SBS Kit v3 according to the manufacturer's instructions.
Ribosome footprints and RNA‐seq analysis
Ribosome footprint analysis was performed with minor changes as previously described (Tuorto et al, 2015). Briefly, sequenced reads from ribosome footprints were trimmed and adaptor sequences were removed. Resulting reads were kept if their size was in 25–35 nt, and further aligned to a depletion reference in a first step, with Bowtie (Ingolia et al, 2012), permitting two mismatches with base quality at least 70, using a seed of length of 23 nt. A reference database for read depletion was established from an exhaustive collection of human tRNA (CCA‐appended sequences from gtrnadb.ucsc.edu), rRNA (arb‐silva, Ensembl ncRNA, NCBI RefSeq, and Biobases), and mitochondrial tRNA and rRNA sequences (Ensembl ncRNA). Remaining reads after depletion were aligned to reference mRNA sequences from RefSeq. Annotation of codon sequence start and end position of this collection of mRNA transcripts was retrieved simultaneously. Total reads for each replicate were between 10.3 and 16.4 million reads, of which 1.4–2.6 million reads mapped to mRNA transcripts. Data analysis was performed using a local Galaxy server and custom Python and R scripts.
Resulting aligned reads containing the annotated codon start site were length‐stratified (25–32 nt), and we counted each read start occurring at 20 nt upstream or downstream of the codon start. Lengths which exhibited three nucleotides periodicity were considered compatible with the subsequent A‐site assignment rationale. A‐site position was assigned according to an offset equal to +15 nt for 26–30 read lengths offset from the 5′ end of the reads (Ingolia, 2010). Reads were assigned to a codon when mapped to −1, 0, +1 relative to the first nucleotide of the codon (Tuorto et al, 2015). Occupancy of A‐site codons was normalized by the frequency of the same codon in the non‐decoded +1, +2, +3 triplets relative to the A‐site, the first 15 codons of each ORF were excluded from this analysis (Lecanda et al, 2016). In addition, read counts per mRNA, RPKM, and standardized metagene density of footprints were determined. For the two latter, reads of at least 25 nt in length were considered.
Sequenced reads from RNA‐seq were filtered and aligned similarly to ribosome footprints except that no superior limit on read length was set. Ribosome occupancy (RO) for differentially translated transcripts were determined as the ratio of number of footprints to number of transcripts from RNA‐seq corresponding to a specific mRNA.
SILAC analysis
HeLa cells were first depleted of Q by growing 3 weeks in DMEM supplemented with 10% dialyzed FBS (Sigma), 2 mM l‐glutamine, and a commercial cocktail of antibiotics (Invitrogen). Then, the cells were shifted to DMEM SILAC‐labeling medium (Sigma D9443) supplemented with 10% dialyzed FBS, 0.105 g/l L‐leucine, 0.084 g/l L‐arginine monohydrochloride, and 0.584 g/l L‐glutamine. 20 nM q, 0.152 g/l 13C6,15N2‐l‐lysine‐hydrochloride, and 0.146 g/l L‐lysine monohydrochloride were added to the cultures according to Fig EV3A. The cells were passed five times resulting in a population doubling higher than 12. Cell pellets were collected after three washes with PBS and snap‐frozen in liquid N2. The proteome was extracted according to Sapcariu et al (2014). Briefly, cell pellets were resuspended in 400 μl of methanol, followed by the addition of 400 μl of water and 400 μl of chloroform. After centrifugation, the formed interface was precipitated and washed once with methanol. The dried pellet was resuspended in 8 M urea and 50 mM ammonium bicarbonate (pH 8.0). After reduction with 5 mM DTT and alkylation with 15 mM iodoacetamide, the proteins were digested in a two‐step digestion with endopeptidase Lys‐C, first digestion in 8 M urea at 37°C (0.4 μg Lys‐C, Wako) followed by dilution to 2 M and digestion overnight (0.4 μg Lys‐C, Wako, 37°C). The resulting peptides were cleaned up on a Sep‐Pak C18 96 well plate (25 mg, Waters). The peptides were measured on a Q‐Exactive Plus mass spectrometer (Thermo Scientific) coupled to an Ultimate 3000 RSLCnano system (Thermo Scientific) in data‐dependent acquisition mode, selecting the top 12 peaks for higher energy collisional dissociation (HCD) fragmentation. A 90‐min gradient (solvent A: 0.1% formic acid; solvent B: HPLC grade acetonitrile in 0.1% formic acid) was applied using an Acclaim PepMap trap column (2 cm × 75 μm i.d., C18, 3 μm, 100 Å, Thermo Scientific) and an Acclaim PepMap RSLC analytical column (50 cm × 75 μm i.d., C18, 2 μm, 100 Å, Thermo Scientific). A volume of 3 μl sample was injected and the peptides eluted with 90 min gradients of 2–35% solvent B at flowrates of 0.3 μl/min. MS1 data acquisition was performed at a resolution of 60,000, using an injection time of 45 ms in the scan range from 375 to 1,500 m/z and MS2 at a resolution of 15,000 with an injection time of 45 ms. The normalized collision energy was set to 28 eV. The mass window for precursor ion selection was set to 1.2 m/z. The recorded spectra were analyzed using the MaxQuant software package (Version 1.6.1.0), and data analysis was done with the R‐software package (Version 3.4.4).
