

Abstract
Argonaute/Piwi proteins can regulate gene expression via RNA degradation and translational regulation using small RNAs as guides. They also promote the establishment of suppressive epigenetic marks on repeat sequences in diverse organisms. In mice, the nuclear Piwi protein MIWI2 and Piwi‐interacting RNAs (piRNAs) are required for DNA methylation of retrotransposon sequences and some other sequences. However, its underlying molecular mechanisms remain unclear. Here, we show that piRNA‐dependent regions are transcribed at the stage when piRNA‐mediated DNA methylation takes place. MIWI2 specifically interacts with RNAs from these regions. In addition, we generated mice with deletion of a retrotransposon sequence either in a representative piRNA‐dependent region or in a piRNA cluster. Both deleted regions were required for the establishment of DNA methylation of the piRNA‐dependent region, indicating that piRNAs determine the target specificity of MIWI2‐mediated DNA methylation. Our results indicate that MIWI2 affects the chromatin state through base‐pairing between piRNAs and nascent RNAs, as observed in other organisms possessing small RNA‐mediated epigenetic regulation.
Keywords: epigenetics, lncRNA, piRNA, Piwi, spermatogenesis
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Development & Differentiation; RNA Biology
Introduction
Gene expression is regulated at multiple steps, including transcription, splicing, RNA export, RNA localization, translation, and RNA degradation, with multiple mechanisms acting together to determine which genes are expressed at each step. Piwi‐interacting RNAs (piRNAs) are 21–30 nt small RNAs that are mainly expressed in the germline (Watanabe & Lin, 2014). piRNAs act together with Piwi family proteins, a subfamily of Argonaute proteins. The Piwi‐piRNA complex binds to target RNAs, including retrotransposon RNA, mRNA, and long non‐coding RNA, via piRNA sequence complementarity (Bagijn et al, 2012; Sienski et al, 2012; Sytnikova et al, 2014; Wasik et al, 2015; Watanabe et al, 2015). The target RNAs are sometimes cleaved by the endonuclease (slicer) activity of Piwi proteins, which leads to degradation of the target RNAs. In addition to post‐transcriptional regulation, nuclear Piwi proteins in mice and flies are essential for the establishment of repressive chromatin marks on some retrotransposon sequences, which is required for normal gametogenesis (Carmell et al, 2007; Klenov et al, 2011).
Although the mechanisms of piRNA‐mediated transcriptional regulation in the fly have been well studied (Sienski et al, 2012, 2015; Huang et al, 2013; Le Thomas et al, 2013; Rozhkov et al, 2013; Yu et al, 2015; Iwasaki et al, 2016; Peng et al, 2016), very little is known about these mechanisms in mammals. There are three Piwi proteins in mice: MIWI, MIWI2, and MILI. MIWI2 and MILI are expressed in prospermatogonia (Aravin et al, 2008; Kuramochi‐Miyagawa et al, 2008), where the genome‐wide establishment of DNA methylation takes place. MIWI2 is present in nuclei and is required for DNA methylation and histone H3 lysine 9 trimethylation (H3K9me3) of some retrotransposons, such as LINE1 (L1) and IAP (Aravin et al, 2007, 2008; Carmell et al, 2007; Kuramochi‐Miyagawa et al, 2008; Shoji et al, 2009; Molaro et al, 2014; Pezic et al, 2014; Manakov et al, 2015; Nagamori et al, 2015; Kojima‐Kita et al, 2016). Because piRNAs expressed at this stage are preferentially mapped to these retrotransposon sequences (Aravin et al, 2008; Kuramochi‐Miyagawa et al, 2008), MIWI2‐bound piRNAs likely function as sequence determinant for the transcriptional repression of these retrotransposons. However, there is little direct evidence that they play this role.
Here, we show that MIWI2 affects chromatin states by targeting RNAs transcribed from piRNA‐dependent regions using piRNAs as a guide. In addition, we report the salient features observed among the piRNA‐dependent regions.
Results
Identification of hypomethylated regions in Mili knockout prospermatogonia
Recent genome‐wide studies have identified genomic targets of piRNA‐mediated epigenetic regulation by comparing DNA methylation (or H3K9me3) pattern between Mili −/− (or Miwi2 −/−) and control mice (Molaro et al, 2014; Pezic et al, 2014; Manakov et al, 2015; Nagamori et al, 2015). These studies used spermatocytes or spermatogonia from 10 days post‐partum (dpp) testes for the analyses despite the occurrence of piRNA‐mediated epigenetic regulation in prospermatogonia of ~16–19 days post‐coitum (dpc) testes (Kato et al, 2007). Given that DNA methylation is dynamically regulated during spermatogonial development (Hammoud et al, 2015; Kubo et al, 2015), some genomic targets might acquire DNA methylation (or H3K9me3) in spermatogonia independently of piRNAs. Such target regions could not be identified using spermatogonium and spermatocyte data in those studies. To identify a more complete set of genomic targets of piRNA‐mediated DNA methylation, we conducted whole‐genome bisulfite sequencing analysis using DNA obtained from 0 to 2 dpp Mili −/− and Mili +/− prospermatogonia (Appendix Supplementary Results and Fig EV1), because genome‐wide de novo DNA methylation has just completed at this stage (Kato et al, 2007). In Mili −/− prospermatogonia, not only MILI‐bound piRNAs but also a large portion of MIWI2‐bound piRNAs are lost, and therefore, de novo DNA methylation of many MIWI2‐dependent regions is compromised (Aravin et al, 2008; Kuramochi‐Miyagawa et al, 2008; Vasiliauskaite et al, 2017). Although global DNA methylation pattern was not changed (Fig EV2A), 6,541 genomic regions were identified to be hypomethylated in Mili −/− prospermatogonia by applying a stringent condition to the output of the Bisulfighter program (Saito et al, 2014). 88.1% of HMRs were 200–3,000 nt in length (Fig EV2B). Most HMRs identified in prospermatogonia were also hypomethylated in Mili −/− spermatocytes (Fig EV2C), although, overall, the extent of hypomethylation of these HMRs seemed to be more severe in prospermatogonia than in spermatocytes (Fig EV2D and E). Conversely, most of the 1,033 hypomethylated regions identified in Mili −/− spermatocytes were also hypomethylated in Mili −/− prospermatogonia (Fig EV2F). Of the 1,033 spermatocyte hypomethylated regions, 828 were overlapped with prospermatogonium HMRs. These results suggest almost the same loci are hypomethylated in Mili −/− prospermatogonia and spermatocytes.
Figure EV1. Statistics for bisulfite sequencing.
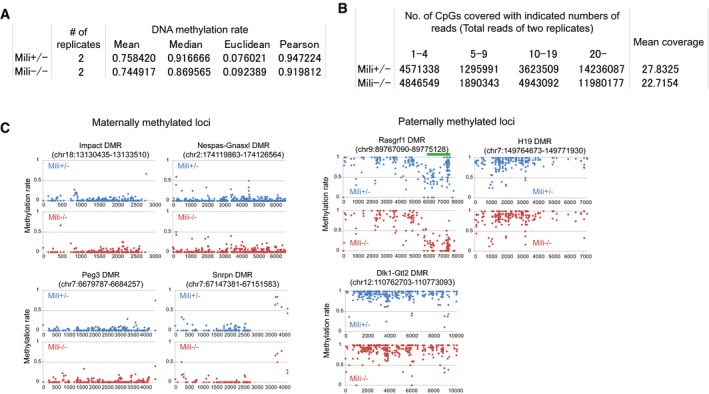
- Statistics for the CpG methylation rate. Pearson correlation and mean Euclidean distance between CpG methylation rates of two biological replicates are shown. Only CpG sites covered with more than 10 mapped reads in both replicates were considered for the analyses. Of 12406548 (Mili +/−) and 8228713 (Mili −/−) CpG sites that were considered for the analyses, 7,650,754 CpG sites were common between Mili +/− and Mili −/−.
- Genomic coverage of bisulfite sequencing. Total read numbers of both biological replicates were used for this analysis. For mean coverage, only CpGs covered with at least one unique sequence read were considered.
- DNA methylation pattern of maternally and paternally methylated loci. Hypomethylated region identified in the Rasgrf1 differentially methylated region (DMR) is indicated by green bar.
Figure EV2. Features of hypomethylated regions identified in Mili −/− prospermatogonia (HMRs).
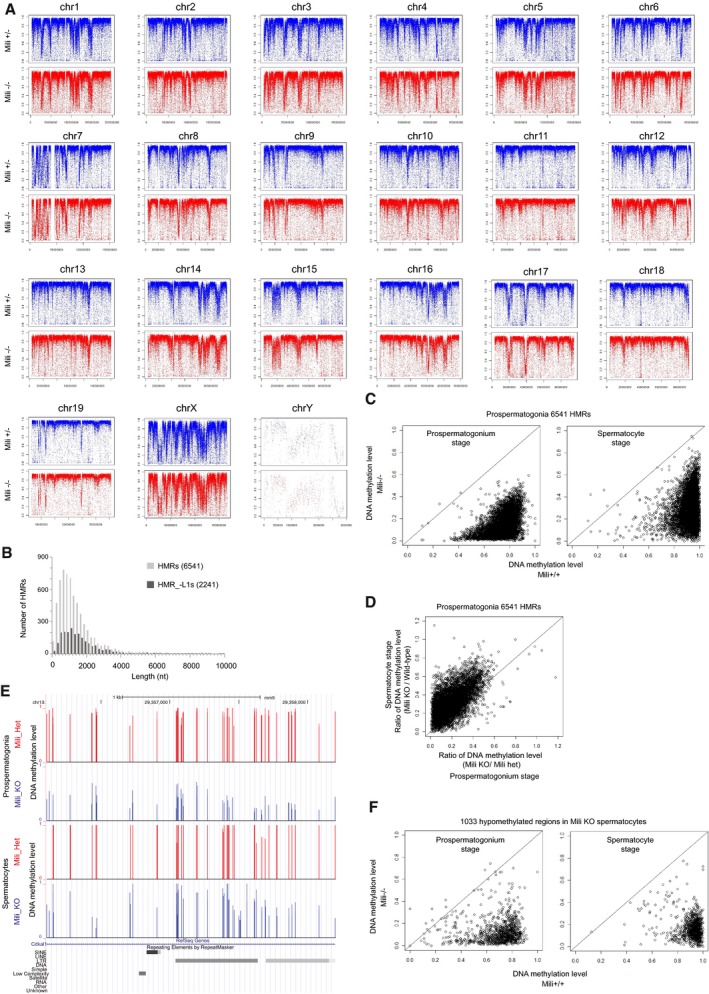
- DNA methylation pattern is not changed in Mili −/− prospermatogonia at chromosome levels. Average CpG DNA methylation levels (2‐kb window) are plotted on each chromosome.
- Length distribution of hypomethylated regions in Mili −/− prospermatogonia (HMRs). The numbers in parentheses represent the numbers of regions examined.
- Most HMRs are kept hypomethylated in Mili −/− spermatocytes. Circles in the charts show 6,541 HMRs. X‐axis (Y‐axis) represents average DNA methylation level in Mili −/− (control) mice. The results in prospermatogonia (top) and spermatocytes (bottom) are shown.
- DNA methylation of HMRs seems to be more severely affected in prospermatogonia compared with spermatocytes. X‐axis (Y‐axis) represents ratio of DNA methylation level between Mili KO and control mice in prospermatogonia (spermatocytes).
- A representative genomic locus that shows clear difference in DNA methylation level between Mili mutant and control mice in prospermatogonia compared with spermatocytes.
- Most hypomethylated regions in Mili KO spermatocyte are also hypomethylated in prospermatogonia. See (D) for information about the chart.
Consistent with studies analyzing epigenetic patterns in spermatogonia and spermatocytes of piRNA pathway mutants (Molaro et al, 2014; Pezic et al, 2014; Manakov et al, 2015; Nagamori et al, 2015), 80.9% of the 6,541 HMRs were located in the intergenic regions (Appendix Fig S1A), and the HMRs frequently included LINE and LTR retrotransposons, with a predominance of young retrotransposons (e.g., L1Md_T, L1Md_A, RLTR10, ETn, IAP, L1Md_Gf, MMERVK10C, and MMERGLN; Appendix Fig S1B–D). Notably, approximately two‐thirds (4,299/6,541) of the HMRs included young L1 retrotransposon sequences that showed > 90% identity with the consensus sequence. Within L1 sequences, the 5′ regions were more frequently associated with the HMRs than internal or 3′ regions (Appendix Fig S1E). This result is in accordance with the studies showing that the 5′ regions, but not internal or 3′ regions, of L1s, are specifically affected in piRNA‐mutant mice (Molaro et al, 2014; Manakov et al, 2015). For subsequent analyses, HMRs were divided into two classes according to the presence (HMR_+L1s, 4,299 regions) or absence (HMR_−L1s, 2,242 regions) of young L1s (Fig EV2A, Appendix Fig S1A, B and F) to allow effective analysis of L1‐free HMRs so that they will not be “buried” by the abundant presence of L1s.
RNA expression in piRNA‐dependently methylated regions
If MIWI2 targets the chromatin of piRNA‐dependent regions via nascent RNAs, RNAs with piRNA‐complementary sequences would be transcribed in these regions. Consistent with this, piRNA‐complementary sequences were specifically observed in HMRs (Appendix Supplementary Results, Fig EV3). A study of DNA methylation analysis in Mili −/− spermatogonia (Molaro et al, 2014) has shown positive correlation between the extent of demethylation in Mili −/− spermatogonia and RNA expression level at the stage of piRNA‐mediated DNA methylation among individual L1 sequences, supporting the view that piRNA‐mediated epigenetic regulation requires RNA expression. However, such correlation has not been observed for non‐L1 retrotransposons, such as IAP, despite their demethylation in Mili −/− spermatogonia (Molaro et al, 2014). This raises a question as to whether HMR_−L1s are transcriptionally active in prospermatogonia.
Figure EV3. HMRs frequently include target sites of MIWI2‐bound piRNAs.
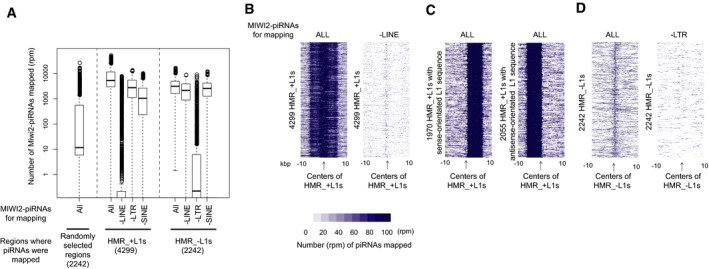
-
ABox plots showing the number of piRNAs mapped to 1‐kb regions spanning the centers of HMRs and randomly selected sites. “–LINE” (–LTR and –SINE): piRNAs derived from LINE (LTR and SINE) were removed before mapping. The numbers in parentheses represent the numbers of regions examined.
-
B–DHeatmap showing the number of piRNAs mapped to HMRs with (“HMR_+L1s”; B and C) or without (“HMR_−L1s”; D) young LINE1 retrotransposons. In (B and D), “–LINE” and “–LTR” on the top of the charts show that piRNAs derived from LINE and LTR sequences, respectively, were removed before mapping. In (C), HMR_+L1s containing sense‐ and antisense‐oriented L1s are shown as separate maps. HMR_+L1s containing both sense‐ and antisense‐orientated L1s were not analyzed.
To examine this, we carried out RNA‐seq analysis using RNA isolated from 16.5 dpc prospermatogonia. Heatmap and metaplot analyses revealed significantly higher levels of RNA expression in HMR_−L1s compared with the surrounding regions despite the high proportions of repeat sequences in HMR_−L1s (Fig 1A and B), which leads to underestimation of the expression level. Although unique RNA‐seq reads were not observed in 10.5% of HMR_−L1s (Appendix Fig S2A), these HMR_−L1s were enriched in repetitive sequences (Appendix Fig S2B). Hence, it appears that more HMR_−L1s actually express RNAs than those where the unique RNA‐seq reads are derived. RNAs expressed from HMR_−L1s seemed to be polyadenylated, because all five individual HMR_−L1‐derived RNAs examined were enriched in oligo (dT) bead‐selected RNAs as compared to non‐polyadenylated RNAs (histone H3.1 mRNA, 18S rRNA, and 7SK RNA; Appendix Fig S2C). Thus, RNA expression at the stage of genome‐wide de novo DNA methylation seems to be a common feature shared among HMRs.
Figure 1. Hypomethylated regions express RNAs potentially targeted by MIWI2‐bound piRNAs.
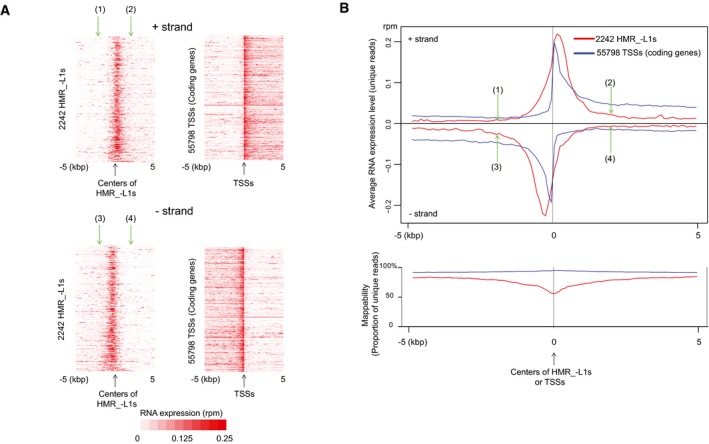
-
A, BHMRs express RNAs. Heatmaps (A) and metaplots (B) show RNA expression levels in 16.5 dpc prospermatogonia. Only uniquely mapped RNA‐seq reads were used for the analyses. Positions (1)–(4) in (A and B) (green arrows) indicate the same positions (±2 kb from the centers). The lower chart in (B) represents mappability defined as the percentage of unique sequences in all possible 50‐nt sequences (see Appendix Fig S2B). TSS isoforms are included in 55,798 TSSs. Low mappability at the centers of HMR_−L1s is due to high occurrence of repeat (retrotransposon) sequences. RPM, reads per million mapped reads.
Hypomethylated regions show specific epigenetic features
The presence of L1 promoter regions in the HMR_+L1s (Fig EV4C, Appendix Fig S1E) and preferential demethylation of promoter regions within retrotransposon sequences in Mili −/− spermatocytes (Molaro et al, 2014) suggested that HMR_−L1s might also harbor promoters that are active at the stage of de novo DNA methylation. Indeed, heatmap and metaplot analyses of the RNA‐seq data of 16.5 dpc prospermatogonia showed a peak of RNA expression at the centers of HMR_−L1s, as observed in transcription start sites (TSSs) of coding genes (Fig 1A and B). Furthermore, plus‐strand RNAs were more highly expressed in the downstream regions of HMR_−L1s [(2) in Fig 1A and B] than in their upstream regions [(1) in Fig 1A and B]; the opposite trend was observed for minus‐strand RNAs [(3) and (4) in Fig 1A and B)]. Again, a similar pattern was observed for TSSs of coding genes (Fig 1A and B).
Figure EV4. A unique epigenetic signature in hypomethylated regions and LINE1 promoters.

- Enrichment of histone marks associated with promoters against input or histone H3 in two representative HMR_−L1s. Y‐axis represents reads per million uniquely mapped reads (RPM). The numbers in parentheses represent the coverage for a whole genome. Only the results of one of the two replicates are shown when there are biological replicates (Appendix Table S1), as the results of the two replicates were similar. The ChIP‐seq data with biological replicates are indicated as “rep1” or “rep2” in the panel. HMR_−L1 shown in the left panel is located in an intergenic region on chromosome 1. HMR_−L1 shown in the right panel is located within the Rasgrf1 differentially methylated region (DMR).
- L1 promoters are active in 15.5 dpc prospermatogonia. ChIP‐seq signals normalized by H3 or input signals are shown. For the analyses of L1Md_Tf and L1Md_A, all the reads were mapped to their consensus sequences. For transcription start sites (TSSs), unique reads were used for mapping, and the average signals across all coding genes are shown.
To investigate the presence of promoters in HMR_−L1s, several histone modifications associated with active promoters (H3K4me1, H3K4me3, H3K9ac, H3K27ac, and H2A.Zac) were examined in 15.5 dpc prospermatogonia using chromatin immunoprecipitation and sequencing (ChIP‐seq) analysis. The results of two representative HMRs are shown in Fig EV4A. Metaplot and heatmap analyses showed clear peaks at the centers of the HMR_−L1s for all of these histone modifications (Fig 2A), suggesting the presence of promoters in many HMR_−L1s. However, the analyses also revealed some differences in the pattern of histone modification between HMR_−L1s and coding gene promoters (Fig 2A). First, HMR_−L1s showed much stronger H2A.Zac signals than coding gene promoters. Second, although the H3K4me3 signal in HMR_−L1s was higher than that in coding gene promoters, the opposite pattern was observed for the H3K4me1 signal. Similar trends (high H2A.Zac and H3K4me3 signals and low H3K4me1 signal as compared to coding gene promoters) were observed with promoters of L1s that are often found in HMR_+L1s (Fig EV4B). Thus, HMRs appear to carry unique epigenetic signatures.