Proteins which are synthesized in −q and +q with the same rate have a log2 ratio of 0 (log2(1) = 0). The top 10% differentially expressed proteins were considered as regulated. Codon numbers for all mRNA transcripts were calculated, using the hg19 CCDS archive from NCBI. The numbers are represented as per thousand of the whole coding sequence. The codon frequencies for the regulated proteins were calculated and compared to the codon frequency of identified unchanged proteins to avoid a bias from the identification preference.
Mice and ethics statement
All mouse husbandry and experiments were carried out at the German Cancer Research Center pathogen‐free animal facility according to applicable laws and regulations. The axenic NMRI mice were held in isolators in a sterile environment. NMRI control mice were fed an autoclaved diet (3307 PML15 Provimi Kliba SA, CH). The queuine‐free diet was based on a previously published chemically defined diet (Marks & Farkas, 1997). All the components were added to the diet in powder form (Altromin, Germany Cat# C 1069), and fresh powder was dissolved in an appropriate amount of sterile water on a daily basis. Tyr‐ethyl ester was replaced with L‐tyrosine.
The addition of q to the feed was done by sterile filtration and UV irradiation of a 100 μM stock of queuine‐hydrochloride in sterile water. The stock was stored at −20°C, and a final concentration of 40 ng/ml was fed the mice.
Dimethyl‐labeling analysis
Protein samples were run on SDS–PAGE and fractionated. Gel pieces were cut out, cysteines were reduced by adding DTT, and then carbamidomethylated using iodoacetamide followed by overnight digestion with trypsin. For quantification, peptides were chemically labeled by stable isotope dimethyl labeling and pooled before LC‐MS analysis according to −q light, +q medium, for two biological replicates per condition. Resulting peptides were loaded on a cartridge trap column, packed with Acclaim PepMap300 C18, 5 μm, 300 Å wide pore (Thermo Scientific), and separated in a 180‐min gradient from 3% to 40% ACN on a nanoEase MZ Peptide analytical column (300 Å, 1.7 μm, 75 μm × 200 mm, Waters). Eluting peptides were analyzed using an online‐coupled Q‐Exactive‐HF‐X mass spectrometer. Data analysis was carried out by MaxQuant (version 1.6.0.16). In total, 24,033 peptides and 2,399 proteins were identified by MSMS based on an FDR cutoff of 0.01 on peptide level and 0.01 on protein level. Quantification was done using a DML duplex approach and DimethLys4;DimethNter4 as isotopically labeled modifications. In total, 2,199 proteins were quantified.
Statistical analysis
Differential expression and RO were assessed using DESeq2 (Anders & Huber, 2010) and RiboDiff (Zhong et al, 2017), respectively. Differences in codon occupancy from unity were determined using an unpaired two‐tailed t‐test. To determine if codon occupancy distribution was significantly different in a condition as compared to a control, it was compared to a basal distribution (calculated by dividing codon occupancy in one replicate by the other replicate) using a Kolmogorov–Smirnov test in R. Error bars represent standard error if not otherwise stated. Heatmap and hierarchical clustering were obtained using ClustVis (http://biit.cs.ut.ee/clustvis/). The significance of quantitative data was tested using unpaired, two‐tailed Student's t‐tests, unless otherwise stated.
Data accessibility
Gene expression and ribosome profiling sequencing data are available from the NCBI GEO database: GSE102315.