Figure 2. Hypomethylated regions show features of active promoters.
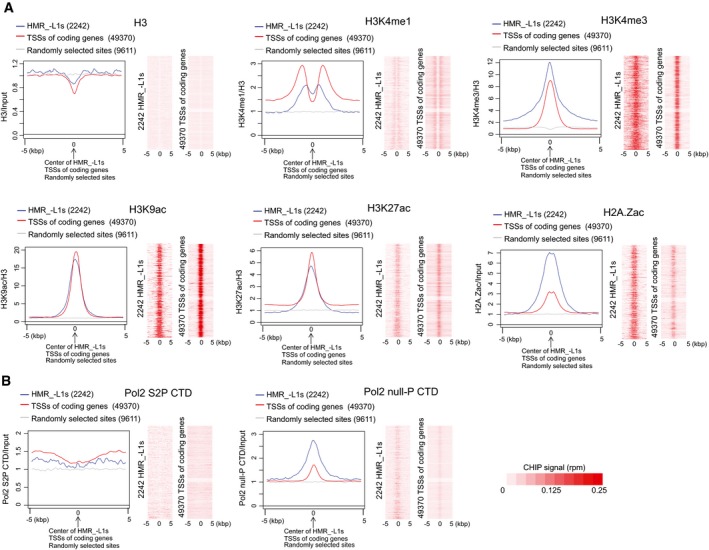
- Chromatin marks associated with promoters are observed in hypomethylated regions without young LINE1 retrotransposons (HMR_−L1s). Metaplots show the average ChIP‐seq signals across HMR_−L1s, transcription start sites (TSSs), and randomly selected sites. The signals were normalized by either H3 or input signal. The results shown in heatmaps were not normalized by H3 or input.
- The initiation form of Pol2 with an unphosphorylated carboxy‐terminal domain (Pol2 null‐P CTD) is highly enriched in HMR_−L1s.
To obtain additional evidence for the presence of active promoters in HMRs, Pol2 ChIP were carried out using 15.5 dpc prospermatogonia. After the initiation of transcription, Pol2 pauses at the promoter‐proximal region, and active elongation starts after the serine residue at position 2 of the carboxy‐terminal domain (CTD) of Pol2 is phosphorylated (Adelman & Lis, 2012; Liu et al, 2015). Therefore, CTD phosphorylation on the serine at position 2 (S2P CTD) is observed only in the elongated form of Pol2, whereas unphosphorylated CTD (null‐P CTD) is mainly observed in the initiation form of Pol2 (Nojima et al, 2015). Consistent with the pattern of promoter‐associated histone modifications (Fig 2A), the null‐P CTD signal peaked at the centers of HMR_−L1s, while the S2P CTD signal increased from the centers toward the sides of HMR_−L1s (Fig 2B). However, compared with coding genes, HMR_−L1s displayed a considerably higher null‐P CTD signal and lower S2P CTD signal (Fig 2B). Similarly, the enrichment of null‐P CTD was observed in L1 promoters that are frequently found in HMR_+L1s (Fig EV4B).
MIWI2 interacts with RNAs transcribed from hypomethylated regions
To investigate whether the RNAs derived from HMR_−L1s are indeed targeted by piRNAs, we examined the RNAs bound to EGFP‐tagged MIWI2 (EGFP‐MIWI2) using BAC transgenic mice expressing EGFP‐MIWI2 from the Miwi2 promoter (Aravin et al, 2007). EGFP‐MIWI2 was immunoprecipitated with an EGFP antibody from lysates extracted from the FACS‐purified 16.5 dpc prospermatogonia. RNAs bound to EGFP in 16.5 dpc prospermatogonia from knock‐in mice expressing EGFP from the Oct4 locus serve as negative controls (Lengner et al, 2007). HMR_−L1‐derived RNAs, but not prospermatogonium‐expressed mRNAs, were significantly enriched in the RNAs bound to EGFP‐MIWI2 (Fig 3A left), whereas no such enrichment was observed in the RNAs bound to EGFP (Fig 3A right). Furthermore, this specific interaction between EGFP‐MIWI2‐ and HMR_−L1‐derived RNAs was not observed in testes from Mili −/− neonatal mice, in which binding of EGFP‐MIWI2 proteins to piRNAs was largely impaired (Fig 3B and Appendix Fig S3A). These results are consistent with our hypothesis that MIWI2 interacts with HMR_−L1‐derived RNAs in a manner dependent on piRNAs.
Figure 3. MIWI2 and piRNAs target RNAs from hypomethylated regions.
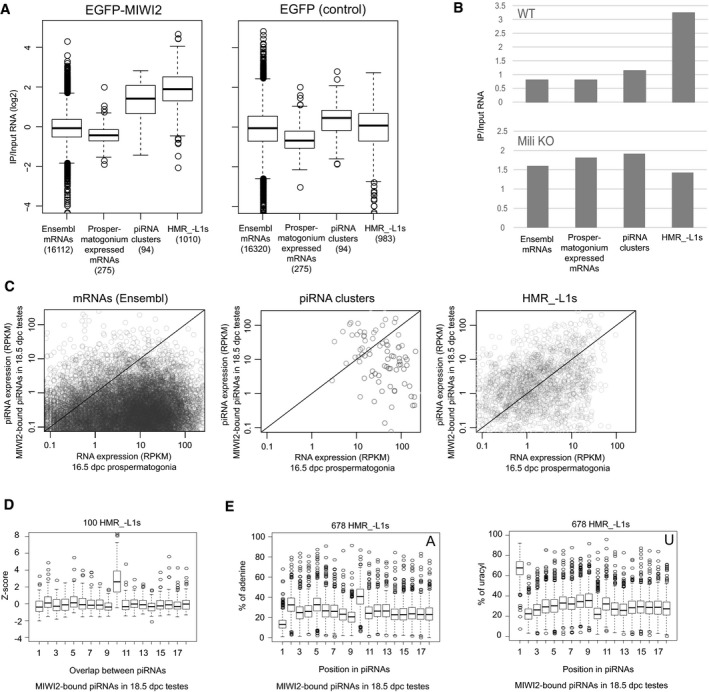
- RNA immunoprecipitation (RIP) analyses of EGFP‐MIWI2 and EGFP. EGFP‐MIWI2 and EGFP were immunoprecipitated from 16.5 dpc prospermatogonia, and long RNAs bound to these proteins and input RNAs were sequenced. RNAs expressed from HMR_−L1s and piRNA clusters, but not Ensembl and prospermatogonium‐expressed mRNAs, are overall enriched by immunoprecipitation (IP) of EGFP‐MIWI2 (left). This enrichment is not observed when EGFP is immunoprecipitated (right). The numbers of mRNAs, piRNA clusters, and HMR_−L1s analyzed were shown in parentheses. Prospermatogonium‐expressed mRNAs are those that are highly and specifically expressed in prospermatogonia. The bottom and top of boxes represent 25% percentile and 75% percentile, respectively. The line in the box represents the median. Circles represent outliers, and the bottom (top) of whiskers represents the minimum (maximum) values except for outliers.
- RIP analyses of EGFP‐MIWI2 in newborn testes from WT and Mili KO mice. RNA bound to EGFP‐MIWI2 and input RNA was sequenced. All RNAs derived from each class was counted for this analysis.
- HMR_−L1s preferentially generate piRNAs. X‐ and Y‐axes represent expression levels of long RNAs (16.5 dpc prospermatogonia) as determined by RNA‐seq and MIWI2‐bound piRNAs (18.5 dpc testes) as determined by small RNA‐seq, respectively. Circles in the charts represent individual mRNAs, piRNA clusters, and HMR_−L1s. More than half of the HMR_−L1s (right) are located in the area of y > x (upper part of the diagonal), whereas most mRNAs (left) are located in the area of y < x (lower part of the diagonal). RPKM, reads per kilobase per million mapped reads.
- Frequent 10‐nt overlap between sense and antisense piRNAs derived from HMR_−L1s. Top 100 HMR_−L1s that generate the largest number of unique piRNAs were analyzed. Frequencies of 1‐ to 18‐nt overlaps were analyzed for each HMR_−L1.
- piRNAs derived from HMR_−L1s are enriched in adenine at their 10th nucleotide. HMR_−L1s generating more than 40 unique piRNAs were analyzed (678 HMR_−L1s). The proportions of piRNAs with adenine (left) and uridine (right) at the indicated positions were analyzed for each HMR_−L1. See (A) for the explanation of box plots.
We found that piRNA precursors were also enriched in the RNAs bound to EGFP‐MIWI2 (Fig 3A). Therefore, the interaction between MIWI2‐ and HMR_−L1‐derived RNAs might reflect the generation of piRNAs from HMR_−L1‐derived RNAs rather than the direct targeting of these RNAs by MIWI2. Consistent with this, 5′ ends of HMR‐derived RNAs bound to EGFP‐MIWI2 were preferentially aligned with 5′ portions of piRNAs from the same HMRs (Appendix Fig S3B and see below for piRNA generation from HMRs). However, given that target RNAs of piRNAs are processed into piRNAs in some cases and piRNAs are preferentially generated from 5′ ends of RNAs sliced by Piwi‐piRNA complexes (Brennecke et al, 2007; Gunawardane et al, 2007; De Fazio et al, 2011; Han et al, 2015; Homolka et al, 2015; Mohn et al, 2015; Yang et al, 2016), these results are nevertheless consistent with the notion that RNAs transcribed from HMRs are targeted by piRNAs.
RNAs transcribed from hypomethylated regions are targeted by piRNAs
To further examine whether transcripts from HMRs are targeted by piRNAs, we analyzed piRNAs biogenesis with regard to these transcripts. In prospermatogonia, after the cleavage of target RNAs by MILI, secondary piRNAs are generated from the 5′ ends of the 3′ cleavage products (Aravin et al, 2007; Kuramochi‐Miyagawa et al, 2008; Shoji et al, 2009; De Fazio et al, 2011). These secondary piRNAs are incorporated into MILI and MIWI2. Because MILI and MIWI2 bind to similar sets of piRNAs in prospermatogonia (Appendix Fig S3C) (Aravin et al, 2008), we reasoned that if HMR_−L1‐derived RNAs are targeted by MIWI2 in nuclei, they would also be targeted by MILI when exported to the cytoplasm, resulting in the generation of secondary piRNAs. Consistent with this, the HMR_−L1s produced a much larger number of piRNAs compared with coding genes showing the same expression level (Fig 3C). In fact, 22 HMR_−L1s were found within the top 95 piRNA clusters that generated the largest number of piRNAs in prospermatogonia (Kuramochi‐Miyagawa et al, 2008), further suggesting that HMR_−L1s preferentially generate piRNAs. We observed piRNA generation in 86% (1,924/2,242) of the HMR_−L1s. However, given that the remaining 14% of HMR_−L1s tended to be shorter in length and were more enriched in repetitive sequences (Appendix Fig S3D and E), it appears that most, if not all, HMR_−L1s produce piRNAs. These results further support targeting of HMR_−L1‐derived RNAs by piRNAs.
We found many HMR_−L1s expressing piRNAs and their precursor RNAs from both strands (Fig EV5A–C). In fact, piRNAs were expressed bidirectionally in all of the top 100 HMR_−L1s showing the highest piRNA expression levels. These observations raised the possibility of a ping‐pong cycle between cis‐natural sense and antisense transcripts (i.e., repeated generation of secondary piRNAs from both strands; Brennecke et al, 2007; Gunawardane et al, 2007). Secondary piRNAs have two specific features: (i) Because cleavage by Piwi proteins occurs between the 10th and 11th position of piRNAs, the 5′ ends of the piRNAs and the newly generated secondary piRNAs overlap by 10 nt; (ii) because the first nucleotide of the majority of primary piRNAs is uridine, the 10th nucleotide of secondary piRNAs is enriched in adenine. These features can be used to reveal the presence of secondary piRNAs. In support of the generation of secondary piRNAs from HMR_−L1s, a frequent 10‐nt overlap between sense and antisense piRNAs and enrichment of adenine at their 10th position were observed among piRNAs from HMR_−L1s (Figs 3D and E, and EV5D). Thus, traces of RNA targeting by piRNAs are found in many HMR_−L1s.
Figure EV5. Ping‐pong cycle within each hypomethylated region.

- Bidirectional piRNA expression from HMR_−L1s. Direction index is defined as indicated. When half (all) of piRNAs are derived from the same strand, direction index is 0.5 (1). Only unique piRNA reads were used for this analysis. HMR_−L1s and mRNAs that generate appreciable level of unique piRNAs (≥ 10 unique piRNAs) were selected for this analysis. We divided them into two groups according to the number of generating unique piRNAs (10–99 and ≥ 100, top and bottom panels) in order to rule out the possibility that the difference is arisen from the number of piRNAs analyzed. The numbers of HMR_−L1s and mRNAs analyzed are shown in parentheses.
- Bidirectional RNA expression from HMR_−L1s. HMR_−L1s, transcription start sites (TSSs), and 2nd exons were divided into two groups according to their RNA expression levels (0.5–5 and 5–50), and they were analyzed separately (top and bottom panels). We considered only unique RNA‐seq reads that were mapped within 2‐kb regions spanning the centers of HMR_−L1s, TSSs, and start sites of 2nd exons (positions −1,000 to 999, when they are considered to be position 0).
- Two representative HMR_−L1s generating sense and antisense piRNAs.
- High frequency of 10‐nt overlap between 5′ ends of uniquely mapped sense and antisense piRNAs from HMR_−L1s. Heatmap shows relative frequencies among 1‐ to 25‐nt overlaps in each HMR_−L1.
RMER4B is required for de novo DNA methylation of the Rasgrf1 locus
These findings led us to examine whether the targeting of HMR_−L1‐derived RNAs by piRNAs is an essential step for piRNA‐mediated DNA methylation. A previous study showed that the piRNA pathway is required for the establishment of DNA methylation of the Rasgrf1 differentially methylated region (DMR; Watanabe et al, 2011b). In the current study, this locus was identified as one of the HMR_−L1s (Figs 4A, EV1C and EV4A). A non‐coding RNA, pit‐RNA is transcribed in the Rasgrf1 DMR, and the RMER4B retrotransposon sequence in pit‐RNA is predicted to be targeted by piRNAs derived from another copy of RMER4B in the chr7 piRNA cluster (Fig 4B; Watanabe et al, 2011b). To test whether targeting of pit‐RNAs by the chr7 cluster‐derived piRNAs was required for DNA methylation of the Rasgrf1 DMR, we generated mice with deletions in key sequences of the Rasgrf1 DMR and chr7 piRNA cluster.
Figure 4. RMER4B sequence in the Rasgrf1 DMR is essential for de novo DNA methylation of the Rasgrf1 DMR .

- DNA methylation status of a portion of the Rasgrf1 differentially methylated region (DMR) in 0–2 dpp Mili +/− and Mili −/− prospermatogonia. Regions corresponding to B, C, D, E, and F are demethylated in Mili −/− prospermatogonia. Vertical bars represent DNA methylation levels at CpG sites determined by whole‐genome bisulfite sequencing (see below). The top dark gray bar represents a portion of the Rasgrf1 gametic DMR (Tomizawa et al, 2011). The CpG cluster is shown. The deleted RMER4B sequence is indicated.
- Deletion of key sequences (circles labeled as ①–③) in the Rasgrf1 DMR and chr7 piRNA cluster. Shown is our proposed mechanism in which piRNAs generated from RMER4B sequence in chr7 piRNA cluster target RMER4B sequence in pit‐RNA for the establishment of DNA methylation of the Rasgrf1 DMR.
- DNA methylation status of the Rasgrf1 DMR in Rasgrf1 ΔRMER4B/ΔRMER4B and control spermatogonia is shown. The regions examined are shown in (A). Black (white) circles represent methylated (unmethylated) CpGs.
- Rasgrf1 RMER4B functions in cis. Wild‐type and mutant alleles of the Rasgrf1 DMR in Rasgrf1 +/ΔRMER4B spermatogonia were analyzed separately.
We first investigated whether the RMER4B sequence in pit‐RNA is required for the establishment of DNA methylation by deleting the entire RMER4B sequence at the pit‐RNA locus in the Rasgrf1 DMR using CRISPR (① in Fig 4B). As expected, spermatogonia of homozygous mice (Rasgrf1 ΔRMER4B/ΔRMER4B) displayed decreased DNA methylation of the Rasgrf1 DMR, and the pattern of demethylation was very similar to that observed in Mili −/− mice (Fig 4A and C). In heterozygous mice (Rasgrf1 +/ΔRMER4B), demethylation was observed only in the mutant allele and not in the wild‐type allele (Fig 4D). These results indicate that the RMER4B sequence is a cis‐element that is essential for the establishment of DNA methylation of the Rasgrf1 DMR.
RMER4B in a chr7 piRNA cluster directs DNA methylation of the Rasgrf1 DMR
We then investigated whether piRNAs derived from the chr7 piRNA cluster are required for DNA methylation of the Rasgrf1 DMR by generating mice with deletion of the promoter region of the chr7 piRNA cluster (② in Fig 4B). To locate the promoter, we first determined the structure of the piRNA precursor of the chr7 piRNA cluster, which is located between Olfr1350 and the Zim2 non‐coding RNA loci (Fig 5A). The putative piRNA precursor was predicted to be a Zim2 transcript with an extended last exon based on the following observations (Fig 5A): (i) Zim2 and the chr7 piRNA cluster are orientated in the same direction; (ii) piRNAs from the chr7 piRNA cluster were generated from Zim2 exons and the downstream region of the last exon, but not from its introns; and (iii) near the 5′ end of the chr7 piRNA cluster, a peak of H3K4me3, a histone mark associated with active promoters, was only observed at the Zim2 promoter region.
Figure 5. chr7 piRNA cluster directs de novo DNA methylation of the Rasgrf1 DMR .

- Structure of the chr7 piRNA cluster. Top gray bars represent a putative piRNA precursor predicted by H3K4me3 ChIP‐seq, RNA‐seq, and small RNA‐seq. Only uniquely mapped reads were used for displaying the sequencing result. For RNA‐seq and small RNA‐seq, only the results of minus strand are shown, because plus strand does not express appreciable level of RNA. A promoter region deleted is shown above the putative piRNA precursor. RPM, read per million.
- Low expression level of piRNAs from chr7 cluster in chr7 piRNA cluster Δpromoter/Δpromoter testes. Unchanged expression of piRNAs from chr10 cluster (bottom) shows chr7 cluster is specifically affected. Only uniquely mapped piRNAs are shown.
- DNA methylation status of the Rasgrf1 differentially methylated region (DMR) in spermatogonia from chr7 piRNA cluster Δpromoter/Δpromoter. See Fig 4A and B for information about the region examined.
To generate mice deficient in piRNAs from the chr7 piRNA cluster, we deleted a 1,350‐bp region spanning the TSS of Zim2 (② in Figs 4B and 5A top). In homozygous mice (chr7 piRNA cluster Δpromoter/Δpromoter), expression of the putative piRNA precursor and mature piRNAs from the chr7 piRNA cluster was reduced to < 10% of the wild‐type levels (Fig 5B, Appendix Fig S4), suggesting that a large portion of piRNAs from this cluster were produced from RNA(s) transcribed from the Zim2 promoter. DNA methylation of the Rasgrf1 DMR was slightly decreased in homozygote spermatogonia (94.3% vs. 68.4%, E1 region in Fig 5C). The milder effect on DNA methylation in this mutant mouse line compared with Mili −/− mice can likely be explained by incomplete loss of the chr7 cluster‐derived piRNAs.
Furthermore, since almost all of the predicted pit‐RNA‐targeting piRNAs were generated from the RMER4B sequence in the chr7 piRNA cluster (Watanabe et al, 2011b), we deleted this sequence (③ in Figs 4B and 5A bottom) to remove as many pit‐RNA‐targeting piRNAs as possible (Fig 6A). In mice homozygous for this deletion (chr7 piRNA cluster ΔRMER4B/ΔRMER4B), we did not find any predicted pit‐RNA‐targeting piRNAs, whereas several such piRNAs were present in wild‐type mice (Fig 6B). The pattern of demethylation in Rasgrf1 DMR of homozygous mice was very similar to those observed in Mili −/− and Rasgrf1 ΔRMER4B/ΔRMER4B mice, although the effect seems to be milder than those in these mice (Figs 4A and C, and 6C). Taken together, these results demonstrate that piRNAs derived from RMER4B in the chr7 piRNA cluster guide the establishment of DNA methylation of the Rasgrf1 DMR by targeting the RMER4B sequence in the Rasgrf1 DMR.