Author contributions
FT, AEE‐M, and FL conceived the study, designed the experiments, and interpreted the results. FT, RL, and CC performed cell cultures and molecular analysis. FT performed ribosome profiling. CL performed statistical and computational analyses. GD performed SILAC measurements. GF and H‐JG performed the mouse analysis and the TEM; MM provided key reagents. FT and FL wrote the manuscript with contributions from the other co‐authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 7
Acknowledgements
We thank Dario Demartin and Nader Alerasool for their contribution during early stages of this project. We also thank Kurt Reifenberg for axenic mice; Larissa Ziegler, Tanja Musch, and Sylvia Kaden for technical support; Felix Bormann for bioinformatics assistance; Hans‐Dieter Gerber and Gerhard Klebe for materials; the DKFZ light microscopy facility for instrument support; the DKFZ Protein Analysis Core Facility and Bernd Heßling for dimethyl‐labeling measurements. We thank and Georg Stoecklin, Manuel Rodríguez‐Paredes, and Thomas G. Hofmann for valuable discussions and comments. We also thank Adele Bourmaud and Celine Jaunty of the Proteome and Genome Research laboratory for their support measuring the proteomic samples. This work was supported by grants from Deutsche Forschungsgemeinschaft (DFG) (SPP1784) to AEM and FL, and DFG TU5371‐1 to FT. Additional support was provided by a DKFZ NCT3.0 Integrative Project in Cancer Research to FL. GD was supported by the PEARL/CPIL grant of the Luxembourg National Research Fund (FNR), and FT was supported by the Institute of Genetics and Biophysics A. Buzzati‐Traverso, C.N.R., Italy.
The EMBO Journal (2018) 37: e99777
See also: https://doi.org/10.15252/embj.2018100405 (September 2018)
References
- Agris PF (2004) Decoding the genome: a modified view. Nucleic Acids Res 32: 223–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agris PF, Eruysal ER, Narendran A, Väre VYP, Vangaveti S, Ranganathan SV (2017) Celebrating wobble decoding: half a century and still much is new. RNA Biol 14: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I (2015) Targeting the translation machinery in cancer. Nat Rev Drug Discov 14: 261–278 [DOI] [PubMed] [Google Scholar]
- Blanchet S, Cornu D, Hatin I, Grosjean H, Bertin P, Namy O (2018) Deciphering the reading of the genetic code by near‐cognate tRNA. Proc Natl Acad Sci USA 115: 3018–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Bukau B, Sommer T (2010) Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 40: 238–252 [DOI] [PubMed] [Google Scholar]
- Chou HJ, Donnard E, Gustafsson HT, Garber M, Rando OJ (2017) Transcriptome‐wide analysis of roles for tRNA modifications in translational regulation. Mol Cell 68: 978–992.e974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick FH (1966) Codon–anticodon pairing: the wobble hypothesis. J Mol Biol 19: 548–555 [DOI] [PubMed] [Google Scholar]
- Durand JM, Dagberg B, Uhlin BE, Bjork GR (2000) Transfer RNA modification, temperature and DNA superhelicity have a common target in the regulatory network of the virulence of Shigella flexneri: the expression of the virF gene. Mol Microbiol 35: 924–935 [DOI] [PubMed] [Google Scholar]
- El Yacoubi B, Bailly M, de Crecy‐Lagard V (2012) Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu Rev Genet 46: 69–95 [DOI] [PubMed] [Google Scholar]
- Farkas WR (1980) Effect of diet on the queuosine family of tRNAs of germ‐free mice. J Biol Chem 255: 6832–6835 [PubMed] [Google Scholar]
- Fergus C, Barnes D, Alqasem MA, Kelly VP (2015) The queuine micronutrient: charting a course from microbe to man. Nutrients 7: 2897–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HD, Klebe G (2012) Concise and efficient syntheses of preQ1 base, Q base, and (ent)‐Q base. Org Biomol Chem 10: 8660–8668 [DOI] [PubMed] [Google Scholar]
- Ghadially FN (1998). Ultrastructural pathology of the cell and matrix, 3rd edn Guildford: Butterworth Scientific Ltd; [Google Scholar]
- Gilbert WV, Bell TA, Schaening C (2016) Messenger RNA modifications: form, distribution, and function. Science 352: 1408–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H, de Crecy‐Lagard V, Marck C (2010) Deciphering synonymous codons in the three domains of life: co‐evolution with specific tRNA modification enzymes. FEBS Lett 584: 252–264 [DOI] [PubMed] [Google Scholar]
- Grosjean H, Westhof E (2016) An integrated, structure‐ and energy‐based view of the genetic code. Nucleic Acids Res 44: 8020–8040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Kasturi P, Bracher A, Loew C, Zheng M, Villella A, Garza D, Hartl FU, Raychaudhuri S (2011) Firefly luciferase mutants as sensors of proteome stress. Nat Methods 8: 879–884 [DOI] [PubMed] [Google Scholar]