Figure 6. RMER4B sequence in the chr7 piRNA cluster is required for de novo DNA methylation of the Rasgrf1 DMR .
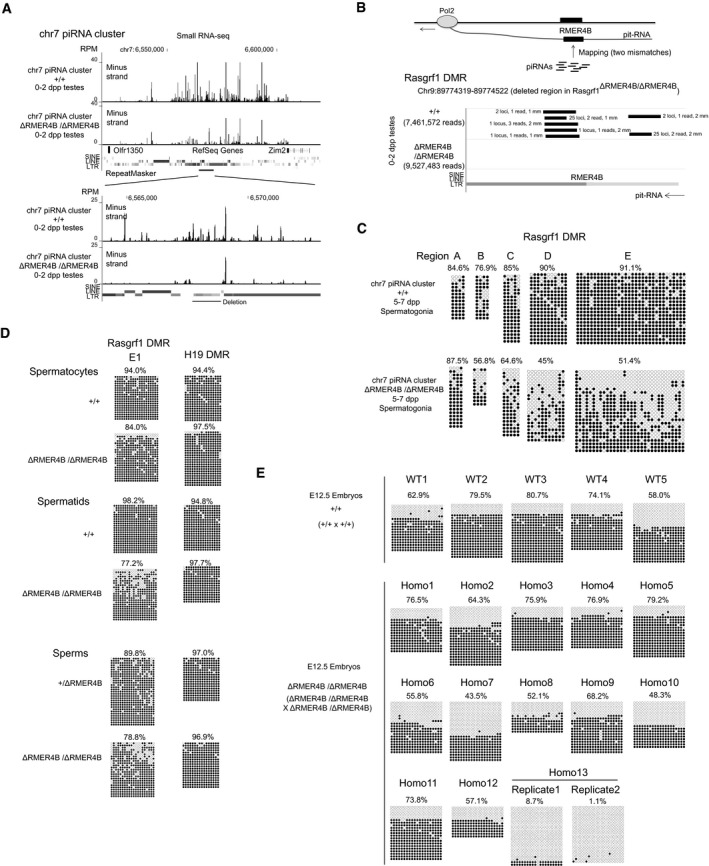
-
ADepletion of piRNAs from chr7 piRNA cluster RMER4B sequence in chr7 piRNA cluster ΔRMER4B/ΔRMER4B mice. Top panel shows overall view of chr7 piRNA cluster, and lower panel shows magnified view of RMER4B region in chr7 piRNA cluster. Only uniquely mapped piRNAs are shown.
-
BDepletion of piRNAs that potentially target pit‐RNA in chr7 piRNA cluster ΔRMER4B/ΔRMER4B mice. piRNAs are mapped to RMER4B region of Rasgrf1 DMR (deleted sequence in Rasgrf1 ΔRMER4B/ΔRMER4B mice) allowing two mismatches. Black bars represent piRNAs that are mapped to this region in antisense direction to pit‐RNA. The numbers flanking the black bars indicate genomic loci where each of these piRNAs is perfectly matched (loci), their read numbers (reads) and mismatches in the alignment between these piRNAs and Rasgrf1 RMER4B sequences (mm). All piRNAs shown here were perfectly matched to chr7 piRNA cluster. The numbers in parentheses show the total number of small RNA reads mapped to the genome.
-
C, DDNA methylation status of the Rasgrf1 differentially methylated region (DMR) in spermatogonia (C), spermatocytes, spermatids, and sperms (D) from chr7 piRNA cluster ΔRMER4B/ΔRMER4B mice. See Fig 4A and B for information about the region examined. Complete DNA methylation of H19 imprinted gene, which is methylated in the male germline, shows high purity of germ cells used for the analyses.
-
EDNA methylation pattern in the progeny of male Rasgrf1 ΔRMER4B/ΔRMER4B mice. DNA methylation status of the Rasgrf1 DMR in E12.5 whole embryos is shown. E12.5 whole embryos were obtained by the crosses shown in the panel. Two independent bisulfite‐treated DNA sample was examined for Homo13 embryo.
Finally, because Rasgrf1 DMR is methylated only in the male germline, and the paternal allele‐specific DNA methylation in the somatic cells, which is transmitted from parents, is responsible for the imprinted expression of Rasgrf1 (Yoon et al, 2002), we investigated whether the defect in DNA methylation of Rasgrf1 DMR in chr7 piRNA cluster ΔRMER4B/ΔRMER4B mice is transmittable to the next generation. In this mutant, we observed lower level of DNA methylation in Rasgrf1 DMR in spermatocytes, spermatids, and sperms of (Fig 6D), although DNA methylation level is restored to some extent. In order to examine whether the decreased level of DNA methylation in Rasgrf1 DMR is transmitted to the next generation, Rasgrf1 DMR DNA methylation was analyzed in 12.5 dpc embryos of the progeny of chr7 piRNA cluster ΔRMER4B/ΔRMER4B mice. In 12 of the 13 embryos examined, Rasgrf1 DMR showed a typical pattern of imprinting locus as observed in wild‐type embryos. Only in one of the embryos, DNA methylation of the Rasgrf1 DMR was almost completely depleted (Fig 6E). These results indicate that defect in de novo DNA methylation of Rasgrf1 DMR in chr7 piRNA cluster ΔRMER4B/ΔRMER4B mice is only infrequently transmitted to the next generation.
Discussion
The mechanism underlying piRNA‐mediated DNA methylation is a fundamental question that merits further investigation. Here, we have reported three major findings that contribute to the understanding of this mechanism in mice. First, using genome‐wide analyses, we revealed that HMRs generate RNAs at the establishment stage of DNA methylation. Second, MIWI2 interacts with these RNAs. This interaction likely occurs in the nucleus, since MIWI2 is mostly a nuclear protein. Third, analysis of mice with deletion of the Rasgrf1 locus and chr7 piRNA cluster provided direct evidence that piRNAs specify targets in MIWI2‐mediated DNA methylation. Thus, the results of this study lend support to the notion that MIWI2 in mice, similar to nuclear Argonaute proteins in other organisms, directs the establishment of suppressive chromatin marks by targeting nascent RNAs using piRNAs as guides.
Recent transgenic mouse study by Nakano et al reported that, using designed zinc‐finger protein, artificial targeting of MIWI2 to specific regions in the genome induces DNA methylation at these loci (Kojima‐Kita et al, 2016). This indicates MIWI2 can act as an effector of de novo DNA methylation. In the naturally occurring situation, does MIWI2 target nascent RNA or genomic DNA? Our results show MIWI2 interaction with HMR‐derived RNAs, although it is not clear whether this interaction occurs directly or indirectly through other MIWI2‐associated proteins, such as GTSF1 (Yoshimura et al, 2018). However, neither can we exclude the possibility that DNA, but not RNA, is targeted by MIWI2. In fact, the observed RNA expression from HMRs is consistent with MIWI2 targeting either nascent RNA or DNA, because the formation of RNA‐DNA hybrid during transcription could promote targeting of the remaining single‐stranded DNA by the MIWI2‐piRNA complex. In Schizosaccharomyces pombe, where siRNA‐mediated epigenetic regulation occurs via nascent RNAs, insertion of siRNA target sites into either introns or exons can cause siRNA‐mediated epigenetic silencing, and the effect is stronger when the target sites are located in exons (Shimada et al, 2016). This is in favor of the nascent RNA model, as splicing is not expected to affect siRNA‐mediated epigenetic silencing if DNA is targeted. It will be interesting to examine whether similar effect is observed in piRNA‐mediated DNA methylation.
Because de novo DNA methyltransferase DNMT3A and DNMT3L specifically bind to unmethylated H3K4, active promoters usually evade establishment of DNA methylation (Bernstein et al, 2005; Ooi et al, 2007). We observed that HMRs contained active promoters associated with epigenetic marks such as H3K9ac, H3K27ac, H2A.Zac, H3K4me1, and H3K4me3. Because the removal of H3K4 methylation mark is a prerequisite for the establishment of DNA methylation of the HMRs, the piRNA pathway may promote DNA methylation by suppression of promoter activity and removal of the H3K4 methylation mark. Further studies are needed to reveal the detailed molecular mechanisms involved in piRNA‐mediated DNA methylation.
Rasgrf1 is one of the three paternally methylated imprinted genes, and imprinted expression of Rasgrf1 is controlled by paternal allele‐specific DNA methylation of DMR (Yoon et al, 2002). Our previous study showed that the piRNA pathway is essential for DNA methylation of part of the Rasgrf1 DMR in the male germline (Watanabe et al, 2011b). Because of the infertility of piRNA pathway mutant mice, it was difficult to determine the impact of demethylation of the Rasgrf1 DMR in these mice on the imprinted expression of Rasgrf1 in the next generation. In the present study, we generated mice deficient in piRNAs targeting the Rasgrf1 DMR (chr7 piRNA cluster ΔRMER4B/ΔRMER4B and chr7 piRNA cluster Δpromoter/Δpromoter) and mice with deletion of the piRNA target sites in Rasgrf1 DMR (Rasgrf1 ΔRMER4B/ΔRMER4B). In these mutant mice, spermatogenesis was normal, and they were fertile (data not shown). Although DNA methylation of the Rasgrf1 DMR was clearly affected in spermatogonia from chr7 piRNA cluster ΔRMER4B/ΔRMER4B mice (Fig 6C and D), this defect becomes milder in sperm. This indicates that the Rasgrf1 DMR from the homozygous mice might acquire DNA methylation during late spermatogenesis. In the next generation, most embryos restored normal pattern of imprinted DNA methylation, and only one of the 13 embryos examined showed almost completely unmethylated Rasgrf DMR (Fig 6E). This indicates that piRNA‐dependent DNA methylation probably has limited impacts on imprinted expression of Rasgrf1 in the next generation.
Although targeting of nascent RNA by small RNAs is a common mechanism in small RNA‐mediated epigenetic regulation, its downstream mechanisms leading to suppression vary largely among organisms (Zhong et al, 2014; Holoch & Moazed, 2015). In Drosophila, Piwi recruits the CG9754 protein, which causes silencing and deposition of repressive epigenetic marks (Sienski et al, 2015; Yu et al, 2015). However, this protein does not exist in mammals, and the degree of similarity in the mechanisms of piRNA‐mediated epigenetic regulation between flies and mice remains unknown. Given that the Miwi2 gene appeared only after divergence of the mammalian lineage (Lim et al, 2013), the mechanisms of piRNA‐mediated epigenetic regulation in mammals and flies might have evolved independently. Alternatively, given the essential role of the piRNA pathway in establishment of the H3K9me3 mark on target retrotransposon sequences in both mice and flies (Sienski et al, 2012; Huang et al, 2013; Le Thomas et al, 2013; Rozhkov et al, 2013; Pezic et al, 2014), the mechanisms in the two species might share a common evolutionary origin and then become diverged. Although Piwi and piRNAs have been identified in many metazoans, piRNA‐mediated epigenetic regulation has thus far only been reported in flies, mice, and Aplysia (Rajasethupathy et al, 2012). Investigation of piRNA‐mediated epigenetic regulation in other species, for example, non‐mammalian vertebrates, could aid in understanding the origin of piRNA‐mediated epigenetic regulation in mammals.
Materials and Methods
Preparation of germ cells
Prospermatogonia were FACS‐isolated using EGFP from EGFP‐MIWI2 transgenic mice or Oct4‐EGFP knock‐in mice (Lengner et al, 2007; Aravin et al, 2008). Spermatogonia were labeled with Alexa 647‐conjugated EpCAM antibody (sc‐53532 AF647, Santa Cruz) for FACS sorting. Spermatocytes were FACS‐isolated using Hoechst 33342 dye as described in the previous paper (Watanabe et al, 2015). Sperms were isolated from cauda epididymis, and sperm DNA was isolated using lysis solution (20 mM Tris–HCl (pH 8), 10 mM dithiothreitol, 150 mM NaCl, 10 mM EDTA (pH8), 1.0% SDS, and 20 μg/ml proteinase K) at 50°C overnight.
Testes were harvested from fetal or neonatal mice, decapsulated, and digested with digestion solution [0.25% trypsin‐EDTA and 25 μg/ml DNaseI in Dulbecco's modified Eagle's medium (DMEM)] at 35°C for 10 min. During digestion, they were gently mixed by inverting tubes several times every 2 min. After digestion, an equal volume of stop solution [20% fetal calf serum (FCS) and 25 μg/ml DNaseI in DMEM] was added. Cells were filtered through a 40‐μm cell strainer and washed once. For EGFP sorting, cells were suspended in FACS solution (25 μg/ml DNaseI, 0.5% FBS, and 1 μg/μl PI in DMEM) and were kept on ice until sorting. For sorting using the EpCAM antibody, cells were suspended in blocking solution (10% FCS and 25 μg/ml DNaseI in DMEM) and kept on ice for 10 min. Alexa 647‐conjugated EpCAM antibody was then added to the cell suspension at a final concentration of 2 μg/ml, and the cells were kept on ice for 1 h, during which the cell suspension was gently mixed every 15 min. After incubation, the cells were washed once, suspended in FACS solution, and sorted using FACSAriaII (BD). The purity of germ cells was estimated by immunocytochemistry using an anti‐TRA98 antibody (ab82527, Abcam), which stains germ cells specifically. The purity was estimated to be 95–99% for EGFP sorting. When cells were sorted using EpCAM, the purity was estimated to be 90–99%.
Whole‐genome Bisulfite sequencing
Prospermatogonia were FACS‐isolated from 0 to 2 dpp Mili +/− and Mili −/− testes using EGFP (Oct4‐EGFP; Kuramochi‐Miyagawa et al, 2004; Lengner et al, 2007). To minimize individual difference, testes from several animals (five to 10) were used for the generation of each library. Bisulfite‐seq libraries were constructed using the PBAT method (Miura et al, 2012). Approximately 100 ng of DNA was used to make each library, and two biological replicates were prepared for each genotype.
Mapping of sequencing reads and calculation of the DNA methylation rate of each CpG site were done by using Bismark (http://www.bioinformatics.babraham.ac.uk/projects/bismark/; Krueger & Andrews, 2011). Information about read numbers is shown in Fig EV1 and Appendix Table S1. HMRs were identified using the ComMet program in the Bisulfighter software package using options –dual and –alpha 20 (http://epigenome.cbrc.jp/bisulfighter; Saito et al, 2014). By using likelihood score > 100 as a cutoff, a total of 6,541 HMRs were identified. For the control, 6,541 genomic regions that had the same length distribution as HMRs were randomly selected from the genome (Appendix Fig S1). See Appendix Supplementary Methods for annotation of HMRs and the other bioinformatic analyses on HMRs.
Bisulfite sequencing analysis of individual loci
The bisulfite reaction was performed using a MethylCode Bisulfite Conversion Kit (Thermo Fisher) in accordance with the manufacturer's instructions. For analyses of spermatogonia, DNA from several animals (two to 10) was used for each bisulfite reaction. For analyses of spermatocytes, spermatids, and sperms, testes from one individual were used. Because Rasgrf1 DMR is usually 100% methylated in these cells, demethylation only in one animal can draw a conclusion that mutated allele is involved in methylation. PCRs were performed with the EpiTaq HS (Takara Clontech) using 5–50 ng of bisulfite‐treated DNA. The primer sequences are listed in Appendix Table S2.
In chr7 piRNA cluster ΔRMER4B/ΔRMER4B sperms, the level of demethylation varies among sperms. We predicted that only sperms with the lowest level of Rasgrf1 DMR methylation convey the defect to the next generation, as de novo DNA methylation again occurred in early embryos. Therefore, we examined Rasgrf1 DMR methylation in as many as 12 mutant embryos [only one embryo showed demethylation (Fig 6E)].
RNA‐seq
Germ cells were FACS‐isolated from 16.5 dpc testes using EGFP (Oct4‐EGFP). Total RNA was isolated using TRIzol (Thermo Fisher). RNA libraries were constructed from rRNA‐depleted RNAs by ScriptSeq Complete Gold Kit Low Input (Illumina) using 500 ng of total RNAs as initial inputs. After sequencing using HiSeq2000, read sequences were mapped to the genome using TopHat software (http://tophat.cbcb.umd.edu/), and reads that were uniquely and perfectly mapped to the genome were used for analyses. Information about read numbers is shown in Appendix Table S1. See Appendix Supplementary Methods for details about RNA‐seq analyses.
Small RNA‐seq
Total RNAs were isolated from 1 dpp testes of chr7 piRNA cluster Δpromoter/Δpromoter, 0–2 dpp testes of chr7 piRNA cluster ΔRMER4B/ΔRMER4B, and control WT mice using TRIzol. Approximately 20 μg of total RNAs was loaded on 15% polyacrylamide‐7 M urea‐TBE gels, and small RNAs ranging in size between 15 and 40 nt were purified. Small RNA libraries were constructed using a TruSeq Small RNA Sample Prep kit (Illumina). After sequencing using HiSeq2000, adapter sequences were removed from the sequencing reads, and small RNAs ranging in size between 18 and 32 nt were extracted. The extracted small RNAs were mapped to the genome and annotated as described previously (Watanabe et al, 2011a). miRNAs and degradation products of abundant non‐coding RNAs (rRNA, tRNA, snRNA, snoRNA, scRNA, srpRNA, and RNA) were removed. Only those that were perfectly and uniquely mapped to the genome were used to display on the genome browser (Figs 5A and B, and 6A). Genome‐matched reads that were not annotated as miRNAs or degradation product of abundant non‐coding RNAs were used for mapping to pit‐RNAs. They were mapped to RMER4B sequence in pit‐RNAs (deleted sequence in Rasgrf1 ΔRMER4B/ΔRMER4B mice) allowing two mismatches using Bowtie. Small RNA reads that were mapped in antisense direction to pit‐RNA were used for displaying in Fig 6B. Information about read numbers is shown in Appendix Table S1.
For MIWI2‐bound piRNAs, published sequences (Pandey et al, 2013) were downloaded from the Sequence Read Archive (GSM1220989, MIWI2 IP from 18 dpc testes). Small RNAs ranging in size between 15 and 32 nt were mapped to the genome, and perfectly matched reads were extracted for analyses. Small RNAs annotated as miRNAs and degradation products of abundant non‐coding RNAs were removed for all the analyses using MIWI2‐bound piRNAs. See Appendix Supplementary Methods for details about bioinformatic analyses of small RNAs.
ChIP‐seq
ChIP was performed using 0.1–0.5 million FACS‐isolated prospermatogonia per experiment. Prospermatogonia were FACS‐isolated from 15.5 dpc EGFP‐MIWI2 mice using EGFP. Fixation, quenching, and washing were performed using a truChIP Chromatin Shearing Reagent kit (Covaris) in accordance with the manufacturer's protocol. To avoid cell loss caused by attachment to the wall of tubes, tubes used for these steps were coated with FCS. After washing, cells were lysed in lysis buffer [10 mM Tris–HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA (pH 8.0), 0.5 mM EGTA (pH 8.0), 0.1% Na‐deoxycholate, 0.5% N‐lauroylsarcosine, 1× protease inhibitor, 1 mM dithiothreitol]. Cell lysates were sonicated using a Covaris Focused‐Ultrasonicator for 15 min using a standard setting for the microTUBES (Covaris) and centrifuged at 21,130 × g for 30 min to clear the lysates. Portions (10%) of the cleared lysates were taken as input chromatin.
To prepare antibody–beads complexes, 15 μl of Dynabeads Protein A (Thermo Fisher) or Protein G (Thermo Fisher) was washed, mixed with 1 μg of antibodies in 200 μl of lysis buffer, and rotated at 4°C for 1 h. The beads were washed using lysis buffer, mixed with the cleared lysates in 200 μl tubes, rotated for 4–12 h at 4°C, and washed six times with wash buffer (0.2% Triton‐X‐100, phosphate‐buffered saline) and once with TE buffer [10 mM Tris–HCl (pH 7.4), 0.1 mM EDTA (pH 8.0)]. The washed beads and the input chromatin were mixed with 100 μl of elution buffer [50 mM Tris–HCl (pH 8.0), 10 mM EDTA (pH 8.0), 1.0% SDS, 1 mg/ml Proteinase K] and incubated at 65°C for at least 6 h. DNA was then isolated using MinElute Reaction Cleanup kit (Qiagen). ChIP‐seq libraries were constructed using ThruPLEX‐FD Prep kit or ThruPLEX DNA‐seq kit (Rubicon Genomics). Antibodies used were anti‐EGFP (ab290, Abcam), anti‐H3 (ab1791, Abcam), anti‐H3K4me1 (C1541094, Diagenode), anti‐H3K4me3 (ab8580, Abcam), anti‐H3K9ac (ab4441, Abcam), anti‐H3K27ac (ab4729, Abcam), anti‐H2A.Zac (ab8895, Abcam), anti‐Pol2 S2P CTD (61083, Active motif), and anti‐Pol2 null‐P CTD (8WG16 ab817, Abcam).