- Guy MP, Young DL, Payea MJ, Zhang X, Kon Y, Dean KM, Grayhack EJ, Mathews DH, Fields S, Phizicky EM (2014) Identification of the determinants of tRNA function and susceptibility to rapid tRNA decay by high‐throughput in vivo analysis. Genes Dev 28: 1721–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada F, Nishimura S (1972) Possible anticodon sequences of tRNA His, tRNA Asm, and tRNA Asp from Escherichia coli B. Universal presence of nucleoside Q in the first position of the anticodons of these transfer ribonucleic acids. Biochemistry 11: 301–308 [DOI] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, Blenis J (2005) mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123: 569–580 [DOI] [PubMed] [Google Scholar]
- Hori H (2014) Methylated nucleosides in tRNA and tRNA methyltransferases. Front Genet 5: 144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igloi GL, Kossel H (1985) Affinity electrophoresis for monitoring terminal phosphorylation and the presence of queuosine in RNA. Application of polyacrylamide containing a covalently bound boronic acid. Nucleic Acids Res 13: 6881–6898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT (2010) Genome‐wide translational profiling by ribosome footprinting. Methods Enzymol 470: 119–142 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS (2011) Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS (2012) The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome‐protected mRNA fragments. Nat Protoc 7: 1534–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492 [DOI] [PubMed] [Google Scholar]
- Kasai H, Nakanishi K, Macfarlane RD, Torgerson DF, Ohashi Z, McCloskey JA, Gross HJ, Nishimura S (1976) Letter: the structure of Q* nucleoside isolated from rabbit liver transfer ribonucleic acid. J Am Chem Soc 98: 5044–5046 [DOI] [PubMed] [Google Scholar]
- Katze JR, Basile B, McCloskey JA (1982) Queuine, a modified base incorporated posttranscriptionally into eukaryotic transfer RNA: wide distribution in nature. Science 216: 55–56 [DOI] [PubMed] [Google Scholar]
- Kilberg MS, Balasubramanian M, Fu L, Shan J (2012) The transcription factor network associated with the amino acid response in mammalian cells. Adv Nutr 3: 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar AA (2016) The “periodic table” of the genetic code: a new way to look at the code and the decoding process. Translation (Austin) 4: e1234431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguesse S, Creppe C, Nedialkova DD, Prevot PP, Borgs L, Huysseune S, Franco B, Duysens G, Krusy N, Lee G, Thelen N, Thiry M, Close P, Chariot A, Malgrange B, Leidel SA, Godin JD, Nguyen L (2015) A dynamic unfolded protein response contributes to the control of cortical neurogenesis. Dev Cell 35: 553–567 [DOI] [PubMed] [Google Scholar]
- Lecanda A, Nilges BS, Sharma P, Nedialkova DD, Schwarz J, Vaquerizas JM, Leidel SA (2016) Dual randomization of oligonucleotides to reduce the bias in ribosome‐profiling libraries. Methods 107: 89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manickam N, Nag N, Abbasi A, Patel K, Farabaugh PJ (2014) Studies of translational misreading in vivo show that the ribosome very efficiently discriminates against most potential errors. RNA 20: 9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks T, Farkas WR (1997) Effects of a diet deficient in tyrosine and queuine on germfree mice. Biochem Biophys Res Commun 230: 233–237 [DOI] [PubMed] [Google Scholar]
- Meier F, Suter B, Grosjean H, Keith G, Kubli E (1985) Queuosine modification of the wobble base in tRNAHis influences ‘in vivo’ decoding properties. EMBO J 4: 823–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RC, Brown KG, Elliott MS (1999) The effect of queuosine on tRNA structure and function. J Biomol Struct Dyn 16: 757–774 [DOI] [PubMed] [Google Scholar]
- Motorin Y, Lyko F, Helm M (2010) 5‐methylcytosine in RNA: detection, enzymatic formation and biological functions. Nucleic Acids Res 38: 1415–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Hartmann M, Schuster I, Bender S, Thuring KL, Helm M, Katze JR, Nellen W, Lyko F, Ehrenhofer‐Murray AE (2015) Dynamic modulation of Dnmt2‐dependent tRNA methylation by the micronutrient queuine. Nucleic Acids Res 43: 10952–10962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabiyouni M, Prakash A, Fedorov A (2013) Vertebrate codon bias indicates a highly GC‐rich ancestral genome. Gene 519: 113–119 [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Gojobori T, Ikemura T (2000) Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res 28: 292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedialkova DD, Leidel SA (2015) Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161: 1606–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa EM, Ribas de Pouplana L (2012) Speeding with control: codon usage, tRNAs, and ribosomes. Trends Genet 28: 574–581 [DOI] [PubMed] [Google Scholar]
- Owenby RK, Stulberg MP, Jacobson KB (1979) Alteration of the Q family of transfer RNAs in adult Drosophila melanogaster as a function of age, nutrition, and genotype. Mech Ageing Dev 11: 91–103 [DOI] [PubMed] [Google Scholar]
- Rakovich T, Boland C, Bernstein I, Chikwana VM, Iwata‐Reuyl D, Kelly VP (2011) Queuosine deficiency in eukaryotes compromises tyrosine production through increased tetrahydrobiopterin oxidation. J Biol Chem 286: 19354–19363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyniers JP, Pleasants JR, Wostmann BS, Katze JR, Farkas WR (1981) Administration of exogenous queuine is essential for the biosynthesis of the queuosine‐containing transfer RNAs in the mouse. J Biol Chem 256: 11591–11594 [PubMed] [Google Scholar]
- Rezgui VA, Tyagi K, Ranjan N, Konevega AL, Mittelstaet J, Rodnina MV, Peter M, Pedrioli PG (2013) tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine‐tune protein translation by promoting ribosome A‐site binding. Proc Natl Acad Sci USA 110: 12289–12294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnina MV, Wintermeyer W (2016) Protein elongation, co‐translational folding and targeting. J Mol Biol 428: 2165–2185 [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: 519–529 [DOI] [PubMed] [Google Scholar]
- Rozov A, Demeshkina N, Westhof E, Yusupov M, Yusupova G (2015) Structural insights into the translational infidelity mechanism. Nat Commun 6: 7251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapcariu SC, Kanashova T, Weindl D, Ghelfi J, Dittmar G, Hiller K (2014) Simultaneous extraction of proteins and metabolites from cells in culture. MethodsX 1: 74–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer M, Pollex T, Hanna K, Lyko F (2009) RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res 37: e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck S, Prinz WA, Thorn KS, Voss C, Walter P (2009) Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol 187: 525–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szanto A, Narkar V, Shen Q, Uray IP, Davies PJ, Nagy L (2004) Retinoid X receptors: X‐ploring their (patho)physiological functions. Cell Death Differ 11(Suppl. 2): S126–S143 [DOI] [PubMed] [Google Scholar]
- Tuorto F, Lyko F (2016) Genome recoding by tRNA modifications. Open Biol 6: 160287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G, Lyko F (2012) RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat Struct Mol Biol 19: 900–905 [DOI] [PubMed] [Google Scholar]
- Tuorto F, Herbst F, Alerasool N, Bender S, Popp O, Federico G, Reitter S, Liebers R, Stoecklin G, Grone HJ, Dittmar G, Glimm H, Lyko F (2015) The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J 34: 2350–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayak M, Pathak C (2010) Queuosine modification of tRNA: its divergent role in cellular machinery. Biosci Rep 30: 135–148 [DOI] [PubMed] [Google Scholar]
- Wang M, Wey S, Zhang Y, Ye R, Lee AS (2009) Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal 11: 2307–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Fujii R, Toyofuku Y, Saito T, Koseki H, Hsu VW, Aoe T (2001) The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J 20: 3082–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CH, Dang Y, Zhou Z, Wu C, Zhao F, Sachs MS, Liu Y (2015) Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co‐translational Protein Folding. Mol Cell 59: 744–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborske JM, DuMont VL, Wallace EW, Pan T, Aquadro CF, Drummond DA (2014) A nutrient‐driven tRNA modification alters translational fidelity and genome‐wide protein coding across an animal genus. PLoS Biol 12: e1002015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R, Yuan Y, de Crecy‐Lagard V (2017) The Escherichia coli COG1738 Member YhhQ Is Involved in 7‐Cyanodeazaguanine (preQ(0)) Transport. Biomolecules 7: E12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Karaletsos T, Drewe P, Sreedharan VT, Kuo D, Singh K, Wendel HG, Ratsch G (2017) RiboDiff: detecting changes of mRNA translation efficiency from ribosome footprints. Bioinformatics 33: 139–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinshteyn B, Gilbert WV (2013) Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet 9: e1003675 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 7
Data Availability Statement
Gene expression and ribosome profiling sequencing data are available from the NCBI GEO database: GSE102315.