Libraries were sequenced using HiSeq2000 (Appendix Table S1 for information about read numbers), and reads were mapped to the genome using Bowtie (http://bowtie-bio.sourceforge.net/index.shtml). For the analyses of HMR_−L1s, TSSs, and randomly selected sites (Figs 2 and EV4), only uniquely mapped reads were considered. For the analyses of L1 sequences (Fig EV4), all genome‐mapped reads were mapped to L1 consensus sequences downloaded from the Repbase (http://www.girinst.org/repbase/) by allowing two mismatches. Metaplot and heatmap analyses were done using 300‐bp window/100‐bp step (Fig 2) or 200‐bp window/200‐bp step conditions (Fig EV4). For metaplot analyses, the average signal across HMR_−L1s (TSSs or randomly selected regions) was calculated in each window, and then, it was divided by the average input (or Histone H3) signal in the same window.
Immunoprecipitation of MIWI2‐bound RNA
Prospermatogonia were FACS‐isolated from 16.5 dpc MIWI2‐EGFP and Oct4‐EGFP mice. Testes from more than 20 fetuses were used for each experiment. Approximately 500,000 prospermatogonia were used for each experiment. Cells were lysed in 250 μl of ice‐cold lysis buffer [50 mM Tris–HCl (pH 7.4), 2.5 mM MgCl2, 100 mM KCl, 0.1% Nonidet P‐40, 1 × complete proteinase inhibitor (Roche), 50 units/ml SUPERase In (Thermo Fisher)]. Cell lysates were sonicated using a Covaris Focused‐ultrasonicator for 6 min using a standard setting for the microTUBES (Covaris). For the analysis of Mili KO background, testes were isolated from 1 to 2 dpp mice. We used five pairs of wild‐type testes and 10 pairs of Mili −/− testes to match the total amount of EGFP‐Miwi2 protein. Testes were lysed in 250 μl of ice‐cold lysis buffer, and 2 μl TURBO™ DNase (Ambion, Cat# AM2239) was added in the lysate and digested at 37°C for 2 min. Cell debris was removed by centrifugation at 21,000 × g for 30 min at 4°C. As an input, 10% of each lysate was taken. In parallel with the lysate preparation, 15 μl of Dynabeads Protein G (Thermo Fisher) was washed using lysis buffer, mixed with 1 μg of anti‐EGFP antibody (ab290, Abcam), and rotated for 1 h at 4°C. After washing, the beads were mixed with the lysate and rotated for 3 h at 4°C. Washing was performed using washing buffer [50 mM Tris–Cl (pH 7.4), 2.5 mM MgCl2, 100 mM KCl, 0.1% Nonidet P‐40] six times at 4°C. RNAs were purified from the beads and the input using TRIzol.
RNA libraries were constructed using a ScriptSeq Complete Gold Kit Low Input (Illumina). The step of rRNA depletion was omitted when constructing libraries from the immunoprecipitated RNAs, because rRNA was mostly cleared during immunoprecipitation. After sequencing using HiSeq2000, reads were mapped to the genome using the TopHat software (http://tophat.cbcb.umd.edu/), and reads that were uniquely and perfectly mapped to the genome were extracted. The extracted sequences were mapped to the abundant non‐coding RNAs (rRNA, tRNA, snRNA, snoRNA, scRNA, srpRNA, and RNA), and reads that were mapped to these sequences were removed. The remaining sequences were used for further analysis, and they were used for the calculation (denominator) of Reads per kilobase per million (RPKM). They were then mapped using Bowtie to HMR_−L1s, Ensembl mRNAs, and 95 prospermatogonium piRNA clusters that generate the largest number of piRNAs (Kuramochi‐Miyagawa et al, 2008). The number of reads mapped to each region and mRNA was counted, and RPKM was calculated. We considered strand information when calculating RPKM values of mRNAs, and reads mapped to the antisense strand of mRNAs were not considered. We selected mRNAs that are highly and specifically expressed in prospermatogonia (prospermatogonium‐expressed mRNAs) from Ensembl mRNAs using RNA‐seq data in 16.5 dpc testis germ and somatic cells (Singh et al, 2013). To select prospermatogonium‐expressed mRNAs, we firstly selected Top 2000 genes that were highly expressed in 16.5 dpc testis germ cells. Then, only genes that were specifically expressed in germ cells were selected (more than five times higher expression in germ cells than in somatic cells).
Generation of mutant mice and animal husbandry
A DNA fragment of Cas9 mRNA with a T7 promoter sequence and DNA fragments of chimeric guide RNAs with a T7 promoter sequence were amplified by PCR using pX330 as a template (Addgene #42230; primer sequences listed in Appendix Table S2). Cas9 mRNA and chimeric guide RNAs were transcribed from the PCR products using an mMESSAGE mMACHINE T7 ULTRA Transcription kit (Ambion) and MEGAshortscript T7 kit (Ambion), respectively. Transcribed RNAs were purified using MEGAclear kit (Ambion). Cas9 mRNA (100 ng/μl) and chimeric guide RNAs (total of 50 ng/μl) were mixed in injection buffer [10 mM Tris–HCl (pH 7.4), 10 mM NaCl, 0.25 mM EDTA (pH 8.0)] and then injected into the cytoplasm of 1‐cell embryos obtained by crossing B6;SJL F1 parents. For generation of mice with mutation in chr7 piRNA clusters, two genomic sites were cut using two sgRNAs to delete genomic regions between the cutting sites. For generation of mice with a mutation in the Rasgrf1 DMR, one genomic site was cut using one sgRNA to delete the flanking genomic regions. Mice with the desired deletions were selected and bred with CD‐1 mice. Deleted genomic regions (mm9) were as follows: Rasgrf1 ΔRMER4B/ΔRMER4B, chr9:89774319‐89774522; chr7 piRNA cluster ΔRMER4B/ΔRMER4B, chr7:6567721‐6568904; chr7 piRNA cluster Δpromoter/Δpromoter, chr7:6614356‐6615705. Genotyping was performed by PCR using the primers listed in Appendix Table S2.
For all experiments in this study, we randomly chose animals used. In addition, mutant animals used in this study did not show any defects in appearance and behavior, and therefore, subjective bias was unlikely to be introduced at the step of selecting animals for experiments.
Mice were kept under husbandry conditions provided by Yale Animal Resource Center. Mouse room was maintained at a constant temperature under a 12‐h light and 12‐h dark cycle. Animal experiments in this study were approved by Yale Institutional Animal Care and Use Committee (IACUC # 2015‐11087) and performed in compliance with ethical regulation.
Data availability
High‐throughput sequencing data are deposited in DDBJ database (DRA004149 and DRA006820).
Author contributions
TW and HL designed the project; TW conducted most experiment and all bioinformatics analysis; XC, ZY, and HQ participated in bisulfite analyses; and XC and HQ did RT–PCR analyses of WT mouse HMR‐derived polyA+ RNAs and RNA IP analyses of EGFP‐MIWI2 mice. TW and HL wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Satomi Miyagawa and Toru Nakano for Mili KO mice, and Yutaka Saito and Toutai Mituyama for help with using the Bisulfighter software package. Sequencing was conducted at Yale Stem Cell Center Genomics Core facility, which was supported by the Connecticut Regenerative Medicine Research Fund and the Li Ka Shing Foundation. This work was supported by NIH grant R37HD42012 and a Mathers Foundation award to H.L.
The EMBO Journal (2018) 37: e95329
Contributor Information
Toshiaki Watanabe, Email: toshwatatoshiakiwatanabe@gmail.com.
Haifan Lin, Email: Haifan.lin@yale.edu.
References
- Adelman K, Lis JT (2012) Promoter‐proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13: 720–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Girard A, Fejes‐Toth K, Hannon GJ (2007) Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 316: 744–747 [DOI] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ (2008) A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31: 785–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagijn MP, Goldstein LD, Sapetschnig A, Weick EM, Bouasker S, Lehrbach NJ, Simard MJ, Miska EA (2012) Function, targets, and evolution of Caenorhabditis elegans piRNAs. Science 337: 574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad‐Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ III, Gingeras TR, Schreiber SL, Lander ES (2005) Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120: 169–181 [DOI] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ (2007) Discrete small RNA‐generating loci as master regulators of transposon activity in Drosophila . Cell 128: 1089–1103 [DOI] [PubMed] [Google Scholar]
- Carmell MA, Girard A, van de Kant HJ, Bourc'his D, Bestor TH, de Rooij DG, Hannon GJ (2007) MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell 12: 503–514 [DOI] [PubMed] [Google Scholar]
- De Fazio S, Bartonicek N, Di Giacomo M, Abreu‐Goodger C, Sankar A, Funaya C, Antony C, Moreira PN, Enright AJ, O'Carroll D (2011) The endonuclease activity of Mili fuels piRNA amplification that silences LINE1 elements. Nature 480: 259–263 [DOI] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC (2007) A slicer‐mediated mechanism for repeat‐associated siRNA 5' end formation in Drosophila . Science 315: 1587–1590 [DOI] [PubMed] [Google Scholar]
- Hammoud SS, Low DH, Yi C, Lee CL, Oatley JM, Payne CJ, Carrell DT, Guccione E, Cairns BR (2015) Transcription and imprinting dynamics in developing postnatal male germline stem cells. Genes Dev 29: 2312–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han BW, Wang W, Li C, Weng Z, Zamore PD (2015) Noncoding RNA. piRNA‐guided transposon cleavage initiates Zucchini‐dependent, phased piRNA production. Science 348: 817–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, Moazed D (2015) RNA‐mediated epigenetic regulation of gene expression. Nat Rev Genet 16: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homolka D, Pandey RR, Goriaux C, Brasset E, Vaury C, Sachidanandam R, Fauvarque MO, Pillai RS (2015) PIWI slicing and RNA elements in precursors instruct directional primary piRNA biogenesis. Cell Rep 12: 418–428 [DOI] [PubMed] [Google Scholar]
- Huang XA, Yin H, Sweeney S, Raha D, Snyder M, Lin H (2013) A major epigenetic programming mechanism guided by piRNAs. Dev Cell 24: 502–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki YW, Murano K, Ishizu H, Shibuya A, Iyoda Y, Siomi MC, Siomi H, Saito K (2016) Piwi modulates chromatin accessibility by regulating multiple factors including histone H1 to repress transposons. Mol Cell 63: 408–419 [DOI] [PubMed] [Google Scholar]
- Kato Y, Kaneda M, Hata K, Kumaki K, Hisano M, Kohara Y, Okano M, Li E, Nozaki M, Sasaki H (2007) Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet 16: 2272–2280 [DOI] [PubMed] [Google Scholar]
- Klenov MS, Sokolova OA, Yakushev EY, Stolyarenko AD, Mikhaleva EA, Lavrov SA, Gvozdev VA (2011) Separation of stem cell maintenance and transposon silencing functions of Piwi protein. Proc Natl Acad Sci USA 108: 18760–18765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima‐Kita K, Kuramochi‐Miyagawa S, Nagamori I, Ogonuki N, Ogura A, Hasuwa H, Akazawa T, Inoue N, Nakano T (2016) MIWI2 as an effector of DNA methylation and gene silencing in embryonic male germ cells. Cell Rep 16: 2819–2828 [DOI] [PubMed] [Google Scholar]
- Krueger F, Andrews SR (2011) Bismark: a flexible aligner and methylation caller for Bisulfite‐Seq applications. Bioinformatics 27: 1571–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo N, Toh H, Shirane K, Shirakawa T, Kobayashi H, Sato T, Sone H, Sato Y, Tomizawa S, Tsurusaki Y, Shibata H, Saitsu H, Suzuki Y, Matsumoto N, Suyama M, Kono T, Ohbo K, Sasaki H (2015) DNA methylation and gene expression dynamics during spermatogonial stem cell differentiation in the early postnatal mouse testis. BMC Genom 16: 624–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramochi‐Miyagawa S, Kimura T, Ijiri TW, Isobe T, Asada N, Fujita Y, Ikawa M, Iwai N, Okabe M, Deng W, Lin H, Matsuda Y, Nakano T (2004) Mili, a mammalian member of piwi family gene, is essential for spermatogenesis. Development 131: 839–849 [DOI] [PubMed] [Google Scholar]
- Kuramochi‐Miyagawa S, Watanabe T, Gotoh K, Totoki Y, Toyoda A, Ikawa M, Asada N, Kojima K, Yamaguchi Y, Ijiri TW, Hata K, Li E, Matsuda Y, Kimura T, Okabe M, Sakaki Y, Sasaki H, Nakano T (2008) DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev 22: 908–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Thomas A, Rogers AK, Webster A, Marinov GK, Liao SE, Perkins EM, Hur JK, Aravin AA, Toth KF (2013) Piwi induces piRNA‐guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev 27: 390–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengner CJ, Camargo FD, Hochedlinger K, Welstead GG, Zaidi S, Gokhale S, Scholer HR, Tomilin A, Jaenisch R (2007) Oct4 expression is not required for mouse somatic stem cell self‐renewal. Cell Stem Cell 1: 403–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SL, Tsend‐Ayush E, Kortschak RD, Jacob R, Ricciardelli C, Oehler MK, Grutzner F (2013) Conservation and expression of PIWI‐interacting RNA pathway genes in male and female adult gonad of amniotes. Biol Reprod 89: 136, 1–13 [DOI] [PubMed] [Google Scholar]
- Liu X, Kraus WL, Bai X (2015) Ready, pause, go: regulation of RNA polymerase II pausing and release by cellular signaling pathways. Trends Biochem Sci 40: 516–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manakov SA, Pezic D, Marinov GK, Pastor WA, Sachidanandam R, Aravin AA (2015) MIWI2 and MILI have differential effects on piRNA biogenesis and DNA methylation. Cell Rep 12: 1234–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura F, Enomoto Y, Dairiki R, Ito T (2012) Amplification‐free whole‐genome bisulfite sequencing by post‐bisulfite adaptor tagging. Nucleic Acids Res 40: e136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Handler D, Brennecke J (2015) Noncoding RNA. piRNA‐guided slicing specifies transcripts for Zucchini‐dependent, phased piRNA biogenesis. Science 348: 812–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molaro A, Falciatori I, Hodges E, Aravin AA, Marran K, Rafii S, McCombie WR, Smith AD, Hannon GJ (2014) Two waves of de novo methylation during mouse germ cell development. Genes Dev 28: 1544–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamori I, Kobayashi H, Shiromoto Y, Nishimura T, Kuramochi‐Miyagawa S, Kono T, Nakano T (2015) Comprehensive DNA methylation analysis of retrotransposons in male germ cells. Cell Rep 12: 1541–1547 [DOI] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Grosso AR, Kimura H, Dye MJ, Dhir S, Carmo‐Fonseca M, Proudfoot NJ (2015) Mammalian NET‐seq reveals genome‐wide nascent transcription coupled to RNA processing. Cell 161: 526–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument‐Bromage H, Tempst P, Lin SP, Allis CD, Cheng X, Bestor TH (2007) DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448: 714–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey RR, Tokuzawa Y, Yang Z, Hayashi E, Ichisaka T, Kajita S, Asano Y, Kunieda T, Sachidanandam R, Chuma S, Yamanaka S, Pillai RS (2013) Tudor domain containing 12 (TDRD12) is essential for secondary PIWI interacting RNA biogenesis in mice. Proc Natl Acad Sci USA 110: 16492–16497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng JC, Valouev A, Liu N, Lin H (2016) Piwi maintains germline stem cells and oogenesis in Drosophila through negative regulation of Polycomb group proteins. Nat Genet 48: 283–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezic D, Manakov SA, Sachidanandam R, Aravin AA (2014) piRNA pathway targets active LINE1 elements to establish the repressive H3K9me3 mark in germ cells. Genes Dev 28: 1410–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasethupathy P, Antonov I, Sheridan R, Frey S, Sander C, Tuschl T, Kandel ER (2012) A role for neuronal piRNAs in the epigenetic control of memory‐related synaptic plasticity. Cell 149: 693–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhkov NV, Hammell M, Hannon GJ (2013) Multiple roles for Piwi in silencing Drosophila transposons. Genes Dev 27: 400–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Tsuji J, Mituyama T (2014) Bisulfighter: accurate detection of methylated cytosines and differentially methylated regions. Nucleic Acids Res 42: e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada Y, Mohn F, Buhler M (2016) The RNA‐induced transcriptional silencing complex targets chromatin exclusively via interacting with nascent transcripts. Genes Dev 30: 2571–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji M, Tanaka T, Hosokawa M, Reuter M, Stark A, Kato Y, Kondoh G, Okawa K, Chujo T, Suzuki T, Hata K, Martin SL, Noce T, Kuramochi‐Miyagawa S, Nakano T, Sasaki H, Pillai RS, Nakatsuji N, Chuma S (2009) The TDRD9‐MIWI2 complex is essential for piRNA‐mediated retrotransposon silencing in the mouse male germline. Dev Cell 17: 775–787 [DOI] [PubMed] [Google Scholar]
- Sienski G, Donertas D, Brennecke J (2012) Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell 151: 964–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienski G, Batki J, Senti KA, Donertas D, Tirian L, Meixner K, Brennecke J (2015) Silencio/CG9754 connects the Piwi‐piRNA complex to the cellular heterochromatin machinery. Genes Dev 29: 2258–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Li AX, Tran DA, Oates N, Kang ER, Wu X, Szabo PE (2013) De novo DNA methylation in the male germ line occurs by default but is excluded at sites of H3K4 methylation. Cell Rep 4: 205–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sytnikova YA, Rahman R, Chirn GW, Clark JP, Lau NC (2014) Transposable element dynamics and PIWI regulation impacts lncRNA and gene expression diversity in Drosophila ovarian cell cultures. Genome Res 24: 1977–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizawa S, Kobayashi H, Watanabe T, Andrews S, Hata K, Kelsey G, Sasaki H (2011) Dynamic stage‐specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non‐CpG methylation in oocytes. Development 138: 811–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliauskaite L, Vitsios D, Berrens RV, Carrieri C, Reik W, Enright AJ, O'Carroll D (2017) A MILI‐independent piRNA biogenesis pathway empowers partial germline reprogramming. Nat Struct Mol Biol 24: 604–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasik KA, Tam OH, Knott SR, Falciatori I, Hammell M, Vagin VV, Hannon GJ (2015) RNF17 blocks promiscuous activity of PIWI proteins in mouse testes. Genes Dev 29: 1403–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Chuma S, Yamamoto Y, Kuramochi‐Miyagawa S, Totoki Y, Toyoda A, Hoki Y, Fujiyama A, Shibata T, Sado T, Noce T, Nakano T, Nakatsuji N, Lin H, Sasaki H (2011a) MITOPLD is a mitochondrial protein essential for nuage formation and piRNA biogenesis in the mouse germline. Dev Cell 20: 364–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Tomizawa S, Mitsuya K, Totoki Y, Yamamoto Y, Kuramochi‐Miyagawa S, Iida N, Hoki Y, Murphy PJ, Toyoda A, Gotoh K, Hiura H, Arima T, Fujiyama A, Sado T, Shibata T, Nakano T, Lin H, Ichiyanagi K, Soloway PD et al (2011b) Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science 332: 848–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Lin H (2014) Posttranscriptional regulation of gene expression by Piwi proteins and piRNAs. Mol Cell 56: 18–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Cheng EC, Zhong M, Lin H (2015) Retrotransposons and pseudogenes regulate mRNAs and lncRNAs via the piRNA pathway in the germline. Genome Res 25: 368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Chen KM, Pandey RR, Homolka D, Reuter M, Janeiro BK, Sachidanandam R, Fauvarque MO, McCarthy AA, Pillai RS (2016) PIWI slicing and EXD1 drive biogenesis of nuclear piRNAs from cytosolic targets of the mouse piRNA pathway. Mol Cell 61: 138–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon BJ, Herman H, Sikora A, Smith LT, Plass C, Soloway PD (2002) Regulation of DNA methylation of Rasgrf1. Nat Genet 30: 92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Watanabe T, Kuramochi‐Miyagawa S, Takemoto N, Shiromoto Y, Kudo A, Kanai‐Azuma M, Tashiro F, Miyazaki S, Katanaya A, Chuma S, Miyazaki JI (2018) Mouse GTSF1 is an essential factor for secondary piRNA biogenesis. EMBO Rep 19: e42054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Gu J, Jin Y, Luo Y, Preall JB, Ma J, Czech B, Hannon GJ (2015) Panoramix enforces piRNA‐dependent cotranscriptional silencing. Science 350: 339–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Du J, Hale CJ, Gallego‐Bartolome J, Feng S, Vashisht AA, Chory J, Wohlschlegel JA, Patel DJ, Jacobsen SE (2014) Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 157: 1050–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
High‐throughput sequencing data are deposited in DDBJ database (DRA004149 and DRA006820).